

Abstract
Proteins of the bromodomain and extra-terminal (BET) family are epigenetics “readers” and promising therapeutic targets for cancer and other human diseases. We describe herein a structure-guided design of 1,4-oxazepines as a new class of BET inhibitors and our subsequent design, synthesis and evaluation of proteolysis-targeting chimeric (PROTAC) small-molecule BET degraders. Our efforts have led to the discovery of extremely potent BET degraders, exemplified by QCA570, which effectively induces degradation of BET proteins and inhibits cell growth in human acute leukemia cell lines even at low picomolar concentrations. QCA570 achieves complete and durable tumor regression in leukemia xenograft models in mice at well-tolerated dose-schedules. QCA570 is the most potent and efficacious BET degrader reported to date.
Graphical Abstract
INTRODUCTION
Bromodomain-containing proteins function as epigenetic “readers”.1–3 Among them, the bromodomain and extra-terminal (BET) family of proteins, which includes BRD2, BRD3, BRD4, and testis-specific BRDT, have emerged as therapeutic targets for cancer, inflammation, HIV infection, CNS disorders, cardiovascular diseases, and male contraception.4–10
All the members of the BET protein family contain two bromodomains (BD1 and BD2) that bind the acetylated lysine residues in histone tails and thus regulate gene transcription. In recent years, potent and specific small-molecule BET inhibitors such as JQ-1,11 I-BET762 (GSK525762A)12 and I-BET15113 which bind to these bromodomains, have been discovered.14–27 A number of BET inhibitors, including I-BET762 (GSK525762),12 OTX-015 (MK-8628),28 and TEN-010 (RO6870810)29, have advanced into clinical development as a new class of therapeutics for the treatment of human cancers.
The Proteolysis Targeting Chimera (PROTAC) concept was formally proposed by Deshaies and Crews in 200130 and the PROTAC strategy has recently gained attention for its promise in the discovery and development of completely new classes of small-molecule therapeutics.31–48 PROTAC degrader molecules are designed to contain a ligand for the target protein of interest tethered to a linker, which is attached to a second ligand capable of binding to and recruiting an E3 ligase complex to ubiquitinate and degrade the target protein. A number of laboratories, including ours, have employed the PROTAC strategy for the design of small-molecule degraders of BET proteins.43–45, 47–49 Recently, we reported the discovery of ZBC246 and ZBC260 as highly potent PROTAC BET degraders that are capable of inducing complete degradation of BET proteins in breast cancer and leukemia cells at low to sub-nanomolar concentrations.44, 45 ZBC246 and ZBC260 are >100-times more potent than the corresponding BET inhibitors in inhibition of cell growth in the majority of leukemia and breast cancer cell lines evaluated.44, 45 These two BET degraders are also very effective in inhibition of tumor growth in breast cancer xenograft models44, 45 and ZBC260 induces tumor regression in acute leukemia xenograft models in mice. Our data on ZBC246 and ZBC260 suggest that PROTAC BET degraders may have a promising therapeutic potential for the treatment of leukemia and solid tumors.
In the present study, we describe the design, synthesis and evaluation of novel PROTAC BET degraders based upon a new class of BET inhibitors that we have designed. Our efforts have yielded the discovery of QCA570 as an extremely potent and highly efficacious BET degrader, capable of degrading BET proteins at low picomolar (pM) concentrations in leukemia cells and achieving complete and durable tumor regression in mice at well-tolerated dose-schedules. QCA570 is the most potent and efficacious BET degrader reported to date.
RESULTS AND DISCUSSION
Structure-guided design of 1,4-oxazepines as a new class of BET inhibitors
Our goal was to discover new classes of BET inhibitors that target BET bromodomains and then use them to design novel BET degraders with improved potency and efficacy.
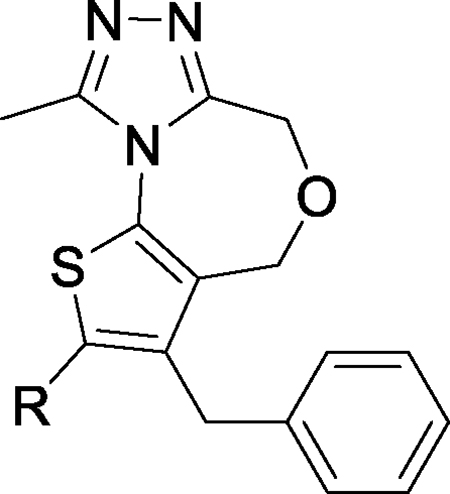
Since BRD4 BD1 domain, but not BRD4 BD2 domain, has been shown to regulate gene expression,50 we focused our design of new classes of BET inhibitors to target the BRD4 BD1 domain. Our modeling showed that 2,9-dimethyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (2, Figure 1) can closely mimic the 2,3,4,9-tetramethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine core in JQ-1 when interacting with the BRD4 BD1 domain protein (Figure 2). We synthesized compound 2 and determined that it binds to BRD4 BD1 protein with a Ki value of 1.61 μM (Table 1) in our fluorescence polarization (FP) assay.26, 27
Figure 1.

Scaffold hopping and structure-based design of 1,4-oxazepines as a new class of BET inhibitors.
Figure 2.

(A) Previously published co-crystal structure of (+)-JQ-1 in complex with the BRD4 BD1 domain protein (PDB ID: 3MXF); (B-D) Predicted binding models of compounds 2, 3 and 22 (QCA276) in a complex with BRD4 BD1. Hydrogen bonds are depicted by cyan dashed lines. Water molecules are shown as red spheres.
Table 1.
Binding affinities of our newly designed BET inhibitors and reference compounds (+)-JQ-1 and OTX-015 to BRD4 BD1 protein, as determined in our FP-based binding assay.
 | |||
|---|---|---|---|
| Compound | R | BRD4 BD1 |
|
| IC50(nM) | Ki(nM) | ||
| 1 (JQ-1) | 22 ± 3 | 5.3 ± 1.0 | |
| 2 | 4400 ± 700 | 1500 ± 200 | |
| 3 | CH3 | 100 ± 10 | 33 ± 4 |
| 4 | -H | 2300 ± 600 | 820 ± 210 |
| 5 | -isopropyl | 9400 ± 600 | 3300 ± 200 |
| 6 | 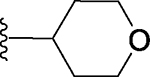 |
8100 ± 700 | 2800 ± 300 |
| 7 | -cyclopropyl | 550 ± 110 | 190 ± 40 |
| 8 | 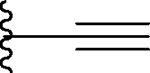 |
210 ± 10 | 70 ± 1 |
| 9 | 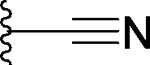 |
720 ± 60 | 250 ± 20 |
| 10 | 45 ± 4 | 13 ± 1 | |
| 11 | 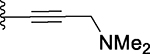 |
270 ± 10 | 92 ± 2 |
| 12 | 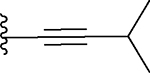 |
44 ± 3 | 13 ± 1 |
| 13 | 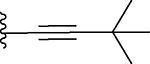 |
200 ± 90 | 68 ± 10 |
| 14 | 18 ± 2 | 3.7 ± 0.7 | |
| 15 | 34 ± 3 | 9.5 ± 1.1 | |
| 16 | 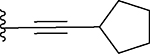 |
63 ± 10 | 20 ± 3 |
| 17 | 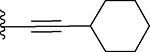 |
200 ± 24 | 66 ± 9 |
| 18 | 200 ± 9 | 68 ± 3 | |
| 19 | 86 ± 11 | 28 ± 4 | |
| 20 | 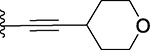 |
34 ± 2 | 9.6 ± 0.9 |
| 21 | 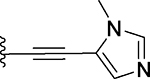 |
50 ± 9 | 15 ± 3 |
| 22 (QCA276) | 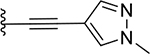 |
10 ± 3 | 2.3 ± 1.0 |
| 23 | 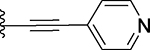 |
14 ± 5 | 4.2 ± 0.8 |
| 24 | 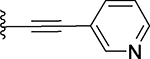 |
11 ± 1 | 2.1 ± 0.3 |
| 25 | 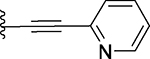 |
13 ± 4 | 2.7 ± 1.2 |
| OTX-015 | 23 ± 1 | 5.7 ± 0.5 | |
The p-chlorophenyl group in JQ-1 is critical for the high binding affinity of JQ-1 to the BRD4 BD1 protein through its interaction with the Trp-Pro-Phe (WPF) shelf in BRD4 BD1 but compound 2 lacks the corresponding p-chlorophenyl group present in JQ-1 (Figure 1). To capture the interactions of the p-chlorophenyl group in JQ-1 with BRD4, we modeled compounds with a substituent at the 3-position of the thiophene in compound 2. Modeling suggested that a benzyl group, but not a phenyl group, at the 3-position of the thiophene, could effectively interact with the WPF shelf in BRD4, and this led to the design of compound 3 (Figure 1). Our FP assay showed that compound 3 binds to BRD4 BD1 protein with a Ki value of 33.0 nM (Table 1) and is therefore >40-times more potent than 2.
In the co-crystal structure of JQ-1 complexed with BDR4 BD1 protein,10, 11 the 2-methyl group on the thiophene group enjoys van der Waals interactions with the Trp81, Gln85 and Leu92 residues in the protein. Accordingly, we performed further modification of 3 to improve its binding affinity to the protein. To assess the contribution of the corresponding methyl group in compound 3 to the BRD4 BD1 binding, we eliminated this methyl group, producing compound 4. In our FP binding assay, compound 4 has a Ki value of 823 nM to the protein, and is thus 25-times less potent than compound 3 (Table 1). The significant reduction in binding affinity of compound 4 to the protein when compared to compound 3 clearly reveals the important contribution of this methyl group to the binding affinity and suggests that further modifications of the methyl group in 3 may improve the binding affinity to BRD4.
Analysis of the co-crystal structure of JQ-1 in a complex with BRD4 BD110, 11 and our modeled structure of compound 3 in a complex with BRD4 BD1 (Figure 2) showed that there is a channel formed by Trp81, Pro82, Gln85, Asp88, Lys91 and Leu92, which is adjacent to the methyl group on the thiophene. We explored this channel by further modification of the methyl group in compound 3.
Replacement of the methyl group in 3 with a hydrophobic and bulkier isopropyl group yielded compound 5, which is >50-times less potent than 3 in binding to BRD4 BD1. Replacement of the methyl by a 4-tetrahydro-2H-pyran group generated compound 6, which is also >50-times less potent than 3 in binding to BRD4 BD1 (Table 1). The reduced binding affinities of 5 and 6 to BRD4 BD1 when compared to that of 3 are consistent with our modeling analysis, which showed that the channel around the methyl group is fairly narrow and cannot accommodate groups much bulkier than the methyl group. We therefore decided to focus on groups smaller than isopropyl or 4-tetrahydro-2H-pyran.
Replacement of the methyl by cyclopropyl gave compound 7, which is >10-times more potent than 5 but still >5-times less potent than 3 in binding to BRD4 BD1 protein. Next, we replaced the methyl group with a linear ethynyl group, producing compound 8. Compound 8 binds to BRD4 BD1 with a Ki value of 70 nM, and is therefore 2-times less potent than compound 3. Changing the ethynyl group to nitrile, a linear and polar group, yielded compound 9, which is 3-times less potent than 8, or 6-times less potent than 3 (Table 1).
The ethynyl group in compound 8 provided us with an opportunity to further explore this channel in BRD4 BD1. Addition of a hydroxyethyl to the ethynyl group gave compound 10, which is 2-times more potent than 3. Replacing the –CH2OH in 10 with a dimethylamino group gave compound 11 which has a Ki value of 91.7 nM and thus a potency similar to that of 3. We synthesized compounds with additional hydrophobic substituents, such as isopropyl, t-butyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl groups on the ethynyl group, yielding 12, 13, 14, 15, 16 and 17, respectively (Table 1). Among these compounds, 14 has a Ki value of 3.7 nM and is thus 10-times more potent than 3.
We next synthesized 18 containing a piperidine substituent, 19 containing a methylpiperidine substituent and 20 with a tetrahydropyran substituent on the ethynyl group. Compounds 18, 19 and 20 bind to BRD4 BD1 with Ki values of 68.4 nM, 27.7 nM and 9.6 nM, respectively. The binding affinities of 19 and 20 are stronger than that of 17 and this shows that a polar atom at the tip of the cyclohexyl substituent in 17 is tolerated well.
Several compounds containing a heteroaromatic substituent on the ethynyl group were synthesized, including 21 containing 1-methyl-1H-imidazole, 22 (QCA276) containing a 1-methyl-1H-pyrazole, and 23, 24 and 25 each containing a pyridine ring in different orientations. With the exception of 21, which has a Ki value of 15.2 nM, compounds 22-25 all have Ki values to BRD4 BD1 protein below 14 nM (Table 1). In our binding assay, OTX-015 has a Ki value of 5.7 nM to BRD4 BD1 protein and compounds 22-25 are thus equally potent binders to BRD4 BD1, as compared to OTX-015.
We further evaluated compound 22 for its binding affinities to BRD4 BD2, and BRD2–3 BD1 and BD2 proteins in our FP assays,24 and the data are presented in Table 2. Compound 22 binds to BRD2 BD1 and BRD3 BD1 with Ki values of 1.7 nM and 2.5 nM, respectively. It also binds to BRD2 BD2, BRD3 BD2 and BRD4 BD2 with high binding affinities, giving Ki values of 8.5 nM, 6.5 nM and 18.5 nM, respectively. Accordingly, compound 22 was selected as the lead BET inhibitor in our subsequent design of a new class of PROTAC BET degraders, described below.
Table 2.
Binding affinities of 22 (QCA276) to recombinant human BRD2, BRD3 and BRD4 BD1 and BD2 proteins as determined in our FP-based assays.
| BRD2 |
BRD3 |
BRD4 |
||||
|---|---|---|---|---|---|---|
| BD1 | BD2 | BD1 | BD2 | BD1 | BD2 | |
| IC50 (nM) | 3.5 ± 0.3 | 40.6 ± 4.6 | 16.4 ± 2.8 | 35.1 ± 8.2 | 10.7 ± 2.8 | 66.6 ± 11.2 |
| Ki (nM) | 1.7 ± 0.1 | 8.5 ± 0.8 | 2.5 ± 0.9 | 6.5 ± 0.9 | 2.3 ± 1.0 | 18.5 ± 5.2 |
Design of a new class of PROTAC BET degraders based upon the BET inhibitor 22 (QCA-276)
Our modeling of 22 (QCA-276) complexed with BRD4 BD1 protein suggested that the 1-methyl-1H-pyrazole group is exposed to the solvent environment, making it a suitable site at which to anchor a chemical link to a ligand for an E3 ligase complex for the design of PROTAC BET degraders.
