

Abstract
Background:
Despite the accessibility of blood, identification of systemic biomarkers associated with cancer progression has been especially challenging. The aim of this study was to determine a difference in baseline serum immune signatures in patients that experienced early pancreatic ductal adenocarcinoma (PDAC) metastasis with patients that did not. We hypothesized that immune mediators would differ in the baseline serum of these patient cohorts. To test this hypothesis, novel approaches of systemic immune analysis were performed.
Methods:
A serum-induced transcriptional assay was used to identify transcriptome signatures. To enable an understanding of the transcriptome data in a global sense, a transcriptome index was calculated for each patient taking into consideration the relationship of up and downregulated transcripts. For each patient, serum cytokine concentrations were also analyzed globally as a cytokine index.
Results:
A transcriptome signature of innate type 1 interferon inflammation was identified in patients that experienced early metastatic progression. Patients without early metastatic progression, had a baseline transcriptome signature of TGFβ/IL10 regulated acute inflammation. The transcriptome index was greater in patients with early metastasis. There was a significant difference in the cytokine index in patients with and without early metastatic progression.
Conclusions:
The association of serum-induced transcriptional signatures with PDAC metastasis is a novel finding. Global assessment of serum cytokine concentrations as a cytokine index is a novel approach to assess systemic cancer immunity.
Impact:
These systemic indices can be assessed in combination with tumor markers to further define subsets of PDAC that will provide insight into effective treatment, progression and outcome.
Keywords: Inflammation, Transcriptome, Cytokine, Pancreatic ductal adenocarcinoma, Metastasis
Introduction
Recent advances in targeted and immune therapy for the treatment of cancer has created an urgency to identify systemic and tumor biomarkers to define carcinoma subsets that correlate with disease progression. An understanding of biomarkers will facilitate the advancement of personalized treatment by identifying which patients will benefit from targeted or immune therapy, monitoring the response to therapy, and predicting disease outcome (1). Once identified, systemic biomarkers will be more universally available for clinical analysis than tumor biomarkers. This study describes the identification of systemic indicators analyzed from serum that associate with pancreatic cancer metastatic progression.
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer with a 5-year survival of only 27%. However, there are some patients that do not experience PDAC progression for years after diagnosis and are long-term survivors (2). Advances in genomic technologies have enabled a molecular understanding of PDAC tumor subsets with different underlying pathology. Through RNA expression profiling, four subtypes of PDAC tumors (squamous, pancreatic progenitor, immunogenic and aberrantly differentiated endocrine/exocrine (ADEX)) have been identified (3–6). Each tumor subset is associated with distinct histopathological characteristics. Interestingly, a squamous subtype and an immunogenic subtype (with a macrophage and T cell co-inhibitory signature) are associated with poor prognosis. Recently, a transcriptome analysis of paired pancreatic tumor and adjacent benign tissues revealed differentially expressed genes belonging to canonical pathways and molecular functions associated with inflammation (7). These data suggest that there are inflammatory related differences in tumor and non-tumor pancreatic tissue.
Despite the accessibility of blood as surrogate biopsy material, identification of systemic biomarkers has been especially challenging. Inflammatory mediators present in serum or plasma have been associated with aggressive disease. For example, tumor necrosis factor (TNF) and IL1β are elevated in advanced breast cancer and with more aggressive carcinomas (8). In aggressive inflammatory breast cancer, inflammatory mediators, IL6 and CXCL8 (IL8), are produced and secreted at high levels (9). Yet, the anti-inflammatory mediator, IL10, has also been elevated and associated with a negative prognosis in multiple cancers (10). Overall, the association of serum cytokine concentrations with cancer progression is difficult to interpret and remains ill-defined. In this study, we used a novel approach to identify systemic inflammatory indicators that associate with PDAC progression. Using baseline pre-treatment serum from PDAC patients with and without early metastatic progression, we analyzed serum-induced transcriptomes (induced in a normal reporter cell) and patient serum cytokine concentrations.
The serum-induced transcriptomes of pancreatic cancer patients with early metastasis (<400 days from diagnosis) were compared with patients that did not develop metastasis or developed metastasis late (>500 days from diagnosis). We hypothesized that at baseline (before treatment), immune mediators would differ in PDAC patients that experience early metastasis (referred to as progressed or P) as compared to patients that do not experience metastasis or experienced metastasis greater than 500 days from diagnosis (referred to as not progressed or N). To sensitively and comprehensively capture these differences, we employed a serum-induced transcriptome analysis (using pre-treatment baseline serum) to induce transcription in a well-controlled reporter cell population consisting of peripheral blood mononuclear cells (PBMCs) obtained from a healthy blood donor. This same serum-based transcriptome assay using PBMCs (obtained from the same PBMC donor as this study) has been used to capture inflammatory signatures in patients with Type 1 diabetes and cervical cancer. In recent onset Type 1 diabetes patients, longitudinal plasma samples identified a signature of high proinflammatory mediators and low anti-inflammatory factors which was evident before the onset of Type 1 diabetes (11,12). For cervical cancer, the transcriptome signature identified an association of innate immune activation with metastatic cervical carcinoma (13).
Results of this study show a baseline serum-induced transcriptome signature of innate type I interferon (IFN) inflammation in PDAC patients that experience early metastasis. This was in contrast to a transcriptome signature of regulated acute inflammation identified in patients that did not experience early metastasis. In order to correlate cytokine concentrations with transcriptome data, a canonical correlation of the transcriptome with serum cytokines was performed. From this correlation, both immune activating and immune regulating transcripts showed significant positive correlation in the not progressed PDAC group. These data were consistent with the transcriptome signature of regulated acute inflammation for this patient cohort. Individual serum cytokine concentrations did not correlate with the transcriptome data. However, when cytokine concentrations were calculated as a cytokine index (i.e. the sum of the concentration of acute inflammatory cytokines divided by the concentration of Th2 cytokines and myeloid differentiation factors), there was a Pearson correlation of <0.05 with the transcriptome index.
While serum/plasma-induced transcriptomes have previously been applied in the cancer field (13), to our knowledge, this is the first analysis of cancer-associated peripheral immunity that describes cancer inflammation/immunity in the context of serum-induced transcription with serum cytokine concentrations. With further study, the serum-induced transcriptome and serum cytokine index have the potential to be used as systemic biomarkers that will contribute to the identification of PDAC subsets that associate with disease progression.
