

Abstract
The centrosome is the main microtubule‐organizing centre. It also organizes a local network of actin filaments. However, the precise function of the actin network at the centrosome is not well understood. Here, we show that increasing densities of actin filaments at the centrosome of lymphocytes are correlated with reduced amounts of microtubules. Furthermore, lymphocyte activation resulted in disassembly of centrosomal actin and an increase in microtubule number. To further investigate the direct crosstalk between actin and microtubules at the centrosome, we performed in vitro reconstitution assays based on (i) purified centrosomes and (ii) on the co‐micropatterning of microtubule seeds and actin filaments. These two assays demonstrated that actin filaments constitute a physical barrier blocking elongation of nascent microtubules. Finally, we showed that cell adhesion and cell spreading lead to lower densities of centrosomal actin, thus resulting in higher microtubule growth. We therefore propose a novel mechanism, by which the number of centrosomal microtubules is regulated by cell adhesion and actin‐network architecture.
Keywords: actin, cell adhesion, centrosome, microtubule
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Immunology
Introduction
The growth of the microtubule network and its architecture regulates cell polarization, migration and numerous key functions in differentiated cells (Mimori‐Kiyosue, 2011; de Forges et al, 2012; Etienne‐Manneville, 2013; Sanchez & Feldman, 2017). Microtubule growth first depends on microtubule nucleation, which is regulated by large complexes serving as microtubule templates and proteins that stabilize early protofilament arrangements (Wieczorek et al, 2015; Roostalu & Surrey, 2017). Then, microtubule elongation becomes regulated by microtubule‐associated proteins and molecular motors acting at the growing end of microtubules (Akhmanova & Steinmetz, 2015). The architecture of the microtubule network—the spatial distribution and orientation of microtubules—is heavily influenced by its biochemical interactions and physical interplay with actin filaments (Rodriguez et al, 2003; Coles & Bradke, 2015; Huber et al, 2015; Colin et al, 2018; Dogterom & Koenderink, 2019). Although the physical cross‐linking of the two networks can occur at any points along microtubule length (Mohan & John, 2015), the sites of intensive crosstalk occur at the growing ends of microtubules (Akhmanova & Steinmetz, 2015; Dogterom & Koenderink, 2019).
The growth of microtubules can also be directed by actin‐based structures (Kaverina et al, 1998; Théry et al, 2006; López et al, 2014). They can force the alignment of microtubules (Elie et al, 2015), resist their progression (Burnette et al, 2007), capture, bundle or stabilize them (Zhou et al, 2002; Hutchins & Wray, 2014), submit them to mechanical forces (Gupton et al, 2002; Fakhri et al, 2014; Robison et al, 2016) or define the limits in space into which they are confined (Katrukha et al, 2017). The actin–microtubule interplay mostly takes place at the cell periphery, because most actin filaments are nucleated at and reorganized into actin‐based structures near the plasma membrane (Blanchoin et al, 2014). We recently have identified a subset of actin filaments that form at the centrosome at the cell centre (Farina et al, 2016). The centrosome is the main microtubule nucleating and organizing centre of the cell and sustains the highest concentration of microtubules in the cell. Centrosomal actin filaments have been shown to be involved in several functions including centrosome anchoring to the nucleus (Obino et al, 2016), centrosome separation in mitosis (Au et al, 2017) and ciliary‐vesicle transport in the early stages of ciliogenesis (Wu et al, 2018). Whether centrosomal actin filaments affect centrosomal microtubules is not yet known.
Here, we investigated how the processes of actin and microtubule growth at the centrosome influence each other. We provide in vivo and in vitro evidence that centrosomal actin network blocks microtubule growth, most likely as a result of physical hindrance. Our results further suggest that the regulation by centrosomal actin filaments restricts microtubule growth in response to cell adhesion.
Results
The centrosomal actin network appears to negatively regulate the microtubule network in B lymphocytes
B‐lymphocyte polarization can be achieved by B‐cell receptor (BCR) activation from binding surface‐tethered cognate antigens and requires the local reduction of centrosomal actin density (Obino et al, 2016). To evaluate how microtubules were affected in resting and activated B lymphocytes, we examined, by fluorescent microscopy of fixed cells, microtubule density throughout the cell in comparison with changes to the density of centrosomal actin filaments (Fig 1A). As expected, B‐lymphocyte activation was associated with a lower density (by 30%) of actin at the centrosome (Obino et al, 2016). It appeared to be also associated with a higher density (by 20%) of microtubules at the centrosome and in the entire cytoplasm (Fig 1B and C). A closer analysis by single cells showed a clear negative correlation between centrosomal actin density and microtubule density in resting (r = −0.44) and activated lymphocytes (r = −0.34) (Fig 1D), suggesting that the interplay between the two networks is not specific to the activation but an intrinsic relationship. Noteworthy, the amount of cortical actin did not vary during the activation (Fig EV1A), and the amount of cortical actin could not be correlated to the amount of microtubules in single cells (Fig EV1B and C), reinforcing the hypothesis of an early regulation at the centrosome. The labelling of actin filaments and microtubules in resting cells revealed the presence at the centrosome of dense actin puncta, from which microtubules were excluded, suggesting they act as a physical barrier through which microtubule cannot grow (Fig 1E).
Figure 1. Cytoskeleton remodelling in B lymphocytes upon antigen stimulation.