Cereblon is an adaptor protein for the cullin 4A RING E3 ligase complex51, and thalidomide51, 52 and lenalidomide53 are known to be cereblon ligands. In our previous studies, we successfully employed thalidomide and lenalidomide in our design of potent and efficacious BET degraders ZBC246 and ZBC260.44, 45 We hence chose to employ thalidomide and lenalidomide as the ligands for cereblon/cullin 4A and compound 22 as the BET inhibitor for the design of new PROTAC BET degraders.
We first synthesized compounds 26–31 as potential BET degraders, by linking one of the nitrogen atoms in the pyrazole group of 22 to the C4 atom of the isoindoline ring in thalidomide (Table 3). As shown in our earlier study,44 the MV4;11, RS4;11 and MOLM-13 acute leukemia cell lines are very responsive to both BET inhibitors and degraders, and accordingly we evaluated our newly synthesized BET degraders for their activity in inhibition of cell growth in these three acute leukemia cell lines, and the data obtained are summarized in Table 3. As a control, we also evaluated the BET inhibitor 22 for its inhibition of cell growth in these leukemia cell lines.
Table 3.
Chemical structures of designed PROTAC BET degraders and their IC50 values in inhibition of cell growth in three acute leukemia cell lines MV4;11, MOLM13 and RS4;11. Cells were treated with compounds for 96 hours and viability was assessed with a Cell Counting Kit-8 (CCK-8) assay.
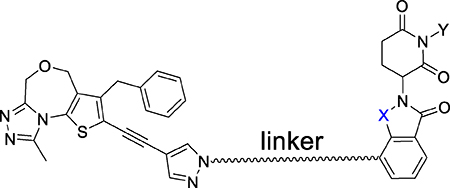 | ||||||
|---|---|---|---|---|---|---|
| Compound | linker | X | Y | IC50 (nM) in cell growth inhibition |
||
| MV4;11 | MOLM13 | RS4;11 | ||||
| 22 (BET inhibitor) | 55.8 ± 7.5 | 207 ± 20 | 173 ± 20 | |||
| 26 |  |
CO | H | >1000 | >1000 | >1000 |
| 27 | 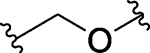 |
CO | H | 19.7 ± 0.76 | 311 | 20.8 ± 1.4 |
| 28 | 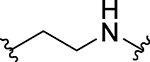 |
CO | H | 0.47 ± 0.11 | 2.23 ± 0.52 | 0.379 ± 0.037 |
| 29 | CO | H | 0.138 ± 0.008 | 0.56 ± 0.1 | 0.038 ± 0.005 | |
| 30 | CO | H | 0.0654 ± 0.007 | 0.19 ± 0.02 | 0.02 ± 0.01 | |
| 31 | CO | H | 0.092 ± 0.007 | 0.19 ± 0.02 | 0.02 ± 0.01 | |
| 32 | CH2 | H | 0.035 ± 0.007 | 0.51 ± 0.09 | 0.025 ± 0.004 | |
| 33 | CH2 | H | 0.051 ± 0.004 | 0.18 ± 0.02 | 0.0012 ± 0.0001 | |
| 34 | CH2 | H | 0.14 ± 0.01 | 0.53 ± 0.18 | 0.081 ± 0.009 | |
| 35 (QCA570) | CH2 | H | 0.0083 ± 0.0012 | 0.062 ± 0.009 | 0.032 ± 0.003 | |
| 36 | CO | H | 0.72 ± 0.09 | 1.6 ± 0.3 | 0.39 ± 0.02 | |
| 37 | CH2 | Me | 25.8 ± 4.4 | 183 ± 25 | 43.7 ± 5.2 | |
| dBET1 | 42 ± 14 | 140 ± 28 | 79 ± 12 | |||
| ARV-825 | 1.05 ± 0.30 | 18.2 ± 4.0 | 3.3 ± 0.7 | |||
| ARV-771 | 0.43 ± 0.12 | 7.45 ± 1.45 | 2.4 ± 0.7 | |||
| ZBC260 (BETd-260) | 0.36 ± 0.28 | 2.2 ± 0.2 | 0.051 ± 0.018 | |||
Consistent with its high binding affinities to BET proteins, the BET inhibitor (22) is an effective inhibitor of cell growth, with IC50 values of 55.8 nM, 207.3 nM and 173.4 nM in the MV4;11, MOLM-13 and RS4;11 cell lines, respectively (Table 3).
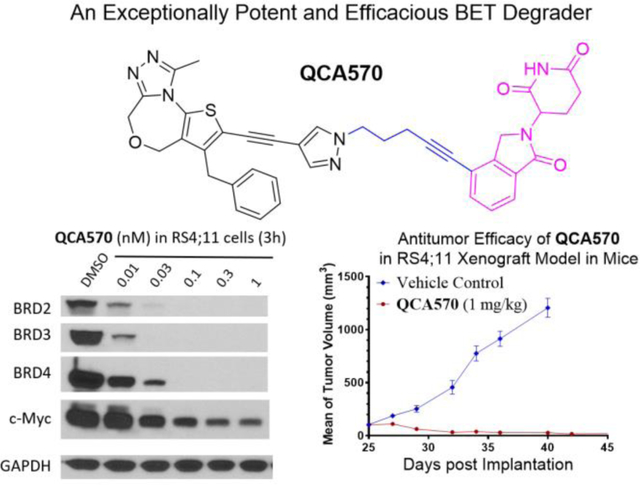
The potential BET degrader (26), in which the pyrazole group in the BET inhibitor portion is directly linked to thalidomide, failed to show significant cell growth inhibitory activity at concentrations up to 1,000 nM in any of these three leukemia cell lines (Table 3). Western blotting analysis consistently showed that at concentrations up to 1,000 nM, compound 26 failed to induce degradation of BRD2, BRD3 and BRD4 proteins in the RS4;11 cells even after a 24 h treatment (Figure 3 and S1).
Figure 3.

Western blotting analysis of BRD2, BRD3, and BRD4 proteins in RS4;11 cells treated with BET inhibitor 22 (QCA276), 26 and BET degraders 27, 28, 29, 30, and 31. RS4;11 cells were treated for 3 h (A) or 24 h (B) with each individual compound at the indicated concentrations. BRD2, BRD3, and BRD4 proteins, c-Myc and cleaved PARP were probed by western blot with specific antibodies and GAPDH was used as the loading control.
Compound 27, which possesses a -CH2O- linker between the pyrazole group in the BET inhibitor portion and the thalidomide, achieves IC50 values of 19.7, 312 and 20.8 nM in the MV4;11, MOLM-13 and RS4;11 cell lines, respectively (Table 3). In contrast to 26, western blotting showed that 27, at concentrations as low as 10 nM, is very effective in inducing degradation of BRD2, BRD3 and BRD4 proteins in the RS4;11 cells in a time- and dose-dependent manner (Figure 3 and S1). Interestingly, 27 induces degradation of BRD2 and BRD3 proteins more effectively than the BRD4 protein (Figure 3 and S1).
Our previous study showed that the nature of the PROTAC linker between the BET inhibitor portion and thalidomide plays a key role in the potency of a BET degrader in degrading BET proteins and therefore, in inhibition of cell growth.44 Accordingly, we synthesized and evaluated a number of BET degraders with linkers longer than that in 27. Compound 28 containing a -(CH2)2NH- linker is a highly potent BET degrader, achieving IC50 values of 0.47 nM, 2.2 nM and 0.38 nM in cell growth inhibition in MV4;11, MOLM-13 and RS4;11 cell lines, respectively (Table 3). Thus 28 is >50-times more potent than 27 in each of these three cell lines.
Increasing the linker length in 28 by one additional methylene yielded 29, which shows further improved potency in cell growth inhibition in each of these three cell lines. Compound 29 achieves IC50 values of 0.14 nM, 0.56 nM and 0.038 nM in cell growth inhibition in MV4;11, MOLM-13 and RS4;11 cell lines respectively, and is therefore 3–10 times more potent than 28.
Increasing the linker length in 29 by an additional methylene led to 30, which is 2–3 times more potent than 29 in cell growth inhibition in each of the three leukemia cell lines. However, a further increase in the linker length in 30 by one more methylene groups, resulting in for example 31, fails to improve the potency in cell growth inhibition in any of the three cell lines. Compounds 30 and 31 in fact have very similar potencies in all the three cell lines, with the data indicating that the linker in 30 is optimal. Indeed, compound 30 achieves extremely high potencies in all three leukemia cell lines with IC50 values of 0.065, 0.19 and 0.02 nM in inhibition of cell growth (Table 3).
We evaluated compounds 27-31 for their ability to induce degradation of BET proteins in the RS4;11 cells (Figure 3 and S1). Our western blotting data showed that 29, and especially 30 and 31 are extremely potent inducers of degradation of BET proteins and are in fact very effective at concentrations as low as 0.1 nM after a 3 h treatment. Consistent with their reduced potencies in inhibition of cell growth in the RS4;11 cell line, compounds 27 and 28 are much less potent than compounds 29, 30 and 31 in inducing degradation of BET proteins.
Since c-Myc protein expression is regulated by BET proteins54, we evaluated the effect of our BET degraders on c-Myc. Consistent with their high potency in inducing complete degradation of BET proteins, the potent BET degraders 30 and 31 are also very effective in reducing the levels of c-Myc protein in in the RS4;11 cells (Figure 3 and S1). For example, both 30 and 31 reduce the levels of c-Myc protein in a 3 h treatment time at concentrations as low as 0.1 nM (Figure 3A). These data further confirm that 30 and 31 are highly potent BET degraders. Moreover, compounds 28, 29, 30, and 31 all potently induce apoptosis at 1 and 10 nM after a 24 h treatment as revealed by cleavage of PARP (Figure 3B), while the inhibitor 22 at 10 μM induces PARP cleavage only marginally.
We next synthesized 32 and 33 by converting the 3-carbonyl group in the thalidomide moiety of 29 and 30 to a methylene group in an effort to further improve potency. Both 32 and 33 have IC50 values in the low picomolar to sub-nanomolar range in cell growth inhibition in these cell leukemia cell lines and are therefore highly potent BET degraders (Table 3).
To further examine the effect of the linking group, we synthesized and evaluated compounds 34 and 35. Changing the NH linking group in 33 to a methylene group yielded 34, which has picomolar IC50 values in inhibition of cell growth in each of the three leukemia cell lines. Replacing the NHCH2 linker in 33 with an ethynyl group gave 35 (QCA570), which inhibits cell growth in MV4;11, MOLM-13 and RS4;11 cell lines with IC50 values of 8.3 pM, 62 pM and 32 pM, respectively (Table 3). Converting the 3-carbonyl group in the thalidomide moiety to a methylene group gave 36, which is 10–100 times less potent than 35.
The amino group of the piperidine-2,6-dione in thalidomide and lenalidomide forms a strong hydrogen bond with cereblon which is revealed in the co-crystal structures.52, 53 Consistently, methylation of the amino group of the piperidine-2,6-dione in thalidomide and lenalidomide completely abolishes their binding to cereblon.37 To facilitate our mechanistic investigations for this class of BET degraders, we synthesized compound 37, in which the amino group of the piperidine-2,6-dione in 35 is methylated. Compound 37 is a fairly potent inhibitor of cell growth in all the three leukemia cell lines but it is >10,000-times less potent than 35 in each of these three leukemia cell lines, supporting that the high potency of 35 as a BET degrader is dependent on binding to cereblon. Compound 37 and the corresponding BET inhibitor 22 have similar IC50 values in inhibition of cell growth in the MV4;11, MOLM-13 and RS4;11 cell lines (Table 3).
We evaluated a number of previously published BET degraders such as dBET1,43 ARV-825,37 ARV-77147 and ZBC26044 for their cell growth inhibitory activity in these three leukemia cell lines (Table 3). Consistent with the previous studies, these BET degraders all potently inhibit cell growth in these leukemia cell lines (Table 3). In direct comparison, 35 is >1,000-times more potent than dBET1 and at least >10-times more potent than ARV-825 and ARV-771 in each of these three cell lines. Although ZBC260 and 35 have similar potencies in the RS4;11 cell line, 35 is >10-times more potent than ZBC260 in the MV4;11 and MOLM-13 cell lines. Based on these data, 35 is the most potent BET degrader reported to date.
We next investigated the ability of 35 to induce degradation of BET proteins in the MV4;11 and RS4;11 cell lines, and its suppression of c-Myc protein expression (Figure 4). At concentrations as low as 10 pM, 35 is very effective in reducing the levels of BRD3 and BRD4 proteins and that of the BRD2 protein at 30–100 pM in both leukemia cell lines with a 3 h treatment (Figure 4A and 4B). Consistently, compound 35, at concentrations as low as 10 pM in the RS4;11 cell line and 30 pM in the MV4;11 cell line, is very effective in reducing the levels of c-Myc protein. The corresponding BET inhibitor (22) fails to reduce the levels of BRD2, BRD3 and BRD4 proteins in the MV4;11 cell line and the levels of BRD2 and BRD4 proteins in the RS4;11 cell line. Interestingly, the level of BRD3 protein was reduced by 22 in the RS4;11 cell line but not in the MV4;11 cell line.
Figure 4.

Western blotting analysis of BRD2, BRD3, BRD4 proteins, c-Myc and cleaved PARP in RS4;11 (A and C) and MV4;11 (B and D) leukemia cells treated with BET degrader 35 (QCA570) and the corresponding BET inhibitor 22 (QCA276). RS4;11 or MV4;11 cells were treated for 3 h (A and B) or 24 h (C and D) at indicated concentrations with 35 or 22. Proteins were probed using specific antibodies.
We investigated the mechanism of action for the degradation of BET proteins induced by 35. Compound 35 at a concentration of 0.5 nM induced complete degradation of BRD2, BRD3 and BRD4 proteins in the RS4;11 cell line with a 3 h treatment time (Figure 5). Degradation of BRD2, BRD3 and BRD4 proteins by 35 is completely blocked by pre-treatment with lenalidomide, a ligand for cereblon; or with MLN4924, an E1 neddylation inhibitor; or MG-132 and carfizomib, two proteasome inhibitors. Therefore, these data clearly demonstrate that 35 induces degradation of BRD2, BRD3 and BRD4 proteins through cereblon-, neddylation- and proteasome-dependent mechanisms, consistent with its PROTAC design.
Figure 5.

Evaluation of the mechanism of action of BET degradation induced by 35 (QCA570). RS4;11 cells were pre-treated for 2 h with DMSO, BET inhibitor 22 (10 μM), cereblon ligand Lenalidomide (10 μM), E1 neddylation inhibitor MLN4924 (1 μM), or proteasome inhibitors MG-132 (20 μM) and Carfilzomib (0.1 μM). Cells were then treated for 3 h with BET degrader 35 (QCA570) at 0.5 nM, a concentration that induces complete degradation of all BET-BRD proteins and downregulation of c-Myc.
