

Abstract
Human murine double minute 2 (MDM2) protein is a primary endogenous cellular inhibitor of the tumor suppressor p53 and has been pursued as an attractive cancer therapeutic target. Several potent, non-peptide small-molecule inhibitors of MDM2 are currently in clinical development. In this paper, we report our design, synthesis and evaluation of small-molecule MDM2 degraders based on the proteolysis targeting chimera (PROTAC) concept. The most promising compound (MD-224) effectively induces rapid degradation of MDM2 at concentrations <1 nM in human leukemia cells. It achieves an IC50 value of 1.5 nM in inhibition of growth of RS4;11 cells and also low nanomolar IC50 values in a panel of acute leukemia cell lines. MD-224 achieves complete and durable tumor regression in vivo in the RS4;11 xenograft tumor model in mice at well tolerated dose-schedules. MD-224 is thus a highly potent and efficacious MDM2 degrader and warrants extensive evaluations as a new class of anticancer agent.
Graphical Abstract
Introduction
The tumor suppressor p53 plays a critical role in the prevention of tumor development.1–3 The TP53 gene, which encodes the p53 protein, is mutated or deleted in about 50% of human cancers, resulting in inactivation of the tumor suppressor function of p534 In human cancers retaining wild-type p53, the tumor suppressor function of p53 can be also suppressed by a variety of mechanisms. One major mechanism of p53 inhibition stems from its primary, endogenous cellular inhibitor, the murine double minute 2 (MDM2) protein. By functioning as an E3 ligase, MDM2 binds to and ubiquitinates p53, leading to efficient p53 degradation.5, 6 The binding of MDM2 to p53 also blocks the interaction of p53 with targeted DNA molecules and transports p53 from the nucleus to the cytoplasm, both of which decrease the transcriptional activity of p53.7 Indeed, over-expression of MDM2 protein has frequently been detected in human cancers carrying wild-type of p538.
Because MDM2 effectively inhibits the tumor suppressor function of p53 through a direct protein-protein interaction, intense efforts have been made to develop small-molecule inhibitors of the MDM2-p53 protein-protein interaction (hereafter called MDM2 inhibitors)9–15. A number of highly potent small-molecule MDM2 inhibitors16, including two discovered in our laboratory11, 13, have been advanced into clinical development for the treatment of human cancers.
Mechanistically, MDM2 inhibitors induce accumulation and transcriptional activation of p53 protein by blocking p53 degradation by MDM2. Since MDM2 is a p53 targeted gene, activation of p53 results in transcription of MDM2 mRNA, leading to robust MDM2 protein accumulation. However, MDM2 protein accumulated in vivo can efficiently and rapidly degrade p53 upon clearance of an MDM2 inhibitor due to the pharmacokinetic effect, and this is predicted to reduce the therapeutic efficacy of the MDM2 inhibitor. Indeed, our previous study has shown that p53 protein is accumulated for only a few hours in xenograft tumor tissues following administration of a single dose of an MDM2 inhibitor13. Furthermore, accumulation of MDM2 protein in normal tissues might have unwanted effects since MDM2 itself is oncogenic. To overcome these potential limitations of MDM2 inhibitors, new strategies are needed to more effectively target MDM2.
The proteolysis targeting chimera (PROTAC) concept17–19 was formally proposed fifteen years ago, with the objective of hijacking the powerful cellular ubiquitin degradation systems to achieve targeted protein degradation. The PROTAC strategy uses a heterobifunctional small-molecule containing a small-molecule ligand binding to the target protein of interest, and another small-molecule ligand binding to an E3 ubiquitin ligase complex, tethered together by a chemical linker to achieve targeted protein degradation. The PROTAC approach has recently gained tremendous momentum due in part to the availability of potent and druglike ligands for the cullin 2 and cullin 4A E3 degradation systems. This strategy has successfully been employed in the design of potent small-molecule PROTAC degraders for a number of proteins, including Bromodomain and Extra-Terminal (BET) proteins20–24, the androgen receptor,25 Bcr-Abl26, 27 and the estrogen-related Receptor α (ERRα)28, among others.
In the present study, we have employed the PROTAC strategy to design small-molecule degraders of MDM2 based upon our previously reported, potent MDM2 inhibitors. This has led to the discovery of MD-224, which induces rapid degradation of MDM2 at low nanomolar concentrations in leukemia cells. MD-224 is a very potent inhibitor of cell growth in a panel of leukemia cell lines carrying wild-type p53 and is capable of achieving complete and durable tumor regression in the RS4;11 xenograft model in mice at well-tolerated dose-schedules. It is therefore a highly potent and promising PROTAC MDM2 degrader.
Results and Discussion
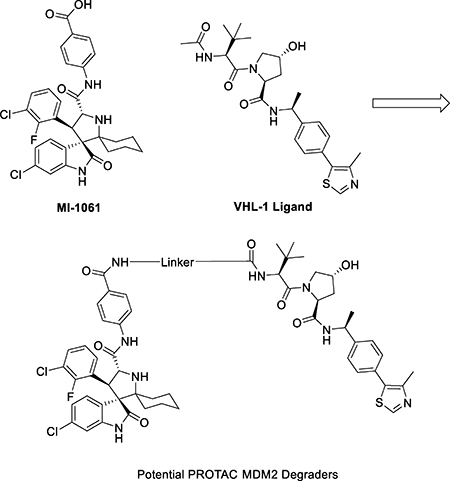
In our previous study, we reported the discovery of MI-1061 as a potent MDM2 inhibitor (Ki= 0.16 nM) 11. MI-1061 effectively activates wild-type p53, and potently inhibits cell growth in cancer cell lines harboring wild-type p53, and strongly suppresses tumor growth in vivo 11. We have therefore employed MI-1061 as a potent MDM2 inhibitor in the design of PROTAC MDM2 degraders.
The design of MI-1061 was based upon one of our first generation MDM2 inhibitors, MI-77301.13 Although we have not obtained a co-crystal structure for MI-1061 complexed with MDM2, we have successfully determined the co-crystal structure of MI-77301 in complex with MDM213 (Figure 1A). Since MI-1061 and MI-77301 are structurally similar, we predicted that in the binding to the MDM2 protein, the carboxylic acid group on the phenyl ring in MI-1061 would be located in a position similar to that of the solvent-exposed 4-hydroxyl group on the cyclohexyl ring in MI-77301 (Figure 1). Accordingly, we proposed to use the carboxylic acid group in MI-1061 as the tethering site for the design of bifunctional PROTAC MDM2 degraders.
Figure 1.

(A) Co-crystal structure of MDM2 in a complex with MI-77301 (Cyan), (PDB ID: 5TRF); (B) Modeled structure of MDM2 complexed with MI-1061 (Yellow); (C) Chemical structures of MDM2 inhibitors MI-77301 and MI-1061.
The design of PROTAC MDM2 degraders requires a small-molecule ligand for an E3 ligase degradation system. Thalidomide and lenalidomide are potent, small-molecule ligands which bind to cereblon,29 which is an adaptor protein in the cullin 4A E3 ligase degradation system. Thalidomide and lenalidomide have been successfully used in the design of PROTAC degraders for BET proteins20, 21 and a number of other proteins30, 31.
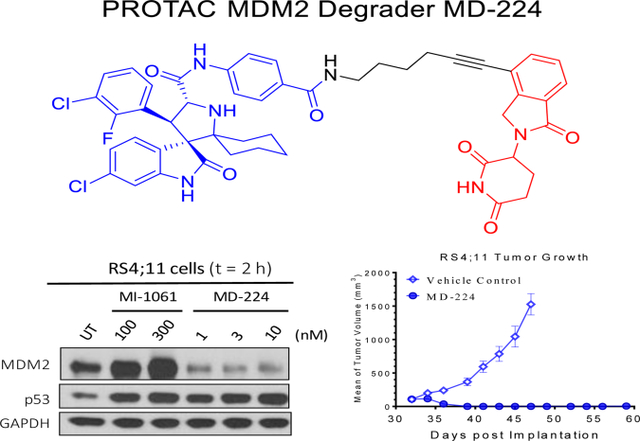
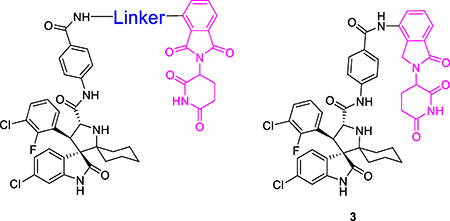
Accordingly, we employed the MDM2 inhibitor MI-1061 and thalidomide/lenalidomide for the design of two initial, putative MDM2 degraders 1 and 2 (Table 1), using linkers similar to those in our previously reported BET degraders ZBC260 and ZBC24622, 23. To facilitate the synthesis of these putative MDM2 degraders, we converted the carboxylic acid group in MI-1061 into an amide, obtaining MI-1242 (Figure 2). Our binding experiments showed that MI-1242 binds to MDM2 with a high affinity (Ki = 2.7 nM), which is weaker than MI-1061 but is consistent with the predicted strong interactions between the negatively charged carboxylic acid in MI-1061 and the Lys94 and His73 in MDM2 protein (Figure 1). However, MI-1242 potently inhibits cell growth in the RS4;11 cell line however with an IC50 value of 89 nM, comparable to the potency of MI-1061, suggesting an enhanced cell permeability for MI-1242 over that of MI-1061.
Table 1.
MDM2 degraders designed with various linkers to pomalidomide.
 | ||
|---|---|---|
| Compound No. | Linker | IC50 (nM) in inhibition of cell growth in the RS4;11 cell line |
| MI-1061 | 141 ± 12 | |
| MI-1242 | 89 ± 12 | |
| 1 | 10 ± 1 | |
| 2 | 7 ± 2 | |
| 3 | Void (no linker) | 68.4 ± 36 |
| 4 |  |
15 ± 2 |
| 5 | 9 ± 2 | |
| 6 | 12 ± 2 | |
| 7 | 22 ± 7 | |
| 8 | 100 ± 27 | |
| 9 | 8 ± 2 | |
| 10 | 5 ± 1 | |
Figure 2.

Design of PROTAC MDM2 degraders using MDM2 inhibitors and cereblon ligands.
We examined the effect of 1 and 2 on MDM2 and p53 proteins in the RS4;11 leukemia cells carrying wild-type p53, with MI-1061 and MI-1242 used as controls. Our data show that consistent with our previous report23, the MDM2 inhibitor MI-1061 is very effective in inducing accumulation of both MDM2 and p53 in a dose-dependent manner (Figure 3). Similarly, MI-1242 is equally effective in inducing accumulation of both MDM2 and p53 in a dose-dependent manner (Figure 3). In contrast, both 1 and 2 are highly potent and effective in inducing accumulation of p53 protein in a dose-dependent manner without increasing the MDM2 protein level (Figure 3). In fact, both 1 and 2 effectively reduce the level of MDM2 protein in the RS4;11 cells over the control treatments, even at concentrations as low as 3 nM. These western blotting data thus suggest that 1 and 2 do not function as regular MDM2 inhibitors but are very potent MDM2 degraders.
Figure 3.

