

Abstract
Vorinostat is a histone deacetylase (HDAC) inhibitor that inhibits cell proliferation and induces apoptosis in solid tumors and is in clinical trials for the treatment of glioblastoma (GBM). The goal of this study was to assess whether hyperpolarized 13C Magnetic Resonance Spectroscopy (MRS) and Magnetic Resonance Spectroscopic Imaging (MRSI) can detect HDAC inhibition in GBM models. First, we confirmed HDAC inhibition in U87 GBM cells and evaluated real-time dynamic metabolic changes using a bioreactor system with live Vorinostat-treated or control cells. We found a significant 40% decrease in the 13C MRS-detectable ratio of hyperpolarized [1-13C]lactate to hyperpolarized [1-13C]pyruvate, [1-13C]Lac/Pyr, and a 37% decrease in the pseudo-rate constant, kPL, for hyperpolarized [1-13C]lactate production, in Vorinostat-treated cells compared to controls. To understand the underlying mechanism for this finding, we assessed the expression and activity of lactate dehydrogenase (LDH), which catalyzes the pyruvate to lactate conversion, its associated cofactor nicotinamide adenine dinucleotide (NADH), the expression of monocarboxylate transporters (MCTs) MCT1 and MCT4, that shuttle pyruvate and lactate in and out of the cell, and intracellular lactate levels. We found that the most likely explanation for our finding that hyperpolarized lactate is reduced in treated cells, is a 30% reduction in intracellular lactate levels that occurs as a result of increased expression of both MCT1 and MCT4 in Vorinostat-treated cells. In vivo 13C MRSI studies of orthotopic tumors in mice also showed a significant 52% decrease in hyperpolarized [1-13C]Lac/Pyr when comparing Vorinostat-treated U87 GBM tumors to controls, and, as in the cell studies, this metabolic finding was associated with increased MCT1 and MCT4 expression in HDAC-inhibited tumors. Thus, the 13C MRSI-detectable decrease in hyperpolarized [1-13C]lactate production could serve as a biomarker of response to HDAC inhibitors.
Keywords: Glioblastoma, HDAC inhibitor, hyperpolarized 13C MRSI
1. INTRODUCTION
Glioblastoma (GBM) is one of the most aggressive and lethal types of brain tumor. In spite of maximum safe surgical resection, radiotherapy, and Temozolomide (TMZ) treatment the median survival of GBM patients is as low as 15 months and the five-year survival rate is only 5.5% (1–3). Thus, novel drugs that improve the overall outcome of GBM patients are urgently needed, and different therapeutic targets are being considered.
Histone deacetylases (HDACs) and histone acetyltransferases (HATs) play key roles in the epigenetic regulation of gene expression via histone deacetylation and acetylation, respectively, and deregulated expression/activity of HDACs and HATs are observed in several cancers (4). Furthermore, preclinical studies have shown that HDAC inhibitors (HDACis) specifically target transformed cells. HDACs are therefore being considered as a potential therapeutic target in a wide variety of cancers including GBM (5,6).
The human HDAC family is comprised of 18 enzymes that are divided into four classes (classes I, II and IV are Zn2+-dependent and class III is Zn2+-independent). Vorinostat or suberoylanilide hydroxamic acid (SAHA), is a small molecule inhibitor of most class I and II HDACs. Pre-clinical studies of Vorinostat have shown its potential in glioma-bearing rodent models. Mice treated with Vorinostat either intravenously (i.v.) or intraperitoneally (i.p.) showed an increase in histone acetylation (acetyl-H3 and acetyl-H4), while proliferation of GL26 murine glioma growth was significantly inhibited (7). Similarly, a study in rats bearing 9L rat glioma led to a significant reduction of tumor size (8) and a single intratumoral injection of Vorinostat doubled the survival of rats bearing GL26 tumors (9). Based on these and other studies, Vorinostat is currently in a phase I/II clinical trial in combination with TMZ and radiation therapy in newly diagnosed GBM patients (NCT00731731) (10).
A standard imaging tool for monitoring of tumor response to drugs in both routine clinical practice and in clinical trials, is magnetic resonance imaging (MRI). However, traditional anatomical MRI using the Response Assessment in Neuro-Oncology (RANO) criteria, can sometimes be hard to interpret especially following treatment when pseudo-progression is frequently observed. To address this challenge, and reduce the variability associated with response assessment, a new standardized Brain Tumor Imaging Protocol (BTIP) was proposed for clinical trials, and recommended for use in routine clinical practice (11,12).
To further support and enhance response monitoring of brain tumors via MR-based approaches, additional magnetic resonance spectroscopy (MRS) metabolic imaging can be used. Proton MRS (1H MRS) is an established technique in clinical practice, and has been shown to enhance noninvasive tumor characterization and grading based on characteristic spectral signatures that distinguish between normal brain and brain tumors (13–15). In particular, total choline-containing metabolites (tCho – comprised of choline, phosphocholine (PC) and glycerophosphocholine (GPC)) are elevated in tumors when compared to normal brain, due primarily to increased PC levels reflecting elevated membrane synthesis in rapidly dividing tumor cells. Conversely, N-acetyl aspartate (NAA), a neuronal marker, is reduced in tumors when compared to normal brain reflecting loss of neuronal tissue within the tumor. Increased levels of lactate are also characteristic of GBM reflecting the aerobic glycolysis that is typical of cancer cells. However, 1H MRS spectra only inform on steady-state metabolite levels and cannot be used to monitor real-time metabolic fluxes.
This limitation can now be addressed by using dissolution Dynamic Nuclear Polarization (DNP) combined with 13C Magnetic Resonance Spectroscopic Imaging (MRSI). This recent technological development improves the signal-to-noise ratio of 13C-containing molecules by more than 10,000-fold allowing monitoring of metabolic fluxes in real time not only in vitro but also non-invasively in vivo. Importantly, metabolic imaging using hyperpolarized 13C MRSI has been shown to provide an early metabolic biomarker of tumor response to treatment in a range of tumor types (16,17). In the case of GBM, our group and others have shown the value of probing the metabolism of pyruvate to lactate for detection of the efficacy of TMZ, Everolimus (an mTOR inhibitor) and voxtalisib (a dual PI3K/mTOR inhibitor), in cells and in vivo in orthotopic rodent models. Following a single dose of TMZ administration to a tumor-bearing rat demonstrated a significant drop in hyperpolarized [1-13C]lactate production from hyperpolarized [1-13C]pyruvate prior to changes in tumor volume (18). This observation was associated with a PKM2-mediated drop in intracellular lactate levels (19). Treatment with Everolimus also showed a reduction in hyperpolarized [1-13C]lactate production prior to a visible reduction in tumor size (20), but in this case the decrease in hyperpolarized [1-13C]lactate was associated with a decrease in expression of lactate dehydrogenase-A (LDH-A), the enzyme that catalyzes the pyruvate to lactate conversion. The decrease in hyperpolarized [1-13C]lactate production was also associated with a decrease in LDH-A expression in the case of voxalisib treatment (21). Moreover, that study showed that the metabolic changes were predictive of increased animal survival even when tumor growth was comparable between control and treated tumors (20–23).
Importantly, the potential value of hyperpolarized [1-13C]pyruvate was successfully demonstrated in the first-in-human clinical trial on prostate cancer patients that exhibited elevated levels of hyperpolarized [1-13C]lactate in tumor regions, and high sensitivity in detecting tumors that were not detectable by standard anatomic MRI or using 1H MRS (24). Hyperpolarized [1-13C]pyruvate has now also been applied to GBM patients (25) and continues to be assessed in clinical trials in brain tumors (NCT03324360). Clinical trials are also ongoing in metastatic prostate (NCT02844647) and cervical (NCT03129776) tumors assessing the presence of tumor. Moreover, clinical trials of response to therapy are currently in phase I/II in breast (NCT03121989) and prostate (NCT02913131) tumors.