A previous study showed that an analogue of lenalidomide can elicit strong antitumor activity by inducing degradation of GSPT1.55 To further investigate the mechanism of action for 35, we examined the effect of 35 on GSPT1 protein in both the MV4;11 and RS4;11 cells. Our western blotting data showed that while 35 is highly potent and effective in inducing degradation of BRD2, BRD3 and BRD4 proteins, it has no effect on GSPT1 protein (SI, Figure S2).
We used flow cytometry to investigate the ability of 35 to induce apoptosis in the MV4;11, MOLM-13 and RS4;11 cell lines, and found that 35 is highly potent and effective in inducing apoptosis in a dose-dependent manner (Figure 6). In both the MOLM-13 and MV4;11 cell lines, it induces >60% of the cells to undergo apoptosis at concentrations as low as 1 nM upon a 24 h treatment (Figure 6A and 6B). It is interesting that 35 has slower kinetics of apoptosis induction in the RS4;11 cell line than that in the MOLM-13 and MV4;11 cell lines. With 24 h treatment time, 35 has a minimal effect at 0.1 nM and induces only 15–30% of the RS4;11 cells to undergo apoptosis at 0.3–10 nM (Figure 6C). However, with a 48 h treatment time, 35 is highly effective in inducing apoptosis at concentrations as low as 0.1 nM; 35 induces >40% of the RS4;11 cells to undergo apoptosis at 0.1 nM (Figure 6D). In direct comparison, the corresponding BET inhibitor 22, at concentrations of 1–10 μM has only a minimal to modest effect in induction of apoptosis in these three cell lines.
Figure 6.

Flow cytometry analysis of apoptosis induction by 35 (QCA570) in MOLM13 (A), MV4;11 (B), and RS4;11 (C and D) leukemia cells. Cells were treated with the BET degrader 35 (QCA570) or the corresponding BET inhibitor (22) at the indicated concentrations for 24 h (A, B, C) or 48 h (D). Apoptosis was assessed by flow cytometry using Annexin V and propidium iodine (PI) double staining.
Consistent with the strong effect on apoptosis induction by 35 determined in the flow cytometry assay, our western blotting analysis showed that it induces robust cleavage of PARP at concentrations as low as 0.1–0.3 nM in the MV4;11 and RS4;11 cell lines after 24 h treatment (Figures 4C and 4D). Also in agreement with our flow cytometry data, 35 induces caspase 3 activation at concentrations as low as 0.3 nM in the MV4;11 cell line. Caspase 3 cleavage was not detected in the RS4;11 cell line with the 24 h treatment, consistent with the slow kinetics in apoptosis induction by 35 in this cell line that was revealed by flow cytometry analysis.
Next, we investigated the effect of 35 in vivo in a pharmacodynamics experiment using RS4;11 xenograft tumors in mice (Figure 7). SCID mice bearing the RS4;11 tumors were administered a single, intravenous dose of 35 at 1 or 5 mg/kg and tumor tissues were harvested for analysis at 1, 6 and 24 h after drug treatment. Western blotting analysis showed that 35 dramatically reduces the levels of BRD2, BRD3 and BRD4 proteins at the 1 h and 6 h time-points at both 1 and 5 mg/kg doses and is more effective in reducing the BRD4 protein level at the 24 h time-point with 5 mg/kg than with 1 mg/kg. Consistent with the profound reduction of BRD2, BRD3 and BRD4 proteins in the RS4;11 tumor tissue, 35 with both 1 and 5 mg/kg treatments reduces c-Myc protein levels dramatically at all the time points examined. Compound 35 is also very effective in inducing cleavage of PARP at the 6 h and 24 h time-points, indicating strong apoptosis induction in the tumor tissue.
Figure 7.

Pharmacodynamic analysis of compound 35 in RS4;11 xenograft tumors. SCID mice bearing RS4;11 tumors were treated with a single intravenous dose of compound 35 at 1 or 5 mg/kg. At the indicated time points (1, 6 and 24 h), mice were sacrificed and tumors were harvested. Tumors were lysed for western blotting analysis of BRD2, BRD3, BRD4, c-Myc, and cleaved PARP. GAPDH was used as the loading control. Two mice were used for each time-point with each mouse bearing one tumor.
We performed tissue distribution analysis of compound 35 in mice bearing RS4;11 xenograft tumors, with the data summarized in Table 4. Our data showed that compound 35 is extensively distributed into all the tissues collected and analyzed. Compound 35 has good exposure in all tissues at 1h time-point. The concentration of 35 is rapidly deceased at 3 hr time-point and becomes undetectable at 6 h time-point, with the exception in the xenograft tumor tissue. Since 35 is capable of inducing rapid degradation of BET proteins in cells even at concentration of <1 nM, our tissue distribution data suggest that a single dose of 35 at 5 mg/kg can achieve sufficient exposure to induce degradation of BET proteins in all tissues.
Table 4.
Tissue distribution of compound 35 (QCA570) in mice bearing RS4;11 xenograft tumors. Tissue distribution of compound 35 in mice bearing RS4;11 xenograft tumors. Each mouse had one tumor and was dosed with a single, intravenous dose of compound 35. Mice were sacrificed at 1, 3 and 6 hr time-points and different tissues were collected for analysis of drug concentration by LC/MS/MS. BLQ, below the limit of quantification.
| Concentrations of QCA570 in plasma (ng/ml) and tissues (ng/g) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| (IV, 5mg/kg) | Mouse 1 (1h) | Mouse 2 (1 h) | Mouse 3 (3 h) | Mouse 4 (3 h) | Mouse 5 (6 h) | Mouse 6 (6h) | Mean (1h) | Mean (3h) | Mean (6 h) |
| plasma | 1575 | 2360 | 47 | 263 | BLQ | BLQ | 1968 | 155 | BLQ |
| Tumor | 642 | 405 | 177 | 199 | 87.9 | 68.7 | 524 | 188 | 78 |
| Liver | 1227 | 1720 | 57 | 209 | BLQ | BLQ | 1473 | 133 | BLQ |
| Heart | 373 | 288 | 17 | 22 | BLQ | BLQ | 331 | 19 | BLQ |
| Kidney | 649 | 675 | 40 | 49 | BLQ | BLQ | 662 | 44 | BLQ |
| Lung | 1041 | 696 | 35 | 89 | BLQ | BLQ | 868 | 62 | BLQ |
| Intestine | 1283 | 1009 | 50 | 92 | BLQ | BLQ | 1146 | 71 | BLQ |
We tested 35 for its anti-tumor efficacy in the RS4;11 and MV4;11 xenograft models and the data are summarized in Figure 8. In the RS4;11 xenograft model, 35 achieved complete and long-lasting tumor regression with doses of 1, 2.5 and 5 mg/kg, 3 times per week for 3 weeks (Figure 8A). Compound 35 at 5 mg/kg weekly was also very effective, achieving complete tumor growth inhibition but not tumor regression (Figure 8A). Compound 35 induced minimal animal weight loss (Figure 8B) and there were no other signs of toxicity in all the treatment groups.
Figure 8.

In vivo anticancer efficacy of BET degrader (35) in the RS4;11 and MV4;11 xenograft models in mice. (A, B) Mice bearing RS4;11 tumors were treated with BET degrader 35 at different dose-schedules. (A) Tumor growth and (B) Animal weights. (C, D) Mice bearing MV4;11 tumors were treated with BET degrader (35) at 1 or 5 mg/kg three times a week for two weeks by intravenous dosing. (C) Tumor growth and (D) Animal weights.
In the MV4;11 xenograft model, 35 achieved complete tumor regression at 1 mg/kg, 3 times per week for 2 weeks (Figure 8C). At 5 mg/kg, 3 times per week for 2 weeks, compound 35 achieves complete and long-lasting tumor regression (Figure 8C). It caused a maximum weight loss of less than 10% in the MV4;11 model (Figure 8D). Taken together, these studies demonstrate that compound 35 is highly efficacious at well-tolerated, dose-schedules.
Chemistry
The synthesis of the BET inhibitor 22 is shown in Scheme 1. Briefly, FmocCl (37) was converted to FmocNCS (38). Cyclization with methyl 4-chloro-3-oxobutanoate followed by bromination with phosphorus(V) oxybromide resulted in Fmoc-protected methyl 2-amino-4-bromothiophene-3-carboxylate (40), and removal of the protecting groups afforded 41. The amino group of 41 was reacted with 2-(OTBDPS) acetic chloride to give the amide (42), which was transformed into the sulfamide (43) using Lawesson’s reagent. A subsequent three-step cyclization generated the triazole (44), whose methyl carboxylate group was reduced, then chlorinated to generate a methylene chloride group, which linked with the unprotected hydroxyl group to form the seven-membered oxygen-containing ring in 46. A benzyl group was introduced into the thiophene through a palladium-catalyzed Suzuki coupling reaction giving the benzyl-substituted thiophene (4). Bromination of 4 gave compound 47 and finally, Sonogashira coupling of 47 with 4-ethynyl-1-methyl-1H-pyrazole gave compound 22 (QCA276).
Scheme 1.

Synthetic route to compound 22 (QCA276).a
aReaction Conditions: (a) KNCS, EtOAc, 0 °C-r.t., 92%; (b) NaH, THF, 0 °C, 62%; (c) POBr3; (d) morpholine, 52% yield over two steps; (e) DIPEA, DCM, 99%; (f) Lawesson’s reagent, dioxane, 57%; (g) NH2NH2•H2O; (h) MeC(OEt)3; (i) AcOH, 58% yield over three steps; (j) LiBH4; (k) SOCl2, 77% yield over two steps; (l) TBAF; (m) NaOt-Bu, 66% yield over two steps; (n) Pd(allyl)Cl2, sSPhos, Na2CO3, H2O, toluene, 77%; (o) NBS, HOAc, 84%; (p) PdCl2(PPh3)2, CuI, THF, Et3N, 92%.
The synthetic route to the BET degrader (35) is shown in Scheme 2. Introduction of 4-ethynyl-1H-pyrazole to compound 47 produced compound 48, alkylation of which with 5-iodopent-1-yne generated 71. Sonogashira coupling of 71 with 3-(4-iodo-1-oxoisoindolin-2-yl)piperidine-2,6-dione gave the final compound 35.
Scheme 2.

Synthesis of BET degrader 35 (QCA570).a
aReaction Conditions: (a) PdCl2(PPh3)2, CuI, THF, Et3N, 70%; (b) K2CO3, DMF, 80%; (c) PdCl2(PPh3)2, CuI, THF, Et3N, 54%.
CONCLUSIONS
In this study, we present our design, synthesis and evaluation of novel PROTAC degraders of BET proteins using a new class of BET inhibitors we have designed and known cereblon ligands. Our study has led to the discovery of a set of extremely potent and highly efficacious BET degraders, exemplified by 35 (QCA570). QCA570 is capable of effectively inducing degradation of BRD2, BRD3 and BRD4 proteins at low picomolar concentrations in human leukemia cell lines and has picomolar IC50 values in inhibition of cell growth in the MV4;11, RS4;11 and MOLM-13 human leukemia cell lines. Significantly, QCA570 achieves complete and long-lasting tumor regression in both the MV4;11 and RS4;11 acute leukemia xenograft models in mice at well-tolerated dose-schedules. Our data demonstrate that QCA570 is the most potent and efficacious BET degrader reported to date and warrants extensive further investigation as a potential therapy for the treatment of acute leukemia and other types of human cancer.
EXPERIMENTAL SECTION
Chemistry. General Methods
Unless otherwise noted, all purchased reagents were used as received without further purification. 1H NMR and 13C NMR spectra were recorded on Bruker Advance 400 MHz and 300 MHz spectrometers. 1H NMR spectra were reported in parts per million (ppm) referenced to 7.26 ppm of CDCl3 or referenced to the center line of a septet at 2.50 ppm of DMSO-d6 or 3.31 ppm of CD3OD. All 13C NMR spectra were reported in ppm and obtained with 1H decoupling. In the reported spectral data, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. MS analyses were carried out on a Waters UPLC-Mass instrument. The final compounds were all purified by C18 reverse phase preparative HPLC column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in CH3CN) as eluents. The purities of all the final compounds were confirmed to be >95% by UPLC-MS or UPLC.
General Procedure for Synthesis of Compound 3 and Compounds 5–7
NBS (716 mg, 4.02 mmol) was added to a solution of compound 4 (0.8 g, 2.68 mmol) in HOAc (25 mL). The reaction mixture was stirred at room temperature for 6 h. If compound 4 was not consumed completely, more NBS was added. All volatiles were removed and the residue was chromatographed on silica gel (DCM and MeOH) to afford compound 47 (950 mg, 84%).
A Shlenk tube was charged with compound 47 (10 mg), the corresponding boronic acid pinacol ester (2.0 eq), Pd2(dba)3 (2.7 mg), PCy3 (1.7 mg), dioxane (1 mL) and Na2CO3 solution (2 M, 0.5 mL) under N2. The tube was then sealed and heated at 100 °C in an oil bath for 4 h. The reaction mixture was extracted with EtOAc and the organic layer was washed with brine, dried and concentrated. The residue was purified through HPLC to afford compound 3 and compounds 5–7.
3-Benzyl-2,9-dimethyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (3)
1H NMR (400 MHz, CDCl3) δ 7.51 – 7.21 (m, 4H), 7.07 (d, J = 7.2 Hz, 2H), 4.82 (s, 2H), 4.72 (s, 2H), 3.88 (s, 2H), 2.96 (s, 3H), 2.52 (s, 3H). ESI-MS [M+H]+: 312.20. UPLC: 4.2 min.
3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (4)
Compound 37 (52 g, 200 mmol) in EtOAc (200 mL) was added dropwise to a suspension of anhydrous KSCN (21.4 g, 2200 mmol) in EtOAc (200 mL) at 0 °C. The reaction mixture was allowed to warm to r.t. and stirred overnight. The reaction mixture was filtered and the filtrate was evaporated under vacuum. NMR analysis of the crude showed a small amount of the solvent EtOAc. The crude product was treated with DCM (100 mL) and hexanes (100 mL) and the resulting solution was evaporated under vacuum to facilitate removal of the small amount of EtOAc. The yield of crude compound 38 was 52 g, 92%.