Western blotting analysis of MDM2 and p53 proteins in RS4;11 cells treated with MDM2 inhibitors MI-1061 and MI-1242, and MDM2 degraders 1 and 2. RS4;11 cells were treated for 2 h with each compound at indicated concentrations, and proteins were probed by specific antibodies. GAPDH, a constitutively expressed housekeeping protein, was used as the loading control. MDM2 and p53 levels were quantified using an imager and were normalized over the GAPDH loading control.
Consistent with their high potency in induction of p53 accumulation, 1 and 2 achieve IC50 values of 10 nM or 7 nM, respectively, in inhibition of cell growth in the RS4;11 acute leukemia cell line in the WST cell growth assay (Table 1), and they are therefore about 10-times more potent than MI-1061 and MI-1242.
Our previous study showed that the length and chemical composition of the linker in our BET protein degraders have a major effect on their cellular potencies in both BET degradation and cell growth inhibition.23 Accordingly, we performed further modifications of the linker in 1 and 2. We evaluated each synthesized compound for its potency in cell growth inhibition in the RS4;11 cell line as an initial assessment of the cellular effect of the linker in our designed MDM2 degraders, and obtained the data summarized in Table 1.
We synthesized compound 3 (Table 1), in which the acid group in MI-1061 is directly coupled to the amino group in lenalidomide, to examine the importance of the linker. Compound 3 has an IC50 value of 108 nM in inhibition of cell growth in the RS4;11 cell line, similar to that for MI-1061 and MI-1242. Our western blotting analysis showed that 3 fails to induce MDM2 degradation in the RS4;11 cells, indicating that this compound works as an MDM2 inhibitor, but not as an MDM2 degrader (Figure 4). Changing the linker from the 4 methylene groups in 1 to 2, 3, 5 and 6 and 7 methylene groups resulted in 4, 5, 6, 7, and 8, respectively. Interestingly, compounds with 2–6 methylene groups in their linker have similar cellular potencies, all within a factor of 2, in inhibition of cell growth, but 8 with 7 methylene groups for the linker is 10 times less potent than 1. Western blotting analysis showed that these compounds all induce degradation of MDM2 and accumulation of p53 in the RS4;11 cells, indicating that they function as MDM2 degraders (Figure 4).
Figure 4.

Western blotting analysis of the effect of potential MDM2 degraders with various linkers on induction of MDM2 and p53 proteins. RS4;11 cells were treated for 2 h with each individual compound at indicated concentrations and proteins were probed by specific antibodies. GAPDH was used as the loading control. MDM2 and p53 levels were quantified using an imager and were normalized over the GAPDH loading control.
We investigated the effect of the linker length in 2 by increasing or decreasing it by one ethylene glycol group, producing 9 and 10, respectively. Compounds 9, 10 and 2 have very similar potencies in inhibition of cell growth in the RS4;11 cell line, with IC50 values of 8, 4.5 and 7 nM, respectively. Western blotting analysis confirmed that these compounds effectively induce degradation of MDM2 and accumulation of p53 in the RS4;11 cells, indicating that they function as MDM2 degraders (Figure 4).
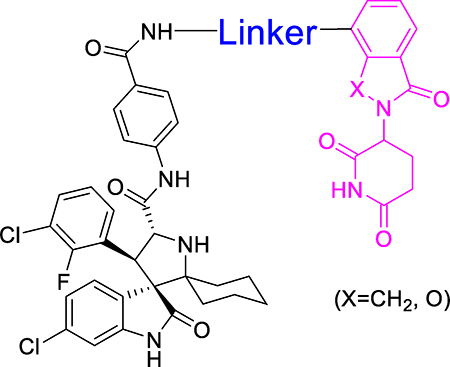
Our previous studies of BET degraders demonstrated that modifications of the cereblon binding portion of the degrader molecule can significantly enhance the degradation of target proteins, cell growth inhibitor activity against cancer cells and in vivo antitumor efficacy23. We therefore performed modifications of the cereblon ligand portion in 1 (Table 2). Changing the NH group in 1 to a CH2 group yielded 11, which has an IC50 value of 29 nM and is therefore 3-times less potent than 1. Conversion of one of the carbonyl groups in 1 into a CH2 group generated 12, which has an IC50 value of 5 nM and is thus twice as potent as 1. Changing the NH linking group in 12 to a CH2 group led to 13 (MD-222), which has an IC50 value of 2.8 nM and is therefore 4-times more potent than 1.
Table 2.
MDM2 degraders with modifications of the cereblon ligand portion in compound 1
 |
|||
|---|---|---|---|
| Compound No. | Linker | X | IC50 (nM) in inhibition of cell growth in the RS4;11 cell line |
| 1 | CO | 10 ± 1 | |
| 11 | CO | 29 ± 8 | |
| 12 | CH2 | 5 ± 5 | |
| 13 (MD-222) | CH2 | 2.8 ± 1.1 | |
| 14 (MD-224) | CH2 | 1.5 ± 0.5 | |
| 15 | CH2 | 3.9 ± 0.4 | |
Compounds 12 and MD-222 have a flexible linker between the MDM2 inhibitor and the cereblon ligand portion. We next investigated the effect of linker rigidification in 12 and MD-222 on cell growth inhibition and MDM2 degradation (Table 2). Conversion of two CH2 groups in MD-222 into an alkyne group resulted in MD-224, which has an IC50 value of 1.5 nM in the RS4;11 cell line and is 2-times more potent than MD-222. Shortening the linker in MD-224 by one CH2 group yielded 15, which has an IC50 value of 3.9 nM and is thus 2-times less potent than MD-224.
The co-crystal structure of lenalidomide in a complex with cereblon (PDB ID: 4CI2)32 suggests that in addition to the linking position in MD-224, two other positions on the isoindolin-1-one ring could be used for linker tethering in the design of PROTAC degraders (Figure 5). Based upon MD-224, we designed and synthesized 16 and 17 (Table 3), which use two other possible tethering positions in the phenyl ring of the cereblon ligand. Compounds 16 and 17 both have IC50 values of 5 nM in inhibition of cell growth in the RS4;11 cell line, which is 3-times less potent than MD-224. Our western blotting data confirmed that 16 and 17 effectively induce degradation of MDM2 and accumulation of p53 in the RS4;11cells, indicating that they function as MDM2 degraders (Figure 6). The data therefore indicate that all three tethering positions in the isoindolin-1-one ring in the cereblon ligand can be used successfully for the design of potent and effective MDM2 degraders.
Figure 5.

Three possible linker tethering positions in the cereblon ligand (shown in red arrows) based upon a previously published co-crystal structure of DDB1-CRBN E3 ubiquitin ligase complexed with lenalidomide (Yellow, PDBID 4CI2).
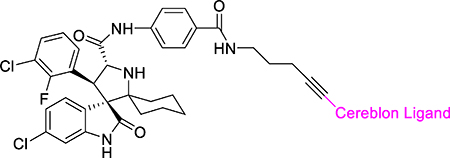
Table 3.
MDM2 degraders linked from different positions of lenalidomide.
 | ||
|---|---|---|
| Compound No. | Cereblon Ligand | IC50 (nM) in inhibition of cell growth in the RS4;11 cell line |
| 14 (MD-224) | 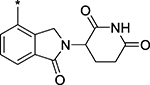 |
1.5 ± 0.5 |
| 16 | 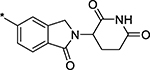 |
5 ± 2 |
| 17 | 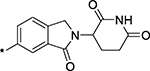 |
5 ± 3 |
Figure 6.

Western blotting analysis of the effect of MDM2 degraders linked from three different tethering sites of lenalidomide on MDM2 and p53 protein levels in RS4;11 cell line. The MDM2 inhibitor MI-1061 and the MDM2 degrader MD-224 were included as controls. RS4;11 cells were treated for 2 h with each individual compound at the indicated concentrations and proteins were probed by specific antibodies. GAPDH was used as the loading control. MDM2 and p53 levels were quantified using an imager and were normalized over the GAPDH loading control.
In addition to the cereblon/Cullin 4A E3 ligase complex, the Von Hippel-Lindau protein 1 (VHL-1)/cullin 2 E3 ligase system has been employed for the design of PROTAC degraders of a number of proteins.25, 33 We investigated if the VHL-1/cullin 2 E3 ligase system can be employed for the successful design of PROTAC MDM2 degraders. In the co-crystal structure of VHL-1 in a complex with a potent, peptidomimetic VHL-1 ligand (Figure 7), the terminal acetyl group is exposed to solvent, making this site suitable for the design of potential PROTAC MDM2 degraders. Indeed, this site has been successfully used for the design of PROTAC BET protein degraders24, 33 and other targets.25, 26, 28 We designed and synthesized three putative MDM2 degraders (18, 19 and 20) using the VHL-1 ligand and MI-1061, with linkers of different lengths and chemical compositions (Table 4).
Figure 7.

A proposed tethering position in the VHL-1 ligand based upon a previously published co-crystal structure of a VHL-1 ligand in complex with pVHL:EloB:EloC (PDB ID: 4W9H)
Table 4.
Potential MDM2 degraders designed using an VHL-1 ligand.
 | ||
|---|---|---|
| Compound No. | Linker | IC50 (nM) in inhibition of cell growth in the RS4;11 cell line |
| MI-1061 | 140 ± 10 | |
| 18 | 1200± 800 | |
| 19 | 500± 140 | |
| 20 | 340 ± 20 | |
In the cell growth assay in the RS4;11 cell line, 18, 19 and 20 display IC50 values of 1.2, 0.5 and 0.3 μM, and thus all are less potent than the corresponding MDM2 inhibitor MI-1061 (Table 4). Western blotting analysis showed that in contrast to 1 and 2, compound 20 fails to reduce the level of MDM2 protein in the RS4;11 cells (Figure 8). Collectively, our data suggest that the VHL-1/Cullin 2 E3 ligase system may not be suited to the design of effective PROTAC MDM2 degraders, although more extensive investigations are needed to confirm this initial finding.
Figure 8.

Western blotting analysis of MDM2, p53, p21 and cleaved PAPR proteins in RS4;11 cells treated with the MDM2 inhibitor MI-1061 and a potential MDM2 degrader 20. RS4;11 cells were treated for 12 h with each individual compound at indicated concentrations and each protein was probed by a specific antibody. GAPDH was used as the loading control. MDM2, p53, p21 and cleaved PARP levels were quantified using an imager and were normalized over the GAPDH loading control.
Further Evaluation of Two Potent PROTAC MDM2 Degraders, MD-222 and MD-224
We performed extensive evaluations to further investigate the mechanism of action and therapeutic potential of MD-224 and MD-222, two potent PROTAC MDM2 degraders, with MI-1061 included as the MDM2 inhibitor control.
We first examined their potency in induction of MDM2 depletion and p53 activation in the RS4;11 and MV4;11 leukemia cell lines. Treatment of the RS4;11 cells with either MD-222 or MD-224 for 2 h effectively induces depletion of MDM2 protein and currently accumulation of p53 protein in a dose-dependent manner (Figure 9A). Consistent with their high potency in inhibition of cell growth, both MD-222 and MD-224 effectively induce marked depletion of MDM2 protein at concentrations ⩽ 1 nM and MD-224 is more potent than MD-222. The MDM2 inhibitor MI-1061 induces accumulation of both MDM2 and p53 proteins, consistent with its mechanism of action as a potent MDM2 inhibitor. Both MD-222 and MD-224 are similarly potent and effective in inducing depletion of MDM2 protein and accumulation of p53 protein in MV4;11 cell line (Figure 9B), when compared to RS4;11 cell line.
Figure 9.