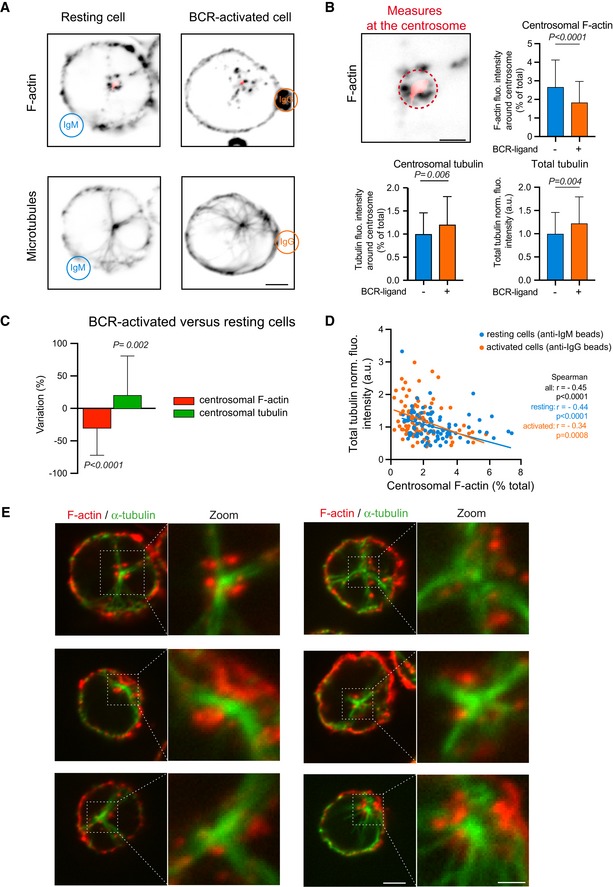
- IIA1.6 B lymphoma cells were stimulated with BCR‐ligand− (anti‐IgM) or BCR‐ligand+ (anti‐IgG) beads for 60 min, fixed and stained for F‐actin (top) and α‐tubulin (bottom). Scale bar: 3 μm.
- Histograms show the quantifications of the polymerized tubulin and F‐actin at the centrosome (dashed outline on the image, values correspond to the fraction of fluorescence in a 2‐micron‐wide area around the centrosome relative to the total fluorescence in the cell) and the total amount of polymerized tubulin (bottom right, values were normalized with respect to the mean of control condition). Measurements were pooled from three independent experiments; anti‐IgM (BCR‐ligand−): n = 88; anti‐IgG (BCR‐ligand+): n = 93. Error bars correspond to standard deviations. P values were calculated with Mann–Whitney test. Scale bar: 2 μm.
- Percentage differences of centrosomal F‐actin and centrosomal microtubule fluorescence intensities in cells stimulated with BCR‐ligand+ beads with respect to cells stimulated with BCR‐ligand− beads. The data set is identical to panel (B). Measurements were pooled from three independent experiments; anti‐IgM (BCR‐ligand−): n = 88; anti‐IgG (BCR‐ligand+): n = 93. Error bars correspond to standard deviations. P values were calculated with one‐sample t‐test (i.e. comparison to a theoretical mean of “0”).
- The graph shows the variations of the total amount of polymerized tubulin per cell with respect to the content of F‐actin at the centrosome in an XY representation of individual measurements. The two lines correspond to linear regressions of the two sets of data relative to cells stimulated with BCR‐ligand+ (activated cells) or BCR‐ligand− (resting cells) beads.
- IIA1.6 B lymphoma cells were fixed and immuno‐stained for F‐actin (red) and α‐tubulin (green). Images show the projection of maximal intensity of three confocal slices spaced by 0.5 μm apart from the centrosome. Scale bars: 2 μm (0.5 μm in the zoomed insets).
Figure EV1. Correlation analysis between centrosomal or cortical actin networks and microtubule content.
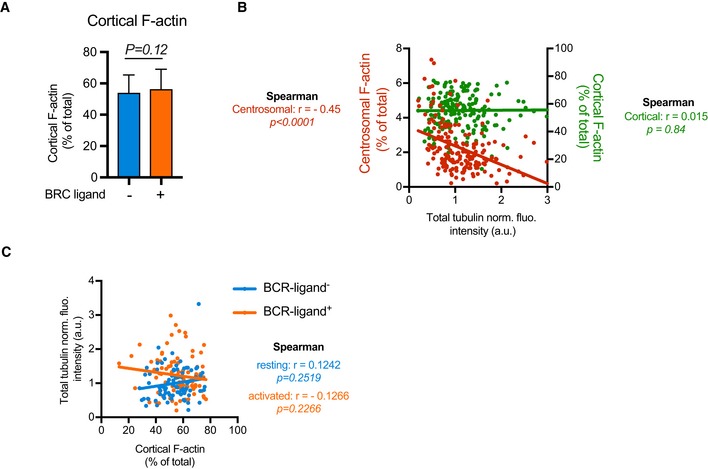
- Histograms show the quantifications of the amount of F‐actin at the cortex of B lymphoma cells. Measurements were pooled from three independent experiments; anti‐IgM (BCR‐ligand−): n = 88; anti‐IgG (BCR‐ligand+): n = 93. Error bars correspond to standard deviations. P values were calculated from Mann–Whitney test.
- Graph shows the variations of the fluorescence intensities of cortical F‐actin (green) or centrosomal F‐actin (red) with respect to the total amount of polymerized tubulin in resting and activated cells (values were normalized with respect to the mean values of all measured cells). The two lines correspond to linear regressions of the two sets of data. The Spearman correlation test indicates the correlation coefficient r and the P value of the significance of the correlation. Only centrosomal actin appeared correlated to the total content of polymerized tubulin.
- The graph shows the variations of the total amount of polymerized tubulin per cell with respect to the content of cortical actin in an XY representation of individual measurements. The two lines correspond to linear regressions of the two sets of data relative to cells stimulated with BCR‐ligand+ (activated cells) or BCR‐ligand− (resting cells) beads. In none of the two cases, the total amount of polymerized tubulin appeared correlated to the proportion of cortical actin filaments.