However, to date, the value of hyperpolarized 13C MRSI for monitoring response to HDAC inhibition has not been investigated. In this study we therefore used 13C MRSI and hyperpolarized [1-13C]pyruvate to monitor GBM response to HDAC inhibition by Vorinostat. We first assessed the effect of Vorinostat in live cells and then investigated orthotopic GBM xenografts in mice. We found that in both cell and animal studies inhibition of HDAC with Vorinostat led to a decrease in hyperpolarized [1-13C]lactate to hyperpolarized [1-13C]pyruvate ratio, [1-13C]Lac/Pyr. Our study highlights the potential of using hyperpolarized 13C MRSI for monitoring HDAC inhibition in GBM patients.
2. MATERIALS AND METHODS
2.1. Cell culture and drug treatment
U87 human GBM cells were supplied by the University of California San Francisco (UCSF) Brain Tumor Research Center Preclinical Therapeutics Core (20,22). Cells were routinely fingerprinted by Cell Line Genetics using single nucleotide polymorphism within 6 months of any study. Cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) supplemented with 10% FBS, 2mM L-glutamine, 100units/mL penicillin, 100μg/mL streptomycin and maintained in 5% CO2 at 37°C.
Vorinostat (SAHA, MK0683) was purchased from Selleckchem. For all in vitro studies Vorinostat was prepared by dissolution in DMSO for a stock concentration of 10mM that was stored at −20°C. Cells were then treated for 48h either with Vorinostat diluted to a final concentration of 10μM (treated cells) or 1:1000 DMSO (controls) in culture medium. The effect of drug on cell number was assessed by seeding ~2×105 cells per flask, allowing the cells to adhere overnight, initiating treatment for 48h with either Vorinostat or DMSO, and then counting the number of cells per flask.
The effect of drug on acetylation (n=2 for both control and Vorinostat-treated) was assessed by immune-precipitation following the previously described method (26) using antibodies specific for the acetyl-H3-Lys9, acetyl-H4-Lys5, histone-H3, and histone-H4 (1:1000) (Cell Signaling Technology).
2.2. MR-compatible bioreactor using hyperpolarized 13C MRS
An MR-compatible live cell bioreactor was used to probe the real-time pyruvate to lactate flux as described previously (23,27). For each bioreactor ~25×106 cells were seeded on ProNectinF microcarrier beads. Cells were allowed to adhere to the beads overnight and then treatment was initiated for 48h. Cell-covered beads were then transferred into a 10mm Nuclear Magnetic Resonance (NMR) tube, assembled with a perfusion system and perfused using DMEM medium with Vorinostat or DMSO at 37°C with a 95% air/5% CO2 overpressure. MRS studies were performed on a 500MHz INOVA (Agilent) NMR spectrometer using a 10mm broadband probe. Cell viability was first confirmed by acquiring proton-decoupled 31P spectra with the following parameters: 30° pulse, 3s relaxation delay, 1024 scans. For dynamic hyperpolarized 13C MRS experiments, 6μl of [1-13C]pyruvate (Sigma Aldrich, USA), containing 15mM of the OX063 trityl radical (Oxford Instruments, UK) and 0.5mM Dotarem (Gd-DOTA, Guerbert, France) were hyperpolarized using HyperSense DNP polarizer (Oxford Instruments, UK) for 1h. Then, hyperpolarized [1-13C]pyruvate was rapidly dissolved in 6mL of isotonic buffer (40mM TRIS HCl, 3μM ethylenediaminetetraacetic acid (EDTA), pH7.8) and injected into the perfusion system within 15s to a final concentration of 5mM hyperpolarized [1-13C]pyruvate. During injection the perfusion system was stopped and 13C data acquisition performed using the following parameters: 5° pulse every 3s over a period of 300s, 40k data points, 20kHz spectral width. The spectra were analyzed using Mnova (Mestrelab Research) (28).
Three approaches were then used to determine changes in hyperpolarized [1-13C]lactate production in response to HDAC inhibition (29,30). First, the maximum intensity of hyperpolarized [1-13C]lactate, (Lacmax), was normalized to maximum intensity of hyperpolarized [1-13C]pyruvate, (Pyrmax), and to cell number, and the average [1-13C]Lacmax/Pyrmax compared in Vorinostat and control samples. Second, the average ratio of the area under the curve (AUC) of hyperpolarized [1-13C]lactate, (LacAUC), to hyperpolarized [1-13C]pyruvate, (PyrAUC), normalized to cell number [1-13C]LacAUC/PyrAUC was compared. Third, the pseudo-rate constant of hyperpolarized [1-13C]lactate production, kPL, was determined as previously described using modified Bloch equations (23) and average values compared in control and Vorinostat-treated samples.
2.3. Mechanistic studies
To assess the reasons for reduced hyperpolarized [1-13C]lactate production in treated cells, studies assessing the expression of LDHs and MCTs, NADH levels and intracellular lactate levels were performed as follows. 1×107 cells per sample were lysed by sonication in cell lysis buffer (Cell signaling). Lysates were loaded based on total cellular protein (~20μg) and run as previously described (29). Immunoblotting was performed using primary antibodies against: MCT1, MCT4, LDH-A, LDH-B (Santa Cruz Biochemistry) and β-actin (Cell Signaling). Blots were scanned; bands quantified using ImageJ software (NIH) and normalized to the loading control (β-actin).
Activities of LDH-A and B were determined using a spectrophotometric assay that monitors the rate of either NADH consumption during LDH-A conversion of pyruvate-to-lactate (LDH-A activity) or NADH production during LDH-B conversion of lactate-to-pyruvate (LDH-B activity) in cell lysates at 340nm as previously described (23). The Lineweaver-Burke plot was used to determine Km and Vmax values for each enzyme.
Concentrations of NADH and the NAD+/NADH ratio were quantified from cell extracts (3×105 cells per sample) using a commercial spectrophotometric kit (Biovision) and monitoring absorbance at 450nm.
To determine extracellular lactate and glucose levels, media samples were taken from control and Vorinostat-treated cells, centrifuged at 13,000rpm and 10% deuterium oxide added to each sample prior to 1H MRS performed as described below. To assess intracellular lactate as well as other intracellular metabolites, cells (~1×107 cells per sample) were extracted using the dual-phase extraction method, as previously described (31). The lyophilized aqueous phase was resuspended in 600μL of deuterium oxide (Cambridge Isotope Laboratories, Andover, MA, USA) and transferred into 5mm NMR tubes for MR studies. 1H MRS experiments were performed on a Bruker 500MHz spectrometer at 297K as described elsewhere (32). Spectra were acquired using the following parameters: 90° pulse, 3s relaxation delay, 8 dummy scans, 16k data points, 256 acquisitions. Proton water pre-saturation was achieved using the ‘zgpr’ sequence. 10mM trimethylsilyl propionate (TSP) dissolved in 600μL of deuterium oxide was used as an external concentration reference and the concentration of metabolites within a sample was determine using PULCON (pulse length-based concentration determination) as described previously (32,33). Metabolites were then integrated using Topspin au programs, ‘quant_calibrate’ and ‘quant’. All detectable metabolites (threonine (Thr), lactate (Lac), alanine (Ala), acetate (Ace), UDP-N-Acetylglucosamine (UDP-GlcNAc), glutamate (Glu), glutamine (Gln), glutathione (GSH), aspartate (Asp), creatine (Cr), phosphocreatine (PCr), Cho, PC, GPC, glycine (Gly) and myo-inositol (m-Ins)), were then quantified using Mnova (MestreLab Research), then peak integrals were corrected for saturation and normalized to cell number (28).