Methods
Study Subjects and Samples
Subjects were recruited through the pancreatic cancer program at the Medical College of Wisconsin, and blood and serum samples were collected into the Pancreatic Cancer Biorepository under an approved Institutional Review Board protocol (PRO00012151; Froedtert Memorial Hospital). Early progressed PDAC patients had radiographic evidence of metastasis within 400 days of diagnosis. Not progressed or late progressed patients had no radiographic evidence of metastasis within 500 days of diagnosis. For patient serum preparation, peripheral blood was drawn into non-anticoagulated tubes and serum was collected, centrifuged, and frozen at −80oC until analyzed.
Serum-induced Transcriptome Analysis
A serum-induced transcription assay was conducted as previously described (12). For this study, commercial cryopreserved PBMCs from a Caucasian HLA-A2 male donor (UPN727, Cellular Technology Ltd., Shaker Heights, OH) were thawed and washed per manufacturer’s protocol. PBMCs were co-cultured with 40% subject serum in RPMI 1640 medium supplemented with 100 U/ml penicillin and 100 µg/ml streptomycin at 37oC in 5% CO2. Cultures were prepared in a Costar 24-well plate (Corning) using 500,000 cells in 500 µl/well. After culture (9 hours), total RNA was extracted using TRIzol reagent (Invitrogen Life Technologies). Using purified total RNA (100 ng), cRNA was synthesized and amplified/labeled using the Affymetrix Express Kit, then fragmented and hybridized to the GeneChip Human Genome U133 plus 2.0 array in accordance with the Affymetrix GeneChip expression analysis technical manual (Affymetrix, Santa Clara, CA). After hybridization, arrays were washed and stained with Affymetrix fluidics protocol FS450_0001 and scanned with a 7G Affymetrix GeneChip Scanner. Image data were analyzed with Affymetrix Expression Console™ 1.1.2 software and normalized with Robust Multichip Analysis (www.bioconductor.org) to determine signal log ratios. The statistical significance of differential gene expression was determined though ANalysis Of VAriation (ANOVA) and false discovery rates (FDR) using Partek Genomics Suite 6.5. Hierarchical clustering was conducted with Genesis (14). Pathway analysis was performed with the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (15) and Integrated Pathway Analysis (IPA) (16). The transcriptome index was calculated by subtracting the sum of the log2 intensity of downregulated transcripts from the sum of the log2 intensity of upregulated transcripts (relative to progressed PDAC). The data generated in this investigation are MIAME compliant (17) and have been deposited in the NCBI Gene Expression Omnibus (18), accessible through GSE107818.
Multiplex Serum Cytokines
Serum was analyzed in duplicate using the Human Cytokine/Human Chemokine Array 65-plex Panel (HD65) (Eve Technologies, Calgary, AB, Canada).
Immunohistochemistry
Histological examinations were performed on standard four-micron thick hematoxylin and eosin (H & E) stained sections of formalin-fixed, paraffin embedded specimens. The pathologic features were evaluated by low power examination of several representative slides from each case by a pathologist (A.C.M.). Immunohistochemistry: Paraffin blocks for immunohistochemical studies were available in all cases. Four-micron sections were stained using a Dako Autostainer Plus according to the manufacturer’s protocol. Slides were dried at 60oC for one hour and deparaffinized. Heat induced epitope retrieval was performed with Dako Envision FLEX target retrieval solution (high pH Tris/EDTA) at 100oC for 20 minutes. The primary antibody for CD8 (C8/144B) was obtained from DAKO, Carpinteria, California. Antibodies were incubated at room temperature for 60 minutes. Signals were detected using a Dako FLEX detection kit. Counterstaining was performed with Envision FLEX hematoxylin for 7 minutes at room temperature. Appropriate positive and negative controls were run concurrently. Immunohistochemical Analysis: Cytoplasmic and membranous expression of immunohistochemical staining was quantified for each case using an Automated Cellular Imaging System III (ACIS III, DAKO) as previously described (19). For each patient, a portion of pancreatic cancer was quantitatively analyzed by scanning a representative whole slide. ACIS software collects individual, overlapping images at 400X, and then the software program tiles these images to create a montage of the entire scanned specimen. The software evaluates each individual 400X image and combines the results into an aggregate quantitative measurement corresponding to the entire tissue specimen. The ACIS system measures the intensity of the staining based on three related color parameters: the color defined by hue, the “darkness” defined as luminosity, and the density of the color defined as saturation. ACIS software for the analysis was programmed by an experienced user-pathologist (A.C.M.) by setting the color-specific thresholds to determine and calculate staining intensity and the ratio of positively stained cells to the entire area of selection. This was used to determine the approximate percentage of positive staining tissue in each specimen.
Statistics
Data were compared using paired and unpaired non-parametric Student’s t test and Pearson correlation. Analyses were calculated with Prism graph pad 6.0 software (GraphPad, San Diego, CA). Canonical correlation analyses between gene expression and cytokines were performed using R package CCA, Version 1.2, published 2014.07.02 (http://www.r-project.org/). A canonical cross correlation was done between the log2 intensity of 66 transcripts that met the statistical cut off of <20% false discovery rate (n=66) and the concentration of 65 serum cytokines for each of the 5 patients in the progressed and not progressed PDAC cohorts.
Results
The serum-induced transcriptome differs in patients with progressed and not progressed PDAC
Patients with resectable or borderline-resectable PDAC (i.e. not locally metastatically advanced and candidates for surgery) were included in this study. All patients were treated with neoadjuvant chemotherapy consisting of chemotherapy or chemotherapy and radiation (Table 1). Previous study of resectable PDAC patients treated with neoadjuvant therapy showed a mean overall survival of 32 months (933 days from diagnosis) (20). For this study, patients with early metastasis (i.e. progressed) were defined as those with radiographic evidence of metastasis within 400 days of diagnosis. Patients with late or no metastatic progression (i.e. not progressed) had no radiographic evidence of metastasis or had radiographic metastatic evidence that appeared 500 days after diagnosis (Tables 1 and 3). We hypothesized that mediators in the serum of patients with progressed and not progressed PDAC would induce unique transcriptional responses in a reporter population of normal PBMCs. A serum-based transcriptome assay extensively used to detect inflammatory signatures in Type 1 diabetes was used to interrogate the hypothesis (11,21,22). Specifically, baseline serum (obtained at diagnosis and prior to treatment) was co-cultured over PBMCs obtained from a standard normal donor. The induced transcription was measured on Affymetrix GeneChip Human Genome U133 Plus 2 arrays. An unsupervised principal component analysis (utilizing the complete unfiltered data) showed distinct clusters between progressed and not progressed samples (Suppl. Fig. 1). Of the greater than 47,000 transcripts interrogated, there were 727 unique transcripts that met thresholds of │log2 ratio│of ≥0.05 (1.2 fold-difference) with a p≤0.01. The heat map in Fig. 1A shows the relative expression of the 727 transcripts in 5 progressed (P) and 5 not progressed (N) PDAC patients. The heat map of the mean fold-difference in expression shows a distinct segregation of up and down regulated transcripts between the 2 patient cohorts (Fig. 1B).