To test the hypothesis that the density of centrosomal actin is driving the reduction in microtubule density, B lymphocytes were treated with actin filament inhibitors (Fig 2A). Treatment with the actin polymerization inhibitors (Arp2/3 inhibitor CK666) or latrunculin A reduced the centrosomal actin density and increased the microtubule density at the centrosome (Fig 2B and C) and throughout the cell (Fig EV2A), thus supporting the hypothesis. Conversely, treatment with the formin inhibitor, SMIFH2, increased centrosomal actin density, by an unknown mechanism possibly related to the actin homeostasis supporting Arp2/3‐based nucleation of actin filament, notably at the centrosome (Farina et al, 2016), when formin is inhibited (Suarez & Kovar, 2016). This increase in centrosomal actin led to a marginally decreased microtubule density at the centrosome and throughout the cell (Figs 2B and C, and EV2A), thus confirming the negative relationship between the two networks. Overall, the analysis of individual cells showed a negative correlation between centrosomal actin filaments and microtubules. The inhibition of formin and Arp2/3 induced higher and lower actin densities at the centrosome, respectively, and thus expanded the range in which the negative correlation could be observed (Fig 2D).
Figure 2. The impact of modulating centrosomal actin network on microtubules in B lymphocytes.
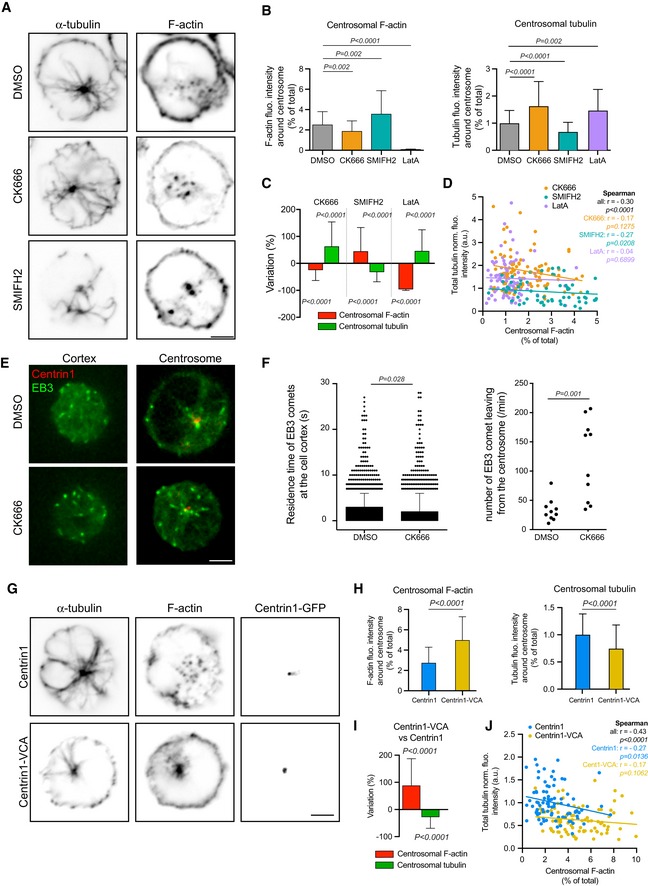
- IIA1.6 B lymphoma cells were treated 45 min with indicated inhibitors (CK666 at 25 μM, SMIFH2 at 25 μM) or DMSO as control prior to being fixed and stained for α‐tubulin (left column) and F‐actin (right column). Scale bar: 3 μm.
- Histograms show the quantifications of the amount of polymerized tubulin (right, values were normalized with respect to the mean of control condition) and F‐actin at the centrosome (left, values correspond to the fraction of fluorescence in a 2‐micron‐wide area around the centrosome relative to the total fluorescence in the cell). Measurements were pooled from three independent experiments; DMSO: n = 91, CK666: n = 82, SMIFH2: n = 74, latrunculin A: n = 96. Error bars correspond to standard deviations. P values were calculated with Mann–Whitney test.
- Percentage differences of centrosomal F‐actin and microtubule fluorescence intensities in cells treated with cytoskeleton inhibitors in comparison with the respective densities in cells treated with DMSO. Error bars represent standard deviations. P values were calculated with one‐sample t‐test (i.e. comparison to a theoretical mean of “0”).
- The graph shows the same measurements as in panel (B) in an XY representation of individual measurements. The three lines correspond to linear regressions of the three sets of data relative to cells treated with each actin drug.
- IIA1.6 B lymphoma cells were transfected to transiently express centrin1‐GFP (red) and EB3‐mCherry (green) and video‐recorded at the contact site with the glass coverslip (left) and at the centrosome (right). Scale bar: 3 μm.
- The duration of EB3‐positive comets’ presence in the bottom plane was measured in DMSO‐ and CK666‐treated cells (left). Error bars correspond to standard deviations. The number of EB3‐positive comets exiting a 2‐μm‐wide centrosomal area was also compared between the two conditions (right). In both cases, P values were calculated with Mann–Whitney test.
- IIA1.6 B lymphoma cells were transfected to transiently express centrin1‐VCA‐GFP (bottom) or centrin1‐GFP (top) as control prior to be fixed and stained for α‐tubulin (left column) and F‐actin (middle column). The GFP signal of centrin1 or centrin1‐VCA is shown in the right column to illustrate the proper centrosome targeting. Scale bar: 3 μm.
- Histograms show the quantifications of the amount of polymerized tubulin (right) and F‐actin at the centrosome (left). Values correspond to the fraction of fluorescence in a 2‐micron‐wide area around the centrosome relative to the total fluorescence in the cell. Measurements were pooled from three independent experiments; centrin1‐GFP: n = 88, centrin1‐VCA‐GFP: n = 87. Error bars represent standard deviations. P values were calculated with Mann–Whitney test.
- Percentage differences of F‐actin and polymerized tubulin fluorescence intensities at the centrosome were compared in cells transfected either with centrin1‐VCA‐GFP or with centrin1‐GFP. Error bars represent standard deviations. P values were calculated with one‐sample t‐test (i.e. comparison to a theoretical mean of “0”).
- The graph shows the variations of the total amount of polymerized tubulin per cell with respect to the content of F‐actin at the centrosome. The two lines correspond to linear regressions of the two sets of data relative to cells transfected with centrin1‐VCA‐GFP or centrin1‐GFP.