2.4. Animal model and drug treatment
All studies were performed under UCSF Institutional Animal Care and Use Committee (IACUC) approval. 6-weeks-old female athymic nude (nu/nu) mice (22–25g) were intracranially injected with 3×105 U87 cells. Tumor size was evaluated starting 7 days after intracranial injection using MRI as described below. Once tumors reached 2–3mm in diameter, a baseline set of anatomical MRI and hyperpolarized 13C MRSI studies was performed (see below) and this time point was considered day zero (D0). Mice were then randomized into control and Vorinostat-treated groups.
For in vivo studies Vorinostat (MW=264.32g/mol) was prepared as a suspension in ORA-plus (Perrigo) by sonication for 30min at room temperature (RT). Mice were treated once daily per os (p.o.) with either Vorinostat (0.189mmol/kg, 100μL, treatment group) or Ora-plus (100μL, control group). All doses for in vivo studies were prepared freshly before administration.
MR studies were then repeated on day 7±2 (D7±2) following treatment initiation. No subsequent MR studies were performed, because control animals reached end-point starting on day 6. All animals were treated and survival monitored until animals needed to be sacrificed per IACUC guidelines.
2.5. In vivo MRI and hyperpolarized 13C MRSI studies
Prior to and during all MR studies animals were anesthetized with a mix of isoflurane in O2 (1.5–2%, 1mL/min). All MR studies were performed on a 14.1T (1000mT/cm gradients) vertical MR system (Agilent Technologies) equipped with a millipede 1H coil for T2-weighted anatomical imaging, or with a dual-tune 1H-13C coil for hyperpolarized 13C MRSI experiments. Anatomical axial T2-weighted images were used to evaluate tumor size and were recorded using a multi-slice spin-echo sequence with the following parameters: echo time (TE)=20ms, repetition time (TR)=1200ms, field of view (FOV)=25×25mm2, matrix=256×256, slice thickness=1.0mm, number of averages (NA)=2.
For hyperpolarized 13C MRSI studies, ~24μL (~32mg) of [1-13C]pyruvate was prepared as previously described (21) and hyperpolarized for ~1h. Hyperpolarized [1-13C]pyruvate was then rapidly dissolved in 4.5mL of buffer (40mM TRIS, 80mM NaOH, 0.1mg/L Na2EDTA) and 300μL of the final 80mM buffered hyperpolarized [1-13C]pyruvate were then injected intravenously through a tail-vein catheter over 12s. 13C spectra were then acquired using a 2D Chemical Shift Imaging (CSI) pulse sequence at 17s post-injection of hyperpolarized [1-13C]pyruvate. The choice of this time point was based on our previously published dynamic study, wherein the average time point at which hyperpolarized [1-13C]lactate was maximal in both normal brain and in U87 tumor voxels was found to be 17s post injection (21). Data acquisition conditions were: TE=0.41ms, TR=66 ms, flip angle=10°, matrix=256×16×16, spectral width=4223 Hz, FOV=20×20mm2, slice thickness=4 mm.
Data analysis was performed as follows. Tumor size was determined by manually contouring the tumor area in each axial slice and calculating the total volume as a sum of the areas multiplied by slice thickness using in-house MR software (34). Tumor size was quantified for each animal at each time point and presented as a percentage of the value at day zero.
Hyperpolarized 13C MRSI data were processed using SIVIC (35). A tumor voxel was chosen after evaluation of all slices such that it contained ~80% of tumor tissue, avoiding necrotic and inhomogeneous regions. Peak integrals corresponding to hyperpolarized [1-13C]pyruvate and hyperpolarized [1-13C]lactate in the tumor voxel were then used to calculate hyperpolarized [1-13C]Lac/Pyr at each time point and the data was then normalized to a voxel in contralateral brain as previously described (21).
2.6. Immunohistochemistry
At the end of MR studies, tumor-bearing brains were immediately removed, fixed in 10% buffered formalin then dehydrated and embedded in wax (Paraplast Plus, McCornick Scientific) prior to sectioning and staining with antibodies against acetyl-H3-Lys9 and acetyl-H4-Lys5 (Cell Signaling Technology), MCT1 and MCT4 (Santa Cruz Biochemistry).
2.7. Statistical analysis
All cell experiments were repeated 3 times unless otherwise stated. In vivo experiments were performed on two groups of animals: control (n=6) and Vorinostat-treated (n=7). All results are presented as mean ± standard deviation. Two-tailed unpaired Student’s t-test was used to determine the statistical significance of differences, and P-value≤0.05 was considered significant. Kaplan-Meier survival curves with Log-rank test was used to compare animal survival.
3. RESULTS
3.1. Vorinostat affects HDAC acetylation and inhibits cell proliferation
To confirm that treatment with Vorinostat resulted in HDAC inhibition in U87 cells, we performed immuno-precipitation western blotting of acetyl-H3 and acetyl-H4 in control and Vorinostat-treated cells. Figure 1A confirms previous work and illustrates that treatment of U87 cells with Vorinostat resulted in an increase in the levels of both acetyl-H3 and acetyl-H4, while total levels of both histones remained unchanged when compared to controls. We also assessed the effect of treatment on cell number and found a significant 48.5% decrease in cell number from 10.3×105±1.7×105 cells per flask in control cells to 5.3×105±1.1×105 cells per flask (P-value<0.0005) in cells treated with Vorinostat (Figure 1B).
Figure 1.
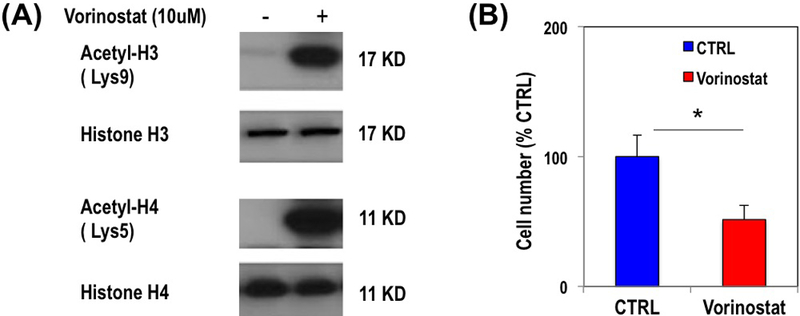
Expression of total and acetylated histones (H3, H4) in control and Vorinostat-treated U87 cells (A). Bar graph illustrating the average change in U87 cell number in response to Vorinostat treatment (B).
3.2. Vorinostat decreases real-time hyperpolarized [1-13C]lactate production in live cells in a bioreactor
To probe the effect of Vorinostat on cellular metabolism we measured real-time lactate production in live U87 cells using hyperpolarized 13C MRS. Figure 2A exhibits hyperpolarized [1-13C]pyruvate conversion to hyperpolarized [1-13C]lactate. Comparison of the temporal evolution of hyperpolarized [1-13C]lactate production in U87 control and Vorinostat-treated cells is illustrated in Figure 2B. Three approaches for quantification of hyperpolarized [1-13C]lactate in Vorinostat-treated and control cells are shown in Figure 2C–E and all showed comparable results. The average ratio of the maximum intensity of hyperpolarized [1-13C]lactate to the maximum intensity of hyperpolarized [1-13C]pyruvate, [1-13C]Lacmax/Pyrmax, showed that production of hyperpolarized [1-13C]lactate was significantly down by 39.8% from 0.012±0.001 in control to 0.007±0.001 (P-value<0.005) in Vorinostat-treated cells (Figure 2C). The average ratio of the area under the curves (AUC) of hyperpolarized [1-13C]lactate to the AUC of hyperpolarized [1-13C]pyruvate, [1-13C]LacAUC/PyrAUC, demonstrated that production of [1-13C]lactate showed a significant 33.2% decrease from 0.021±0.001 in control to 0.014±0.001 (P-value<0.0005) in Vorinostat-treated cells (Figure 2D). Finally, the pseudo-rate constant of hyperpolarized [1-13C]pyruvate-to-lactate conversion, kPL, showed a significant 37.4% decrease from 0.216±0.015s−1/109 cells in controls to 0.135±0.036s−1/109 cells (P-value<0.001) following Vorinostat treatment (Figure 2E). Collectively, our data therefore indicate that Vorinostat treatment is associated with a change in reduced lactate production that is detectable in real-time using hyperpolarized 13C MRS.