A solution of methyl 4-chloro-3-oxobutanoate (30.1 g, 200 mmol, 1.1 eq) in THF (50 mL) was added dropwise to a suspension of NaH (60% in mineral oil, 8.8 g, 220 mmol, 1.2 eq) in THF (200 mL) at 0 °C. After the addition was complete, the reaction mixture was allowed to warm to r.t. and stirred for 20 min. Then the reaction mixture was cooled to 0 °C and a solution of crude FmocNCS (52 g, 184 mmol) in THF (50 mL) was added dropwise. After the addition was complete, the reaction mixture was quenched at 0 °C by the addition of 50 mL of saturated NH4Cl solution and 50 mL of H2O. Then the majority of the solvent was evaporated under vacuum when, at this time point a precipitate started to form. Then 150 mL H2O and 150 mL EtOAc were added and the mixture was stirred at room temperature until it became a suspension. The crude mixture was filtered and °Ccasional stirring of the filter cake was used to speed up the filtration process. The filter cake was further rinsed with H2O (100 mL) and EtOAc (100 mL). Again the filter cake was stirred °Ccasionally during the washing process to facilitate filtration. The filter cake (56.3 g) was transferred to a flask. This crude product was further dried under high vacuum overnight to remove H2O and the organic solvent. After drying, the crude compound (39) weighed 44.9 g (62% yield) as light yellow solid. 1H NMR (400 MHz, CDCl3) δ 11.90 (s, 1H), 7.81 (d, J = 6.5 Hz, 2H), 7.62 (d, J = 7.4 Hz, 2H), 7.48–7.38 (m, 2H), 7.37 (d, J = 6.7 Hz, 2H), 4.59 (d, J = 6.9 Hz, 2H), 4.37–4.29 (m, 1H), 3.94 (s, 2H), 3.66 (s, 2H).
POBr3 (39 g, 137 mmol, 1.2 eq) was added to a suspension of compound 39, methyl 2-((((9H-fluorene-9-yl)methoxy)carbonyl)amino)-4-oxo-4,5-dihydrothiophene-3-carboxylate (44.9 g, 114 mmol) in dioxane (250 mL) and the reaction mixture was heated to 100 °C. The reaction was monitored by TLC which showed all the starting material to have been consumed in less than 1 h. The reaction mixture was cooled and poured into the mixture of ice-water. The reaction mixture was extracted with EtOAc (3×300 mL). The combined organic layers were washed with brine twice, dried over Na2SO4 and concentrated. The crude product compound (40) was used directly in the next step.
The residue was dissolved in DCM (75 mL) and the reaction mixture was cooled to 0 °C. Morpholine (52 mL, 5 eq) was added slowly and the reaction mixture was allowed to warm to room temperature then stirred overnight. The reaction mixture was filtered and rinsed with a small amount of Et2O. The filtrate was evaporated under vacuum and the residue was chromatographed on silica gel (pure DCM) to afford 2-amino-4-bromothiophene-3-carboxylate (41), as off-white solid (14 g, 52% over two steps).
2-((tert-Butyldiphenylsilyl)oxy)acetic acid (6 g, 19 mmol) was dissolved in DCM (60 mL) and the solution was cooled to 0 °C under N2. Oxalyl chloride (1.3 eq, 25 mmol, 2.1 mL) was added followed by the addition of DMF (0.1 mL). The mixture was allowed to warm to r.t. and stirred for a further 1 h. All the volatiles were removed under vacuum and the residue was dissolved in DCM (10 mL). This solution was added to a solution of compound 41 (2.36 g, 10 mmol) in DCM (60 mL) and DIPEA (5.2 mL, 30 mmol) at 0 °C under N2. The reaction mixture was allowed to warm to r.t. and stirred for 1 h prior to being quenched with saturated NaHCO3 and extracted with DCM (3×50 mL). The combined organic layer was washed with H2O and then, dried with Na2SO4, filtered and then concentrated. The oil was chromatographed on silica gel (1:16 to 1:8 EtOAc/hexanes) to give compound 42 as an oil: (5.3 g, 99%).
Lawesson’s reagent (2.4 g, 6 mmol, 0.6 eq) was added to a solution of compound 42 (5.5 g, 10 mmol) in dioxane (40 mL) and the reaction mixture was heated at reflux. The reaction was monitored by TLC and all the starting material was consumed in 4–6 h. The reaction mixture was cooled and diluted with H2O and EtOAc. After extraction, the organic layers were combined and washed with brine twice. The organic layer was dried and removed under vacuum. The residue was chromatographed on silica gel (1:16 to 1:8 EtOAc /hexanes) to give compound 43 as an oil: (3.2 g, 57%).
Hydrazine monohydrate (0.55 mL, 11.4 mmol, 2 eq) was added at r.t. to a solution of compound 43 (3.2 g, 5.7 mmol) in THF (20 mL). The reaction mixture was stirred for 1 h and then concentrated in vacuum. The residue was taken up in DCM and washed with H2O and brine. The organic layer was separated, dried and concentrated. The residue was taken up in EtOH (10 mL) and THF (2 mL), and triethyl orthoacetate (3.1 mL, 3 eq) was added. The reaction mixture was heated at reflux for 1 h. All volatiles were removed under vacuum and the residue was treated with AcOH (20 mL). The reaction mixture was heated at reflux for 1 h prior to the removal of the solvent under vacuum. The residue was treated with EtOAc, washed with 1M NaOH, saturated NaHCO3, and brine. The organic layer was dried and concentrated. The residue was chromatographed on silica gel (1:2 EtOAc/hexanes followed by EtOAc, then DCM/MeOH 15:1) to give compound 44 (1.9 g, 58%).
A solution of LiBH4 (2 M in THF, 3.3 mL, 6.6 mmol, 2 eq). MeOH (2 mL) was added at 0 °C to a solution of compound 44 (1.9 g, 3.3 mmol) in THF (20 mL) and the reaction mixture was allowed to warm to r.t. then stirred for 12 h. All volatiles were removed and the residue was taken up in EtOAc. The organic layer was washed with H2O and brine prior to being dried and concentrated. The residue was dissolved in DCM (20 mL) and cooled to 0 °C. Thionyl chloride (0.72 mL, 9.9 mmol, 3 eq) was added and the reaction mixture was allowed to warm to r.t. After 1 h, all the volatiles were removed and the residue was taken up in EtOAc and washed with 1M Na2CO3 and brine, dried and concentrated to give crude compound 45 (1.75 g).
A solution of TBAF (3.1 mL, 3.1 mmol, 1M in THF) was added to a solution of compound 45 (1.75 g, 3.1 mmol) in THF (10 mL). The solution was stirred for 1 h prior to being added to a heated solution of t-BuONa (595 mg, 6.2 mmol, 2 eq) in t-BuOH (40 mL) at 80 °C. The reaction mixture was stirred for 5 min and then cooled. All the solvent was removed under vacuum and the residue was taken up in EtOAc and H2O. The organic layer was washed with brine, dried and concentrated. The residue was purified by HPLC to afford the TFA salt of the compound 46 (667 mg, 51% over four steps).
A solution of Na2CO3 (2 M in H2O, 10 mL) and H2O (10 mL) was added to a solution of compound 46 (1.0 g, 3.5 mmol), Pd(ally)Cl2 (10% mmol), sodium 2’-dicyclohexylphosphino-2,6-dimethoxy-1,1’-biphenyl-3-sulfonate hydrate, (sSPhos) (20% mmol), and potassium benzyltrifluoroborate (1.39 g, 7.0 mmol) in toluene (25 mL). The reaction mixture was stirred at room temperature for 30 min and then heated to 110 °C and stirred for 3–4 h. All volatiles were removed and the residue was dissolved in EtOAc. The organic layer was washed with water and brine prior to being dried and concentrated. The residue was chromatographed on silica gel (DCM and MeOH) to afford compound 4, (800 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.40 – 7.30 (m, 3H), 7.20 (d, J = 7.2 Hz, 2H), 6.82 (s, 1H), 4.79 (s, 2H), 4.73 (s, 2H), 3.88 (s, 2H), 2.75 (s, 3H). Retention time: 4.37 min; ESI-MS [M+H]+: 298.10. UPLC: 3.4 min.
3-Benzyl-2-isopropyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (5)
1H NMR (400 MHz, MeOD) δ 7.30 (d, J = 7.4 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.13 (d, J = 7.1 Hz, 2H), 4.72 (s, 2H), 4.66 (s, 2H), 4.00 (s, 2H), 3.50–3.43 (m, 1H), 2.81 (s, 3H), 1.34 (d, J = 6.8 Hz, 6H). ESI-MS: 340.02. UPLC: 5.9 min.
3-Benzyl-9-methyl-2-(tetrahydro-2H-pyran-4-yl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4c][1,4]oxazepine (6)
1H NMR (400 MHz, MeOD) δ 7.30 (t, J = 7.4 Hz, 2H), 7.22 (t, J = 7.3 Hz, 1H), 7.13 (d, J = 7.1 Hz, 2H), 4.71 (s, 2H), 4.66 (s, 2H), 4.03 (s, 2H), 4.01 – 3.95 (m, 1H), 3.65 – 3.42 (m, 3H), 3.40–3.35 (m, 1H), 2.78 (s, 3H), 1.97 – 1.69 (m, 4H). ESI-MS: 382.09. UPLC: 4.9 min.
3-Benzyl-2-(tert-butyl)-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (7)
1H NMR (400 MHz, MeOD) δ 7.30 (t, J = 7.6 Hz, 3H), 7.22 (d, J = 6.8 Hz, 1H), 7.18 (d, J = 7.7 Hz, 2H), 4.73 (s, 2H), 4.68 (s, 2H), 4.09 (s, 2H), 2.79 (d, J = 0.6 Hz, 3H), 2.22 – 2.10 (m, 1H), 1.11 (q, J = 5.0 Hz, 2H), 0.79 (q, J = 4.9 Hz, 2H). ESI-MS: M+H 338.18.
General Procedure for Synthesis of Compounds 8, 10–25
The corresponding alkyne (2.0 eq), Pd(PPh3)2Cl2 (4.2 mg), CuI (2.3 mg), THF (1 mL) and Et3N (1 mL) were added under N2 to a flask charged with 47 (20 mg, 0.05 mmol). The reaction mixture was stirred at 60 °C for 18 h. The reaction mixture was extracted with EtOAc and the organic layer was washed with brine, dried and concentrated. The residue was purified through HPLC to afford compound 8, 10–25.
3-Benzyl-2-ethynyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (8)
was obtained by coupling of 47 with TMS-acetylene followed by treatment with TBAF to remove the TMS group. 1H NMR (400 MHz, MeOD) δ 7.37 – 7.16 (m, 5H), 4.72 (s, 2H), 4.69 (s, 2H), 4.22 (s, 1H), 4.10 (s, 2H), 2.76 (s, 3H). ESI-MS: 322.20. UPLC: 4.9 min.
3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine-2-carbonitrile (9)
A flask was charged with compound 47 (20 mg, 0.05 mmol), K3Fe(CN)6 (7 mg), CuI (2 mg), 1-methyl-1H-imidazole (0.5 mL) under N2. The reaction mixture was stirred at 140 °C for 18 h. The reaction mixture was extracted with EtOAc and the organic layer was washed with brine, dried and concentrated. The residue was purified through HPLC to afford compound 9 (45% yield). 1H NMR (400 MHz, MeOD) δ 7.35 (t, J = 7.3 Hz, 2H), 7.28 (d, J = 7.3 Hz, 1H), 7.27 – 7.20 (m, 2H), 4.77 (s, 2H), 4.76 (s, 2H), 4.18 (s, 2H), 2.79 (s, 3H). ESI-MS: 323.20.
4-(3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)but-3-yn-1-ol (10)
1H NMR (400 MHz, MeOD) δ 7.29 (t, J = 7.5 Hz, 2H), 7.25 – 7.15 (m, 3H), 4.72 (s, 2H), 4.69 (s, 2H), 4.06 (s, 2H), 3.75 (t, J = 6.5 Hz, 2H), 2.75–2.70 (m, 5H). ESI-MS: 366.11.
3-(3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)-N,N-dimethylprop-2-yn-1-amine (11)
1H NMR (400 MHz, MeOD) δ = 7.29 (dd, J = 7.0 Hz, J = 7.6 Hz, 2H), 7.21 (d, J = 7.0 Hz, 1H), 7.17 (d, J = 7.6 Hz, 2H), 4.73 (s, 2H), 4.70 (s, 2H), 4.41 (s, 2H), 4.11 (s, 2H), 2.94 (s, 6H), 2.77 (s, 3H), 13C NMR (100 MHz, MeOD) 146.4, 138.9, 132.6, 132.3, 129.9, 129.2, 127.9, 116.1, 87.0, 81.9, 69.0, 62.8, 48.4, 42.8, 34.6, 12.5. ESI-MS: 379.33. UPLC: 1.6 min.
3-Benzyl-9-methyl-2-(3-methylbut-1-yn-1-yl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]-oxazepine (12)
1H NMR (400 MHz, MeOD) δ = 7.31–7.22 (m, 2H), 7.24–7.15 (m, 3H), 4.70 (s, 2H), 4.69 (s, 2H), 4.00 (s, 2H), 2.90–2.82 (m, 1H), 2.75 (s, 3H), 1.26 (d, J = 7.2 Hz, 6H), 13C NMR (100 MHz, MeOD) 142.9, 139.5, 132.4, 129.7, 129.33, 129.30, 127.7, 119.7, 105.5, 71.9, 69.2, 62.7, 34.5, 23.1, 22.7, 12.4. ESI-MS: 364.33. UPLC: 4.88 min.
3-Benzyl-2-(3,3-dimethylbut-1-yn-1-yl)-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]- oxazepine (13)
1H NMR (400 MHz, MeOD) δ = 7.32–7.23 (m, 2H), 7.23–7.17 (m, 3H), 4.71 (s, 2H), 4.70 (s, 2H), 4.00 (s, 2H), 2.75 (s, 3H), 1.32 (s, 9H), 13C NMR (100 MHz, MeOD) 142.9, 139.6, 132.4, 129.7, 129.3, 129.1, 127.7, 119.6, 108.4, 71.5, 69.2, 62.7, 34.5, 31.0, 29.6, 12.4. ESI-MS: 378.38. UPLC: 5.24 min.
3-Benzyl-2-(cyclopropylethynyl)-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (14)
1H NMR (400 MHz, MeOD) δ = 7.32–7.23 (m, 2H), 7.23–7.15 (m, 3H), 4.69 (s, 2H), 4.67 (s, 2H), 4.00 (s, 2H), 2.74 (s, 3H), 1.62–1.51 (m, 1H), 1.02–0.91 (m, 2H), 0.81–0.73 (m, 2H), 13C NMR (100 MHz, MeOD) 155.4, 153.0, 143.1, 139.6, 132.2, 129.7, 129.3, 127.7, 119.8, 103.8, 69.0, 67.4, 62.7, 34.5, 12.4, 9.6, 1.0. ESI-MS: 362.40. UPLC: 4.77 min.
3-Benzyl-2-(cyclobutylethynyl)-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (15)
1H NMR (400 MHz, MeOD) δ = 7.30–7.22 (m, 2H), 7.24–7.14 (m, 3H), 4.70 (s, 2H), 4.69 (s, 2H), 4.00 (s, 2H), 3.33–3.28 (m, 1H), 2.76 (s, 3H), 2.42–2.30 (m, 2H), 2.26–2.12 (m, 2H), 2.25–1.90 (m, 2H), ESI-MS: 376.42. UPLC: 5.41 min.