Figure EV2. Variations of total and local actin and tubulin content in cells expressing centrin1‐VCA.
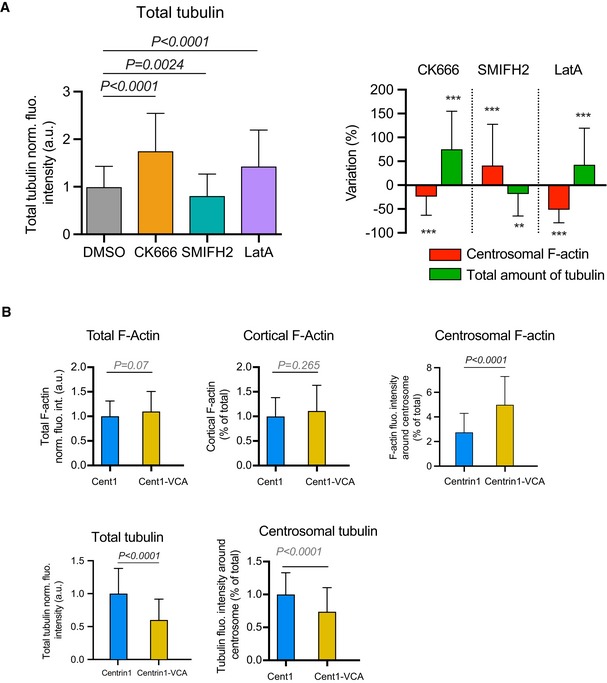
- Histograms on the left show the quantifications of the total amount of polymerized tubulin (values were normalized with respect to the mean of control condition). Error bars correspond to standard deviations. P values were calculated with Mann–Whitney test. Histograms on the right show the percentage differences of centrosomal F‐actin as compared to the variations of the total microtubule fluorescence intensities in cells treated with cytoskeleton inhibitors in comparison with the respective densities in cells treated with DMSO. Error bars represent standard deviations. P values were calculated with one‐sample t‐test (i.e. comparison to a theoretical mean of “0”). The same data set was used for the two types of quantification (and for Fig 2B–D): measurements were pooled from three independent experiments; DMSO: n = 91, CK666: n = 82, SMIFH2: n = 74, latrunculin A: n = 96.
- Histograms show the quantifications of the amount F‐actin in the entire cell (top left), at the cell cortex (top middle) and at the centrosome (top left) as well as the amount of polymerized tubulin in the entire cell (bottom left) and at the centrosome (bottom right). Values at the centrosome correspond to the fraction of fluorescence in a 2‐micron‐wide area around the centrosome relative to the total fluorescence in the cell. Measurements were pooled from three independent experiments; centrin1‐GFP: n = 88, centrin1‐VCA‐GFP: n = 87. Error bars represent standard deviations.
Noteworthy, local perturbations to the actin network could have affected other actin networks in the same cell by a process of actin‐network homeostasis that operates throughout the cell (Burke et al, 2014; Suarez et al, 2014; Suarez & Kovar, 2016). Therefore, an increase in actin density at the centrosome could have been offset by a corresponding decrease in actin density elsewhere in the cell (e.g. in cytoplasmic and cortical networks). To evaluate this effect, we measured the impact of CK666 on the growth of microtubules at the centrosome and along the cortex by quantifying the dynamics of EB3‐mCherry, which labelled microtubule plus ends (Fig 2E, Movie EV1). We found no major difference in the residency time of EB3 comets, and therefore in the microtubule growth, at the cortex (Fig 2F), suggesting that the changes in the cortical actin induced by Arp2/3 inhibition were not responsible for the overall increase in microtubule number. By contrast, treatment with CK666 significantly increased the number of microtubules growing out of the centrosome (Fig 2F), confirming the involvement of centrosomal actin in this regulation.
To assess more directly the role of centrosomal actin filaments, we next examined B lymphocytes which expressed a fusion protein (centrin1‐VCA‐GFP; Obino et al, 2016) that promotes actin filament nucleation at the centrosome specifically (Fig 2G). Hence, the expression of centrin1‐VCA‐GFP strongly increased the density of centrosomal actin filaments and decreased the microtubule density at the centrosome and throughout the cell demonstrating the specific role of actin filaments at the centrosome in the negative regulation of the microtubule network (Figs 2H–J and EV2B).
The centrosomal actin network perturbs the elaboration of the microtubule network in vitro
A limitation to the interpretation of the B‐lymphocyte experiments was that on top of the influence of subcellular actin networks on each other, actin and microtubule networks share numerous signalling pathways (Dogterom & Koenderink, 2019). It was therefore not possible to distinguish purely sterical effects at the centrosome from the modulation of cross‐signalling pathways. To circumvent this limitation, we used an in vitro model that reconstituted actin and microtubule networks from actin monomers and tubulin dimers incubated in the presence of a centrosome labelled with centrin1‐GFP. In this model and as expected (Farina et al, 2016), 25% of the centrosomes (i.e. centrin1‐GFP‐positive puncta) were associated with actin and microtubule networks (Fig 3A). Among those centrosomes, the actin density per centrosome was negatively correlated with the number of microtubules per centrosome (Fig 3B). Actin filament density at the centrosome was then altered by incubating centrosomes in different concentrations of free actin monomers, with tubulin dimer concentration kept constant (Fig 3C). Consistent with the hypothesis, higher actin concentrations were associated with lower microtubule numbers per centrosome (Fig 3D). Moreover, the highest actin concentration almost completely inhibited microtubule growth (Fig 3D). These changes were not due to the interference of dense actin networks with microtubule growth rate, which did not seem to depend on the density of actin network (Fig 3E). They were neither due to the removal of microtubule nucleation complexes from the centrosome since the intensity of gamma‐tubulin staining appeared independent on the presence of actin filaments (Fig 3F). These results from in vitro experiments rather suggest that actin filaments perturb the early stages of microtubule elongation at the centrosome. Therefore, it is plausible that in B‐lymphocyte experiments, the centrosomal actin network had direct and antagonistic effects on the microtubule network emanating from the centrosome.