Table 1.
Clinical Treatment and Progression
| Patient Number |
Age | Disease Burden |
Neoadjuvant/ Induction Chemotherapy |
Chemoradiation | Therapy Completed |
Surgical Resection |
Days to Metastatic Progression |
| PI | 62 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | 249 |
| P2 | 71 | R | None | Capecitabine XRT | Yes | No | 84 |
| P3 | 53 | R | FOLFIRINOX | None | Yes | Yes | 307 |
| P4 | 67 | BR | FOLFIRI | None | Unknown | No | 84 |
| P5 | 50 | R | FOLFIRINOX | None | Yes | Yes | 398 |
| P6 | 73 | BR | Gem/Nab-P | Gem XRT | Yes | Yes | 308 |
| P7 | 71 | BR | FOLFIRINOX | Gem XRT | Yes | No | 150 |
| P8 | 60 | BR | FOLFIRINOX | None | No | No | 70 |
| P9 | 84 | BR | None | Capecitabine XRT | Yes | No | 91 |
| P10 | 80 | R | None | Gem XRT | Yes | No | 97 |
| P11 | 81 | BR | Gem/Nab-P | Gem XRT | Yes | No | 130 |
| P12 | 83 | R | None | Gem XRT | Yes | Yes | 201 |
| P13 | 60 | BR | FOLFIRINOX | Gem XRT | Yes | No | 203 |
| P14 | 63 | BR | None | Gem XRT | Yes | No | 209 |
| P15 | 63 | BR | FOLFIRINOX | Capecitabine XRT | Yes | No | 209 |
| P16 | 84 | BR | FOLFIRINOX | Gem XRT | Yes | No | 343 |
| P17 | 59 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | 358 |
| N1 | 70 | R | None | Gem XRT | Yes | Yes | None |
| N2 | 52 | R | None | Gem XRT | Yes | Yes | None |
| N3 | 60 | R | FOLFIRINOX | None | Yes | Yes | None |
| N4 | 66 | R | FOLFIRI | None | Yes | Yes | 1841 |
| N5 | 56 | R | None | Gem XRT | Yes | Yes | 1092 |
| N6 | 58 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | None |
| N7 | 66 | R | FOLFIRINOX | None | Yes | Yes | None |
| N8 | 73 | R | None | Capecitabine XRT | Yes | Yes | None |
| N9 | 70 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | 545 |
| N10 | 66 | R | None | Capecitabine XRT | Yes | Yes | 678 |
| N11 | 72 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | 803 |
| N12 | 64 | R | FOLFIRINOX | None | Yes | Yes | 1187 |
| N13 | 73 | BR | FOLFOX | Gem XRT | Yes | Yes | None |
| N14 | 65 | BR | FOLFIRINOX | Capecitabine XRT | Yes | Yes | 1614 |
| N15 | 65 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | None |
| N16 | 70 | BR | FOLFIRINOX | Gem XRT | Yes | Yes | None |
| N17 | 74 | R | None | Gem XRT | Yes | Yes | None |
| N18 | 86 | R | None | Gem XRT | Yes | Yes | None |
P = Progressed, N = Not progressed, R = Resectable, BR = Borderline resectable, XRT = Radiation therapy, Gem = Gemcitabine, Nab-P = Nab-paclitaxel
Table 3.
Transcriptome Indices, Cytokine Index and Tumor CD8+ T Cell Density
| Patient Number and Days to Progression | Transcriptome Index n=727 | Inflammatory Transcriptome Index n=194 | Cytokine Index |
CD8+ T Cell Density | |
| P1 | 249 | 207 | 80 | 1.51 | 0.66 |
| P2 | 84 | 142 | 41 | 1.42 | |
| P3 | 307 | 206 | 60 | 0.79 | 0.65 |
| P4 | 84 | 14 | −18 | 3.15 | |
| P5 | 398 | 79 | 17 | 1.30 | 0.28 |
| P6 | 308 | 0.39 | 1.49 | ||
| P7 | 150 | 1.69 | |||
| P8 | 70 | 5.28 | |||
| P9 | 91 | 3.00 | |||
| P10 | 97 | 2.41 | |||
| P11 | 130 | 0.42 | |||
| P12 | 201 | 3.43 | |||
| P13 | 203 | 1.90 | |||
| P14 | 209 | 0.94 | |||
| P15 | 209 | 4.42 | |||
| P16 | 343 | 4.78 | |||
| P17 | 358 | 2.29 | |||
| P mean | 130 | 36 | 2.3 | 0.77 | |
| N1 | None | −174 | −51 | 5.08 | 1.55 |
| N2 | None | −69 | −22 | 2.65 | 9.52 |
| N3 | None | −150 | −44 | 3.30 | 1.86 |
| N4 | 1841 | −134 | −33 | 3.08 | 0.16 |
| N5 | 1092 | −120 | −29 | 9.30 | 2.69 |
| N6 | None | 3.22 | 1.44 | ||
| N7 | None | 1.33 | 0.52 | ||
| N8 | None | 1.41 | 1.56 | ||
| N9 | 545 | 2.61 | |||
| N10 | 678 | 2.84 | |||
| N11 | 803 | 2.44 | |||
| N12 | 1187 | 4.44 | |||
| N13 | None | 2.81 | |||
| N14 | 1614 | 11.00 | |||
| N15 | None | 2.40 | |||
| N16 | None | 6.87 | |||
| N17 | None | 1.61 | |||
| N18 | None | 3.03 | |||
| N mean | −130 | −36 | 3.90 | 2.41 | |
|
Average of Cls normal controls (n=10) |
2.47 | ||||
P = Progressed and N = Not progressed
Figure 1. Serum-induced transcriptome.