3-Benzyl-2-(cyclopentylethynyl)-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (16)
1H NMR (400 MHz, MeOD) δ = 7.31–7.22 (m, 2H), 7.21–7.15 (m, 3H), 4.70 (s, 2H), 4.68 (s, 2H), 4.00 (s, 2H), 2.97–2.90 (m, 1H), 2.75 (s, 3H), 2.05–1.96 (m, 2H), 1.80–1.58 (m, 6H), 13C NMR (100 MHz, MeOD) 155.4, 153.1, 142.8, 139.6, 132.3, 129.7, 129.3, 129.0, 127.7, 119.9, 104.7, 72.2, 69.1, 62.7, 34.8, 34.5, 32.2, 25.9, 12.4. ESI-MS: 390.37. UPLC: 5.50 min.
3-Benzyl-2-(cyclohexylethynyl)-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (17)
1H NMR (400 MHz, MeOD) δ = 7.32–7.28 (m, 2H), 7.23–7.15 (m, 3H), 4.70 (s, 2H), 4.68 (s, 2H), 4.03 (s, 2H), 2.77–2.68 (m, 1H), 2.74 (s, 3H), 1.91–1.86 (m, 2H), 1.78–1.67 (m, 2H), 1.61–1.48 (m, 3H), 1.46–1.34 (m, 3H), 13C NMR (100 MHz, MeOD) 155.3, 153.1, 142.9, 139.6, 132.3, 129.7, 129.32, 129.28, 127.7, 119.7, 104.3, 72.9, 69.1, 62.7, 34.5, 33.5, 31.0, 26.9, 25.6, 12.4. ESI-MS: 404.48. UPLC: 6.40 min.
3-Benzyl-9-methyl-2-(piperidin-4-ylethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]-oxazepine (18)
1H NMR (400 MHz, DMSO) δ = 7.35–7.27 (m, 2H), 7.25–7.17 (m, 3H), 4.74 (s, 2H), 4.69 (s, 2H), 3.99 (s, 2H), 3.22–3.15 (m, 2H), 3.12–2.97 (m, 3H), 2.73 (s, 3H), 2.66 (brs, 1H), 2.08–2.01 (m, 2H), 1.82–1.69 (m, 2H), ESI-MS: 405.23. UPLC: 2.3 min.
3-Benzyl-9-methyl-2-((1-methylpiperidin-4-yl)ethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (19)
1H NMR (400 MHz, MeOD) δ = 7.36–7.22 (m, 2H), 7.22–7.15 (m, 3H), 4.70 (s, 2H), 4.67 (s, 2H), 4.04 (s, 2H), 3.57–3.50 (m, 2H), 3.09–2.93 (m, 3H), 2.87 (s, 3H), 2.72 (s, 3H), 2.09–2.01 (m, 2H), 1.96–1.83 (m, 2H), ESI-MS: 419.51. UPLC: 1.77 min.
3-Benzyl-9-methyl-2-((tetrahydro-2H-pyran-4-yl)ethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (20)
1H NMR (400 MHz, MeOD) δ = 7.32–7.24 (m, 2H), 7.23–7.16 (m, 3H), 4.71 (s, 2H), 4.69 (s, 2H), 4.04 (s, 2H), 3.89–3.80 (m, 2H), 3.58–3.48 (m, 2H), 3.03–2.93 (m, 1H), 2.76 (s, 3H), 1.95–1.85 (m, 2H), 1.75–1.62 (m, 2H), 13C NMR (100 MHz, MeOD) 155.4, 153.1, 143.3, 139.4, 132.4, 129.8, 129.5, 129.2, 127.7, 119.2, 102.4, 73.6, 69.2, 67.2, 62.7, 34.5, 33.1, 28.2, 12.4. ESI-MS: 406.40. UPLC: 4.27 min.
3-Benzyl-9-methyl-2-((1-methyl-1H-imidazol-5-yl)ethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (21)
1H NMR (400 MHz, MeOD) δ = 7.32–7.24 (m, 2H), 7.23–7.16 (m, 3H), 4.71 (s, 2H), 4.69 (s, 2H), 4.04 (s, 2H), 3.89–3.80 (m, 2H), 3.58–3.48 (m, 2H), 3.03–2.93 (m, 1H), 2.76 (s, 3H), 1.95–1.85 (m, 2H), 1.75–1.62 (m, 2H), 13C NMR (100 MHz, MeOD) 155.4, 153.1, 143.3, 139.4, 132.4, 129.8, 129.5, 129.2, 127.7, 119.2, 102.4, 73.6, 69.2, 67.2, 62.7, 34.5, 33.1, 28.2, 12.4. ESI-MS: 406.40. UPLC: 4.27 min.
3-Benzyl-9-methyl-2-((1-methyl-1H-pyrazol-4-yl)ethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (22)
1H NMR (400 MHz, MeOD) δ = 7.87 (s, 1H), 7.63 (s, 1H), 7.32–7.24 (m, 2H), 7.24–7.16 (m, 3H), 4.70 (s, 2H), 4.68 (s, 2H), 4.07 (s, 2H), 3.89 (s, 3H), 2.73 (s, 3H), 13C NMR (100 MHz, MeOD) 141.9, 141.4, 138.2, 133.8, 130.7, 129.4, 128.4, 127.9, 126.3, 117.5, 102.0, 88.5, 80.2, 67.5, 61.4, 37.8, 33.3, 11.1. ESI-MS: 402.38. UPLC: 3.27 min.
3-Benzyl-9-methyl-2-(pyridin-4-ylethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (23)
1H NMR (400 MHz, MeOD) δ 8.77 (d, J = 6.4 Hz, 1H), 7.98 (d, J = 6.4 Hz, 1H), 7.31 (dd, J = 8.0 Hz, J = 7.2 Hz, 2H), 7.29 – 7.19 (m, 3H), 4.78–4.73 (m, 4H), 4.23 (s, 2H), 2.79 (s, 3H). ESI-MS: M+H 399.41. UPLC: 3.52 min.
3-Benzyl-9-methyl-2-(pyridin-3-ylethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (24)
1H NMR (400 MHz, MeOD) δ 8.88–8.66 (m, 2H), 8.25 (d, J = 8.0 Hz, 1H), 7.76 – 7.70 (m, 1H), 7.31 (dd, J = 7.4 Hz, J = 7.3 Hz, 2H), 7.27 – 7.18 (m, 3H), 4.76 (s, 2H), 4.75 (s, 2H), 4.18 (s, 2H), 2.80 (s, 3H). ESI-MS: M+H 399.44. UPLC: 3.50 min.
3-Benzyl-9-methyl-2-(pyridin-2-ylethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (25)
1H NMR (400 MHz, MeOD) δ = 8.57 (d, J = 5.2 Hz, 1H), 7.89 (dd, J = 7.6 Hz, J = 8.0 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.44 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.35–7.17 (m, 5H), 4.73 (s, 2H), 4.72 (s, 2H), 4.20 (s, 2H), 2.76 (s, 3H), ESI-MS: 399.31. UPLC: 3.44 min.
4-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (26)
4-Ethynyl-1Hpyrazole (9.2 mg, 2 eq), Pd(PPh3)2Cl2 (4.2 mg), CuI (2.3 mg), THF (1 mL) and Et3N (1 mL) were added under N2 to a flask charged with 47 (20 mg, 0.05 mmol). The reaction mixture was stirred at 60 °C for 8 h. The reaction mixture was extracted with EtOAc and the organic layer was washed with brine, dried and concentrated. The residue was purified through HPLC to afford 2-((1H-pyrazol-4-yl)ethynyl)-3-benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (48) in 70% yield. 1H NMR (400 MHz, DMSO-d6) δ = 13.3 (brs, 1H), 8.19 (s, 1H), 7.78 (s, 1H), 7.32 (dd, J = 7.6 Hz, J = 7.2 Hz, 2H), 7.28 – 7.17 (m, 3H), 4.71 (s, 2H), 4.69 (s, 2H), 4.04 (s, 2H), 2.65 (s, 3H), 13C NMR (100 MHz, DMSO-d6) 163.1, 150.6, 141.7, 138.4, 129.74, 129.65, 128.8, 128.3, 126.6, 115.8, 100.3, 89.7, 80.8, 67.6, 62.0, 33.3, 12.3. ESI-MS: 388.22.
2-(2,6-Dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (49) (14 mg, 1 eq), Et3N (0.1 mL) and DMSO (2 mL) were added to a flask charged with compound 48 (20 mg, 0.05 mmol). The reaction mixture was stirred at 120 °C for 8 h. The reaction mixture was extracted with EtOAc and the organic layer was washed with brine, dried and concentrated. The residue was purified through HPLC to afford compound 26 (20% yield).
4-((4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)methoxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (27)
Compound 51 2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione (274 mg, 1.0 mmol) in DMF (2 mL) was added to a solution of compound 50 (1H-pyrazol-1-yl) methyl methanesulfonate (193 mg, 1.1 mmol KHCO3 (200 mg) and KI (10 mg). The reaction mixture was stirred at 90 °C for 12 h prior to being taken up in EtOAc and H2O. The organic layer was separated, dried, and evaporated. The residue was purified by chromatography to afford 4-((1H-pyrazol-1-yl)methoxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione 52 (180 mg, 51%). ESI-MS: 355.12.
N-Iodosuccinimide (113 mg) was added to a solution of 52 (180 mg, 0.5 mmol) in AcOH (2 mL). The reaction was stirred for 1 h prior to being concentrated. The residue was purified by HPLC to afford 2-(2,6-dioxopiperidin-3-yl)-4-((4-iodo-1H-pyrazol-1-yl)methoxy)isoindoline-1,3-dione (53). ESI-MS: 481.02.
CuI (1 mg), Pd(Ph3P)2Cl2 (3.5 mg), 53 (33 mg, 0.069 mmol), and ethynyltrimethylsilane (20 mg), THF (2 mL) and Et3N (0.5 mL) was placed in a Schlenk tube. The reaction mixture was heated at 50°C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by chromatography (DCM:MeOH 9:1) to afford crude product, which was dissolved in THF and a solution of TBAF in THF (1M, 0.1 mL) was added. After 5 min, the reaction mixture was evaporated and the residue was purified by HPLC to afford 2-(2,6-dioxopiperidin-3-yl)-4-((4-ethynyl-1H-pyrazol-1-yl)methoxy)isoindoline-1,3-dione (54) (17 mg, 65%). ESI-MS: 379.12.
CuI (0.45 mg), Pd2(dba)3 (1.38 mg), HPtBu3BF4 (1.39 mg), 54 (19 mg, 0.05 mmol), and 47 (10 mg, 0.03 mmol), THF(2 mL) and HNiPr2 (0.1 mL) were placed in a Schlenk tube. The reaction mixture was heated at 60–70 °C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by HPLC to afford compound 27 (6.5 mg, 32% yield). 1H NMR (400 MHz, MeOD) δ = 8.19 (s, 1H), 7.91 (s, 1H), 7.79–7.73 (m, 1H), 7.71 (s, 1H), 7.70–7.56 (m, 2H), 7.30–7.26 (m, 2H), 7.23–7.16 (m, 3H), 6.29 (s, 2H), 5.15–5.04 (m, 1H), 4.71 (s, 2H), 4.69 (s, 2H), 4.08 (s, 2H), 2.77–2.64 (m, 2H), 2.74 (s, 3H), 2.16–2.06 (m, 2H). ESI-MS: 674.22.
General Procedure for Synthesis of Compounds 28–31
MsCl (1 mL, 12.6 mmol) was added at 0 °C to a solution of compound 55 4-iodo-1H-pyrazole (2.4 g, 12 mmol) and triethylamine (1.85 mL, 13mmol) in DCM (20 mL). The reaction mixture was allowed to warm to r.t. and stirred for a further 1 h. The reaction mixture was quenched with a saturated NH4Cl solution and extracted with DCM. The organic layer was separated, washed with brine, dried, and evaporated. The residue was dissolved in CH3CN (70 mL) and tert-butyl(4-hydroxybutyl) carbamate (1.89 g, 10 mmol) and Cs2CO3 (3.9 g, 12 mmol) were added. The reaction mixture was heated to reflux for 12 h. After the reaction was cooled, the mixture was filtered and the filtrate was evaporated. The residue was taken up in EtOAc and H2O. The organic layer was separated, washed with brine, dried, and evaporated. The residue was purified by chromatography (EtOAc/hexanes:1:2) to afford crude tert-butyl(4-(4-iodo-1H-pyrazol-1-yl)butyl)carbamate (2.3 g, 53%), which was treated with DCM (5 mL) and TFA (5 mL). This reaction mixture was stirred for 12 h. All the volatiles were removed under vacuum and the residue was subject to HPLC purification to afford 4-(4-iodo-1H-pyrazol-1-yl)butan-1-amine (56).
DIPEA (0.52 mL, 3 mmol) was added to a solution of the TFA salt of compound 56 (378 mg, 1 mmol) and 2-(2, 6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (276 mg, 1 mmol) in DMF (1 mL). The reaction mixture was heated at 90 °C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was subject to HPLC purification to afford compound 57, 2-(2, 6-dioxopiperidin-3-yl)-4-((4-(4-iodo-1H-pyrazol-1-yl)butyl)amino)isoindoline-1,3-dione (122 mg, 23% yield).
CuI (5.3 mg), Pd(Ph3P)2Cl2 (20 mg), compound 57 (100 mg, 0.2 mmol), ethynyltrimethylsilane (39.2 mg, 0.4 mmol), THF (4 mL) and Et3N (1 mL) were placed in a Schlenk tube. The reaction mixture was heated at 60 °C for 12 h and then cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by chromatography (EtOAc) to afford crude product, which was dissolved in THF and a solution of TBAF in THF (1M, 0.2 mL) was added. After 5 min, the reaction mixture was evaporated and the residue was subjected to HPLC purification to afford 2-(2,6-dioxopiperidin-3-yl)-4-((4-(4-ethynyl-1H-pyrazol-1-yl)butyl)-amino)isoindoline-1,3-dione (58) (50mg, 60% yield). ESI-MS: 420.13.
CuI (3.8 mg), Pd(Ph3P)2Cl2 (7 mg), compound 58 (20 mg, 0.05 mmol), compound 47 (42 mg, 0.1 mmol), THF (2 mL) and Et3N (0. 5 mL) were placed in a Schlenk tube. The reaction mixture was heated at 70 °C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was subjected to HPLC purification to afford compound 30. Following the procedures used to prepare compound 30 from tert-butyl(4-hydroxybutyl) carbamate, compounds 28, 29 and 31 with different alkynyl chains were obtained in same methods.