Figure 3. Assembly of microtubules and F‐actin on isolated centrosomes.

- Two sets of representative images showing fluorescent microtubules and F‐actin assembled from isolated centrosomes. Centrosomes were isolated from Jurkat cells expressing centrin1‐GFP. Upper and lower lines show F‐actin and microtubules radiating from two distinct centrosomes with low (top) and high (bottom) densities of F‐actin. Scale bars: 10 μm.
- The graph shows the number of microtubules per centrosome relative to the density of actin filaments. Inset shows F‐actin at the centrosome with a FIRE look‐up table and a 20‐μm‐wide circle in which F‐actin fluorescence intensity is measured. Measurements were pooled from five independent experiments; n = 50.
- Microtubules (top line) and F‐actin (bottom line) assembly from isolated centrosomes in the presence of increasing concentration of monomeric actin (from left to right). Scale bar: 20 μm.
- The graph shows the number of microtubules per centrosome in response to increasing concentrations of monomeric actin. Data were pooled from two independent experiments; 0 μM: n = 21; 0.3 μM: n = 17; 0.5 μM: n = 17; 1.0 μM: n = 17. ****P < 0.001 Mann–Whitney test.
- The image shows the density of F‐actin (in the presence of 1 μM of actin monomers) at the centrosome colour‐coded with the FIRE look‐up table and the definition of central, peripheral and distal regions corresponding to decreasing concentrations of F‐actin. Scale bar: 20 μm. The graph shows the measurements of microtubule growth rate in each region. Data were pooled from three independent experiments: central zone: n = 58, peripheral: n = 104, outside: n = 61; n. s. means no statistical difference between the data set according to Mann–Whitney test.
- The graphs show the various intensities of centrosome immuno‐staining with antibodies against gamma‐tubulin on the same coverslip depending on the presence/absence of F‐actin (left) or on the amount of F‐actin (right). Data were pooled from three independent experiments. Left graph: without F‐actin n = 69, with F‐actin n = 26, right graph n = 26. n.s. means no statistical difference between the data set according to Mann–Whitney test.
To further explore the dynamics of the interaction between the centrosomal actin network and the microtubule network, the in vitro model was manipulated by sequential addition of the network components. By incubating with tubulin dimers first, microtubules formed in the absence of actin filaments (Fig 4A and B). When actin monomers were introduced afterwards (together with tubulin dimers to maintain the tubulin dimer concentration), the number of microtubules increased on all centrosomes, irrespective of whether centrosomes triggered the formation of actin filaments or not (Fig 4C). An explanation for this unexpected observation was that the addition of new tubulin dimers increased the effective concentration of free tubulins. Furthermore, not all centrosomes were capable of nucleating actin filaments, and there was no difference in the microtubule numbers per centrosome between those centrosomes with and those without actin filaments (Fig 4C). This suggested that in this model, the stability of preassembled microtubules may not be sensitive to actin filaments that form at the microtubule ends proximal to the centrosome, and newly assembled microtubules could form in spaces along pre‐existing microtubules or in spaces created from depolymerized microtubules.
Figure 4. Blockage of microtubule growth by F‐actin on isolated centrosomes.
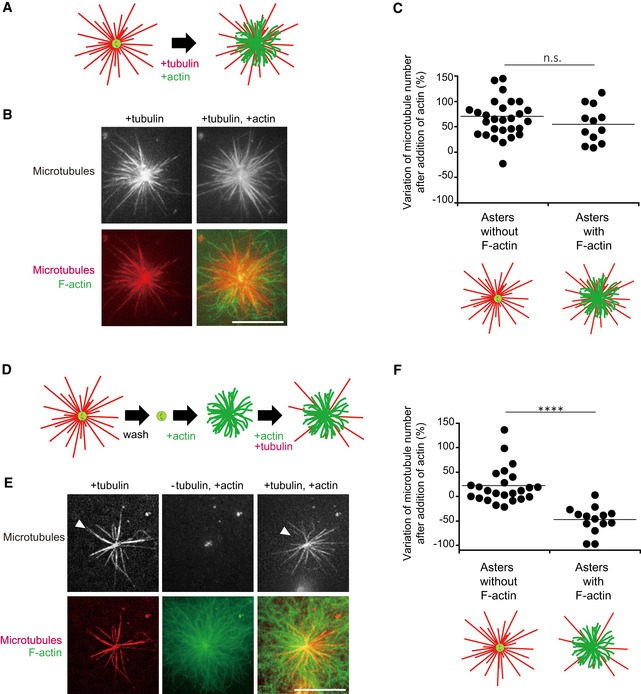
- Schematic illustration of the first dynamic assay: sequential addition of tubulin followed by tubulin and actin on isolated centrosomes.
- Representative images showing microtubules (top line) and the merged images of F‐actin and microtubules (bottom line) for the two steps of the assay; in the presence of tubulin only (left column) and in the presence of tubulin and actin (right column). Scale bar: 10 μm.
- Quantification of the differences in the number of microtubules per centrosome between the two stages of the experiment described above on centrosomes capable (first condition), or not (second condition), to grow F‐actin. Data were collected from a single experiment; asters without F‐actin: n = 29; asters with F‐actin: n = 13. Data were analysed using Mann–Whitney test.
- Schematic illustration of the second dynamic assay: tubulin is added to measure centrosome nucleation capacity and washed out. Then, actin is added followed by actin and tubulin.
- Representative images showing microtubules (top line) and the merged images of F‐actin and microtubule (bottom line) during the three steps of the assay; in the presence of tubulin only (left column), in the absence of tubulin and presence of actin (middle column) and in the presence of tubulin and actin (right column). Scale bar: 10 μm. Arrowheads indicate microtubules unable to re‐grow after assembly of F‐actin.