Western blotting analysis for MDM2 and p53 protein levels after treatment with the MDM2 inhibitor MI-1061 and the MDM2 degraders MD-222 and MD-224 in (A) RS4;11 and (B) MV-4–11 cell lines. RS4;11 or MV-4–11 cells were treated with each individual compound at the indicated concentrations for 2 h and proteins were probed by specific antibodies. GAPDH was used as the loading control. MDM2 and p53 protein levels were quantified using an imager and were normalized over the GAPDH loading control.
As bona fide PROTAC degrader molecules, depletion of MDM2 protein by MD-224 and MD-222 should require their binding to cereblon/Cullin 4 E3 ligase complex through their lenalidomide segment. Accordingly, we predicted that excessive amounts of lenalidomide should effectively block the MDM2 degradation and reduce p53 activation induced by MD-224 and MD-222 and should have no effect on the upregulation of MDM2 and p53 proteins induced by the MDM2 inhibitor MI-1061. Western blotting analysis showed that lenalidomide indeed effectively blocks MDM2 degradation and accumulation of p53 protein induced by MD-224 and MD-222 and has no effect on the MDM2 and p53 accumulation induced by the MDM2 inhibitor MI-1061 (Figure 10A).
Figure 10.

Investigation of activity dependence of the MDM2 degraders MD-222 and MD-224 and the MDM2 inhibitor MI-1061 on cereblon (CRBN)-binding, proteasome and neddylation. (A). Western blotting analysis of MDM2 and p53 proteins in RS4;11 cells. RS4;11 cells were treated with the MDM2 inhibitor MI-1061, the MDM2 degrader MD-222 or MD-224 for 2 h in the presence or absence of excess lenalidomide. MDM2, p53 and GAPDH (loading control) proteins were probed with specific antibodies. (B, C). Cell growth inhibition activity of the MDM2 inhibitor MI-1061 and the MDM2 degraders MD-222 and MD-224 in the absence or presence of lenalidomide in the RS4;11 cell line. Cells were treated for 4 days with MI-1061, MD-222 or MD-224 alone or in combination with indicated concentrations of lenalidomide for 4 days and cell viability was determined by a WST-8 assay. (D). Western blotting analysis of MDM2 and p53 proteins in RS4;11 cells. RS4;11 cells were treated with DMSO, MG-132 (10μM), PR-171 (1μM) or MLN4924 (3μM) for 4 hours, followed by treatment with DMSO control, MDM2 inhibitor MI-1061, or the MDM2 degrader MD-224 for additional 2 h. MDM2, p53 and GAPDH (loading control) proteins were probed with specific antibodies.
Since lenalidomide can effectively block MDM2 degradation induced by MD-224 and MD-222, we predicted that lenalidomide would be effective in reducing the potent cell growth inhibitory activity of these MDM2 degraders but would have no effect on the activity of the MDM2 inhibitor MI-1061. Indeed, pretreatment with lenalidomide greatly reduces the cell growth inhibitory activity of MD-222 and MD-224 to the same level observed for the MDM2 inhibitor MI-1061 (Figure 10B-C). These data indicate that when the binding of MD-222 or MD-224 to cereblon is effectively blocked by lenalidomide, both MD-222 and MD-224 behave as an MDM2 inhibitor in cells. As expected, pretreatment with lenalidomide has no significant effect on the cell growth inhibitory activity of MI-1061. These data clearly show that the superior cellular potency of MD-222 or MD-224 compared to MI-1061 is achieved through the efficient MDM2 degradation by MD-222 or MD-224. In the MV4;11 AML cell line, pretreatment with lenalidomide also effectively reduces the cell growth inhibitory activity of MD-222 or MD-224 and has no effect on the activity of MI-1061. (Figure S1). Pre-incubated with proteasome inhibitors MD-132 and PR- 171 or a neddylation inhibitor MLN4924 in RS4;11 cells can block MDM2 degradation induced by MD-224 (Figure 10d). These data showed that the MDM2 degradation was significantly reduced by proteasome inhibitor and neddylation inhibitor, indicating that the MDM2 degradation by MD-224 is proteasome and neddylation-dependent.
Together, these data showed that MD-224 and MD-222 are highly effective and potent in induction of MDM2 degradation and accumulation of p53 in both RS4;11 and MV4;11 cell lines. MD-224 is >10–50 times more potent than the MDM2 inhibitor, MI-1061, in induction of p53 activation and in inhibition of cell growth in the RS4;11 and MV4;11 cell lines. The induction of MDM2 degradation by MD-224 can be effectively blocked by the cereblon ligand lenalidomide, a proteasome inhibitor and a neddylation inhibitor. These data establish that MD-224 is a highly potent, effective and bona fide PROTAC MDM2 degrader.
To further investigate the mechanism of action of MDM2 degradation induced by MD-224, we synthesized two control compounds 21 and 22 (Figure 11A). Compound 21 is an enantiomer of MD-224 with all the chiral centers in the MDM2 inhibitor portion inverted. Our data showed that compound 21 fails to induce any MDM2 degradation in the RS4;11 cell line (Figure 11B), and has a minimal activity in inhibition of cell growth in the RS4;11 cell line (Figure 11C). The amino group of the glutarimide in lenalidomide forms a strong hydrogen bond with cereblon as shown in the co-crystal structure29, 32 and methylation of the amino group of the glutarimide in lenalidomide completely abrogates the binding to cereblon21, 32. We therefore synthesized 22 by methylation of the amino group of the glutarimide in MD-224 as an additional control compound. Compound 22 was found to be completely inactive in inducing MDM2 degradation (Figure 11B) and is >100-times less potent than MD-224 in inhibition of cell growth in the RS4;11 cells (Figure 11C). These data firmly establish that the induced degradation of MDM2 by MD-224 requires its strong binding to MDM2 and to cereblon, consistent with its PROTAC design.
Figure 11.

Control compounds 21 and 22 fail to induce MDM2 degradation and show much weaker activity in cell growth inhibition in the RS4;11 cell line: (A) Chemical structures of 21 and 22; (B) Western blotting analysis of p53 and MDM2 proteins in RS4;11 cells treated with MI-1061, MD-224, 21 and 22; (C) Cell growth inhibition of MI-1061, MD-224, 21 and 22 in the RS4;11 cell line.
Since p53 is a powerful transcription factor, we used qRT-PCR analysis to examine the transcriptional activation of p53 by MD-224, MD-222 and MI-1061 (Figure 12). A 6 h treatment with the MDM2 inhibitor MI-1061 effectively induces marked transcriptional upregulation of p53 target genes in the RS4;11 cell line, including MDM2, the cell cycle regulator gene p21, and pro-apoptotic PUMA gene but not TP53, which is the gene encoding p53 (Figure 12). MD-224 and MD-222 are >10 times more potent than MI-1061 in induction of transcriptional upregulation of these p53 target genes but have no effect on TP53 itself in RS4;11 cells (Figure 12). Despite the robust upregulation of MDM2 mRNA by MD-224 and MD-222, the MDM2 protein is effectively depleted by both MD-224 and MD-222, indicating that they are highly efficient degraders of the MDM2 protein. Very similar data were obtained in the MV4;11 cell line for MD-224, MD-222 and MI-1061 (Figure S2), when compared to the data obtained in the RS4;11 cell line.
Figure 12.

qRT-PCR analysis mRNA levels of p53 target genes and TP53 after treatment with the MDM2 inhibitor, MI-1061 and the MDM2 degraders MD-222 and MD-224 in RS4;11 cells. RS4;11 cells were treated for 6 hr and mRNA levels of MDM2, p21, TP53, and PUMA were analyzed by qRT-PCR.
Because both the MDM2 degrader MD-224 and the MDM2 inhibitor MI-1061 effectively induce upregulation of PUMA, a potent pro-apoptotic gene, we investigated induction of apoptosis by both compounds in the RS4;11 cells by flow cytometry (Figure 13). While both compounds are effective in inducing apoptosis in a dose-dependent manner upon a 24 h treatment, MD-224 is >10-times more potent than MI-1061. In fact, MD-224 induces robust apoptosis at ⩽10 nM, consistent with its high potency in induction of p53 activation and of PUMA mRNA upregulation.
Figure 13.

Flow cytometric analysis of apoptosis induction using Annexin V/ PI double staining after treatment with the MDM2 inhibitor, MI-1061, and the MDM2 degrader MD-224 in RS4;11 cells.
We further evaluated MD-222, MD-224 and MI-1061 in additional human acute leukemia cell lines containing wild-type p53 and mutated p53 for their growth inhibitory activity and obtained the data shown in Table 5.
Table 5.
Cell growth inhibition activity of MI-1061, MD-222 and MD-224 in a panel of human AML or ALL cell lines.
| Cell line | Leukemia Type | p53 status | IC50 (nM) in cell growth inhibition assay | ||
|---|---|---|---|---|---|
| MI-1061 | MD-222 | MD-224 | |||
| MOLM-13 | AML | WT | 76.8 ± 40 | 11.7 ± 4 | 7.3 ± 3 |
| MOLM-14 | AML | WT | 143.3 ± 64 | 22.5 ± 8 | 10.5 ± 3 |
| SIG-M5 | AML | WT | 105.6 ± 73 | 29.9 ± 16 | 19.8 ± 11 |
| ML-2 | AML | WT | 150.7 ± 51 | 11.5 ± 6 | 4.4 ± 2 |
| OCL-AML-5 | AML | WT | 374.5 ± 143 | 58.2 ± 29 | 33.1 ± 18 |
| Mono-Mac-6 | AML | MUT | >10 μM | >10 μM | >10 μM |
| KG-1 | AML | MUT | >10 μM | >10 μM | >10 μM |
Both MI-1061 and MD-224 potently inhibit cell growth in these human acute leukemia cell lines with wild-type p53 but MD-224 is much more potent than MI-1061. MD-224 has IC50 values of 4.4–33.1 nM in these cell lines and is >10-times more potent than MI-1061. Both MI-1061 and MD-224 are highly selective over leukemia cell lines with mutated p53, displaying IC50 >10 μM, indicating that the cell growth inhibition by both MDM2 inhibitors and degraders depends upon wild-type p53.
We next investigated the pharmacodynamic (PD) effect of MD-224 in vivo in mice bearing the RS4;11 xenograft tumor (Figure 14). A single, intravenous dose of MD-224 effectively depletes MDM2 protein at the 3 h time point, with the effect persisting for >24 h (Figure 14). MD-224 induces strong upregulation of p53 and p21 at the 3 h time-point in the tumor tissue with the effect lasting for >24 h. PARP cleavage is evident at the 24 h time point, indicating induction of apoptosis. The PD data thus show that by degradation of MDM2 protein, a single dose of the MDM2 degrader MD-224 can achieve robust and sustained MDM2 degradation and p53 upregulation for >24 h. Our previous data showed that a single dose of our potent MDM2 inhibitor MI-77301 can upregulate p53 protein for only a few hours from tumor tissue in mice13, likely due to robust accumulation of MDM2 protein, which can bind to and rapidly degrade p53 once the MDM2 inhibitor is cleared from the tumor tissue. The sustained p53 protein accumulation by MD-224 suggests that infrequent (e.g. weekly) administration may be sufficient to achieve strong antitumor activity.
Figure 14.