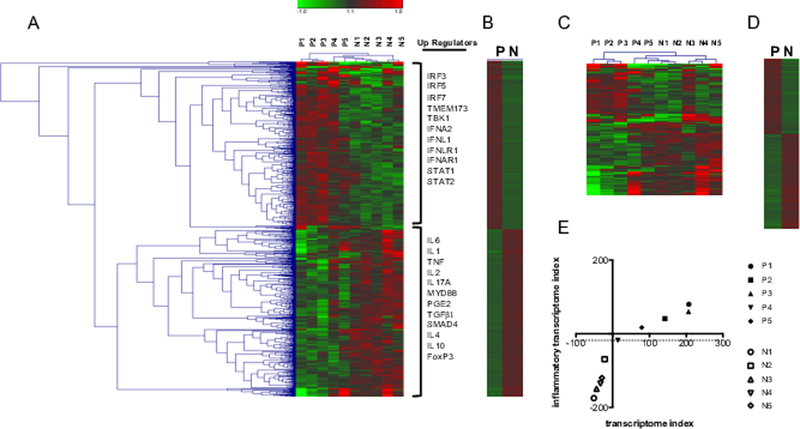
(A) Heatmap of transcripts with 1.2-fold difference and p≤0.01 showing upstream (up) regulators as identified in IPA. n=727 (B) Heatmap of the mean log2 intensity of transcripts shown in A. (C) Heatmap of inflammation-related transcripts as identified in IPA and DAVID with 1.2-fold difference and p≤0.01. n=194. (D) Heatmap of the mean log2 intensity of transcripts shown in C. (E) Graph showing segregation of progressed and not progressed PDAC patient cohorts based on the transcriptome and inflammatory transcriptome indices.
The 727 unique transcripts were uploaded into Database for Annotation, Visualization and Integrated Discovery (DAVID) and Integrated Pathways Analysis (IPA) for ontological analysis. Putative upstream regulators (up regulators) of gene transcription were identified in IPA (Fig. 1A and Table 2). Identification of upstream regulators was of particular interest as we hypothesized that mediators/regulators in patient serum are responsible for regulating transcription in normal donor PBMCs. Upregulated transcripts in progressed patients included those associated with activated innate immunity. Specifically, upstream regulators included IFN regulatory factors (IRF3, IRF5, IRF7), stimulator of IFN genes (STING (TMEM173)), tank binding kinase (TBK), type 1 IFN (IFNA2), type III IFN (IFNL1 or IL29), IFN receptor subunits (IFNLR1, and IFNAR1), and transcription factors associated with IFN signaling (STAT1 and STAT2) (Fig. 1A and Table 2). In contrast to the upstream regulators identified in progressed PDAC patients, the upstream regulators of upregulated transcripts in not progressed PDAC was one of acute inflammation and TGFβ1/IL10 immune regulation. For this patient cohort, there were upstream regulators associated with acute inflammation (IL6, IL1, TNF, IL2, IL17A, MYD88) as well as mediators associated with immune regulation (PGE2, TGFβ, SMAD4, IL4, IL10, and FoxP3) (Fig. 1A and Table 2). In summary, progressed patients have a serum-induced transcriptome signature consistent with activated innate inflammation, whereas not progressed patients have a serum-induced transcriptome signature consistent with acute inflammation and TGFβ and IL10 immune regulation.
Table 2.
Upstream Regulators of the Upregulated Transcripts (n=727) Identified in Top Canonical Inflammatory Associated Pathways: IPA
| Progressed Upregulated n=364 | Not Progressed Upregulated n=363 | |||
| Top Canonical Pathways | p-value | Top Canonical Pathways | p-value | |
| Pathogenesis of MS | 1.31E-05 | Role of 17A in Psoriasis | 7.81E-05 | |
| EIF2 Signaling | 3.02E-05 | Histamine Degradation | 2.57E-03 | |
| Role of Hypercytokinemia/Hyperchemokinemia in the Pathogenesis of Influenza | 8.83E-04 | Inhibition of Angiogenesis by TSP1 | 5.44E-03 | |
| IL-9 Signaling | 1.36E-03 | Neuregulin Signaling | 6.43E-03 | |
| Communication between Innate and Adaptive Immunity | 3.11E-03 | |||
| Upstream Regulators | p-value | Upstream Regulators | p-value | |
| SAMSN1 | 1.36E-10 | Lipopolysaccharide | 8.29E-09 | |
| IFNLR1 | 9.87E-09 | TNF | 2.21E-08 | |
| STAT1 | 1.33E-09 | PGE2 | 1.68E-07 | |
| TBK1 | 1.35E-09 | CSF2 | 1.84E-07 | |
| IRF3 | 1.42E-09 | PD98059 | 3.33E-07 | |
| TLR3 | 1.46E-10 | Immunoglobulin | 5.28E-07 | |
| Salmonella enterica serotype | 1.92E-09 | IL2 | 9.43E-06 | |
| IFNA2 | 5.05E-09 | U0126 | 1.65E-06 | |
| TLR9 | 6.48E-09 | TGFB1 | 2.15E-06 | |
| STAT2 | 7.92E-09 | MYD88 | 2.70E-06 | |
| PTPRJ | 7.92E-09 | IL4 | 3.40E-06 | |
| IRF5 | 8.35E-09 | Diethylstilbestrol | 1.25E-05 | |
| DOCK8 | 1.01E-08 | EGR1 | 1.40E-05 | |
| IRF7 | 1.18E-08 | EGF | 1.46E-05 | |
| TMEM173 | 1.48E-08 | 9,10-Dimethyl-1,2-benzanthracene | 2.15E-05 | |
| TREM1 | 1.97E-08 | ATP-gamma-S | 2.17E-05 | |
| TLR4 | 2.07E-08 | Tretinoin | 2.28E-05 | |
| IFNAR1 | 2.16E-08 | LY294002 | 2.53E-05 | |
| IFNL1 | 5.57E-08 | Tributyrin | 2.74E-05 | |
| ELANE | 7.47E-05 | |||
| IL17A | 3.53E-05 | |||
| IL10 | 4.14E-05 | |||
| SMAD4 | 4.31E-05 | |||
| Methylprenisolone | 4.68E-05 | |||
| IL6 | 4.89E-05 | |||
| IL1 | 4.97E-05 | |||
| FoxP3 | 5.12E-05 | |||
| Alpha catenin | 5.56E-05 | |||
To understand a global relationship of these genes, a transcriptome index was calculated as a method to ascribe a numerical value to the upregulated or downregulated transcripts depicted in the heat map in Fig. 1A. Relative to progressed patients, the transcriptome index was calculated as the sum of the log2 intensity of all downregulated transcripts subtracted from the sum of the log2 intensity of all upregulated transcripts. Patients in the progressed cohort had transcriptome indices greater than 0 and patients in the not progressed patient cohort had transcriptome indices less than 0 (Table 3). Thus, the transcriptome index showed a numerical segregation of the patient cohorts.