Figure 2.
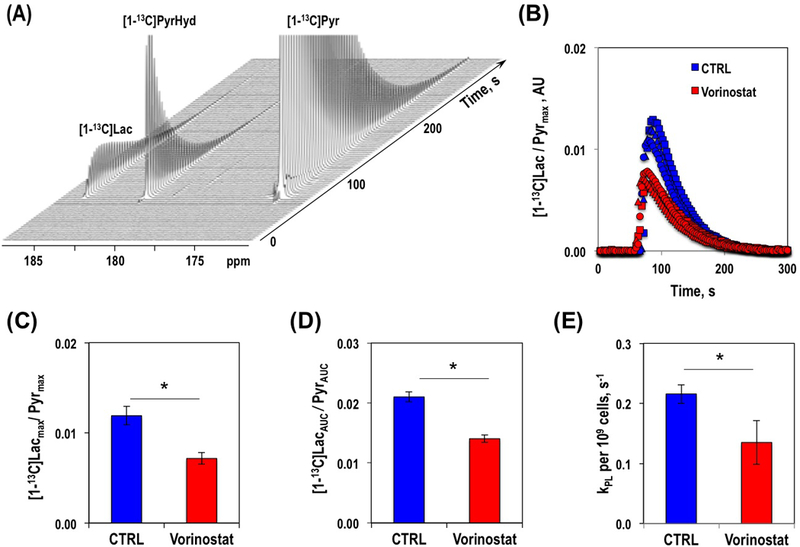
Hyperpolarized 13C MR spectra over time illustrating hyperpolarized [1-13C]pyruvate to hyperpolarized [1-13C]lactate conversion in U87 cells (A). Temporal evolution of hyperpolarized [1-13C]lactate levels in U87 control and Vorinostat-treated cells (B). Quantification of hyperpolarized [1-13C]lactate production, as described in Methods section, [1-13C]Lacmax/Pyrmax, (C) [1-13C]LacAUC/PyrAUC (D) and kPL (E) in U87 control and Vorinostat-treated cells.
3.3. Vorinostat treatment increases MCT1 and MCT4 expression and decreases the steady-state intracellular lactate pool in U87 cells
In order to explain the observed changes in hyperpolarized [1-13C]lactate production in U87 GBM cells we further analyzed the factors that might affect it. Previous studies have shown that the level of hyperpolarized [1-13C]lactate produced from injected hyperpolarized [1-13C]pyruvate depends on the following factors: 1) expression and activity of LDH; 2) levels of the cofactors NAD+ and NADH; 3) expression of MCT1 and MCT4, transporters that shuttle pyruvate and lactate in and out of the cell; 4) level of the intracellular lactate pool (36,37). We therefore analyzed these different factors.
We observed that Vorinostat treatment did not significantly affect the expression of LDH-A (1.51±0.26(AU) for control and 1.61±0.26(AU) for Vorinostat-treated cells, P-value=0.59) in U87 cells when compared to controls (Figure 3A). The expression of LDH-B, the enzyme that catalyzes the conversion of lactate-to-pyruvate was also unaffected by Vorinostat treatment (2.40±0.47(AU) for control and 2.45±0.18(AU) for Vorinostat-treated cells, P-value=0.86; Figure 3A). To further rule out the role of LDH in reduced hyperpolarized lactate production, we also compared the activities of LDH-A and LDH-B. The Km values for both enzymes were unchanged by treatment (for LDH-A 0.9±0.4μmolsPyr/106cells in control cells and 1.0±0.7μmolsPyr/106cells in treated cells, P-value=0.83; for LDH-B 1.4±0.6μmolsPyr/106 cells for control and 1.1±0.5μmolsPyr/106cells for treated, P-value=0.61), and although the Vmax for both enzymes increased (from 29.8±7.9 to 85.9±36.6μmolNADH/sec/106cells, P-value=0.05 for LDH-A and from 6.2±1.1 to 12.2±3.3μmolNADH/sec/106cells, P-value=0.03 for LDH-B), when considering the ratio of Vmax values this was not significantly different between the control and Vorinostat-treated cells (5.0±2.1 for controls to 7.9±4.9 for Vorinostat-treated cells, P-value=0.34; Figure 3B). We also did not detect significant differences in the levels of NADH (0.012±0.004fmol/106cells for control and 0.010±0.003fmol/106cells for Vorinostat-treated cells, n=4, P-value=0.42) or NAD+/NADH (12.7±2.1 for control and 9.5±1.9 for Vorinostat-treated cells, n=4, P-value=0.07), in Vorinostat-treated cells when compared to controls (Figure 3C).
Figure 3.
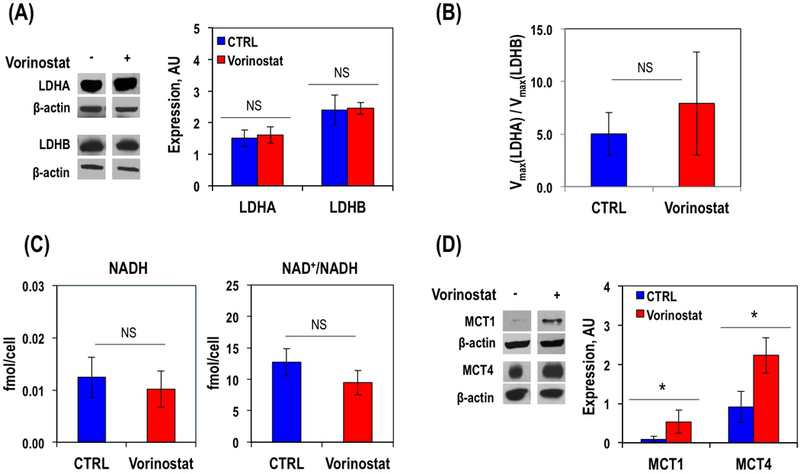
Representative western blot and its quantification for LDH enzymes expression in U87 GBM cells (A). LDH activity quantification presented as ratio of Vmax(LDH-A) to Vmax(LDH-B) (B). Spectrophotometric quantification of NADH levels and NAD+/NADH ratios in U87 GBM cell (C). Representative western blot and its quantification for expression of MCT1 and MCT4 transporters in U87 cells (D).
In contrast, when we assessed expression of the MCTs (Figure 3D) we found a significant increase in both transporters following Vorinostat treatment: MCT1 by 338.2% (0.120±0.099(AU) for control and 0.523±0.263(AU) in Vorinostat-treated cells) and MCT4 by 143.7% (0.916±0.397(AU) for control and 2.231±0.451(AU) in Vorinostat-treated cells), (P-value<0.05 for both). We then determined the levels of intracellular lactate using 1H MRS of cell extracts (Figure 4), and found that it dropped significantly by 29.6% from 1.56±0.09fmol/cell in control cells to 1.09±0.06fmol/cell in Vorinostat-treated cells (P-value<0.005), consistent with the elevated expression of MCTs that would export lactate from the cells. Also consistent with the elevated expression of MCTs, analysis of media samples exhibited a significant 54.6% increase in extracellular lactate concentration from 623.8±209.6fmol/cell in control to 997.3±110.5fmol/cell in Vorinostat-treated cells (P-value<0.03). Surprisingly, analysis of the media samples also showed a significant increase in glucose uptake in the treated cells compared to controls increasing from 405.1±37.8fmol/cell in control to 606±111.3fmol/cell. Other intracellular metabolites detectable in the 1H MR spectra were also altered following treatment (Table 1), but except for lactate, none of these metabolites would serve to explain the change in hyperpolarized lactate production observed in our studies.