Pharmacodynamic (PD) analysis of MDM2, p53, p21 and PARP cleavage protein levels by western blotting after a single dose of MD-224 administered in the RS4;11 xenograft mouse model. A single dose of MD-224 (25 mg/kg, IV) effectively induces MDM2 degradation, p53 activation and PAPR cleavage in the xenograft RS4;11 tumors.
Based upon these promising PD data, we tested MD-224 for its antitumor activity in the RS4;11 xenograft model in two experiments. In the first experiment (Figure 15), we tested MI- 1061 at a near maximum tolerated dose (100 mg/kg, oral, daily dosing) and 25 mg/kg of MD-224, administered intravenously. The treatments started on day 26 when the average tumor volumes reached 100 mm3 with 5 mice/tumors in each group. The MDM2 inhibitor MI-1061 effectively retards tumor growth over the control but MD-224 can achieve much stronger antitumor activity than MI-1061 (Figure 15A). MD-224 at a 25 mg/kg weekly, intravenous dose, or at a dose of 10 mg/kg three times per week has similar antitumor activity as MI-1061 at 100 mg/kg, 5 days a week via oral gavage. MD-224 at 25 mg/kg, three times a week, can regress the tumors by 50%. MI-1061 and MD-224 cause no significant weight loss or other signs of toxicity in this experiment.
Figure 15.

In vivo antitumor activity of MI-1061 and MD-224 in the RS4;11 xenograft model in mice: (A) Tumor growth. (B) Average tumor volume at the end of administration.
In the second experiment, we investigated if MD-224 at higher doses or more frequent dosing schedules can achieve complete tumor regression in the RS4;11 model (Figure 16). Treatments were initiated when the tumor volume reached 100 mm3 at day 33. It was found that MD-224 at 25 mg/kg, daily, 5 days a week for 2 weeks or at 50 mg/kg, every other day for three weeks achieves complete tumor regression. Consistent with its strong and sustained p53 activation following a single dose in RS4;11 tumor tissues, MD-224 with weekly dosing at 50 mg/kg achieves complete tumor growth inhibition (Figure 16A) while causing no weight loss or any other signs of toxicity in mice (Figure 16B). These data establish that MD-224 is highly efficacious against the RS4;11 xenograft tumors even with weekly dosing and can achieve complete tumor regression with either daily or every other day (three times per week) dosing schedules.
Figure 16.

In vivo antitumor activity of MD-224 in the RS4;11 xenograft model: (A) Tumor volume during the administration; (B) Monitoring animal weight as an indication of tolerability. MD-224 effectively induced complete tumor regression or strong tumor growth inhibition in RS4;11 xenograft models at well tolerated dose-schedules.
Taken together, these two efficacy experiments clearly demonstrate that the MDM2 degrader MD-224 can achieve complete tumor regression at well tolerated dose-schedules and is much more efficacious than the MDM2 inhibitor MI-1061.
Synthesis
The synthesis of MDM2 degraders containing thalidomide or lenalidomide is shown in Scheme 1. The intermediates (24a-24i) were synthesized by a nucleophilic substitution reaction with 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (23) and the Boc protecting groups were removed under acidic conditions. The intermediate (24j) was synthesized by reductive amination reaction with lenalidomide following with removing Boc protecting group under acidic conditions. MDM2 degraders 1, 10 and 12 were obtained by an amide condensation reaction with MI-1061.
Scheme 1.

Reaction conditions: (a) DIPEA, DMF, 80 °C; (b) NaBH(OAc)3, HOAc, DCE; (c) TFA, DCM, rt; (d) HATU, DIPEA, DMF, rt.
The synthesis of MDM2 degraders 11, 13–17, 21 and 22 is shown in Scheme 2. Intermediates 26a-26f were synthesized by Sonogashira coupling reactions following with removing Boc protecting groups under acidic conditions from 25a-25e. The triple bond in 26b was then reduced by H2 with Pd/C (Palladium on carbon) to form the intermediate 27. MDM2 degraders 11, 13–17, 21 and 22 were then obtained by an amide condensation reaction with MI-1061 and its enantiomer.
Scheme 2.

Reaction conditions: (a) Pd(PPh3)2Cl2, CuI, Et3N, DMF, 80 °C; (b) TFA, DCM; rt (c) H2 (1 atm), Pd/C, EtOH, rt (d) HATU, DIPEA, DMF, rt.
The synthesis of MDM2 degraders containing a VHL ligand is shown in Scheme 3. The VHL ligands with different linkers (28a-28c) were synthesized as described previously.33 Compounds 18, 19 and 20 were then obtained by an amide condensation reaction with MI-1061.
Scheme 3.