- Quantification of the differences in the number of microtubules per centrosome between the first and last steps of the experiment described above (panels D and E) on centrosomes capable (first condition), or not (second condition), to grow F‐actin. Data were pooled from two independent experiments; asters without F‐actin: n = 24; asters with F‐actin: n = 13. ****P < 0.001 Student's t‐test.
In a second experiment, tubulin dimers were initially added to quantify the number of microtubules per centrosome and, in effect, to select those centrosomes with the capability to nucleate microtubules. The tubulin dimers and microtubules were then removed by rinsing the centrosomes in buffer. Actin monomers were then added, followed by tubulin dimers again (Fig 4D–F). For those centrosomes devoid of actin filaments, the microtubule number was not significantly different between the initial and final stages of the experiment (Fig 4F). By contrast, for centrosomes which nucleated actin filaments, the microtubule number was significantly reduced at the final stage compared to the initial stage (Fig 4F). This effect was not due to actin filaments impact on the centrosome itself since the number of microtubules was not reduced if actin filaments were disassembled prior to microtubule regrowth (Fig EV3). These experiments confirmed that microtubule regrowth was impaired in the presence of pre‐existing actin filaments.
Figure EV3. Impact of actin filament growth on the number of microtubule nucleation sites in isolated centrosomes.
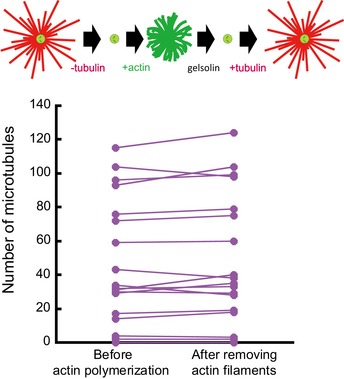
These data are associated with the set of experiments shown in Fig 4D–F and aimed at testing the impact of actin filament growth on the composition and structure of isolated centrosomes. Top line shows a schematic illustration of the dynamic assay: tubulin is added to measure centrosome nucleation capacity and washed out. Then, actin is added and later disassembled with gelsolin. This step of actin assembly/disassembly is followed by the addition of tubulin to measure again centrosome nucleation capacity. The amount of microtubule per centrosome is compared between the first and the last step of tubulin addition. No significant difference could be detected (Student's paired t‐test).
Actin filaments block microtubule growth in a biochemical model
In the above in vitro model, only 25% of the isolated centrosomes had the capability of nucleating microtubules, reflecting the difficulties in centrosome purification. Despite the optimization steps to improve the quality of the centriole (Gogendeau et al, 2015), the isolation step results in centrosome with more or less fragmented peri‐centriolar material. As a consequence, the investigation of their nucleation capacities was informative but intrinsically biased. Therefore, to directly test steric competition between actin and microtubules during the first stages of microtubule growth, we combined two distinct biochemical assays in which short microtubule seeds and actin nucleators were grafted onto the same microfabricated spot on a planar surface in vitro (Reymann et al, 2010; Portran et al, 2013) (Fig 5A).
Figure 5. Reconstitution of the interplay between F‐actin and microtubules on micropatterns.
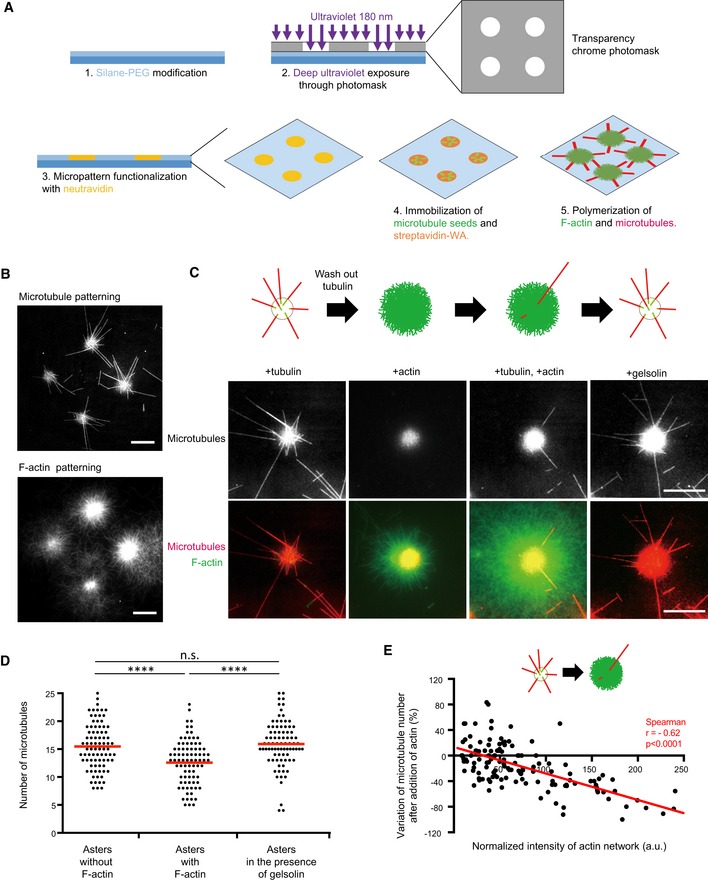
- Schematic illustration of the micropatterning method used to graft microtubule seeds (green) via neutravidin (yellow) and F‐actin‐nucleation‐promoting complexes (streptavidin‐WA) (orange) on 8‐micron‐wide discoidal micropatterns. A glass coverslip (deep blue) coated with polyethyleneglycol (PEG) (light blue) was placed in contact with a transparency photomask and exposed to deep UV light. The exposed coverslip was then immersed with neutravidin to fix biotinylated microtubule seeds (green) on exposed regions. Streptavidin‐WA was immobilized on microtubule seeds via their interaction with biotin. Tubulin dimers and actin monomers were then added to allow filaments elongation.