Figure 4.
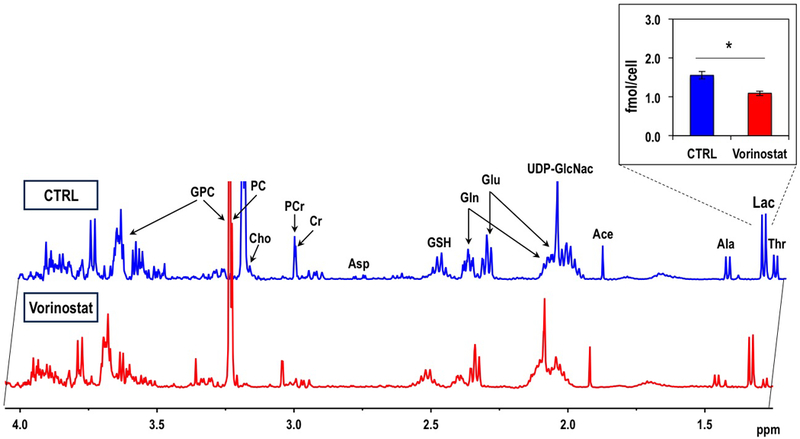
Typical 1H MRS intracellular steady-state metabolite spectra of the extracts of control and Vorinostat-treated U87 cells. Quantified intracellular lactate concentrations are shown in the insert.
Table 1.
Intracellular metabolites in control and Vorinostat-treated U87 cells.
| CTRL | Vorinostat | P-value | |
|---|---|---|---|
| Thr | 0.65±0.08 | 0.24±0.06 | 0.003 |
| Lac | 1.56±0.09 | 1.09±0.06 | 0.004 |
| Ala | 0.58±0.01 | 0.35±0.02 | 0.001 |
| Ace | 0.45± 0.10 | 0.44± 0.05 | 0.862 |
| UDP-GlcNac | 14.67± 1.35 | 9.33± 0.56 | 0.011 |
| Glu | 3.42±0.66 | 3.03±0.19 | 0.42 |
| Gln | 2.72±0.46 | 1.50±0.08 | 0.04 |
| GSH | 3.21±0.49 | 2.81±0.63 | 0.439 |
| Asp | 0.20±0.03 | 0.09±0.01 | 0.011 |
| Cr | 0.36±0.03 | 0.23±0.02 | 0.003 |
| PCr | 0.52±0.08 | 0.30±0.01 | 0.033 |
| Cho | 0.05±0.01 | 0.08±0.02 | 0.104 |
| PC | 1.13±0.10 | 0.79±0.09 | 0.012 |
| GPC | 3.23±0.44 | 2.84±0.18 | 0.257 |
| Gly | 2.13±0.75 | 1.65±0.31 | 0.385 |
| m-Ins | 2.58±0.21 | 2.09±0.37 | 0.132 |
3.4. Vorinostat decreases hyperpolarized [1-13C]Lac/Pyr and tumor growth, while increasing MCT expression and animal survival in vivo
To further investigate the effect of Vorinostat we followed with hyperpolarized 13C MRSI studies on orthotopic U87 xenografts. Tumor growth was monitored starting 7 days post intracranial injection using anatomical T2-weighted MRI. All animals developed MR detectable tumors by 7±2 days post-implantation and were allowed to grow until they reached a size considered sufficient for hyperpolarized 13C MRSI studies (2×2×4mm3) at 14±3 days post-implantation. This time point was considered D0 and treatment was initiated at this time.
Anatomical axial T2-weighted image of a representative control mouse and corresponding hyperpolarized 13C MRSI spectrum from the tumor voxel acquired at D0 is illustrated in Figure 5A. The individual and average hyperpolarized [1-13C]Lac/Pyr for control and Vorinostat-treated tumors at D7±2 are shown in Figures 5B and 5C respectively. Similar to the findings in cells, we detected that the hyperpolarized [1-13C]Lac/Pyr measured on D7±2 was significantly lower by 52.2% (P-value=0.006) in Vorinostat-treated U87 tumors (1.50±0.44) when compared to control tumors (3.15±0.94). Consistent with this finding, Figure 5D shows the heatmaps of hyperpolarized [1-13C]Lac/Pyr at D0 and D7±2 illustrating the differences between control and Vorinostat-treated groups.
Figure 5.
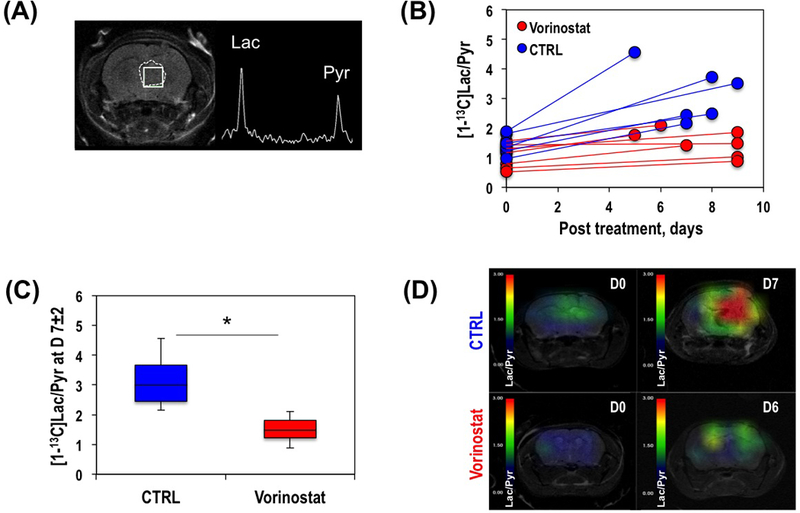
Anatomical axial T2-weighted image of a representative mouse showing tumor area (dashed line) and tumor voxel (solid line), and corresponding hyperpolarized 13C MRSI spectrum from the tumor voxel (A). Individual hyperpolarized [1-13C]Lac/Pyr at D7±2 in control and Vorinostat-treated tumors (B). Average hyperpolarized [1-13C]Lac/Pyr at D7±2 (C). Hyperpolarized [1-13C]Lac/Pyr heat map at D0 (top) and D7±2 (bottom) of control (left) and Vorinostat-treated (right) mice bearing U87 tumor (D).
We also analyzed the effect of Vorinostat on tumor growth and survival probability. Figure 6A illustrates anatomical T2-weighted images of control and Vorinostat-treated U87 tumor-bearing mice. Figure 6B exhibits individual tumor sizes over time for control and Vorinostat-treated tumors. At D0, prior to a random assignment of animals into two treatment groups, the average tumor size was 0.03±0.01cm3. Figure 6C illustrates the average change in tumor size and demonstrates that a significant 55.6% (P-value<0.002) inhibition of tumor growth was observed by D7±2 in Vorinostat-treated animals when compared to the growth of control tumors. Furthermore, comparing the survival probabilities of control and treated animals (Figure 6D), our results indicate that treatment with Vorinostat led to a significantly increased animal survival (Χ2-value=23.4, P-value<0.00001).