Reaction conditions: (a) HATU, DIPEA, DMF, rt; (b) TFA, DCM, rt; (c) HATU, DIPEA, DMF, rt.
Conclusion
In this study, we report our design, synthesis and evaluation of the first-in-class PROTAC small-molecule MDM2 degraders. Our study shows that by employing our previous reported potent MDM2 inhibitor MI-1061 and cereblon ligands thalidomide and lenalidomide, we have successfully obtained highly potent and efficacious MDM2 degraders, as exemplified by MD-224. MD-224 achieves rapid MDM2 protein degradation even at concentrations below 1 nM with a 2 h treatment in leukemia cell lines and is highly effective in inducing activation of p53 in leukemia cells carrying wild-type p53. MD-224 is highly potent in inhibition of cell growth and in induction of apoptosis in p53 wild-type leukemia cells and is >10–100 times more potent than the MDM2 inhibitor MI-1061. A single dose of MD-224 effectively induces MDM2 degradation and p53 activation in the RS4;11 leukemia xenograft tissue with the effect persisting for > 24 h. Consequently, MD-224 is highly efficacious and is capable of achieving complete and long-lasting tumor regression in the RS4;11 xenograft model in mice at well tolerated dose-schedules. Collectively, our data demonstrate that MD-224 is a highly potent, efficacious, and promising MDM2 degrader which warrants further evaluation as a potential new therapy for the treatment of human acute leukemia and other types of human cancer.
Experimental Section General
Unless otherwise noted, all purchased reagents were used as received without further purification. 1H NMR and 13C NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer. 1H NMR spectra are reported in parts per million (ppm) downfield from tetramethylsilane (TMS). In reported spectral data, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. MS analyses were carried out with a Waters UPLC-mass spectrometer. The final compounds were all purified by C18 reverse phase preparative HPLC column with solvent A (0.1% TFA in water) and solvent B (0.1% TFA in MeCN) as eluents. The purity of all the final compounds was confirmed to be >95% by UPLC-MS or UPLC.
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (1)
Reaction 1
23 (200 mg, 0.73 mmol) was added to a solution of tert-butyl (4-aminobutyl)carbamate (207 mg, 1.1 mmol) and DIEA (0.6 mL, 4 mmol) in DMF (2 mL). The reaction was heated to 90 °C for 12 h, then the mixture was cooled to room temperature and loaded onto Celite. The crude residue was purified by reverse phase C18 column to afford tert-butyl (4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butyl)-carbamate without further purification. The product was then dissolved in 5 mL DCM and 1 mL TFA was added to the reaction. The reaction was stirred for 2 h at room temperature until no starting material could be detected by HPLC. The solvent was removed to yield 24a (107 mg, yield: 43%) as a yellow solid.
Reaction 2
HATU (13.3 mg, 1.2 eq.) and N,N-diisopropylethylamine (0.026 mL, 0.15 mmol) were stirred with a solution of MI-1061 (20 mg, 0.029 mmol) in 0.5 mL DMF. After 10 min, 24a (0.35 mL, 0.1 M in DMSO) was added to the reaction. After 30 min, the solvent was removed and the crude was dissolved in 3:1 MeOH/water, acidified with TFA and purified by reverse-phase preparative HPLC. The purified fractions were combined, concentrated in vacuo, re-dissolved in water, frozen and lyophilized to give 1 (18.5 mg, Yield: 71%) as a yellow powder. LC-MS(ESI) m/z (M +H)+: 908.32; calcd for C47H45Cl2FN7O7 (M +H)+ : 908.27; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.79 – 7.68 (m, 3H), 7.63 – 7.58 (m, 2H), 7.53 (dd, J = 8.3, 2.4 Hz, 1H), 7.49 (ddd, J = 8.5, 7.1, 1.4 Hz, 1H), 7.39 – 7.32 (m, 1H), 7.17 (t, J = 8.1 Hz, 1H), 7.11 (dd, J = 8.2, 2.0 Hz, 1H), 7.02 (d, J = 8.6 Hz, 1H), 6.98 (dd, J = 6.8, 2.8 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.30 (d, J = 10.7 Hz, 1H), 5.03 (dd, J = 12.6, 5.5 Hz, 1H), 4.96 (d, J = 10.8 Hz, 1H), 3.44 – 3.34 (m, 4H), 2.92 – 2.79 (m, 2H), 2.78 – 2.65 (m, 2H), 2.17 (d, J = 14.1 Hz, 1H), 2.13 – 2.05 (m, 1H), 2.02 – 1.87 (m, 3H), 1.81 – 1.69 (m, 6H), 1.62 – 1.48 (m, 1H), 1.27 – 1.16 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)-ethyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (2)
2 (20.8 mg, Yield: 75% from MI-1061) was obtained as yellow powder using the same synthetic strategy described for 1 with 24b.
LC-MS(ESI) m/z (M +H)+: 968.35; calcd for C49H49Cl2FN7O9 (M +H)+ : 968.30; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.79 – 7.66 (m, 3H), 7.59 – 7.47 (m, 3H), 7.40 – 7.27 (m, 2H), 7.19 (t, J = 8.1 Hz, 1H), 7.10 (dd, J = 8.2, 1.8 Hz, 1H), 6.95 – 6.83 (m, 2H), 6.79 (d, J = 1.9 Hz, 1H), 5.40 (d, J = 10.9 Hz, 1H), 5.03 – 4.96 (m, 3H), 3.73 – 3.62 (m, 8H), 3.57 – 3.50 (m, 2H), 3.42 – 3.35 (m, 2H), 2.94 (d, J = 8.0 Hz, 1H), 2.85 – 2.57 (m, 3H), 2.21 (d, J = 13.6 Hz, 1H), 2.07 – 1.87 (m, 4H), 1.76 (d, J = 11.6 Hz, 2H), 1.53 (dd, J = 26.9, 13.3 Hz, 1H), 1.31 – 1.10 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(4-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (3)
2 (3.5 mg, Yield: 17% from MI-1061) was obtained as white powder using the same condensation method for 1 with lenalidomide.
LC-MS(ESI) m/z (M +H)+: 823.45; calcd for C43H38Cl2FN6O6 (M +H)+ : 823.22; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 8.05 – 7.91 (m, 2H), 7.84 – 7.63 (m, 5H), 7.60 (t, J = 7.7 Hz, 1H), 7.54 (dd, J = 8.3, 2.5 Hz, 1H), 7.37 (t, J = 7.5 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.12 (dd, J = 8.3, 2.0 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 5.18 (dd, J = 13.3, 5.1 Hz, 2H), 4.98 – 4.93 (m, 1H), 4.55 (d, J = 2.0 Hz, 2H), 3.14 – 3.02 (m, 1H), 2.97 – 2.87 (m, 1H), 2.83 – 2.75 (m, 1H), 2.57 – 2.43 (m, 1H), 2.24 – 2.16 (m, 1H), 2.15 – 2.06 (m, 1H), 2.01 – 1.85 (m, 3H), 1.78 (t, J = 15.1 Hz, 2H), 1.62 (d, J = 13.2 Hz, 1H), 1.44 – 1.30 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (4)
4 (10.9 mg, Yield: 43% from MI-1061) was obtained as a yellow powder using the synthetic strategy described for 1 with 24c.
LC-MS(ESI) m/z (M +H)+: 880.21; calcd for C45H41Cl2FN7O7 (M +H)+ : 880.24; >98% purity.
1H NMR (400 MHz, MeOH-A) δ 7.85 – 7.75 (m, 2H), 7.74 – 7.61 (m, 3H), 7.53 (dd, J = 8.6, 7.1 Hz, 1H), 7.45 (dd, J = 8.2, 2.4 Hz, 1H), 7.25 (t, J = 7.4 Hz, 1H), 7.19 (d, J = 8.5 Hz, 1H), 7.13 – 6.98 (m, 3H), 6.74 (d, J = 2.0 Hz, 1H), 5.05 (dd, J = 12.4, 5.5 Hz, 1H), 4.83 – 4.63 (m, 2H), 3.69 – 3.52 (m, 4H), 2.93 – 2.59 (m, 4H), 2.25 – 1.90 (m, 5H), 1.79 – 1.71 (m, 2H), 1.62 – 1.59 (m, 1H), 1.15 – 0.99 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (5)
5 (10.9 mg, Yield: 69% from MI-1061) was obtained as yellow powder using the same synthetic strategy described for 1 with 24d.
LC-MS(ESI) m/z (M +H)+: 894.24; calcd for C46H43Cl2FN7O7 (M +H)+ : 894.26; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.87 – 7.74 (m, 2H), 7.72 (t, J = 7.4 Hz, 1H), 7.67 – 7.57 (m, 2H), 7.58 – 7.44 (m, 2H), 7.37 (t, J = 7.6 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.12 (dd, J = 8.2, 2.0 Hz, 1H), 7.08 – 6.94 (m, 2H), 6.79 (d, J = 2.0 Hz, 1H), 5.31 (d, J = 10.8 Hz, 2H), 5.07 – 4.93 (m, 2H), 3.55 – 3.37 (m, 5H), 2.93 – 2.77 (m, 3H), 2.78 – 2.60 (m, 1H), 2.24 – 2.13 (m, 1H), 2.12 – 2.01 (m, 1H), 2.03 – 1.90 (m, 5H), 1.84 – 1.71 (m, 3H), 1.54 (d, J = 14.0 Hz, 1H), 1.26 (d, J = 21.4 Hz, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (6)
6 (8.6 mg, Yield: 33% from MI-1061) was obtained as a yellow powder using the same synthetic strategy described for 1 with 24e.
LC-MS(ESI) m/z (M +H)+: 922.30; calcd for C48H47Cl2FN7O7 (M +H)+: 922.29; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.79 – 7.67 (m, 3H), 7.63 – 7.57 (m, 2H), 7.57 – 7.46 (m, 2H), 7.37 – 7.30 (m, 1H), 7.16 (t, J = 7.9 Hz, 1H), 7.10 (dd, J = 8.2, 1.9 Hz, 1H), 7.02 (d, J = 8.6 Hz, 1H), 6.98 (dd, J = 6.7, 4.1 Hz, 1H), 6.78 (d, J = 1.9 Hz, 1H), 5.22 (d, J = 9.8 Hz, 1H), 5.04 – 4.98 (m, 1H), 4.95 (dd, J = 10.6, 2.9 Hz, 1H), 3.41 – 3.33 (m, 4H), 2.90 – 2.61 (m, 4H), 2.17 – 2.04 (m, 2H), 1.97 – 1.82 (m, 3H), 1.78 – 1.49 (m, 9H), 1.24 – 1.13 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (7)
7 (14.3 mg, Yield: 53% from MI-1061) was obtained as a yellow powder using the same synthetic strategy described for 1 with 24f.
LC-MS(ESI) m/z (M +H)+: 936.27; calcd for C49H49Cl2FN7O7 (M +H)+ : 936.31; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.76 (d, J = 8.7 Hz, 2H), 7.71 (t, J = 6.7 Hz, 1H), 7.60 (d, J = 8.7 Hz, 2H), 7.56 – 7.47 (m, 2H), 7.36 (t, J = 7.0 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.11 (dd, J = 8.2, 1.9 Hz, 1H), 7.02 – 6.97 (m, 2H), 6.79 (d, J = 1.9 Hz, 1H), 5.33 (d, J = 10.9 Hz, 1H), 5.03 (dd, J = 12.4, 5.3 Hz, 1H), 4.97 (d, J = 10.9 Hz, 1H), 3.40 – 3.27 (m, 4H), 2.93 – 2.65 (m, 4H), 2.19 (d, J = 11.6 Hz, 1H), 2.15 – 2.06 (m, 1H), 2.04 – 1.89 (m, 3H), 1.78 (d, J = 11.8 Hz, 2H), 1.71 – 1.59 (m, 4H), 1.55 – 1.38 (m, 5H), 1.27 – 1.18 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (8)
8 (16.3 mg, Yield: 60% from MI-1061) was obtained as yellow powder using the same synthetic strategy described for 1 with 24g.
LC-MS(ESI) m/z (M +H)+: 950.29; calcd for C50H51Cl2FN7O7 (M +H)+ : 950.32; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.80 – 7.74 (m, 2H), 7.74 – 7.68 (m, 1H), 7.64 – 7.59 (m, 2H), 7.55 – 7.47 (m, 2H), 7.37 – 7.30 (m, 1H), 7.16 (t, J = 8.1 Hz, 1H), 7.10 (dd, J = 8.2, 1.9 Hz, 1H), 7.01 (s, 1H), 6.99 (d, J = 2.2 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.27 (d, J = 10.6 Hz, 1H), 5.03 (dd, J = 12.6, 5.4 Hz, 1H), 4.95 (d, J = 10.7 Hz, 1H), 3.38 – 3.27 (m, 4H), 2.90 – 2.64 (m, 4H), 2.20 – 2.05 (m, 2H), 2.00 – 1.84 (m, 3H), 1.83 – 1.71 (m, 2H), 1.67 – 1.53 (m, 5H), 1.46 – 1.35 (m, 6H), 1.25 – 1.13 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (9)
9 (15.9 mg, Yield: 60% from MI-1061) was obtained as a yellow powder using the same synthetic strategy described for 1 with 24h.