Analysis of the transcriptome index with an inflammatory transcriptome index segregates the PDAC progressed and not progressed patient cohorts
Inflammation is a hallmark of cancer (23), yet markers of immune inflammation are difficult to interpret. To analyze transcripts in the context of inflammation, genes listed in IPA and DAVID belonging to ontology categories of Inflammatory Response, Cytokine and Cytokine-Cytokine Receptor Interaction were identified (Suppl. Table 1). Within the original 727 transcripts, there were 194 related to inflammation. The heatmap in Fig. 1C shows the relative transcript expression of these inflammatory genes for each patient. While there is variation of transcript expression between individuals within the progressed and not progressed patient cohorts, calculation of the mean fold difference showed a distinct segregation of gene expression between the 2 groups (Fig. 1D).
Similar to the transcriptome index calculated from 727 transcripts, an inflammatory transcriptome index was calculated from 194 inflammatory-associated transcripts. Analogous to the transcriptome signature, each of the progressed patients had an inflammatory transcriptome index greater than that of not progressed patients (Table 3). When the data from the transcriptome and inflammatory indices were analyzed together, the progressed and not progressed patient cohorts segregated (Fig. 1E). These data suggest that inflammation is a factor associated with disease progression.
To validate the results of the microarray analysis, quantitative real-time PCR was done from cDNA made from the PBMCs (same donor PBMCs as used for the microarray analysis) following incubation with patient serum. Supplementary Fig. 2 shows a correlation of the log2 intensity (Suppl. Fig. 2A) with PCR amplification (fold change based on ΔΔCT value) (Suppl. Fig. 2B) for several genes.
Serum cytokine and transcriptome indices segregate PDAC progressed and not progressed patient cohorts
Inflammatory (IL6, CXCL8, TNF, IL1β) and regulatory (IL10, IL1RA) serum cytokine concentrations are elevated in patients with aggressive cancers (10), but to date, there are no validated serum mediators as markers of cancer progression. For this study, we hypothesized that serum immune mediators (cytokines, chemokines and growth factors) analyzed in the context of each other would reflect a state of systemic immunity that would correlate with disease progression. For this assay, baseline (pre-treatment) serum was analyzed using a luminex-based platform.
To understand serum-mediators in context with the transcriptome, a canonical correlation between the log2 intensity of individual transcripts and individual serum cytokine concentrations (pg/ml) was done. For this analysis, the transcriptome statistical stringency was increased to include a false discovery rate of <20%. These criteria reduced the transcriptome list from 727 to 66 transcripts. The log2 intensity of each of the 66 transcripts was correlated with the 65 serum cytokine concentrations analyzed for each patient. Fig. 2 shows the correlation of transcriptome data (X) with serum cytokine concentrations (Y) in the progressed patient cohort (Fig. 2A) and the not progressed patient cohort (Fig. 2B). Values are translated into colors from blue (negative correlation) to red (positive correlation) to generate a correlation map. The maps show distinct differences in the progressed and not progressed patient groups. From the list of 66 transcripts subjected to canonical correlation, those that correlated significantly (Cor p ≥ 0.94 or Cor p ≤ -0.94) with cytokine concentrations in progressed and not progressed PDAC patient cohorts are listed in Table 4.
Figure 2. Canonical correlation of the log2 intensity transcriptome values and cytokine concentrations.
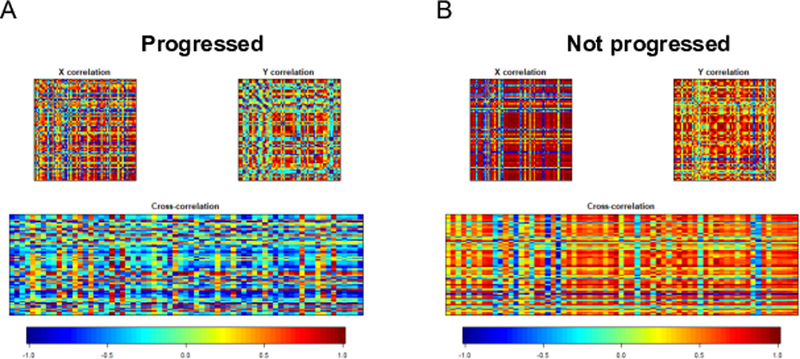
(A) Correlation matrices for progressed PDAC: X variables (upper-left panel, the transcript log2 intensity), Y variables (upper-right panel, cytokine concentrations (pg/ml)), cross-correlation X x Y (bottom panel). (B) Correlation matrices for not progressed PDAC. X variables (upper-left panel, the transcript log2 intensity), Y variables (upper-right panel, cytokine concentrations), cross-correlation X x Y (bottom panel). Values are translated into colors from blue (negative correlation) to red (positive correlation).
Table 4.
Canonical Correlation of FDR<20% Transcript Log2 intensity (n=66) with Serum Cytokine Concentrations (n=65)
| Progressed PDAC |
|
Negative correlation Electron transfer flavoprotein dehydrogenase (ETFDH) −0.97 SMG7 −0.94 |
| Positive correlation |
| Thioredoxin (TXN) 0.94 |
| Not progressed PDAC |
| Negative correlation |
| Cyclin dependent kinase1 (CDK1) −0.97 |
| Positive correlation |
| SAM and SH3 domain-containing protein 1 (SASH1) 0.99 |
| Epiregulin (EREG) 0.98 |
| Thrombomodulin (THBD) 0.98 |
| Erythrocyte membrane protein band 4.1 like 3 (EPB41L3) 0.98 |
| Myoferlin (MYOF) 0.98 |
| Thrombospondin 1 (THBS1) 0.98 |
| DMX like 2 (DMXL2) 0.98 |
| Macrophage expressed 1 (MPEG1) 0.98 |
| Alanyl aminopeptidase (ANPEP) 0.98 |
| Mitochondrial translocator assembly and maintenance homolog (TAMM41) 0.98 |
| Ras homolog family member U (RHOU) 0.97 |
| CXC motif chemokine ligand 5 (CXCL5) 0.97 |
| Fc fragment of IgA receptor (FCAR) 0.97 |
| Cluster of differentiation 14 (CD14) 0.97 |
| Integrin subunit beta 8 (ITGB8) 0.97 |
| ATP-dependent helicase II (XRCC5) 0.97 |
| CD93 0.96 |
| Versican (VCAN) 0.96 |
| Thioredoxin (TXN) 0.95 |
| C-type lectin domain containing 5A (CLEC5A) 0.95 |
| Tyrosine protein kinase HCK (HCK) 0.95 |
| Interleukin 24 (IL24) 0.95 |
| Complement C5A receptor 1 (C5AR1) 0.94 |
| Neuropilin 1 (NRP1) 0.94 |
| SMAD family member 4 (SMAD4) 0.94 |
Interestingly, in the progressed group, there were 2 genes related to mitochondrial metabolic function that significantly correlated with serum cytokines (Table 4). With reference to the cytokine signature, the electron transfer flavoprotein dehydrogenase gene (ETFDH) was negatively correlated and thioredoxin (TXN) was positively correlated. Both transcripts were upregulated in the progressed group when compared to the not progressed group with a mean fold difference of 1.48 for ETFDH and 1.61 for TXN. ETFDH is a mitochondrial enzyme involved in energy production by fat and protein catabolism, and thioredoxin is a redox active protein important in the regulation of mitochondrial membrane potential. Recent studies have reported that metabolic reprogramming may enable PDAC cells to maintain adequate intracellular nutrient levels despite limited supplies (24,25). Furthermore, autophagy, microlipophagy and electron transport chain activity were reported to be critical for survival of tumor initiating cells (26). Together these data suggest that metabolic states may play a role in progression.