4-((2-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (28)
1H NMR (400 MHz, CDCl3) δ = 7.97 (brs, 1H), 7.71 (s, 1H), 7.58 (s, 1H), 7.46 (dd, J = 7.0 Hz, J = 7.8 Hz, 1H), 7.30 (dd, J = 6.8 Hz, J = 7.2 Hz, 1H), 7.23 (d, J = 6.8 Hz, 1H), 7.18 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 7.0 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 6.49 (s, 1H), 4.94–4.83 (m, 1H), 4.73 (s, 2H), 4.60 (s, 2H), 4.35 (t, J = 5.8 Hz, 2H), 4.02 (s, 2H), 3.83–2.76 (m, 2H), 2.79–2.66 (m, 4H), 2.73 (s, 3H), ESI-MS: 687.37. UPLC: 3.74 min.
4-((3-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2yl)ethynyl)-1H-pyrazol-1-yl)propyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (29)
1H NMR (400 MHz, CDCl3) δ = 7.97 (brs, 1H), 7.71 (s, 1H), 7.58 (s, 1H), 7.46 (dd, J = 7.0 Hz, J = 7.8 Hz, 1H), 7.30 (dd, J = 6.8 Hz, J = 7.2 Hz, 1H), 7.23 (d, J = 6.8 Hz, 1H), 7.18 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 7.0 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 6.49 (s, 1H), 4.94–4.83 (m, 1H), 4.73 (s, 2H), 4.60 (s, 2H), 4.35 (t, J = 5.8 Hz, 2H), 4.02 (s, 2H), 3.83–2.76 (m, 2H), 2.79–2.66 (m, 4H), 2.73 (s, 3H), ESI-MS: 701.40. UPLC: 4.01 min.
4-((4-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2yl)ethynyl)-1H-pyrazol-1-yl)butyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (30)
1H NMR (400 MHz, DMSO-d6) δ = 11.09 (brs, 1H), 8.19 (s, 1H), 7.75 (s, 1H), 7.55 (dd, J = 7.2 Hz, J = 6.8 Hz, 1H), 7.31 (dd, J = 7.6 Hz, J = 7.2 Hz, 2H), 7.27–7.15 (m, 3H), 7.08 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 6.8 Hz, 1H), 6.61 (brs, 1H), 5.08–5.00 (m, 1H), 4.71 (s, 2H), 4.69 (s, 2H), 4.17 (t, J = 6.8 Hz, 2H), 4.03 (s, 2H), 2.94–2.81 (m, 1H), 2.65 (s, 3H), 2.65–2.50 (m, 2H), 2.50–2.43 (m, 2H), 2.05–1.97 (m, 2H), 1.90–1.79 (m, 2H), 1.55–1.44 (m, 2H), 13C NMR (100 MHz, DMSO-d6) 172.9, 170.2, 168.9, 167.4, 115.1, 150.6, 146.4, 141.8, 141.5, 138.3, 136.3, 133.7, 132.3, 129.8, 129.7, 128.8, 128.3, 126.6, 117.3, 115.7, 110.5, 109.2, 100.6, 89.3, 80.8, 67.6, 62.0, 51.2, 48.6, 41.3, 33.3, 31.1, 27.0, 25.7, 22.2, 12.3. ESI-MS: 715.39. UPLC: 4.31 min.
4-((5-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)pentyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (31)
1H NMR (400 MHz, CDCl3) δ = 8.23 (brs, 1H), 7.71 (s, 1H), 7.61 (s, 1H), 7.48 (dd, J = 7.2 Hz, J = 6.8 Hz, 1H), 7.33 (dd, J = 7.2 Hz, J = 7.6 Hz, 2H), 7.23 (d, J = 7.6 Hz, 1H), 7.18 (d, J = 6.8 Hz, 2H), 7.09 (d, J = 6.8 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 4.95–4.83 (m, 1H), 4.80 (s, 2H), 4.68 (s, 2H), 4.17 (t, J = 7.0 Hz, 2H), 4.04 (s, 2H), 3.30–3.20 (m, 2H), 2.95–2.65 (m, 3H), 2.88 (s, 3H), 2.19–2.05 (m, 1H), 2.00–1.85 (m, 2H), 1.78–1.61 (m, 2H), 1.50–1.33 (m, 2H), ESI-MS: 729.40. UPLC: 4.33 min.
General Procedure for Synthesis of Compounds 32 and 33
Compound 60, 4-chlorobutan-1-ol (3.3 g, 1.3eq), Cs2CO3 (16.4 g, 60 mmol), and NaI (600 mg) were added to a solution of compound 59 and 4-iodo-1H-pyrazole (3.88 g, 20 mmol) in CH3CN (140 mL) was added The reaction mixture was heated at 50 °C for 12 h. The reaction mixture was filtered and the filtrate was evaporated. The residue was purified by chromatography (EtOAc/hexanes: 1:1 to EtOAc) to afford compound 61, 4-(4-iodo-1H-pyrazol-1-yl) butan-1-ol (4 g, 75%).
The SO3·pyridine complex (7.1 g, 45 mmol) was added to a solution of compound 61 (4 g, 15 mmol) in DMSO (24 mL) and Et3N (16 mL). The reaction mixture was stirred for 3 h prior to being quenched with H2O. The reaction mixture was extracted with EtOAc. The organic layer was separated, washed with brine, dried, and evaporated. The residue was purified by chromatography (EtOAc/hexanes: 1:2 to EtOAc) to afford compound 62, 4-(4-iodo-1H-pyrazol-1-yl)butanal (2.8 g, 73%).1H NMR (400 MHz, CDCl3) δ 9.76 (s, 1H), 7.52 (s, 1H), 7.44 (s, 1H), 4.20 (t, J = 6.7 Hz, 2H), 2.48 (t, J = 6.9 Hz, 2H), 2.18–2.10 (m, 2H).
AcOH (0.06 mL) was added to a solution of compound 62 (526 mg, 2 mmol) and compound 63 (lenalidomide) (520 mg, 2 mmol) in DCE (20 mL). The reaction was stirred for 20 min prior to the addition of NaHB(OAc)3 (848 mg). The reaction mixture was stirred for 12 h prior to being quenched with H2O. The reaction mixture was extracted with DCM. The organic layer was separated, washed with brine, dried, and evaporated. The residue was purified by HPLC to afford 3-(4-((4-(4-iodo-1H-pyrazol-1-yl)butyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (64) (420 mg, 38%). 1H NMR (400 MHz, MeOD) δ 7.73–7.70 (m, 1H), 7.50–7.45 (m, 1H), 7.32–7.25 (m, 1H), 7.10–7.05 (m, 1H), 6.80–6.75 (m, 1H), 5.16–5.06 (m, 1H), 4.28–4.20 (m, 2H), 4.22–4.12 (m, 2H), 3.24–3.20 (m, 2H), 2.84–2.80 (m, 2H), 2.48–2.40 (m, 1H), 2.20–2.15 (m, 1H), 1.99–1.89 (m, 2H), 1.63–1.58 (m, 2H). ESI-MS: 508.95.
CuI (9.5 mg), Pd(Ph3P)2Cl2 (35 mg), compound 64 (267 mg, 0.5 mmol), and ethynyltrimethylsilane (98 mg, 1 mmol), THF (4 mL) and Et3N (1 mL) were placed in a Schlenk tube. The reaction mixture was heated at 40 °C for 12 h then cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by chromatography (EtOAc) to afford crude product (215 mg, 90%), which was dissolved in THF and a solution of TBAF in THF (1 M, 0.45 mL) was added. After 5 minutes, the reaction mixture was evaporated and the residue was purified by chromatography (EtOAc) to afford crude product, which was further purified by HPLC to afford 3-(4-((4-(4-ethynyl-1H-pyrazol-1-yl)butyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione compound (65) (100 mg, 55% yield). ESI-MS: 406.24.
CuI (1.9 mg), Pd2(dba)3 (18.3 mg), HPtBu3BF4 (11.6 mg), compound 65 (81 mg, 0.2 mmol), and compound 47 (39 mg, 0.1 mmol), THF(4 mL) and HNiPr2 (0.14 mL) were placed in a Schlenk tube. The reaction mixture was heated at 40 °C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by by HPLC to afford compound 33. Following the procedures used to prepare compound 33 from 4-chlorobutan-1-ol, compounds 32 with different alkynyl chains were obtained in same methods.
3-(4-((3-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)propyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (32)
1H NMR (400 MHz, DMSO-d6) δ = 11.01 (brs, 1H), 8.21 (s, 1H), 7.77 (s, 1H), 7.31 (dd, J = 7.2 Hz, J = 7.2 Hz, 2H), 7.30–7.18 (m, 4H), 6.94 (d, J = 6.8 Hz, 1H), 6.70 (d, J = 7.6 Hz, 1H), 6.53 (brs, 1H), 5.15–5.07 (m, 1H), 4.71 (s, 2H), 4.69 (s, 2H), 4.25 (t, J = 7.0 Hz, 2H), 4.22–4.08 (m, 2H), 4.03 (s, 2H), 3.15–3.06 (m, 2H), 2.65 (s, 3H), 2.68–2.55 (m, 1H), 2.14–2.05 (m, 2H), ESI-MS: 687.42. UPLC: 3.59 min.
3-(4-((4-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)butyl)amino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (33)
1H NMR (400 MHz, MeOD) δ = 7.93 (s, 1H), 7.66 (s, 1H), 7.33–7.25 (m, 2H), 7.22–7.16 (m, 3H), 7.06 (d, J = 7.2 Hz, 1H), 6.79 (d, J = 8.0 Hz, 1H), 5.19–5.10 (m, 1H), 4.71 (s, 2H), 4.69 (s, 2H), 4.27 (t, J = 4.0 Hz, 2H), 4.23–4.18 (m, 2H), 4.08 (s, 2H), 3.16–3.11 (m, 2H), 2.74 (s, 3H), 2.53–2.38 (m, 2H), 2.22–2.11 (m, 2H), 2.05–1.92 (m, 2H), 1.67–1.55 (m, 2H), ESI-MS: 701.43. UPLC: 3.79 min.
3-(4-(5-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)pentyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (34)
CuI (5.3 mg), Pd(Ph3P)2Cl2 (20 mg), compound 66 3-(4-bromo-1-oxoisoindolin-2-yl)piperidine-2,6-dione (100 mg, 0.31 mmol), and 1-(pent-4-yn-1-yl)-1H-pyrazole (50 mg, 0.37 mmol), DMF (4 mL) and Et3N (1 mL) were placed in a Schlenk tube. The reaction mixture was heated at 80 °C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by chromatography (MeOH/DCM) to afford the desired product (67), 3-(4-(5-(1H-pyrazol-1-yl)pent-1-yn-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (82 mg, 70% yield). ESI-MS: 377.15.
10% Pd/C was added to a solution of the compound 67 (100 mg, 0.266 mmol) in MeOH (2 mL). The reaction was stirred under an H2 balloon for 4 h and then was filtered. The organic solvent was removed to afford compound 68, 3-(4-(5-(1H-pyrazol-1-yl)pentyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (97 mg, 95%).
NIS (56 mg) was added to compound 68 (100 mg, 0.26 mmol) in acetic acid (2 mL). The reaction was stirred for 6 h prior to being concentrated. The residue was purified by HPLC to afford compound 69 3-(4-(5-(4-iodo-1H-pyrazol-1-yl)pentyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (118 mg, 90%). ESI-MS: 507.19.
CuI (5.3 mg), Pd(Ph3P)2Cl2 (20 mg), compound 69 (101 mg, 0.2 mmol), ethynyltrimethylsilane (39.2 mg, 0.4 mmol), THF (4 mL) and Et3N (1 mL) were placed in a Schlenk tube. The reaction mixture was heated at 70 °C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was purified by chromatography (EtOAc) to afford a crude product, which was dissolved in THF and a solution of TBAF in THF (1M, 0.2 mL) was added. After 5 min, the reaction mixture was evaporated and the residue was subjected to HPLC purification to afford compound 70, 3-(4-(5-(4-ethynyl-1H-pyrazol-1-yl)pentyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (44 mg, 55% yield). ESI-MS: 405.19.
CuI (3.8 mg), Pd(Ph3P)2Cl2 (7 mg), compound 47 (20 mg, 0.05 mmol), compound 70 (40 mg, 0.1 mmol), THF (2 mL) and Et3N (0. 5 mL) were placed in a Schlenk tube. The reaction mixture was heated at 70°C for 12 h. The reaction mixture was cooled and treated with EtOAc and brine. The organic layer was separated, dried, and evaporated. The residue was subjected to HPLC purification to afford compound 34 (35.7 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δ = 8.01 (brs, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.68 (s, 1H), 7.58 (s, 1H), 7.43 (dd, J = 7.2 Hz, J = 7.6 Hz, 1H), 7.40–7.34 (m, 2H), 7.32 (dd, J = 7.6 Hz, J = 6.8 Hz, 2H), 7.18 (d, J = 7.6 Hz, 1H), 5.33–5.22 (m, 1H), 4.79 (s, 2H), 4.67 (s, 2H), 4.49–4.22 (m, 2H), 4.14 (t, J = 7.0 Hz, 2H), 4.05 (s, 2H), 2.93–2.73 (m, 4H), 2.86 (s, 3H), 2.69–2.55 (m, 1H), 1.97–1.83 (m, 2H), 1.75–1.62 (m, 2H), 1.49–1.23 (m, 2H), ESI-MS: 700.40. UPLC: 5.43 min.
3-(4-(5-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)pent-1-yn-1-yl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (35)
K2CO3 (4.0 mmol) was added to a solution of compound 48 (0.6 g, 1.55 mmol) and 5-iodopent-1-yne (776 mg, 4.0 mmol) in DMF (10 mL). The reaction mixture was stirred at 80 °C for 4 h. All volatiles were removed and the residue was chromatographed on silica gel (DCM and MeOH) to afford compound 71, 3-benzyl-9-methyl-2-((1-(pent-4-yn-1-yl)-1H-pyrazol-4-yl)ethynyl)-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepine (560 mg, 80%).
Pd(PPh3)2Cl2 (0.124 mmol), CuI (0.062 mmol) and Et3N (2.5 mL) were added to a solution of compound 71 (0.56 g, 1.24 mmol) and compound 72 3-(4-iodo-1-oxoisoindolin-2-yl) piperidine-2,6-dione (600 mg, 1.62 mmol) in DMF (5 mL). The reaction mixture was degassed completely and stirred at 80 °C for 6 h. All volatiles were removed and the residue was subjected to HPLC purification to afford the compound 35 (QCA570) (509 mg, 59% yield). 1H NMR (400 MHz, DMSO-d6) δ = 11.00 (brs, 1H), 8.23 (s, 1H), 7.75 (s, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.51 (dd, J = 8.0 Hz, J = 7.6 Hz, 1H), 7.31 (dd, J = 7.6 Hz, J = 7.2 Hz, 2H), 7.26–7.17 (m, 3H), 5.18–5.10 (m, 1H), 4.72 (s, 2H), 4.69 (s, 2H), 4.53–4.30 (m, 2H), 4.28 (t, J = 5.8 Hz, 2H), 4.03 (s, 2H), 3.00–2.85 (m, 1H), 2.66 (s, 3H), 2.66–2.53 (m, 1H), 2.55–2.40 (m, 3H), 2.14–2.05 (m, 2H), 2.05–1.98 (m, 1H), 13C NMR (100 MHz, DMSO-d6) 172.9, 171.0, 167.7, 153.2, 150.6, 144.0, 141.8, 141.6, 138.3, 134.2, 133.9, 133.7, 132.0, 129.8, 128.8, 128.6, 128.3, 126.6, 122.8, 118.7, 115.7, 100.8, 94.9, 89.2, 80.8, 77.0, 67.6, 62.0, 51.7, 50.7, 47.1, 31.3, 28.7, 22.5, 16.2, 12.3. ESI-MS: 696.41. UPLC: 4.00 min.