LC-MS(ESI) m/z (M +H)+: 924.20; calcd for C47H45Cl2FN7O8 (M +H)+ : 924.27; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.76 – 7.68 (m, 3H), 7.61 – 7.51 (m, 3H), 7.47 – 7.39 (m, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.19 (t, J = 8.1 Hz, 1H), 7.11 (dd, J = 8.2, 0.9 Hz, 1H), 7.01 (d, J = 8.6 Hz, 1H), 6.91 (t, J = 7.2 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.39 (d, J = 10.9 Hz, 1H), 5.03 – 4.92 (m, 3H), 3.69 (dt, J = 10.5, 5.0 Hz, 4H), 3.58 – 3.50 (m, 2H), 3.45 (t, J = 4.9 Hz, 2H), 3.03 – 2.53 (m, 4H), 2.22 (d, J = 13.4 Hz, 1H), 2.09 – 1.86 (m, 4H), 1.78 (d, J = 12.1 Hz, 2H), 1.54 (dd, J = 27.2, 13.5 Hz, 1H), 1.29 – 1.12 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((2-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)-ethyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (10)
10 was (11.8 mg, Yield: 41% from MI-1061) obtained as a yellow powder using the same synthetic strategy described for 1 with 24i.
LC-MS(ESI) m/z (M +H)+: 1012.30; calcd for C51H53Cl2FN7O10 (M +H)+ : 1012.32; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.76 (d, J = 8.7 Hz, 2H), 7.72 (t, J = 6.6 Hz, 1H), 7.58 (d, J = 8.7 Hz, 2H), 7.53 (dd, J = 8.2, 2.3 Hz, 1H), 7.49 – 7.43 (m, 1H), 7.36 (t, J = 7.0 Hz, 1H), 7.19 (t, J = 8.1 Hz, 1H), 7.11 (dd, J = 8.2, 1.8 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 6.79 (d, J = 1.9 Hz, 1H), 5.37 (d, J = 10.9 Hz, 1H), 5.02 (ddd, J = 12.3, 5.4, 1.3 Hz, 1H), 4.97 (d, J = 11.0 Hz, 1H), 3.68 – 3.60 (m, 12H), 3.52 (t, J = 5.3 Hz, 2H), 3.42 (t, J = 5.2 Hz, 2H), 2.95 – 2.87 (m, 1H), 2.86 – 2.79 (m, 1H), 2.76 – 2.61 (m, 2H), 2.21 (d, J = 13.4 Hz, 1H), 2.13 – 2.05 (m, 1H), 2.00 – 1.88 (m, 3H), 1.77 (d, J = 11.7 Hz, 2H), 1.53 (dd, J = 27.1, 13.2 Hz, 1H), 1.29 – 1.19 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)pentyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (11)
11 (18.2 mg, Yield: 58% from MI-1061) was obtained as a white powder using the same synthetic strategy described for MD-222 with 26a.
LC-MS(ESI) m/z (M +H)+: 907.27; calcd for C48H46Cl2FN6O7 (M +H)+ : 907.28; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.75 – 7.68 (m, 3H), 7.66 – 7.63 (m, 2H), 7.63 – 7.57 (m, 3H), 7.54 (dd, J = 8.2, 2.2 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 7.19 (t, J = 8.0 Hz, 1H), 7.11 (dd, J = 8.2, 1.4 Hz, 1H), 6.80 (d, J = 1.5 Hz, 1H), 5.38 (d, J = 10.9 Hz, 1H), 5.09 (dd, J = 12.6, 5.4 Hz, 1H), 4.99 (d, J = 11.0 Hz, 1H), 3.34 (t, J = 7.0 Hz, 2H), 3.11 (dd, J = 17.7, 10.1 Hz, 2H), 2.98 – 2.79 (m, 2H), 2.79 – 2.62 (m, 2H), 2.22 (d, J = 13.9 Hz, 1H), 2.10 (dd, J = 8.6, 3.5 Hz, 1H), 2.04 – 1.91 (m, 3H), 1.80 – 1.51 (m, 7H), 1.44 (dd, J = 15.0, 8.0 Hz, 2H), 1.31 – 1.19 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(4-((4-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)butyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (12)
Reaction 1
Lenalidomide (73 mg, 0.28 mmol) was added to a solution of tert-butyl (4-hydroxybutyl)-carbamate (65 mg, 0.34 mmol) in 3 mL DCE and stirred for 30 min. Then sodium triacetoxyborohydride (120 mg, 0.56 mmol) was added to the mixture and reaction was stirred at room temperature overnight. The reaction was quenched by adding saturated sodium bicarbonate solution (30 mL), and the product was extracted with DCM (3×30 mL) and the combined organic solution was washed by brine. The crude product was obtained by removing the solvent in vacuum and was then dissolved in 10 mL DCM and 2 mL TFA. The reaction was stirred for 30 min and the solvent was removed in vacuo. The residue was purified by reverse-phase chromatography over a C18 column to yield 24j as a colorless oil.
Reaction 2
HATU (13.3 mg, 1.2 eq.) and N,N-diisopropylethylamine (0.026 mL, 0.15 mmol) were added to a solution of MI-1061 (20 mg, 0.029 mmol) in 0.5 mL DMF and stirred. After 10 min, 24j (0.35 mL, 0.1 M in DMSO) was added to the reaction. After 30 min, the solvent was removed and the crude was dissolved in 3:1 MeOH/water, acidified with TFA and purified by reverse-phase preparative HPLC. The purified fractions were combined, concentrated in vacuo, redissolved in water, frozen and lyophilized to yield 12 (15.1mg, yield: 58%) as a white powder.
LC-MS(ESI) m/z (M +H)+: 894.30; calcd for C47H47Cl2FN7O6 (M +H)+ : 894.29; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.80 – 7.66 (m, 3H), 7.66 – 7.49 (m, 3H), 7.42 – 7.34 (m, 1H), 7.32 – 7.24 (m, 1H), 7.19 (t, J = 8.1 Hz, 1H), 7.12 (dd, J = 8.2, 2.0 Hz, 1H), 7.04 (d, J = 7.5 Hz, 1H), 6.94 – 6.67 (m, 2H), 5.34 (d, J = 10.9 Hz, 1H), 5.12 (dd, J = 13.3, 5.1 Hz, 1H), 4.98 (d, J = 11.0 Hz, 1H), 4.41 – 4.11 (m, 2H), 3.47 – 3.35 (m, 2H), 3.29 – 3.19 (m, 2H), 2.90 (ddd, J = 18.4, 13.3, 5.6 Hz, 2H), 2.78 (ddd, J = 17.6, 4.9, 2.6 Hz, 1H), 2.43 (dddd, J = 17.1, 14.0, 10.3, 3.6 Hz, 1H), 2.28 – 2.10 (m, 2H), 2.08 – 1.89 (m, 3H), 1.89 – 1.61 (m, 6H), 1.52 (t, J = 13.7 Hz, 1H), 1.30 – 1.19 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (13)
Reaction 1
Cul (50mg, 0.25 mmol) and Pd(Ph3P)2Cl2 (90 mg, 0.13 mmol) were added to a solution of tert-butyl pent-4-yn-1-ylcarbamate (236 mg, 1.29 mmol), 25b (400mg, 1.29 mmol) in Et3N (3mL) and DMF (3 mL). The mixture was stirred at 80 °C under an N2 atmosphere overnight. The reaction mixture was poured into a saturated aqueous solution of NH4Cl and after separation of the organic layer the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography to afford 26b as a white solid (350 mg, Yield: 64%).
Reaction 2
Pd/C (20mg) was added to a solution of 26b (210 mg, 0.5 mmol) in EtOH (5 mL). The reaction was stirred under hydrogen atmosphere for 2 h. Then the mixture was filtered through Celite and the solvent was removed in vacuo. The residue was dissolved in 10 mL DCM and 2 mL TFA. The reaction was stirred for 30 min and then the solvent was removed in vacuo. The residue was purified by reverse-phase chromatography over C18 column to give 27 as colorless oil (180 mg, Yield: 85%)
Reaction 3
HATU (13.3 mg, 1.2 eq.) and N,N-diisopropylethylamine (0.026 mL, 0.15 mmol) were added to a solution of MI-1061 (20 mg, 0.029 mmol) in 0.5 mL DMF and stirred. After 10 min, 27 (0.35 mL, 0.1 M in DMSO) was added to the reaction. After 30 min, the solvent was removed and the crude was dissolved in 3:1 MeOH/water, acidified with TFA and purified by reverse-phase preparative HPLC. The purified fractions were combined, concentrated in vacuo, re-dissolved in water, frozen and lyophilized to give 13 (17.5 mg, Yield: 68%) as a white powder.
LC-MS(ESI) m/z (M +H)+: 893.19; calcd for C48H48Cl2FN6O6 (M +H)+ : 893.30; >98% purity.
1H NMR (400 MHz, MeOD) δ 7.78 – 7.66 (m, 3H), 7.66 – 7.56 (m, 3H), 7.53 (dd, J = 8.2, 2.5 Hz, 1H), 7.47 – 7.38 (m, 2H), 7.38 – 7.32 (m, 1H), 7.17 (t, J = 8.1 Hz, 1H), 7.11 (dd, J = 8.2, 2.0 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.29 (d, J = 10.7 Hz, 1H), 5.14 (dd, J = 13.3, 5.2 Hz, 1H), 4.97 (d, J = 10.8 Hz, 1H), 4.46 (dd, J = 5.7, 2.5 Hz, 2H), 3.41 – 3.33 (m, 2H), 2.96 – 2.64 (m, 5H), 2.50 (qdd, J = 13.3, 4.6, 2.5 Hz, 1H), 2.22 – 2.09 (m, 2H), 2.02 – 1.84 (m, 3H), 1.79 – 1.48 (m, 7H), 1.48 – 1.35 (m, 2H), 1.22 (td, J = 13.7, 4.0 Hz, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)pent-4-yn-1-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (14)
HATU (13.3 mg, 1.2 eq.) and N,N-diisopropylethylamine (0.026 mL, 0.15 mmol) were added to a solution of MI-1061 (20 mg, 0.029 mmol) in 0.5 mL DMF and stirred. After 10 min, 26b (0.35 mL, 0.1 M in DMSO) was added to the reaction. After 30 min, the solvent was removed and the crude was dissolved in 3:1 MeOH/water, acidified with TFA and purified by reverse-phase preparative HPLC. The purified fractions were combined, concentrated in vacuo, re-dissolved in water, frozen and lyophilized to give MD-224 (8.7 mg, Yield: 34%) as a white powder.
LC-MS(ESI) m/z (M +2H)2+: 445.30; calcd for C48H45Cl2FN6O6 (M +2H)2+ : 445.14; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.83 – 7.62 (m, 4H), 7.56 (dd, J = 8.3, 2.5 Hz, 1H), 7.54 – 7.47 (m, 2H), 7.47 – 7.32 (m, 3H), 7.19 (ddd, J = 8.9, 7.6, 3.3 Hz, 1H), 7.12 (dd, J = 8.2, 2.0 Hz, 1H), 6.80 (d, J = 1.9 Hz, 1H), 5.30 (d, J = 10.8 Hz, 1H), 5.13 (ddd, J = 13.0, 7.4, 5.2 Hz, 1H), 4.98 (d, J = 10.9 Hz, 1H), 4.49 – 4.25 (m, 2H), 3.57 (t, J = 6.6 Hz, 2H), 2.97 – 2.83 (m, 2H), 2.83 – 2.73 (m, 1H), 2.68 – 2.42 (m, 3H), 2.28 – 2.13 (m, 2H), 2.13 – 1.84 (m, 5H), 1.84 – 1.68 (m, 2H), 1.57 (ddd, J = 16.3, 6.2, 3.0 Hz, 1H), 1.34 – 1.19 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(4-((4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)but-3-yn-1-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (15)
15 (16.9 mg, Yield: 67% from MI-1061) was obtained as a white powder using the same synthetic strategy described for MD-224 with 26c.
LC-MS(ESI) m/z (M +H)+: 875.36; calcd for C47H42Cl2FN6O6 (M +H)+ : 875.25; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.89 – 7.80 (m, 2H), 7.76 – 7.69 (m, 2H), 7.69 – 7.61 (m, 2H), 7.59 (dt, J = 7.7, 0.8 Hz, 1H), 7.56 (dt, J = 8.3, 2.6 Hz, 1H), 7.47 (t, J = 7.7 Hz, 1H), 7.42 – 7.30 (m, 1H), 7.18 (td, J = 8.1, 1.1 Hz, 1H), 7.11 (dt, J = 8.2, 1.7 Hz, 1H), 6.80 (d, J = 1.9 Hz, 1H), 5.43 – 5.27 (m, 1H), 5.12 – 4.97 (m, 2H), 4.32 (d, J = 7.8 Hz, 2H), 3.73 – 3.56 (m, J = 6.4 Hz, 2H), 3.02 – 2.76 (m, 4H), 2.67 (ddt, J = 21.0, 17.7, 3.4 Hz, 1H), 2.20 (d, J = 13.9 Hz, 1H), 2.12 – 1.85 (m, 5H), 1.78 (d, J = 12.9 Hz, 2H), 1.63 – 1.49 (m, 1H), 1.31 – 1.16 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)pent-4-yn-1-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (16)
16 (18.1 mg, Yield: 71% from MI-1061) was obtained using the same synthetic strategy described for MD-224 with 26d.
LC-MS(ESI) m/z (M +H)+:889.38; calcd for C48H44Cl2FN6O6 (M +H)+ 889.27; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.82 – 7.74 (m, 2H), 7.71 (ddd, J = 6.4, 4.0, 1.5 Hz, 1H), 7.66 (dd, J = 7.9, 4.2 Hz, 1H), 7.61 – 7.46 (m, 4H), 7.38 (dt, J = 9.2, 8.4 Hz, 2H), 7.19 (t, J = 8.0 Hz, 1H), 7.15 – 7.06 (m, 1H), 6.80 (d, J = 1.9 Hz, 1H), 5.35 (dd, J = 10.8, 8.6 Hz, 1H), 5.24 – 5.08 (m, 1H), 4.96 (dd, J = 11.0, 3.8 Hz, 1H), 4.55 – 4.28 (m, 2H), 3.66 – 3.44 (m, 2H), 2.97 – 2.84 (m, 2H), 2.84 – 2.72 (m, 1H), 2.55 (t, J = 6.4 Hz, 2H), 2.53 – 2.40 (m, 1H), 2.27 – 2.10 (m, 2H), 2.04 – 1.87 (m, 5H), 1.78 (d, J = 12.3 Hz, 2H), 1.62 – 1.43 (m, 1H), 1.30 – 1.15 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-3-oxoisoindolin-5-yl)pent-4-yn-1-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (17)
17 (15.4 mg, Yield: 60% from MI-1061) was obtained using the same synthetic strategy described for MD-224 with 26e.