Figure 6.
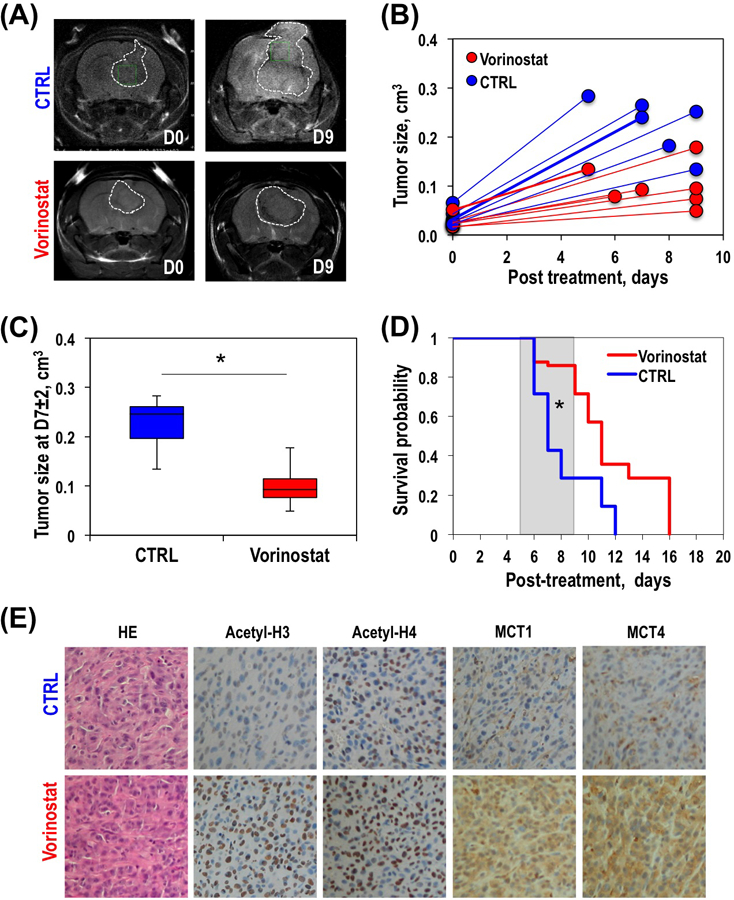
Anatomical T2-weighted images of a representative control (top panel) and Vorinostat-treated (bottom panel) U87 tumor-bearing mice at D0 (left) and D7±2 (right) (A). Temporal evolution of individual (B) and average at D7±2 (C) tumor size of control and Vorinostat-treated U87 tumors. Kaplan-Meier survival plot of U87-bearing control and Vorinostat-treated mice (D). Immunohistochemical analysis of control and Vorinostat-treated U87 tumors. Expression of acetyl-H3, acetyl-H4, MCT1 and MCT4 for each treatment group at the end of the study (E). All histological images are the same magnification (20x).
In order to confirm that our observations were associated with treatment, and that Vorinostat led to HDAC inhibition in vivo, we performed immunohistochemical staining to determine levels of acetyl-H3 and acetyl-H4 in control and Vorinostat-treated tumor samples. Figure 6E illustrates increased levels of both acetyl-H3 and acetyl-H4 in treated tumors when compared to controls confirming drug action in vivo. We also confirmed that our mechanistic findings in cells were relevant to the in vivo setting by assessing the expression of MCT1 and MCT4. As expected, Vorinostat treatment resulted in a higher expression of both transporters (Figure 6E). We also wanted to assess whether our metabolic findings were correlated with tumor size. We found no significant relationship between hyperpolarized [1-13C]Lac/Pyr and tumor size on D0 (r=0.11, P-value=0.712) consistent with the random assignment of our animals into their treatment group. On D7±2 we found a trend to a slight correlation, consistent with the simultaneous effect of treatment on both metabolism and size, but this did not reach statistical significance (r=0.53, P-value=0.063) pointing to the added value of the metabolic information.
DISCUSSION
The development and in vivo implementation of dissolution DNP combined with 13C MRSI has opened the door to noninvasive monitoring of real-time cellular metabolism. Most notably, monitoring the conversion of hyperpolarized [1-13C]pyruvate to hyperpolarized [1-13C]lactate has recently been implemented in the clinic (24,25,38). Importantly, previous studies have shown that this metabolic reaction can serve as an early biomarker of response to therapy in a range of tumor types including in orthotopic intracranial GBM models (17,18,20,21,38,39). However, the effect of HDAC inhibitors had not been previously investigated using this emerging imaging method. In this study we therefore investigated HDAC inhibition by Vorinostat in GBM models performing studies first in live cells and then in vivo in orthotopic GBM tumors in mice. In both cells and tumors we found that response to therapy, as assessed by histone acetylation that was accompanied by a drop in cell proliferation and inhibition of tumor growth and enhanced animal survival, are associated with a drop in hyperpolarized [1-13C]lactate production.
Our findings with regard to the efficacy of Vorinostat are in agreement with previous studies performed in human, mouse and rat glioma cells as well as in vivo in a syngeneic rat glioma model and in a human U87 orthotopic mouse model (4,7–9). These earlier studies showed that for all species Vorinostat inhibited the growth of cells in culture and that Vorinostat treatment of glioma-bearing rodents was highly effective in enhancing animal survival (7,8). More importantly, a phase II study of Vorinostat monotherapy was well tolerated in patients with recurrent GBM and had modest single-agent activity. Accordingly, further clinical studies of the HDAC inhibitor, either alone or in combination with other chemotherapy or/and radiotherapy, are underway (40–43).
The observation that GBM response to Vorinostat treatment is associated with a significant drop in hyperpolarized [1-13C]lactate production is similar to the findings in a range of tumor types in which response to both chemotherapy and targeted therapies led to a drop in [1-13C]lactate production (17,44). Since our in vivo studies use a CSI sequence, partial volume effects could be a concern. However, it should be noted that our maximum gradient strength is 1000mT/cm. The gradient strength for 4mm slice thickness and 10kHz Shinnar Le Roux excitation pulse is therefore 2.336mT/cm, resulting in no more than a 20% mismatch in the slice selection for lactate and pyruvate signals.
Previous work has shown that the drop in hyperpolarized lactate can be due, as mentioned above, to a range of different factors including LDH expression, NADH levels, expression of MCTs and intracellular lactate levels (20,21,45). In this study, the most likely explanation for the drop in hyperpolarized lactate is the significant decrease in intracellular lactate levels observed in our treated cells. Our finding that intracellular lactate is reduced following HDAC inhibition in vitro is consistent with a previous study where reduced lactate levels were detected by 1H MRS in response to short-term treatment with Vorinostat of orthotopic 9L rat glioma in rat model (8).
When considering the reasons for the drop in intracellular lactate, others have reported a drop in glucose uptake and lactate production in HDAC-inhibited hepatocellular carcinoma (46). However, in our cells glucose uptake was, surprisingly, increased likely ruling out a drop in glycolytic rate as an explanation for the reduced intracellular lactate levels observed in our treated cells. A more plausible explanation for the decreased intracellular lactate level is the increased expression of MCT1 and MCT4 transporters observed following treatment in both cells and tumors. Lactate homeostasis is balanced not only by its synthesis, but also by efflux through the cell membrane via the MCTs. MCTs have different affinities for lactate and pyruvate. MCT1 is mostly associated with pyruvate uptake into the cell, while MCT4 is mostly associated with lactate export out of the cell, but both transporters can contribute to lactate export and their increase can therefore explain our observed increase in extracellular lactate levels and decrease in intracellular lactate.