In the not progressed group, there was a positive correlation of transcripts related to activated inflammation (i.e. SASH1, CXCL5, FCAR, CD14, VCAN, CLEC5A, C5AR1, and IL1R). There were also positive correlations of cytokine concentrations with transcripts associated with TGFβ activation and signaling (i.e. SMAD4 and ITGB8) in the not progressed patient cohort. These data are consistent with the transcriptome data suggesting not progressed patients have a regulated acute inflammatory signature (Table 2).
A cytokine index (CI) was calculated as a strategy to globally assess serum cytokine concentrations. The CI was calculated as the sum of the concentrations of acute phase cytokines (IL1α, IL1β, IL6, IL17A, IL28A, IFNA2, IFNγ, and TNFα) divided by the sum of concentrations of T helper Type 2 (Th2) cytokines and myeloid growth factors (IL4, IL5, IL10, IL13, GM-CSF, FLT3L, and G-CSF). A Pearson correlation of the CI with the transcriptome index was significant at p=0.0332. (R2=0.452) (Figure 3A and Suppl. Table 2). There was no significance observed when individual serum cytokine concentrations were correlated with the transcriptome index (Suppl. Table 2). Figure 3 shows separation of the progressed and not progressed patient cohorts when the CI was graphed with the transcriptome index (Fig. 3A) and inflammatory transcriptome index (Fig. 3B). In order to substantiate the CI as an indicator for PDAC metastatic progression, it was calculated from baseline serum from additional patients (i.e. progressed (n=17) and not progressed (n=18)) (Table 3). From this validation cohort, the CI showed statistical significance (p=0.0487) between the progressed and not progressed PDAC cohorts (Fig. 3C). The mean CI of the progressed group was 2.3 and the mean CI of the not progressed group was 3.9 (Table 3). It is important to note that some progressed patients had a CI greater than 3.9 (P8, P15, and P16) and some not progressed patients had a CI less than 2.3 (N7, N8 and N17). These data suggest that a higher CI may be a favorable indicator of delayed PDAC progression only in some patients as the CI did not completely segregate the two patient cohorts. Furthermore, this index requires validation with larger cohorts of PDAC early and late metastatically progressed patients.
Figure 3. Serum cytokine analysis.
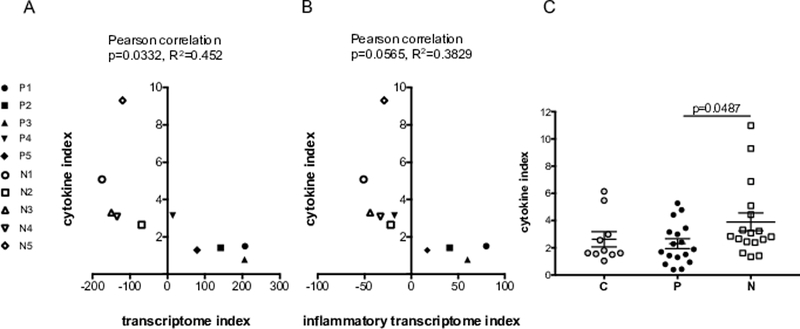
Segregation of patient cohorts when the cytokine index is graphed with the transcriptome index (A) and inflammatory transcriptome index (B). (C) The cytokine index with standard error of the mean (SEM) of progressed and not progressed PDAC patient cohorts. P = Progressed, N = Not progressed and C = Normal controls.
Few CD8+ T cells are detected in tumors from both patient cohorts
Infiltration of CD3+CD8+ cells in the tumor core (CT) and invasive margins (IM) (referred to as the immunoscore) has been shown to correlate with overall survival in multiple carcinomas including bladder, breast, colorectal, esophagus, head and neck, liver, lung, melanoma and ovarian (27). Often there is only 5–20% cellularity within the extracellular matrix-rich stroma of pancreatic cancer (28). Despite the presence of patchy disease, a better disease-specific survival and overall survival has been associated with the presence of CD8+ T cells in the PDAC tumor core and CD3+ T cells in the tumor core and invasive margin (29). High tumor infiltration of both CD4+ and CD8+ T cells also correlated with a significant increase in survival (30). Thus, for this study, it was of interest to determine if CD8+ T cells could be detected in the resected tumors of our PDAC patients. Available tumor samples were analyzed in 4 progressed patients (P1, P3, P5 and P6) and 8 not progressed patients (N1-N8). Tumor sections were stained with anti-CD8, and the percentage of CD8+ T cells was determined in multiple regions of the tissue. The CD8+ T cell density score represents the average percent of CD8+ T cells from each case (Table 3). Fig. 4 shows the CD8+ T cell density graphed against the inflammatory transcriptome index (Fig. 4A) and the cytokine index (Fig. 4B). Figure 4C shows representative tumor sections stained with hematoxylin/eosin (panel 1) and anti-CD8 (panel 2) From these limited data, a few interesting observations were made. Two of the not progressed patients had few T cells detected in tumor (N4 and N7) (Table 3). Patient N4 relapsed with metastasis 1824 days since diagnosis and patient N7 is beyond 1000 days without relapse (Tables 1 and 3). The average number of CD8+ T cells in patient P6 was greater than patients N4 and N7 (Table 3). Patient P6 relapsed 308 days from diagnosis. It could be that PDAC T cell infiltration is a useful indicator of PDAC progression, but it may not serve as a predictor of PDAC progression for all patients. This hypothesis requires further testing.