LC-MS(ESI) m/z (M +H)+:889.32; calcd for C48H44Cl2FN6O6 (M +H)+ 889.27; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.83 – 7.76 (m, 2H), 7.76 – 7.68 (m, 2H), 7.63 – 7.57 (m, 3H), 7.54 (dd, J = 8.3, 2.5 Hz, 1H), 7.49 (dd, J = 7.9, 1.0 Hz, 1H), 7.41 – 7.34 (m, 1H), 7.19 (t, J = 8.0 Hz, 1H), 7.12 (dd, J = 8.2, 1.9 Hz, 1H), 6.80 (d, J = 1.9 Hz, 1H), 5.36 (d, J = 10.9 Hz, 1H), 5.15 (ddd, J = 13.3, 5.1, 1.5 Hz, 1H), 4.97 (d, J = 11.0 Hz, 1H), 4.57 – 4.34 (m, 2H), 3.53 (t, J = 6.9 Hz, 2H), 2.90 (ddd, J = 15.3, 10.3, 3.6 Hz, 2H), 2.78 (ddd, J = 17.6, 4.5, 2.3 Hz, 1H), 2.59 – 2.43 (m, 3H), 2.26 – 2.12 (m, 2H), 2.04 – 1.86 (m, 5H), 1.78 (d, J = 11.9 Hz, 2H), 1.53 (dd, J = 27.2, 13.4 Hz, 1H), 1.30 – 1.18 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((5-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (18)
28a-28c were synthesized as previously reported.20 Then 18 (21.3 mg, Yield: 67% from MI-1061) was synthesized with the same synthetic strategy described for 13 with 28a.
LC-MS(ESI) m/z (M +H)+:1107.41, 5.765 min; calcd for C58H56Cl2FN8O7S (M +H)+ : 1107.41; >95% purity.
1H NMR (400 MHz, MeOH-d4) δ 9.01 (s, 1H), 7.88 – 7.74 (m, 2H), 7.72 (t, J = 6.5 Hz, 1H), 7.69 – 7.60 (m, 2H), 7.53 (dd, J = 8.4, 2.6 Hz, 1H), 7.47 – 7.30 (m, 5H), 7.19 (t, J = 8.1 Hz, 1H), 7.14 – 7.05 (m, 1H), 6.80 (d, J = 1.9 Hz, 1H), 5.38 (d, J = 11.0 Hz, 1H), 5.02 – 4.96 (m, 2H), 4.65 – 4.51 (m, 2H), 4.43 (s, 1H), 3.88 (d, J = 11.2 Hz, 1H), 3.74 (dd, J = 11.0, 3.9 Hz, 1H), 3.37 (t, J = 6.5 Hz, 2H), 2.95 (d, J = 8.1 Hz, 1H), 2.49 (s, 3H), 2.37 – 2.12 (m, 4H), 2.06 – 1.84 (m, 4H), 1.78 (d, J = 11.8 Hz, 2H), 1.72 – 1.59 (m, 4H), 1.55 – 1.42 (m, 4H), 1.36 – 1.14 (m, 2H), 1.03 (s, 9H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-((2-(2-(3-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropoxy)ethoxy)ethyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (19)
19 (25.0 mg, Yield: 75% from MI-1061) was obtained using the same synthetic strategy described for 18 with 28b.
LC-MS(ESI) m/z (M +H)+:1167.64, 5.656 min; calcd for C60H70Cl2FN8O9S (M +H)+: 1167.44; >95% purity.
1H NMR (400 MHz, MeOH-d4) δ 8.97 (s, 1H), 7.86 – 7.76 (m, 2H), 7.71 (dd, J = 10.4, 3.9 Hz, 1H), 7.66 – 7.58 (m, 2H), 7.53 (dd, J = 8.2, 2.5 Hz, 1H), 7.47 – 7.30 (m, 5H), 7.19 (t, J = 8.1 Hz, 1H), 7.10 (dd, J = 8.2, 1.9 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.38 (d, J = 10.9 Hz, 1H), 5.00 – 4.95 (m, 2H), 4.64 (s, 1H), 4.57 (dd, J = 11.9, 4.6 Hz, 1H), 4.42 (s, 1H), 3.86 (d, J = 11.2 Hz, 1H), 3.76 – 3.53 (m, 11H), 2.94 (d, J = 8.9 Hz, 1H), 2.62 – 2.40 (m, 5H), 2.26 – 2.09 (m, 2H), 2.04 – 1.86 (m, 4H), 1.78 (d, J = 11.9 Hz, 2H), 1.63 – 1.38 (m, 4H), 1.32 – 1.17 (m, 2H), 1.01 (s, 9H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-Chloro-2-fluorophenyl)-N-(4-(((S)-14-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-15,15-dimethyl-12-oxo-3,6,9-trioxa-13-azahexadecyl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (20)
20 (23.9 mg, Yield: 69% from MI-1061) was obtained using the same synthetic strategy described for 18 with 28c.
LC-MS(ESI) m/z (M +2H)2+:606.31, calcd for C62H75Cl2FN8O10S (M +2H)2+: 606.23; >95% purity.
NMR (400 MHz, MeOH-d4) δ 8.94 (s, 1H), 7.87 – 7.76 (m, 2H), 7.76 – 7.66 (m, 1H), 7.66 – 7.57 (m, 2H), 7.54 (dd, J = 8.2, 2.5 Hz, 1H), 7.46 – 7.32 (m, 5H), 7.18 (t, J = 8.1 Hz, 1H), 7.11 (dd, J = 8.2, 2.0 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.36 (d, J = 10.9 Hz, 1H), 5.04 – 4.95 (m, 2H), 4.64 (s, 1H), 4.57 (dd, J = 9.9, 6.7 Hz, 1H), 4.43 (d, J = 1.8 Hz, 1H), 3.86 (d, J = 11.0 Hz, 1H), 3.76 – 3.53 (m, 15H), 2.94 (d, J = 8.7 Hz, 1H), 2.62 – 2.36 (m, 5H), 2.28 – 2.07 (m, 2H), 2.03 – 1.83 (m, 4H), 1.78 (d, J = 12.1 Hz, 2H), 1.61 – 1.44 (m, 4H), 1.29 – 1.18 (m, 2H), 1.02 (s, 9H).
(3’S,4’R,5’S)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(4-((5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pent-4-yn-1-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (21)
21 (27.9 mg, Yield: 87%) was obtained as white powder using the same synthetic strategy described for MD-224 from the enantiomer of MI-106111, with 26b.
LC-MS(ESI) m/z (M +H)+: 903.35; calcd for C49H46Cl2FN6O6 (M +H)+ : 903.28; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.75 – 7.68 (m, 3H), 7.64 (ddd, J = 12.7, 7.6, 1.1 Hz, 1H), 7.56 (dd, J = 8.2, 2.6 Hz, 1H), 7.52 – 7.46 (m, 2H), 7.46 – 7.42 (m, 1H), 7.41 – 7.31 (m, 2H), 7.19 (tdd, J = 8.2, 3.5, 1.1 Hz, 1H), 7.12 (dd, J = 8.2, 2.0 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 5.36 (dd, J = 10.8, 1.4 Hz, 1H), 5.13 (ddd, J = 13.1, 7.8, 5.1 Hz, 1H), 4.99 (dd, J = 10.9, 1.7 Hz, 1H), 4.41 – 4.31 (m, 2H), 3.57 (t, J = 6.6 Hz, 2H), 2.98 – 2.84 (m, 2H), 2.84 – 2.70 (m, 1H), 2.62 – 2.43 (m, 3H), 2.29 – 2.11 (m, 2H), 2.05 – 1.87 (m, 5H), 1.79 (d, J = 13.2 Hz, 2H), 1.62 – 1.48 (m, 1H), 1.25 (t, J = 13.5 Hz, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(4-((5-(2-(1-methyl-2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pent-4-yn-1-yl)carbamoyl)phenyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (22)
22 (26 mg, Yield: 98% from MI-1061) was obtained as a white powder using the same synthetic strategy described for MD-224 with 26f.
LC-MS(ESI) m/z (M +H)+: 903.35; calcd for C49H46Cl2FN6O6 (M +H)+ : 903.28; >98% purity.
1H NMR (400 MHz, MeOH-d4) δ 7.76 – 7.68 (m, 3H), 7.68 – 7.62 (m, 1H), 7.56 (ddd, J = 8.3, 2.7, 1.3 Hz, 1H), 7.53 – 7.44 (m, 3H), 7.44 – 7.33 (m, 2H), 7.23 – 7.15 (m, 1H), 7.11 (dd, J = 8.2, 2.0 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 5.32 (d, J = 10.8 Hz, 1H), 5.15 (dt, J = 13.5, 4.7 Hz, 1H), 4.98 (d, J = 10.8 Hz, 1H), 4.41 – 4.27 (m, 2H), 3.57 (t, J = 6.7 Hz, 2H), 3.09 (d, J = 5.9 Hz, 3H), 2.96 – 2.89 (m, 2H), 2.58 (t, J = 6.5 Hz, 2H), 2.55 – 2.38 (m, 1H), 2.29 – 2.08 (m, 2H), 2.04 – 1.87 (m, 5H), 1.78 (d, J = 13.2 Hz, 2H), 1.62 – 1.49 (m, 1H), 1.33 – 1.14 (m, 3H).
Determination of Binding Affinities to MDM2 Proteins
The binding affinity of MDM2 inhibitors or degraders to MDM2 protein was determined by an optimized, sensitive and quantitative fluorescence polarization-based (FP-based) binding assay, as described previously. A recombinant human MDM2 protein (residues 1–118) and a FAM tagged p53-based peptide as the fluorescent probe were used in the binding assays.13
Cell Growth Inhibition, Apoptosis Analysis, and Western Blotting Assays
The human acute leukemia RS4;11 cell line (CRL-1873) was purchased from the American Type Culture Collection (ATCC). In all experiments, RS4;11 human leukemia cells were used within three months of thawing fresh vials. RS4;11 cells were cultured in RPMI 1640 media supplemented with 10% FBS and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere containing 5% CO2.
In cell growth experiments, cells were seeded in 96-well cell culture plates at a density of 10000–20000 cells/well in 100 μL of culture medium. Each compound tested was serially diluted in the appropriate medium, and 100 μL of the diluted solution containing the tested compound was added to the appropriate wells of the cell plate. After addition of the tested compound, the cells were incubated for 4 days at 37 °C in an atmosphere of 5% CO2. Cell growth was evaluated by a lactate dehydrogenase-based WST-8 assay (Dojindo Molecular Technologies) using a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). The WST-8 reagent was added to the plate, incubated for at least 1 h, and read at 450 nm. The readings were normalized to the DMSO-treated cells, and the IC50 was calculated by nonlinear regression analysis using GraphPad Prism 6 software.
For Western blot analysis, 2 × 106 cells/well were treated with compounds at the indicated concentrations for various times. Cells were collected and lysed in RIPA buffer containing protease inhibitors. An amount of 20 μg of lysate was run in each lane of a PAGE-SDS and blotted into PVDF membranes. Antibodies for immunoblotting were MDM2 (SMP14) and p53 (DO-1) purchased from Santa Cruz.
For apoptosis assays, apoptosis in RS4;11 cells was analyzed by flow cytometry with propidium iodide (PI)/Annexin V-FLUOS double staining (Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit, Invitrogen) following manufacturer’s protocols after treatments for 24 hours as indicated.
Efficacy and Pharmacodynamics Studies in the RS4;11 Xenograft Model in Mice
To develop xenograft tumors, 5 × 106 RS4;11 cells with 50% Matrigel were injected subcutaneously on the dorsal side of severe combined immunodeficient (SCID) mice, obtained from Charles River Co., one tumor per mouse. When tumors reached ~100 mm3, mice were randomly assigned to treatment and vehicle control groups. Animals were monitored daily for any signs of toxicity, weighed 2–3 times per week during the treatment and weighed at least weekly after the treatment ended. Tumor size was measured 2–3 times per week by electronic calipers during the treatment period and at least weekly after the treatment was ended. Tumor volume was calculated as V = L*W2/2, where L is the length and W is the width of the tumor. For pharmacodynamic analysis, resected RS4;11 xenograft tumor tissues were ground into powder in liquid nitrogen and lysed in lysis buffer (1% CHAPS, 150 mM NaCl, 20 mM Tris-HCl, 1 mM. EDTA, 1 mM EGTA, and COMPLETE proteinase inhibitor (Roche)). Whole tumor lysates were separated on 4–20% Novex gels. The separated proteins were transferred to a polyvinylidene difluoride membrane for immunoblotting. The following antibodies were used: antibodies for MDM2 (SMP14), p53 (DO-1), GAPDH (FL-335) from Santa Cruz Biotechnology; p21 (12D1), PARP (46D11), and caspase-3 (8G10) from Cell Signaling Technology. The secondary antibody used was horseradish peroxidase conjugated goat anti-rabbit (Thermo Scientific). The BIO-RAD Clarity Western Enhanced Chemiluminescence Substrates and HyBlot Chemiluminescence film were used for signal development and detection using a SRX-101A tabletop processor (Konica Minolta).
All the animal experiments were performed under an approved animal protocol (Protocol ID: PRO00007499, PI, Shaomeng Wang) by the Institutional Animal Care & Use Committee of the University of Michigan.
Molecular modeling