The finding that extracellular lactate increases with HDAC inhibition and inhibition of tumor growth is somewhat surprising, as typically elevated lactate production is associated with tumor aggressiveness and has been considered a therapeutic target (47). Interestingly however, Latham and co-authors have found that extracellular lactate itself can serve to inhibit HDACs. Thus lactate export might serve to reinforce the effect of HDAC inhibitor treatment (48).
The decrease in intracellular lactate would explain the reduced hyperpolarized [1-13C]lactate production as demonstrated by others who have shown that the size of lactate pool impacts hyperpolarized lactate levels (36,37). This finding is also in line with a previous report indicating that increased MCT4-mediated lactate efflux was associated with lower production of hyperpolarized [1-13C]lactate (49).
With regard to the mechanistic link between HDAC acetylation and the expression of MCTs, previous studies in different tumor types have established that HDAC inhibition results in increased expression of multiple genes (7,48) and Lai and co-authors (50) have shown that MCT1 expression is modulated by its promoter acetylation. Nonetheless, further studies are needed to establish this connection in our system.
Previous work with other types of therapy has shown that the metabolic changes in pyruvate conversion to lactate occurred early, preceded any changes in tumor size, and were prognostic of animal survival (20,21). Here, the effect of Vorinostat on tumor size was so rapid that the metabolic changes were detected simultaneously with the changes in tumor size. Whether this is also the case in the clinical setting, or whether the metabolic changes in the clinic might proceed tumor shrinkage remains to be seen. Nonetheless, our studies highlight the value of the hyperpolarized 13C MRSI approach as a complementary noninvasive metabolic imaging method for assessing whether a tumor is responsive to HDAC inhibition. As such, this method could help improve the treatment of GBM patients and their outcome.
Acknowledgments
The authors acknowledge M. VanCriekinge and S. Subramaniam for their technical support. The authors also acknowledge support from the hyperpolarized MRI Technology Resource Center (P41EB013598 D. B. Vigneron).
Funding: National Institute of Health P01 CA118816 (SMR and ROP)
Abbreviation used:
- Ace
acetate
- Ala
alanine
- Asp
aspartate
- Cho
choline
- Cr
creatine
- CTRL
control
- DMEM
Dullbecco’s modifird Eagle’s medium
- DMSO
dimethylsulfoxide
- DNP
Dynamic Nuclear Polarization
- EDTA
ethylenediaminetetraacetic acid
- FOV
field of view
- GBM
glioblastoma
- Gly
glycine
- GPC
glycerophosphocholine
- HAT
Histone acetyltransferase
- HDAC
Histone Deacetylase
- kPL
pseudo-rate constant of pyruvate to lactate conversion
- Lac
Lactate
- LDH
lactate dehydrogenase
- MCT
monocarboxylate transporter
- m-Ins
myo-inositol
- MRSI
magnetic resonance spectroscopic imaging
- NAA
N-acetylaspartate
- NAD+/NADH
nicotinamide adenine dinucleotide
- NS
no statistically significant difference
- PC
phosphocholine
- PCr
phosphocreatine
- Pyr
pyruvate
- SAHA
suberoylanilide hydroxamic acid
- TE
echo time
- TMZ
Temozolomide
- TR
repetition time
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
REFERENCES
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–996. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoornd MJB, Janzer RC. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology 2009;10:459–466. [DOI] [PubMed] [Google Scholar]
- 3.Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol 2016;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015;20(3):3898–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bezecny P Histone deacetylase inhibitors in glioblastoma: pre-clinical and ciinical experience. Med Oncol 2014;2014(31):985. [DOI] [PubMed] [Google Scholar]
- 6.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009;27(32):5459–5468. [DOI] [PubMed] [Google Scholar]
- 7.Yin D, Ong JM, Hu J, Desmond JC, Kawamata N, Konda BM, Black KL, Koeffler HP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin Cancer Res 2007;13(3):1045–1052. [DOI] [PubMed] [Google Scholar]
- 8.Wei L, Hong S, Yoon Y, Hwang SN, Park JC, Zhang Z, Olson JJ, Hu XP, Shim H. Early prediction of response to Vorinostat in an orthotopic glioma rat model. NMR Biomed 2012;25(9):1104–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eyüpoglu I, Hahnen E, Buslei R, Siebzehnrübl F, Savaskan N, Lüders M, Tränkle C, Wick W, Weller M, Fahlbusch R, Blümcke I. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J Neurochem 2005;93(4):992–999. [DOI] [PubMed] [Google Scholar]
- 10. Vorinostat, Temozolomide, and Radiation Therapy in Treating Patients With Newly Diagnosed Glioblastoma Multiforme https://clinicaltrials.gov/ct2/show/NCT00731731.
- 11.Eisele SC, Wen PY, Lee EQ. Assessment of Brain Tumor Response: RANO and Its Offspring. Current Treatment Options in Oncology 2016;17:35. [DOI] [PubMed] [Google Scholar]
- 12.Ellingson BM, Bendszus M, Boxerman J, Barboriak D, Erickson BJ, Smits M, Nelson SJ, Gerstner E, Alexander B, Goldmacher G, Wick W, Vogelbaum M, Weller M, Galanis E, Kalpathy-Cramer J, Shankar L, Jacobs P, Pope WB, Yang D, Chung C, Knopp MV, Cha S, van den Bent MJ, Chang S, Al Yung WK WK, Cloughesy TF, Wen PY, Gilbert MR, Committee tJBTDDCISS, Whitney A, Sandak D, Musella A, Haynes C, Wallace M, Arons DF, Kingston A, Sul J, Krainak D. Consensus recommendations for a standardized Brain Tumor Imaging Protocol in clinical trials. Neuro Oncol 2015;17(9):1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majósa C, Julià-Sapéb M, Alonsoa J, Serrallongaa M, Aguileraa C, Acebesc JJ, Arúsb C, Gilia J. Brain Tumor Classification by Proton MR Spectroscopy: Comparison of Diagnostic Accuracy at Short and Long TE. Am J Neuroradiol 2004;25(10):1696–1704. [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Ma L, Wang Q, Zheng X, Wu C, Xu BN. Role of magnetic resonance spectroscopy for the differentiation of recurrent glioma from radiation necrosis: a systematic review and meta-analysis. Eur J Radiol 2014;83(12):2181–2189. [DOI] [PubMed] [Google Scholar]
- 15.Nelson SJ. Multivoxel Magnetic Resonance Spectroscopy of Brain Tumors. Mol Cancer Ther 2003;2:497. [PubMed] [Google Scholar]
- 16.Chaumeil MM, Najac C, Ronen SM. Studies of Metabolism Using (13)C MRS of Hyperpolarized Probes. Methods Enzymol 2015;561:1–71. [DOI] [PubMed] [Google Scholar]
- 17.Najac C, Ronen SM. MR Molecular Imaging of Brain Cancer Metabolism Using Hyperpolarized 13C Magnetic Resonance Spectroscopy. Top Magn Reson Imaging 2016;25(5):187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park I, Bok R, Ozawa T, Phillips JJ, James CD, Vigneron DB, Ronen SM, Nelson SJ. Detection of early response to temozolomide treatment in brain tumors using hyperpolarized 13C MR metabolic imaging. J Magn Reson Imaging 2011;33(6):1284–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park I, Mukherjee J, Ito M, Chaumeil MM, Jalbert LE, Gaensler K, Ronen SM, Nelson SJ, Pieper RO. Changes in Pyruvate Metabolism Detected by Magnetic Resonance Imaging Are Linked to DNA Damage and Serve as a Sensor of Temozolomide Response in Glioblastoma Cells. Cancer Res 2014. [DOI] [PMC free article] [PubMed]