- Representative images of microtubules (top) and F‐actin (bottom) growth from micropatterns. Scale bars: 20 μm.
- Schematic illustration of the assay on micropatterned substrate. Tubulin was first added alone to measure the nucleation capacity of each micropattern, and then washed out. Later on, actin was added followed by actin and tubulin. Finally, actin was rinsed out and gelsolin was added to fully disassemble F‐actin. Representative images showing microtubules (top line) and the merged images of actin filaments and microtubules (bottom line) during the four steps of the assay; in the presence of tubulin only, in the absence of tubulin and presence of actin, in the presence of tubulin and actin, and finally in the presence of tubulin and gelsolin but in the absence of actin (from left to right). Scale bars: 10 μm.
- Quantification of the number of microtubules per micropattern in the presence of tubulin only (left), actin and tubulin (middle) and tubulin only after actin filament disassembly (right). Data were pooled from 2 independent experiments; n = 133. ****P < 0.001 Student's t‐test.
- The graph shows the same measurements as in panel (E) in an XY representation of individual measurements. It illustrates the differences in the number of microtubules per micropattern between the first to the second step (tubulin only versus actin and tubulin together) with respect to the density of F‐actin per micropattern.
In the biochemical model, the addition of free tubulin dimers and actin monomers led to the growth of both actin filaments and microtubules from each micropattern (Fig 5B). As with the in vitro model above, the micropatterns were treated according to the following sequence: addition of tubulin dimers and growth of microtubules; microtubule count; wash; addition of actin monomers and growth of actin filaments; addition of tubulin dimers and microtubule regrowth (Fig 5C). The model showed again that microtubule formation was perturbed by the presence of actin filaments (Fig 5D). Interestingly, the addition of gelsolin to promote the disassembly of actin filaments overcame the perturbation, indicating that the nucleation of actin filaments did not detach microtubule seeds (Fig EV4) but blocked their elongation (Fig 5C and D). Moreover, the relative density of actin was negatively correlated with microtubule numbers (Fig 5E). Therefore, given the absence of signalling pathways or cross‐linking proteins, the actin filaments physically blocked microtubule growth, and the denser the actin network, the stronger the barrier.
Figure EV4. Impact of actin filament growth on the number of microtubule seeds on micropatterns.
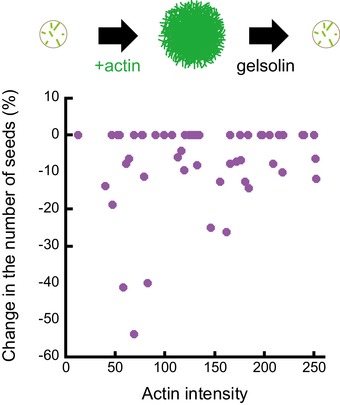
These data are associated with the set of experiments shown in Fig 5C–E and aimed at testing the impact of actin filament growth on the presence of microtubule seeds on the micropatterned region. Top line shows a schematic illustration of the dynamic assay: the number of microtubule seeds was counted on each micropattern, then actin was added to trigger actin filament assembly on the micropattern, then actin filaments were disassembled with gelsolin and the amount of microtubule seeds was counted again. The graph shows the variation of the amount of seeds before and after the growth of actin filaments depending on the density of the actin network. Dense networks did not seem to remove more seeds than sparse actin networks.
Actin filament density at the centrosome is negatively affected by the degree of cell spreading
The experiments above supported the model in which actin filaments perturb the formation of microtubules at the centrosome by forming a physical barrier. This led us to investigate how actin density at the centrosome is regulated in living cells. We have previously shown that with B lymphocyte forming an immune synapse with antigen‐presenting cells, actin nucleation is decreased at the centrosome (Obino et al, 2016). Because immune synapses are enriched for actin and adhesion molecules such as integrins (Carrasco et al, 2004; Bretou et al, 2016), we hypothesized that the actin filament density at the centrosome is inversely related to the degree of cell adhesion and spreading because actin nucleating structures compete for available actin monomers in the cell (Suarez & Kovar, 2016). Hence, minimal cell spreading permits a high amount of actin filaments to form at the centrosome, thus perturbing microtubule growth, whereas extensive cell spreading sequesters most of the available actin monomers, reducing the number of actin filaments at the centrosome and thus favouring microtubule growth (Fig 6A).
Figure 6. Modulation of microtubule growth by cell spreading and centrosomal actin filaments.
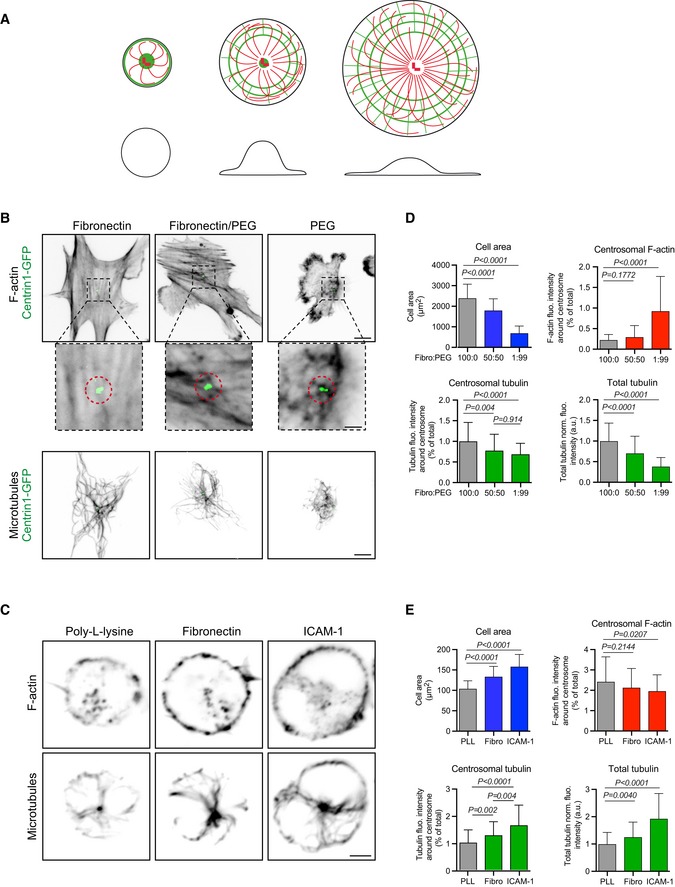
- Schematic illustration of our model according to which cell spreading sequesters monomeric actin to the cortex and thereby enables the centrosome to grow more microtubules. Drawings show top (top line) and side views (bottom line) of cells with increased spreading from left to right. Actin filaments are in green; microtubules are in red.