Figure 4. Immunohistochemical analysis of CD8+ T cells in PDAC tumor.
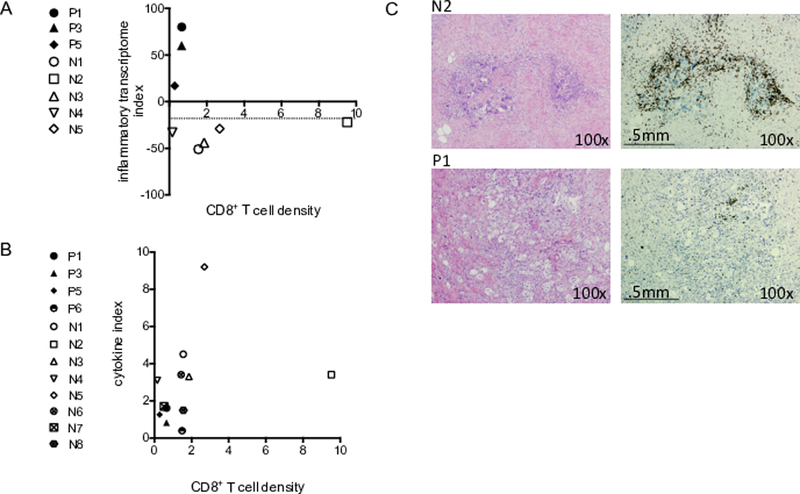
Graph of CD8+ T cell density with the inflammatory transcriptome index (A) and cytokine index (B). P = Progressed and N = Not progressed. (C) A representation of tumor tissue sections stained with hematoxylin and eosin (panel 1) and anti-CD8 (panel 2). N2 is not progressed patient 2 (top) and P1 is progressed patient 1 (bottom).
Discussion
There are multiple indicators that associate with the development and progression of pancreatic cancer. The presence of somatic mutations in tumor cells (e.g. KRAS, TP53, CDKN2A, SMAD4, RNF43, ARID1A, TGFβR2, GNAS, RREB1 and PBRM1) (4), tumor RNA expression profiles (3–6), tumor immune cell infiltration (27,29,30) and metabolic function (24–26) all contribute to identifying more and less aggressive subsets of PDAC. The main objective of this study was to query the systemic response in PDAC to further enhance knowledge of PDAC disease progression.
Identification of systemic indicators such as inflammatory mediators present in the serum and their association with PDAC progression has been a difficult task. In this study, serum was analyzed using novel strategies that allowed for a global analysis of indicators. An in-depth analysis of a serum-induced transcriptome and serum cytokine concentrations was conducted on a pilot sample of patients with pancreatic ductal adenocarcinoma in order to investigate these associations with disease progression. Remarkably, despite the limitation in sample size, the baseline serum-induced transcriptome showed a difference in immune activation and immune regulation in PDAC patients that do and do not develop early metastasis (Figs. 1A and B). Ontological analysis of the baseline serum-induced transcriptome from progressed patients that experience early metastasis showed putative upstream regulators consistent with induction of type I IFN production through the stimulator of IFN genes pathway (STING). The presence of activators of innate immunity in this patient cohort is counter intuitive since activation of the STING pathway and the secretion of type I IFNs, inflammatory cytokines and chemokines promote maturation of dendritic cells which prime cancer antigen-specific T cells to produce an adaptive anti-cancer response (31). It is possible that these immune upstream regulators are present in response to innate activation signals, but there is immune dysregulation that prevents activation of Th1 adaptive immunity in this patient cohort. Utilizing the same serum-induced transcriptome assay and the same reporter PBMC cells, an innate immune activation signature was also identified in patients with metastatic cervical carcinoma (13). Remarkably, and similar to progressed PDAC patients, the transcriptome upstream regulators for metastatic cervical cancer patients included IRF5, IRF7, IFNL1, IFNA2 and IFNL1.
While the transcriptome data is striking, this method of analysis is costly and may not be clinically feasible. The ability to identify alternative inflammatory states from serum cytokine analysis is of great clinical relevance. To date, serum cytokine analysis has shown that both immune stimulatory and immunosuppressive cytokines are present in the serum of patients with multiple aggressive cancers (10). Most notably, both high concentrations of the acute inflammatory cytokine IL6, as well as the inflammation regulatory cytokine IL10, are associated with poor outcome. Our data showed no correlation of individual cytokines with the transcriptome index (Suppl. Table 2). Using the upstream regulators identified from the transcriptional data as a guide, cytokines were clustered into acute inflammatory cytokines and regulatory cytokines and myeloid differentiation factors to mathematically calculate a cytokine index. A Pearson correlation of the cytokine index with the transcriptome index yielded a correlation of p<0.05 (R2=0.452) (Figure 3A). Analysis from a larger cohort of 17 progressed and 18 not progressed patients, yielded a mean cytokine index that was statistically significant between the progressed and not progressed patient cohorts (Fig. 3C and Table 3). It is important to note that not every patient in the progressed cohort had a low cytokine index and not every patient in the not progressed cohort had a high cytokine index. With further study, the cytokine index may be shown to be an indicator of favorable outcome in some patients within a particular subset of PDAC that is yet to be defined.
To date, most of the information defining subsets of PDAC is derived from tumor. Often, tumor samples are not available and mutational, transcriptional, immunogenic and metabolic tumor parameters cannot be assessed. Therefore, further investigation of systemic indicators of PDAC progression is of critical importance. However, when tumor is available, it is conceivable that more and less aggressive subsets of PDAC can be further defined by combining tumor analysis with systemic analysis. The subtype of tumor (i.e. squamous, pancreatic progenitor, immunogenic and ADEX), tumor immunogenicity (i.e. detection of tumor infiltrating CD3+, CD8+ and CD4+ T cells), tumor mitochondrial metabolism, and the systemic inflammatory profile (serum-induced transcriptome or cytokine index) may need to be assessed together in order to identify specific subsets of PDAC with a predictable outcome.
In summary, both the transcriptome index and the cytokine index consider multiple immune-associated genes or cytokines that reflect a global state of systemic inflammation. The ability to segregate PDAC patients that experience early metastasis from those that do not through analysis of baseline cytokine concentrations and baseline transcriptome indices is conceptually novel. An in-depth analysis of this pilot sample of patients has introduced the serum-induced transcriptome and the cytokine index as potential indicators of PDAC progression. Further study of these systemic indicators alone or in combination with tumor characteristics should lead to a better understanding of subsets of PDAC and their association with progression and survival.