- 20.Chaumeil MM, Ozawa T, Park I, Scott K, James CD, Nelson SJ, Ronen SM. Hyperpolarized 13C MR spectroscopic imaging can be used to monitor Everolimus treatment in vivo in an orthotopic rodent model of glioblastoma. Neuroimage 2012;59(1):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radoul M, Chaumeil MM, Eriksson P, Wang AS, Phillips JJ, Ronen SM. MR Studies of Glioblastoma Models Treated with Dual PI3K/mTOR Inhibitor and Temozolomide:Metabolic Changes Are Associated with Enhanced Survival. Mol Cancer Ther 2016;15(5):1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatesh HS, Chaumeil MM, Ward CS, Haas-Kogan DA, James CD, Ronen SM. Reduced phosphocholine and hyperpolarized lactate provide magnetic resonance biomarkers of PI3K/Akt/mTOR inhibition in glioblastoma. Neuro Oncol 2012;14(3):315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward CS, Venkatesh HS, Chaumeil MM, Brandes AH, Vancriekinge M, Dafni H, Subramaniam S, Nelson SJ, Vigneron DB, Kurhanewicz J, James CD, Haas-Kogan DA, Ronen SM. Noninvasive detection of target modulation following phosphatidylinositol 3-kinase inhibition using hyperpolarized 13C magnetic resonance spectroscopy. Cancer Res 2010;70(4):1296–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PEZ, Harzstark AJ, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, Reed G, Carvajal L, Small EJ, Munster P, Weinberg VK, Ardenkjaer-Larsen JH, Chen AP, Hurd RE, Odegardstuen LI, Robb FJ, Tropp J, Murray J, A. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1–13C]Pyruvate. Science Translational Medicine 2013;5(198):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park I, Larson PEZ, Gordon JW, Carvajal L, Chen HY, Bok R, Van Criekinge M, Ferrone M, Slater JB, Xu D, Kurhanewicz J, Vigneron DB, Chang S, Nelson SJ. Development of methods and feasibility of using hyperpolarized carbon-13 imaging data for evaluating brain metabolism in patient studies. Magn Reson Med 2018; doi: 10.1002/mrm.27077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida M, Kijima M, Akita M, Beppu T. Potent and Specific Inhibition of Mammalian Histone Deacetylase Both in Vivo and in Vitro by Trichostatin A. J Biol Chem 1990;265:17174–17179. [PubMed] [Google Scholar]
- 27.Lodi A, Woods SM, Ronen SM. Treatment with the MEK inhibitor U0126 induces decreased hyperpolarized pyruvate to lactate conversion in breast, but not prostate, cancer cells. NMR Biomed 2013;26(3):299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mnova http://mestrelab.com/.
- 29.Ward CS, Eriksson P, Izquierdo-Garcia JL, Brandes AH, Ronen SM. HDAC inhibition induces increased choline uptake and elevated phosphocholine levels in MCF7 breast cancer cells. PLOS one 2013;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viswanath P, Najac C, Izquierdo-Garcia JL, Pankov A, Hong C, Eriksson P, Costello JF, Pieper RO, Ronen SM. Mutant IDH1 expression is associated with down-regulation of monocarboxylate transporters. Oncotarget 2016;7(23):34942–34955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tyagi RK, Azrad A, Degani H, Salomon Y. Simultaneous extraction of cellular lipids and water-soluble metabolites: evaluation by NMR spectroscopy. Magn Reson Med 1996;35:194–200. [DOI] [PubMed] [Google Scholar]
- 32.Wider G, Dreier L. Measuring protein concentrations by NMR spectroscopy. JACS 2006;128:2571–2576. [DOI] [PubMed] [Google Scholar]
- 33.Goldoni L, Beringhelli T, Rocchia W, Realini N, Piomelli D. A simple and accurate protocol for absolute polar metabolite quantification in cell cultures using quantitative nuclear magnetic resonance. Analytical Biochemistry 2016;501:26–34. [DOI] [PubMed] [Google Scholar]
- 34.Nelson SJ. Analysis of volume MRI and MR spectroscopic imaging data for the evaluation of patients with brain tumors. Magn Reson Med 2001;46(2):228–239. [DOI] [PubMed] [Google Scholar]
- 35.Crane JC, Olson MP, Nelson SJ. SIVIC: Open-Source, Standards-Based Software for DICOM MR Spectroscopy Workflows. Int J Biomed Imaging 2013. [DOI] [PMC free article] [PubMed]
- 36.Brindle KM, Bohndiek SE, Gallagher FA, Kettunen MI. Tumor imaging using hyperpolarized 13C magnetic resonance spectroscopy. Magn Reson Med 2011;66(2):505–519. [DOI] [PubMed] [Google Scholar]
- 37.Witney TH, Kettunen MI, Brindle KM. Kinetic modeling of hyperpolarized 13C label exchange between pyruvate and lactate in tumor cells. J Biol Chem 2011;286(28):24572–24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park I, Larson PEZ, Gordon J, Carvajal L, Chen HY, VanCriekinge M, Bok R, Crane JC, Elkhaled A, Phillips J, Slater JB, Ferrone M, Kurhanewicz J, Vigneron D, Chang S, Nelson SJ. Dynamic Hyperpolarized 13C Metabolic Imaging of Patients with Brain Tumors. ISMRM 2017;0555. [Google Scholar]
- 39.Chaumeil MM, Lupo JM, Ronen SM. Magnetic Resonance (MR) Metabolic Imaging in Glioma. Brain Pathol 2015;25(6):769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.ClinicalTrials.gov Vorinostat in Treating Patients With Progressive or Recurrent Glioblastoma Multiforme NCT00238303.
- 41.ClinicalTrials.gov Vorinostat, Temozolomide, and Radiation Therapy in Treating Patients With Newly Diagnosed Glioblastoma Multiforme NCT00731731.
- 42.ClinicalTrials.gov A Phase I Trial of Vorinostat in Combination With Bevacizumab & Irinotecan in Recurrent Glioblastoma NCT00762255.
- 43.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, Richon VM, Scheithauer B, Giannini C, Flynn PJ, Moore DFJ, Zwiebel J, Buckner JC. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 2009;27(19):3262–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaumeil MM, Najac C, Ronen SM. Studies of Metabolism Using (13)C MRS of Hyperpolarized Probes. Methods Enzymol 2015;561:1–71. [DOI] [PubMed] [Google Scholar]
- 45.Witney TH, Kettunen MI, Brindle KM. Kinetic modeling of hyperpolarized 13C label exchange between pyruvate and lactate in tumor cells. J Biol Chem 2011;286(28):24572–24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Jin X, Yan Y, Shao Y, Pan Y, Roberts LR, Zhang J, Huang H, Jiang J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Scientific Reports 2017;7:43864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gillies RJ, Gatenby RA. Metabolism and Its Sequelae in Cancer Evolution and Therapy. Cancer J 2015;21(2):88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Latham T, Mackay L, Sproul D, Karim M, Jayne Culley, Harrison DJ, Hayward L, Langridge-Smith P, Gilbert N, Ramsahoye BH. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Research 2012;40(11):4794–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keshari KR, Sriram R, Koelsch BL, Van Criekinge M, Wilson DM, Kurhanewicz J, Wang ZJ. Hyperpolarized 13C-pyruvate magnetic resonance reveals rapid lactate export in metastatic renal cell carcinomas. Cancer Res 2013;73(2):529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai Q, Du W, Wu L, Wang X, Li X, Qu X, Wu X, Dong F, Yao R, Fan H. H3K9ac and HDAC2 Activity Are Involved in the Expression of Monocarboxylate Transporter 1 in Oligodendrocyte. Frontiers in Molecular Neuroscience;10:376. [DOI] [PMC free article] [PubMed] [Google Scholar]