Co-crystal structures of MDM2/MI-77301 (PDB entry: 5TRF) was used to model the binding poses of MI-1061 with MDM2. We extracted chain A of MDM2 from both crystal structures and used the “protonate 3D” module in MOE34 to protonate MDM2 at the physiological pH = 7.0 condition. The structure of MI-1061 was drawn and optimized using the MOE program. The GOLD35–37 program (version 5.2) was used in the docking simulation. The binding site was centered at Ile99 in MDM2 with a radius of 13 A to cover the entire binding pocket for both MDM2 structures. The default parameters for the genetic algorithm (GA) run and the GoldScore were used to generate and evaluate the docked poses. After analyzing the twenty binding poses of MI-1061 to MDM2, we selected the highest ranked pose for each MDM2 structure as the binding model of MI-1061. Figures were prepared using the PyMOL program (www.pymol.org).
Supporting Information Available
Effect of lenalidomide on the activity of MDM2 inhibitor and degraders in MV4;11 cells, qRT-PCR analysis mRNA levels of p53 target genes and TP53 after treatment with the MDM2 inhibitor, MI-1061 and the MDM2 degraders MD-222 and MD-224 in MV4;11 cells, 1H NMR spectrum for MDM2 degrader MD-224, UPLC-MS results for MDM2 degrader MD-224, PDB coordinates of a computational models of MI-1224 in a complex with MDM2, and molecular string file. The Supporting Information is available free of charge on the ACS Publications website:
Supplementary Material
ACKNOWLEDGMENT
We are grateful for the financial support from the National Cancer Institute, NIH (1R01CA208267 to S. Wang), Oncopia Therapeutics LLC (to S. Wang) and the University of Michigan Rogel Cancer Center support grant from the National Cancer Institute, NIH (P30 CA046592).
Funding Sources
This study was supported in part by the National Cancer Institute, NIH (R01CA219345 to S. Wang), and the P30 Cancer Center Core grant (CA046592) from the National Cancer Institute, NIH and Oncopia Therapeutics LLC (to S. Wang).
ABBREVIATIONS USED
- MDM2
Human murine double minute 2
- PROTAC
proteolysis targeting chimera
- BET
Bromodomain and Extra-Terminal
- ERRα
estrogen-related Receptor α
- VHL-1
Von Hippel-Lindau protein 1
- CRBN
cereblon
- PUMA
p53 upregulated modulator of apoptosis
- PD
pharmacodynamics
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- HOAc
acetic acid
- DCE
1,2-dichloroethane
- TFA
trifluoroacetic acid
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- DCM
Dichloromethane
- TMS
tetramethylsilane
- Pd/C
Palladium on carbon
Footnotes
The University of Michigan has filed a number of patent applications on these MDM2 degraders reported in this study, which have been licensed to Oncopia Therapeutics, LLC. S. Wang, Y Li, J. Yang, A. Aguilar, D. McEachern are co-inventors on one or more of these patents, and receive royalties on these patents from the University of Michigan. S. Wang is a co-founder and a paid consultant of Oncopia Therapeutics, LLC. The University of Michigan and S. Wang own stock in Oncopia.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: Molecular formula string file (CSV) and PDB coordinates of homology model of MI-1061 in complex with MDM2 protein.
Figure S1 showing the evaluation of the effect of cereblon ligand lenalidomide on the activity of MDM2 inhibitor and degraders in MV4;11 cells; Figure S2 showing the qRT-PCR analysis mRNA levels of p53 target genes and TP53 after treatment with the MDM2 inhibitor, MI-1061 and the MDM2 degraders MD-222 and MD-224 in MV4;11 cells; Figure S3 showing the 1H NMR spectrum for MD-224. Figure S4 showing the UPLC-MS results for MD-224.
References
- 1.Wade M; Li YC; Wahl GM MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013, 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown CJ; Lain S; Verma CS; Fersht AR; Lane DP Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009, 9, 862–873. [DOI] [PubMed] [Google Scholar]
- 3.Stiewe T The p53 family in differentiation and tumorigenesis. Nat Rev Cancer 2007, 7, 165–168. [DOI] [PubMed] [Google Scholar]
- 4.Hainaut P; Hollstein M p53 and Human cancer: the first ten thousand mutations. Adv Cancer Res 2000, 77, 81–137. [DOI] [PubMed] [Google Scholar]
- 5.Freedman DA; Wu L; Levine AJ Functions of the MDM2 oncoprotein. Cell Mol Life Sci 1999, 55, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu X; Bayle JH; Olson D; Levine AJ The p53-mdm-2 autoregulatory feedback loop. Gene Dev 1993, 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- 7.Juven-Gershon T; Oren M Mdm2: the ups and downs. Mol Med 1999, 5, 71–83. [PMC free article] [PubMed] [Google Scholar]
- 8.Momand J; Zambetti GP; Olson DC; George D; Levine AJ The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- 9.Ding Q; Zhang Z; Liu JJ; Jiang N; Zhang J; Ross TM; Chu XJ; Bartkovitz D; Podlaski F; Janson C; Tovar C; Filipovic ZM; Higgins B; Glenn K; Packman K; Vassilev LT; Graves B Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 2013, 56, 5979–5983. [DOI] [PubMed] [Google Scholar]
- 10.Holzer P; Masuya K; Furet P; Kallen J; Valat-Stachyra T; Ferretti S; Berghausen J; Bouisset-Leonard M; Buschmann N; Pissot-Soldermann C; Rynn C; Ruetz S; Stutz S; Chene P; Jeay S; Gessier F Discovery of a dihydroisoquinolinone derivative (NVP-CGM097): A highly potent and selective MDM2 inhibitor undergoing Phase 1 clinical trials in p53wt tumors. J Med Chem 2015, 58, 6348–6358. [DOI] [PubMed] [Google Scholar]
- 11.Aguilar A; Lu J; Liu L; Du D; Bernard D; McEachern D; Przybranowski S; Li X; Luo R; Wen B; Sun D; Wang H; Wen J; Wang G; Zhai Y; Guo M; Yang D; Wang S Discovery of 4-((3’R,4’S,5’R)-6”-chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2”-oxodispiro[ cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamido)bicyclo[2.2.2]octane −1-carboxylic Acid (AA-115/APG-115): A potent and orally active murine double minute 2 (MDM2) inhibitor in clinical development. J Med Chem 2017, 60, 2819–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vu B; Wovkulich P; Pizzolato G; Lovey A; Ding Q; Jiang N; Liu JJ; Zhao C; Glenn K; Wen Y; Tovar C; Packman K; Vassilev L; Graves B Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett 2013, 4, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang S; Sun W; Zhao Y; McEachern D; Meaux I; Barriere C; Stuckey JA; Meagher JL; Bai L; Liu L; Hoffman-Luca CG; Lu J; Shangary S; Yu S; Bernard D; Aguilar A; Dos-Santos O; Besret L; Guerif S; Pannier P; Gorge-Bernat D; Debussche L SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res 2014, 74, 5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun D; Li Z; Rew Y; Gribble M; Bartberger MD; Beck HP; Canon J; Chen A; Chen X; Chow D; Deignan J; Duquette J; Eksterowicz J; Fisher B; Fox BM; Fu J; Gonzalez AZ; Gonzalez-Lopez De Turiso F; Houze JB; Huang X; Jiang M; Jin L; Kayser F; Liu JJ; Lo MC; Long AM; Lucas B; McGee LR; McIntosh J; Mihalic J; Oliner JD; Osgood T; Peterson ML; Roveto P; Saiki AY; Shaffer P; Toteva M; Wang Y; Wang YC; Wortman S; Yakowec P; Yan X; Ye Q; Yu D; Yu M; Zhao X; Zhou J; Zhu J; Olson SH; Medina JC Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem 2014, 57, 1454–1472. [DOI] [PubMed] [Google Scholar]
- 15.Vassilev LT; Vu BT; Graves B; Carvajal D; Podlaski F; Filipovic Z; Kong N; Kammlott U; Lukacs C; Klein C; Fotouhi N; Liu EA In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y; Aguilar A; Bernard D; Wang S Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem 2015, 58, 1038–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crews CM; Georg G; Wang S Inducing protein degradation as a therapeutic strategy. J Med Chem 2016, 59, 5129–5130. [DOI] [PubMed] [Google Scholar]
- 19.Neklesa TK; Winkler JD; Crews CM Targeted protein degradation by PROTACs. Pharmacol Therapeut 2017, 174, 138–144. [DOI] [PubMed] [Google Scholar]
- 20.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai L; Zhou B; Yang CY; Ji J; McEachern D; Przybranowski S; Jiang H; Hu J; Xu F; Zhao Y; Liu L; Fernandez-Salas E; Xu J; Dou Y; Wen B; Sun D; Meagher J; Stuckey J; Hayes DF; Li S; Ellis MJ; Wang S Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res 2017, 77, 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou B; Hu J; Xu F; Chen Z; Bai L; Fernandez-Salas E; Lin M; Liu L; Yang CY; Zhao Y; McEachern D; Przybranowski S; Wen B; Sun D; Wang S Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem 2018, 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AM; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neklesa TK; Jin MZ; Crew AP; Rossi AK; Willard RR; Dong HQ; Siu K; Wang J; Gordon DA; Chen X; Ferraro C; Crews CM; Coleman K; Winkler JD ARV-330: Androgen receptor PROTAC degrader for prostate cancer. J Clin Oncol 2016, 34. [Google Scholar]
- 26.Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angewandte Chemie (International ed. in English) 2016, 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demizu Y; Shibata N; Hattori T; Ohoka N; Motoi H; Misawa T; Shoda T; Naito M; Kurihara M Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorganic & Medicinal Chemistry Letters 2016, 26, 4865–4869. [DOI] [PubMed] [Google Scholar]
- 28.Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in vivo protein knockdown by small-molecule PROTACs. Nature Chemical Biology 2015, 11, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mori T; Ito T; Liu S; Ando H; Sakamoto S; Yamaguchi Y; Tokunaga E; Shibata N; Handa H; Hakoshima T Structural basis of thalidomide enantiomer binding to cereblon. Sci Rep 2018, 8, 1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Remillard D; Buckley DL; Paulk J; Brien GL; Sonnett M; Seo HS; Dastjerdi S; Wuhr M; Dhe-Paganon S; Armstrong SA; Bradner JE Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angewandte Chemie (International ed. in English) 2017, 56, 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robb CM; Contreras JI; Kour S; Taylor MA; Abid M; Sonawane YA; Zahid M; Murry DJ; Natarajan A; Rana S Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun 2017, 53, 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischer ES; Bohm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith RE; Harper JW; Jenkins JL; Thoma NH Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zengerle M; Chan KH; Ciulli A Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chemical Biology 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.