4-(5-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)pent-1-yn-1-yl)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (36)
was obtained by the coupling of compound 71 with 2-(2,6-dioxopiperidin-3-yl)-4-iodoisoindoline-1,3-dione. The synthetic procedure is the same as was used for compound 35. 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.85 (s, 1H), 7.82 (d, J = 6.4 Hz, 1H), 7.77–7.66 (m, 3H), 7.33–7.25 (m, 2H), 7.17 (d, J = 7.6 Hz, 1H), 4.97 (dd, J = 12.0, 5.0 Hz, 1H), 4.77 (s, 2H), 4.64 (s, 2H), 4.56–4.48 (m, 2H), 4.04 (s, 2H), 2.89–2.70 (m, 6H), 2.50–2.43 (m, 2H), 2.25–2.17 (m, 4H). ESI-MS: 710.18.
3-(4-(5-(4-((3-Benzyl-9-methyl-4H,6H-thieno[2,3-e][1,2,4]triazolo[3,4-c][1,4]oxazepin-2-yl)ethynyl)-1H-pyrazol-1-yl)pent-1-yn-1-yl)-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione (37)
was obtained by coupling of compound 71 with 3-(4-iodo-1-oxoisoindolin-2-yl)-1-methyl-piperidine-2,6-dione. The synthetic procedure is the same as was used for compound 35. 1H NMR (400 MHz, CDCl3) δ = 7.83 (d, J = 7.6 Hz, 1H), 7.66 (s, 1H), 7.63 (s, 1H), 7.57 (d, J = 6.8 Hz, 1H), 7.44 (dd, J = 7.6 Hz, J = 7.6 Hz, 1H), 7.30 (dd, J = 7.2 Hz, J = 7.6 Hz, 2H), 7.24 (d, J = 7.2 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 5.26–5.15 (m, 1H), 4.74 (s, 2H), 4.61 (s, 2H), 4.52–4.32 (m, 2H), 4.30 (t, J = 6.8 Hz, 2H), 4.04 (s, 2H), 3.19 (s, 3H), 3.06–2.77 (m, 4H), 2.75 (s, 3H), 2.48 (t, J = 7.0 Hz, 2H), 2.24–2.12 (m, 2H), ESI-MS: 710.40. UPLC: 4.37 min.
Molecular modeling
The co-crystal structure of BRD4 BD1/(+)-JQ-1 (PDB entry: 3MXF)11 was used to model the binding poses of compounds 2, 3 and 22 with BRD4 BD1. The chain A of BRD4 BD1 from the crystal structure was extracted and the missing side chain of K141 was rebuilt using the MOE56 program. For the amino acids where different conformations were determined, the rotamer A conformations were selected. Protons were then added to BRD4 BD1 using the “protonate 3D” module in MOE by setting at the pH 7.0 condition. All water molecules from the crystal structure were saved. Structures of the compounds were constructed and optimized using MOE. Fifteen binding poses of each compound to BRD4 BD1 were generated by the GOLD program (version 5.0.2)57, 58 in which the highest ranked pose was selected as the binding model for each compound. In the docking simulation, we set the binding site centered at F129 in BRD4 BD1 with a radius of 12 Å. In the binding site, water molecules W9, W12, W18, W15, W33 and W209 of BRD4 BD1 were included during the docking simulations with the flags on. PLP fitness function in Gold 5.0.2 was used to evaluate the docked poses and default parameters for the genetic algorithm (GA) run were used. The top pose of each compound ranked by PLP was selected as its binding mode to BRD4 BD1. Figures were prepared using the PyMOL program (www.pymol.org).
Binding Affinities of Compounds to BET proteins
Human BRD2 BD1 (residues 72–205), BRD2 BD2 (residues 349–460), BRD3 BD1 (residues 24–144), BRD3 BD2 (residues 306–417), BRD4 BD1 (residues 44–168), BRD4 BD2 (residues 333–460) recombinant proteins were used in the binding assays. A fluorescence polarization (FP) binding assay was used to determine the binding affinities of the BET inhibitors to BRD2 (BD1 and BD2), BRD3 (BD1 and BD2), and BRD4 (BD1 and BD2) proteins, as described previously.26, 27
Cell Growth Inhibition, Apoptosis Analysis and Western Blotting
RS4;11 (CRL-1873) and MV4;11 (CRL-9591) human acute leukemia cell lines were purchased from the American Type Culture Collection. The MOLM13 (ACC554) human acute leukemia cell line was purchased from the DSMZ German cell bank. All the experiments were performed within two months of thawing fresh vials of the cells. RS4;11 and MOLM-13 cells were cultured in RPMI-1640 and MV4;11 cells were cultured in IMDM media supplemented with 10% FBS and 50 U/ml penicillin-50 μg/ml streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2 in air.
For cell growth experiments, compounds were diluted in the corresponding medium and then 3-fold serially diluted in a 96-well tissue culture plate to a final volume of 100 μl/well. 20,000 cells/well in 100 μl were added to each well containing compound to a final volume of 200 μl/well. Cells were incubated for 4 days at 37 °C in an atmosphere of 5% CO2. Cell growth was evaluated utilizing a lactate dehydrogenase-based WST-8 assay (Dojindo Molecular Technologies, MD). An adequate volume of WST-8 reagent was added to each well, incubated for at least 1 h in the cell culture incubator, and read at 450 nm in a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). The readings were normalized to the DMSO-treated cells and fitted using a nonlinear regression analysis with the GraphPad Prism 6 software to obtain the IC50 value for each compound.
Apoptosis induced by the BET degrader (35) and BET inhibitor (22) was evaluated using flow cytometry. Cells were treated with compounds at indicated concentrations for 24 h or 48 h, collected, washed with cold PBS, and stained with Annexin V Alexa Fluor™ 488 and Propidium Iodide (PI) following manufacturer’s instructions (ThermoFisher Scientific, CA). Stained cells were analyzed in the Attune NxT Flow Cytometer (ThermoFisher Scientific).
For Western blot analysis, 1.5–2×106 cells/well were plated in 12 well-plates and treated with compounds at the indicated concentrations and times. Cells were collected, washed with cold PBS, and lysed in cold RIPA buffer containing Halt protease inhibitors. Twenty micrograms of lysate were run in each lane of 4–20% or 4–12% Novex gels and blotted into Polyvinylidene Difluoride membranes. Antibodies for immunoblotting were rabbit polyclonal antibodies for BRD2 (A302–583A), BRD3 (A302–368A) and BRD4 (A301–985A100) purchased from Bethyl Laboratories (Montgomery, TX,); c-Myc (D84C12), cleaved PARP (D64E10), and cleaved Caspase 3 (D3E9) from Cell Signaling Technology (Danvers, MA); GSPT1 polyclonal antibody (ab126090) from Abcam (Cambridge, MA) and HRP-GAPDH from Santa Cruz Biotechnology (Dallas, TX). The BIO-RAD Clarity Western Enhanced Chemiluminescence Substrate was used for signal development. Blots were imaged in an iBright Imaging System (ThermoFisher).
Efficacy and Pharmacodynamics Studies in the RS4;11 and MV4;11 Xenograft Models in Mice
All animal experiments were performed under the guidelines of the University of Michigan Committee for Use and Care of Animals and using an approved animal protocol (PI, Shaomeng Wang).
Xenograft tumors were established by injecting 5×106 RS4;11 or MV4;11 cells in 50% Matrigel subcutaneously on the dorsal side of severe combined immunodeficient (SCID) mice, obtained from Charles River, one tumor per mouse. When tumors reached ~100 mm3, mice were randomly assigned to treatment and vehicle control groups. Animals were monitored daily for any signs of toxicity and weighed 2–3 times per week during the treatment period and at least weekly after the treatment ended. Tumor size was measured utilizing electronic calipers 2–3 times per week during the treatment period and at least weekly after the treatment ended. Tumor volume was calculated as V = L × W2/2, where L is the length and W is the width of the tumor.
For pharmacodynamic analysis, resected control and treated RS4;11 xenograft tumor tissues were ground into powder in liquid nitrogen and lysed in CST lysis buffer with halt proteinase inhibitors. Twenty micrograms of whole tumor clarified lysates were separated on 4–20% or 4–12% Novex gels. Western blots were performed as detailed in the previous section.
Methods for tissue distribution study in RS4;11 tumor-bearing mice
Female SCID mice bearing RS4;11 xenograft tumors were dosed with a single intravenous dose of compound 35 (QCA570) at 5 mg/kg. QCA570 was dissolved in the vehicle containing 20% (v/v) PCP and 80% (v/v) phosphate-buffered saline (PBS). The dosed animals were sacrificed at 1, 3 and 6 hr for collecting different tissue and plasma samples. Isolated RS4;11 tumor samples, small intestinal, kidney, liver, heart and lung were immediately frozen and placed in tared Precellys® 2 mL hard tissue tubes with homogenizing ceramic beads 16859 (Cayman Chemical, Ann Arbor, MI), weighed, then stored at −80°C for homogenization and analysis. Blood samples (400 μL) from each SCID mice were collected in 1.5 mL microfuge tubes containing Heparin and centrifuged at 15000 rpm for 10 min. Then the plasmas were also stored at −80 °C until analysis.
To precipitate the proteins in mice plasma, 160 μL of acetonitrile containing 50 ng/mL of an internal standard was added to 40 μL of mouse plasma samples and vortexed for 10 minutes. The extracts were centrifuged at 15000 rpm and the supernatant was transferred to the autosampler vials for LC-MS/MS analysis. Blank mouse plasma was used to prepare spiked calibration standards. The concentrations of the calibration standards were 2, 10, 50, 100, 250, 500 and 2000 ng/mL. Tumor and different tissue samples were homogenized by Precellys tissue homogenizer with the addition of a diluent solution, with a ratio of 5:1 (volume to 20% Acetonitrile in water) (mL) to weight of tissue (g). Then the homogenized tissue solution was treated using the same procedure as that for the plasma samples to extract the compound for LC-MS/MS analysis.
Plasma and tissue concentrations of the compound were determined by the LC−MS/MS method developed and validated for this study. The LC−MS/MS method consisted of a Shimadzu LC-20AD HPLC system (Kyoto, Japan), and chromatographic separation of the tested compound was achieved using a Waters XBridageTM C18 column (50 × 2.1 mm, 3.5 μm) at 25 ᵒC. 5 μL of the supernatant was injected. The flow rate of gradient elution was 0.4 mL/min with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in Acetonitrile). An AB Sciex QTrap 5500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. Protonated molecular ions and its respective ion products were monitored at the transitions of m/z 696.2 > 678.2 for QCA570 and 455.2>425.2 for the internal standard. We adjusted the instrument settings to maximize analytical sensitivity and specificity of detection. Total analysis time per sample was 5.7 min. Data were processed with software Analyst (version 1.6).
Supplementary Material
ACKNOWLEDGMENT
We are grateful for the financial support from the Breast Cancer Research Foundation (to S. Wang), the Prostate Cancer Foundation (to S. Wang), the National Cancer Institute, NIH (R01CA215758 to S. Wang), and the University of Michigan Comprehensive Cancer Center support grant from the National Cancer Institute, NIH (P30 CA046592).
Funding Sources
This study was supported in part by the Breast Cancer Research Foundation (to S. Wang), the Prostate Cancer Foundation (to S. Wang), the National Cancer Institute, NIH (R01CA215758 to S. Wang), and the P30 Cancer Center Core grant (CA046592) from the National Cancer Institute, NIH.
ABBREVIATIONS USED
- BD1, BD2
Bromodomain 1 and 2
- BRD2
Bromodomain-containing protein 2
- BRD3
Bromodomain-containing protein 3
- BRD4
Bromodomain-containing protein 4
- HIV
human immunodeficiency virus
- CNS
Central nervous system
- WPF
Trp81, Pro82 and Phe83
- PARP
poly ADP ribose polymerase
- FP
fluorescence polarization
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- PD
Pharmacodynamics
- GSPT1
G1 To S Phase Transition 1
- PROTAC
Proteolysis Targeting Chimera
- SCID mice
Severe Combined Immunodeficiency mice
- TBDPS
tert-Butyldiphenylsilyl
Footnotes
The University of Michigan has filed a number of patent applications on these BET inhibitors and degraders reported in this study, which have been licensed to Oncopia Therapeutics, LLC. S. Wang, H. Yang, Q. Chong, B. Zhou, C.-Y. Yang, F. Xu, J. Hu, Z. Chen, E. Fernandez-Salas, L. Bai, and D. McEachern are co-inventors on one or more of these patents, and receive royalties on these patents from the University of Michigan. S. Wang is a co-founder and a paid consultant of Oncopia Therapeutics, LLC. The University of Michigan and S. Wang own stock in Oncopia.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Western blotting analysis of BRD2, BRD3, and BRD4 proteins in RS4;11 cells treated with BET inhibitor 22, compound 26 and BET degraders 27, 28, 29, 30, and 31; 1H NMR spectrum of compound 35; Western blotting analysis of GSPT1 protein in RS4;11 and MV4;11 cells treated with BET inhibitor 22 and BET degrader 35 (QCA570); 1H NMR and 13C NMR spectra and UPLC-MS chromatogram of compound 35 (PDF).
Molecular String files for all the final target compounds (CSV).
PDB coordinates of computational models of compounds 2, 3 and 22 (QCA276) in a complex with BRD4 BD1 (PDB).