Supplementary Material
Acknowledgments
Funding:JA Gershan was supported by the Advancing a Healthier Wisconsin Program and the Medical College of Wisconsin Cancer Center, the Midwest Athletes for Childhood Cancer (MACC) fund, and the Children’s Hospital of Wisconsin Children’s Research Institute. MJ Hessner was supported by the Juvenile Diabetes Research Foundation International (grants 2-SRA-2015–109-Q-R and 3-SRA-2018–478-S-B to MJH); National Institutes of Health (R56DK108802 to MJH, DP3DK098161 to MJH and 1-UL1-RR031973 and The Clinical and Translational Science Institute of Southeast Wisconsin. This research was supported in part by a Seed Grant – FP9214 from the A Healthier Wisconsin Endowment to the MCW Cancer Center to MB Dwinell.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
Ethics approval: Human blood and serum samples were collected into the Pancreatic Cancer Biorepository under an approved Institutional Review Board protocol (PRO00012151; Froedtert Memorial Hospital).
Availability of data and material: The microarray data generated in this investigation has been deposited in the NCBI Gene Expression Omnibus, accessible through GSE107818. Data generated or analyzed for this study are included in this published article.
References
- 1.Gulley JL, Berzofsky JA, Butler MO, Cesano A, Fox BA, Gnjatic S, et al. Immunotherapy biomarkers 2016: overcoming the barriers. Journal for immunotherapy of cancer 2017;5(1):29–017-0225–6 doi 10.1186/s40425-017-0225-6 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paniccia A, Hosokawa P, Henderson W, Schulick RD, Edil BH, McCarter MD, et al. Characteristics of 10-Year Survivors of Pancreatic Ductal Adenocarcinoma. JAMA surgery 2015;150(8):701–10 doi 10.1001/jamasurg.2015.0668 [doi]. [DOI] [PubMed] [Google Scholar]
- 3.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 4.andrew_aguirre@dfci.harvard.edu CGARNEa, Network CGAR. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32(2):185–203.e13 doi 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47(10):1168–78 doi 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17(4):500–3 doi 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mao Y, Shen J, Lu Y, Lin K, Wang H, Li Y, et al. RNA sequencing analyses reveal novel differentially expressed genes and pathways in pancreatic cancer. Oncotarget 2017;8(26):42537–47 doi 10.18632/oncotarget.16451 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldberg JE, Schwertfeger KL. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Current Drug Targets 2010;11(9):1133–46 doi BSP/CDT/E-Pub/00110 [pii]. [DOI] [PubMed] [Google Scholar]
- 9.Mohamed MM, El-Ghonaimy EA, Nouh MA, Schneider RJ, Sloane BF, El-Shinawi M. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. The international journal of biochemistry & cell biology 2014;46:138–47 doi 10.1016/j.biocel.2013.11.015 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. The LancetOncology 2013;14(6):e218–28 doi 10.1016/S1470-2045(12)70582-X [doi]. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. Journal of immunology (Baltimore, Md: 1950) 2008;180(3):1929–37 doi 180/3/1929 [pii]. [DOI] [PubMed] [Google Scholar]
- 12.Levy H, Wang X, Kaldunski M, Jia S, Kramer J, Pavletich SJ, et al. Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes. Genes and immunity 2012;13(8):593–604 doi 10.1038/gene.2012.41 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palatnik A, Ye S, Kendziorski C, Iden M, Zigman JS, Hessner MJ, et al. Identification of a serum-induced transcriptional signature associated with metastatic cervical cancer. PloS one 2017;12(8):e0181242 doi 10.1371/journal.pone.0181242 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics (Oxford, England) 2002;18(1):207–8. [DOI] [PubMed] [Google Scholar]
- 15.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome biology 2003;4(5):P3. [PubMed] [Google Scholar]
- 16.Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (Oxford, England) 2014;30(4):523–30 doi 10.1093/bioinformatics/btt703 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nature genetics 2001;29(4):365–71 doi 10.1038/ng1201-365 [doi]. [DOI] [PubMed] [Google Scholar]
- 18.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research 2002;30(1):207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency--histologic and immunohistochemical analyses of 16 cases. Human pathology 2015;46(9):1306–14 doi 10.1016/j.humpath.2015.05.011 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christians KK, Heimler JW, George B, Ritch PS, Erickson BA, Johnston F, et al. Survival of patients with resectable pancreatic cancer who received neoadjuvant therapy. Surgery 2016;159(3):893–900 doi 10.1016/j.surg.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 21.Cabrera SM, Chen YG, Hagopian WA, Hessner MJ. Blood-based signatures in type 1 diabetes. Diabetologia 2016;59(3):414–25 doi 10.1007/s00125-015-3843-x [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YG, Cabrera SM, Jia S, Kaldunski ML, Kramer J, Cheong S, et al. Molecular signatures differentiate immune states in type 1 diabetic families. Diabetes 2014;63(11):3960–73 doi 10.2337/db14-0214 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009;30(7):1073–81 doi 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 24.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015;524(7565):361–5 doi 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perera RM, Bardeesy N. Pancreatic Cancer Metabolism: Breaking It Down to Build It Back Up. Cancer Discov 2015;5(12):1247–61 doi 10.1158/2159-8290.CD-15-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014;514(7524):628–32 doi 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirilovsky A, Marliot F, El Sissy C, Haicheur N, Galon J, Pages F. Rational bases for the use of the Immunoscore in routine clinical settings as a prognostic and predictive biomarker in cancer patients. International immunology 2016;28(8):373–82 doi 10.1093/intimm/dxw021 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer J 2012;18(6):492–501 doi 10.1097/PPO.0b013e31827459b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tahkola K, Mecklin JP, Wirta EV, Ahtiainen M, Helminen O, Böhm J, et al. High immune cell score predicts improved survival in pancreatic cancer. Virchows Arch 2018;472(4):653–65 doi 10.1007/s00428-018-2297-1. [DOI] [PubMed] [Google Scholar]
- 30.Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 2017;8:15095 doi 10.1038/ncomms15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corrales L, McWhirter SM, Dubensky TW Jr., Gajewski TF. The host STING pathway at the interface of cancer and immunity. The Journal of clinical investigation 2016;126(7):2404–11 doi 10.1172/JCI86892 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.