REFERENCES
- 1.Tamkun JW; Deuring R; Scott MP; Kissinger M; Pattatucci AM; Kaufman TC; Kennison JA Brahma: A Regulator of Drosophila Homeotic Genes Structurally Related to the Yeast Transcriptional Activator SNF2/SWl2 Cell 1992, 68, 561–572. [DOI] [PubMed] [Google Scholar]
- 2.Dhalluin C; Carlson JE; Zeng L; He C; Aggarwal AK; Zhou MM Structure and Ligand of a Histone Acetyltransferase Bromodomain. Nature 1999, 399, 491–496. [DOI] [PubMed] [Google Scholar]
- 3.Patel DJ; Wang Z Readout of Epigenetic Modifications. Annu. Rev. Biochem 2013, 82, 81–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belkina AC; Denis GV BET Domain Co-regulators in Obesity, Inflammation and Cancer. Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arrowsmith CH; Bountra C; Fish PV; Lee K; Schapira M Epigenetic Protein Families: A New Frontier for Drug Discovery. Nat. Rev. Drug Discov 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- 6.Chaidos A; Caputo V; Karadimitris A Inhibition of Bromodomain and Extra-Terminal Proteins (BET) as a Potential Therapeutic Approach in Haematological Malignancies: Emerging Preclinical and Clinical Evidence. Ther. Adv. Hematol 2015, 6, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao Y; Liang L; Huang M; Qiu Q; Zeng S; Shi M; Zou Y; Ye Y; Yang X; Xu H Bromodomain and Extra-Terminal Domain Bromodomain Inhibition Prevents Synovial Inflammation via Blocking IκB Kinase–Dependent NF-κB Activation in Rheumatoid Fibroblast-like Synoviocytes. Rheumatology 2015, 55, 173–184. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee C; Archin N; Michaels D; Belkina AC; Denis GV; Bradner J; Sebastiani P; Margolis DM; Montano M BET Bromodomain Inhibition as a Novel Strategy for Reactivation of HIV-1. J. Leukoc. Biol 2012, 92, 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magistri M; Velmeshev D; Makhmutova M; Patel P; C Sartor G; Volmar C-H; Wahlestedt C; Ali Faghihi M The BET-Bromodomain Inhibitor JQ1 Reduces Inflammation and Tau Phosphorylation at Ser396 in the Brain of the 3xTg Model of Alzheimer’s Disease. Curr. Alzheimer Res 2016, 13, 985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matzuk MM; McKeown MR; Filippakopoulos P; Li Q; Ma L; Agno JE; Lemieux ME; Picaud S; Yu RN; Qi J; Knapp S; Bradner JE Small-Molecule Inhibition of BRDT for Male Contraception. Cell 2012, 150, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective Inhibition of BET Bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicodeme E; Jeffrey KL; Schaefer U; Beinke S; Dewell S; Chung CW; Chandwani R; Marazzi I; Wilson P; Coste H; White J; Kirilovsky J; Rice CM; Lora JM; Prinjha RK; Lee K; Tarakhovsky A Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dawson MA; Prinjha RK; Dittmann A; Giotopoulos G; Bantscheff M; Chan WI; Robson SC; Chung CW; Hopf C; Savitski MM; Huthmacher C; Gudgin E; Lugo D; Beinke S; Chapman TD; Roberts EJ; Soden PE; Auger KR; Mirguet O; Doehner K; Delwel R; Burnett AK; Jeffrey P; Drewes G; Lee K; Huntly BJ; Kouzarides T Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature 2011, 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filippakopoulos P; Knapp S Targeting Bromodomains: Epigenetic Readers of Lysine Ccetylation. Nat. Rev. Drug Discov 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- 15.Chung CW; Coste H; White JH; Mirguet O; Wilde J; Gosmini RL; Delves C; Magny SM; Woodward R; Hughes SA; Boursier EV; Flynn H; Bouillot AM; Bamborough P; Brusq JM; Gellibert FJ; Jones EJ; Riou AM; Homes P; Martin SL; Uings IJ; Toum J; Clement CA; Boullay AB; Grimley RL; Blandel FM; Prinjha RK; Lee K; Kirilovsky J; Nicodeme E Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem 2011, 54, 3827–3838. [DOI] [PubMed] [Google Scholar]
- 16.Gehling VS; Hewitt MC; Vaswani RG; Leblanc Y; Cote A; Nasveschuk CG; Taylor AM; Harmange JC; Audia JE; Pardo E; Joshi S; Sandy P; Mertz JA; Sims RJ 3rd; Bergeron L; Bryant BM; Bellon S; Poy F; Jayaram H; Sankaranarayanan R; Yellapantula S; Bangalore Srinivasamurthy N; Birudukota S; Albrecht BK Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors. ACS Med. Chem. Lett 2013, 4, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G; Plotnikov AN; Rusinova E; Shen T; Morohashi K; Joshua J; Zeng L; Mujtaba S; Ohlmeyer M; Zhou MM Structure-guided Design of Potent Diazobenzene Inhibitors for the BET Bromodomains. J. Med. Chem 2013, 56, 9251–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor AM; Vaswani RG; Gehling VS; Hewitt MC; Leblanc Y; Audia JE; Bellon S; Cummings RT; Cote A; Harmange JC; Jayaram H; Joshi S; Lora JM; Mertz JA; Neiss A; Pardo E; Nasveschuk CG; Poy F; Sandy P; Setser JW; Sims RJ 3rd; Tang Y; Albrecht BK Discovery of Benzotriazolo[4,3-d][1,4]diazepines as Orally Active Inhibitors of BET Bromodomains. ACS Med. Chem. Lett 2016, 7, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue X; Zhang Y; Liu Z; Song M; Xing Y; Xiang Q; Wang Z; Tu Z; Zhou Y; Ding K; Xu Y Discovery of Benzo[cd]indol-2(1H)-ones as Potent and Specific BET Bromodomain Inhibitors: Structure-Based Virtual Screening, Optimization, and Biological Evaluation. J. Med. Chem 2016, 59, 1565–1579. [DOI] [PubMed] [Google Scholar]
- 20.Ayoub AM; Hawk LML; Herzig RJ; Jiang J; Wisniewski AJ; Gee CT; Zhao P; Zhu JY; Berndt N; Offei-Addo NK; Scott TG; Qi J; Bradner JE; Ward TR; Schonbrunn E; Georg GI; Pomerantz WCK BET Bromodomain Inhibitors with One-Step Synthesis Discovered from Virtual Screen. J. Med. Chem 2017, 60, 4805–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Millan DS; Kayser-Bricker KJ; Martin MW; Talbot AC; Schiller SER; Herbertz T; Williams GL; Luke GP; Hubbs S; Alvarez Morales MA; Cardillo D; Troccolo P; Mendes RL; McKinnon C Design and Optimization of Benzopiperazines as Potent Inhibitors of BET Bromodomains. ACS Med. Chem. Lett 2017, 8, 847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharp PP; Garnier JM; Hatfaludi T; Xu Z; Segal D; Jarman KE; Jousset H; Garnham A; Feutrill JT; Cuzzupe A; Hall P; Taylor S; Walkley CR; Tyler D; Dawson MA; Czabotar P; Wilks AF; Glaser S; Huang DCS; Burns CJ Design, Synthesis, and Biological Activity of 1,2,3-Triazolobenzodiazepine BET Bromodomain Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1298–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L; Pratt JK; Soltwedel T; Sheppard GS; Fidanze SD; Liu D; Hasvold LA; Mantei RA; Holms JH; McClellan WJ; Wendt MD; Wada C; Frey R; Hansen TM; Hubbard R; Park CH; Li L; Magoc TJ; Albert DH; Lin X; Warder SE; Kovar P; Huang X; Wilcox D; Wang R; Rajaraman G; Petros AM; Hutchins CW; Panchal SC; Sun C; Elmore SW; Shen Y; Kati WM; McDaniel KF Fragment-Based, Structure-Enabled Discovery of Novel Pyridones and Pyridone Macrocycles as Potent Bromodomain and Extra-Terminal Domain (BET) Family Bromodomain Inhibitors. J. Med. Chem 2017, 60, 3828–3850. [DOI] [PubMed] [Google Scholar]
- 24.Law RP; Atkinson SJ; Bamborough P; Chung CW; Demont EH; Gordon LJ; Lindon M; Prinjha RK; Watson AJB; Hirst DJ Discovery of Tetrahydroquinoxalines as Bromodomain and Extra-Terminal Domain (BET) Inhibitors with Selectivity for the Second Bromodomain. J. Med. Chem 2018, 61, 4317–4334. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M; Zhang Y; Song M; Xue X; Wang J; Wang C; Zhang C; Li C; Xiang Q; Zou L; Wu X; Wu C; Dong B; Xue W; Zhou Y; Chen H; Wu D; Ding K; Xu Y Structure-Based Discovery and Optimization of Benzo[d]isoxazole Derivatives as Potent and Selective BET Inhibitors for Potential Treatment of Castration-Resistant Prostate Cancer (CRPC). J. Med. Chem 2018, 61, 3037–3058. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y; Bai L; Liu L; McEachern D; Stuckey JA; Meagher JL; Yang CY; Ran X; Zhou B; Hu Y; Li X; Wen B; Zhao T; Li S; Sun D; Wang S Structure-Based Discovery of 4-(6-Methoxy-2-methyl-4-(quinolin-4-yl)-9H-pyrimido[4,5-b]indol-7-yl)-3,5-dimethylisoxazole (CD161) as a Potent and Orally Bioavailable BET Bromodomain Inhibitor. J. Med. Chem 2017, 60, 3887–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran X; Zhao Y; Liu L; Bai L; Yang C-Y; Zhou B; Meagher JL; Chinnaswamy K; Stuckey JA; Wang S Structure-based Design of γ-Carboline Analogues as Potent and Specific BET Bromodomain Inhibitors. J. Med. Chem 2015, 58, 4927–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coudé M-M; Braun T; Berrou J; Dupont M; Bertrand S; Masse A; Raffoux E; Itzykson R; Delord M; Riveiro ME BET Inhibitor OTX015 Targets BRD2 and BRD4 and Decreases c-MYC in Acute Leukemia Cells. Oncotarget 2015, 6, 17698–17712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith SG; Sanchez R; Zhou MM Privileged Diazepine Compounds and Their Emergence as Bromodomain Inhibitors. Chem. & Biol 2014, 21, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raina K; Crews CM Chemical Inducers of Targeted Protein Degradation. J. Biol. Chem 2010, 285, 11057–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tinworth CP; Lithgow H; Churcher I Small Molecule-mediated Protein Knockdown as a New Approach to Drug Discovery. MedChemComm 2016, 7, 2206–2216. [Google Scholar]
- 33.Toure M; Crews CM Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- 34.Ottis P; Crews CM Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol 2017, 12, 892–898. [DOI] [PubMed] [Google Scholar]
- 35.Salami J; Crews CM Waste Disposal-An Attractive Strategy for Cancer Therapy. Science 2017, 355, 1163–1167. [DOI] [PubMed] [Google Scholar]
- 36.Walczak MJ; Petzold G; Thoma NH Targeted Protein Degradation: You Can Glue It Too! Nat. Chem. Biol 2017, 13, 452–453. [DOI] [PubMed] [Google Scholar]
- 37.Gustafson JL; Neklesa TK; Cox CS; Roth AG; Buckley DL; Tae HS; Sundberg TB; Stagg DB; Hines J; McDonnell DP; Norris JD; Crews CM Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed 2015, 54, 9659–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cyrus K; Wehenkel M; Choi EY; Lee H; Swanson H; Kim KB Jostling for Position: Optimizing Linker Location in the Design of Estrogen Receptor-Targeting PROTACs. ChemMedChem 2010, 5, 979–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Demizu Y; Okuhira K; Motoi H; Ohno A; Shoda T; Fukuhara K; Okuda H; Naito M; Kurihara M Design and Synthesis of Estrogen Receptor Degradation Inducer Dased on a Protein Knockdown Strategy. Bioorg. Med. Chem. Lett 2012, 22, 1793–1796. [DOI] [PubMed] [Google Scholar]
- 40.Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol 2015, 11, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demizu Y; Shibata N; Hattori T; Ohoka N; Motoi H; Misawa T; Shoda T; Naito M; Kurihara M Development of BCR-ABL Degradation Inducers via the Conjugation of an Imatinib Derivative and a cIAP1 Ligand. Bioorg. Med. Chem. Lett 2016, 26, 4865–4869. [DOI] [PubMed] [Google Scholar]
- 42.Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed 2016, 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in vivo Target Protein Degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou B; Hu J; Xu F; Chen Z; Bai L; Fernandez-Salas E; Lin M; Liu L; Yang CY; Zhao Y; McEachern D; Przybranowski S; Wen B; Sun D; Wang S Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J. Med. Chem 2018, 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai L; Zhou B; Yang CY; Ji J; McEachern D; Przybranowski S; Jiang H; Hu J; Xu F; Zhao Y; Liu L; Fernandez-Salas E; Xu J; Dou Y; Wen B; Sun D; Meagher J; Stuckey J; Hayes DF; Li S; Ellis MJ; Wang S Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winter GE; Mayer A; Buckley DL; Erb MA; Roderick JE; Vittori S; Reyes JM; di Iulio J; Souza A; Ott CJ; Roberts JM; Zeid R; Scott TG; Paulk J; Lachance K; Olson CM; Dastjerdi S; Bauer S; Lin CY; Gray NS; Kelliher MA; Churchman LS; Bradner JE BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AM; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zengerle M; Chan KH; Ciulli A Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemi. & Biol 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Picaud S; Wells C; Felletar I; Brotherton D; Martin S; Savitsky P; Diez-Dacal B; Philpott M; Bountra C; Lingard H; Fedorov O; Muller S; Brennan PE; Knapp S; Filippakopoulos P RVX-208, an Inhibitor of BET Transcriptional Regulators with Selectivity for the Second Bromodomain. Proc. Natl. Acad. Sci 2013, 110, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito T; Ando H; Suzuki T; Ogura T; Hotta K; Imamura Y; Yamaguchi Y; Handa H Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- 52.Fischer ES; Böhm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chamberlain PP; Lopez-Girona A; Miller K; Carmel G; Pagarigan B; Chie-Leon B; Rychak E; Corral LG; Ren YJ; Wang M Structure of the Human Cereblon–DDB1–Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat. Struct. Mol. Biol 2014, 21, 803–809. [DOI] [PubMed] [Google Scholar]
- 54.Delmore JE; Issa GC; Lemieux ME; Rahl PB; Shi J; Jacobs HM; Kastritis E; Gilpatrick T; Paranal RM; Qi J; Chesi M; Schinzel AC; McKeown MR; Heffernan TP; Vakoc CR; Bergsagel PL; Ghobrial IM; Richardson PG; Young RA; Hahn WC; Anderson KC; Kung AL; Bradner JE; Mitsiades CS BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matyskiela ME; Lu G; Ito T; Pagarigan B; Lu CC; Miller K; Fang W; Wang NY; Nguyen D; Houston J; Carmel G; Tran T; Riley M; Nosaka L; Lander GC; Gaidarova S; Xu S; Ruchelman AL; Handa H; Carmichael J; Daniel TO; Cathers BE; Lopez-Girona A; Chamberlain PP A Novel Cereblon Modulator Recruits GSPT1 to the CRL4(CRBN) Ibiquitin Ligase. Nature 2016, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- 56.MOE. Chemical Computing Group: Montreal, Quebec, Canada. [Google Scholar]
- 57.Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- 58.Verdonk ML; Cole JC; Hartshorn MJ; Murray CW; Taylor RD Improved Protein-Ligand Docking Using GOLD. Proteins 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.