- RPE1 cells stably expressing centrin1‐GFP were plated for 3 h on coverslips coated with different ratios (100:0; 50:50 or 1:99) of fibronectin and PLL‐PEG prior to fixation and staining for F‐actin (top line and magnified views around centrosome below. Scale bars: 10 μm and 2 μm, respectively) and α‐tubulin (bottom line. Scale bar: 10 μm).
- IIA1.6 B lymphoma cells were plated for 60 min on poly‐L‐lysine, fibronectin or ICAM‐1‐coated cover slides prior to be fixed and stained for F‐actin (top line) and α‐tubulin (bottom line). Scale bar: 3 μm.
- Quantification of the area occupied by RPE1 cells on the substrate (top left), F‐actin content at the centrosome (top right), polymerized tubulin at the centrosome (bottom left) and in the entire cell (bottom right) for the three conditions of cell adhesion described in (B). Measurements came from three independent experiments with more than 60 analysed cells in each. Error bars represent standard deviations. F‐actin and microtubule contents were compared using Mann–Whitney test, and variations of the cell area were compared using unpaired t‐test.
- Quantification of the area occupied by B lymphoma cells on the substrate (top left), F‐actin content at the centrosome (top right), polymerized tubulin at the centrosome (bottom left) and in the entire cell (bottom right) for the three conditions of cell adhesion described in (D). Measurements came from three independent experiments with more than 80 analysed cells in each. Error bars represent standard deviations. F‐actin and microtubule contents were compared using Mann–Whitney test, and variations of the cell area were compared using unpaired t‐test.
For highly adherent RPE1 cells, three states of cell spreading (low, medium and high) were dictated by the degree of substrate adhesiveness (by tuning fibronectin concentration in PEG; Fig 6B). For low‐adherent B lymphocytes, three states of cell adhesion and spreading were dictated by plating on poly‐L‐lysine, fibronectin and ICAM‐1 (Carrasco et al, 2004) (Fig 6C). For both cell types, the degree of cell adhesion and/or spreading (i.e. the area occupied by the cell on the substrate) was negatively correlated with centrosomal actin density and positively correlated with the density of microtubules at the centrosome and throughout the cell (Fig 6D and E). Although these results do not indicate the exact mechanism by which cell spreading modulates the amount of microtubules, and notably do not exclude the possibility that microtubules were stabilized by contact with focal adhesions (Byron et al, 2015; Bouchet et al, 2016), they support a model in which microtubule growth from the centrosome is modulated by the adhesion state of the cell via the degree to which actin filaments are prevented from forming at the centrosome.
Discussion
Actin is the most abundant protein in the cytoplasm and as such has long been considered as a major contaminant of centrosome proteomic studies (Bornens & Moudjou, 1999; Andersen et al, 2003). However, actin filaments have been directly observed at the poles of mitotic spindles (Stevenson et al, 2001; Chodagam et al, 2005) and at the centrosome of several cell types in interphase (Farina et al, 2016; Obino et al, 2016; Au et al, 2017). Centrosomal actin filaments have been shown to anchor the centrosome to the nucleus (Bornens, 1977; Burakov & Nadezhdina, 2013; Obino et al, 2016), support the transport of vesicles during ciliogenesis (Assis et al, 2017; Wu et al, 2018), connect basal bodies to the actin cortex in ciliated cells (Pan et al, 2007; Antoniades et al, 2014; Walentek et al, 2016; Mahuzier et al, 2018) and power centrosome splitting in prophase (Uzbekov et al, 2002; Wang et al, 2008; Au et al, 2017).
The results of our study identify a new function for actin filaments at the centrosome. Noteworthy, in the lymphocytes we analysed, actin filaments formed dense clouds in close proximity to the centrosome, i.e. within a micrometre from the centre of the microtubule array, but did not seem to colocalize with the centrosome (Fig 1E), raising some doubts about the actual origin of these filaments. It is still unclear whether actin filaments were nucleated at the centrosome, which would be consistent with the localization of Arp2/3 at the centrosome (Farina et al, 2016) or at endosomes, where WASH also triggers actin filament assembly (Derivery et al, 2009), which were later gathered around the centrosome. Regardless of their actual origin, we propose a model in which these centrosomal, or peri‐centrosomal, actin filaments provide a conduit through which changes to actin networks at the cell periphery modulate the formation and growth of microtubules emanating from the centrosome. The centrosomal actin filaments primarily perturb the formation of microtubules by physically blocking the early stages of their elongation. Although the actin networks adopted quite different architectures in cells and in reconstituted experiments in vitro, i.e. tiny clouds and radial array, respectively, in both cases the density of the network blocked microtubule elongation. Although we cannot exclude that other mechanisms, such as shared signalling pathways or competition for common resources, support the negative impact of centrosomal actin filaments on microtubules in cells, we favoured the interpretation based on the role of physical constraints since they exist in cells and were proven to be capable to block microtubule growth in our in vitro assays. But these physical constraints may not be the only mechanism co‐regulating the two networks at the centrosome. Noteworthy, these results add to pre‐existing body of evidences showing that physical constraints imposed by actin filaments (Huber et al, 2015) can limit microtubule growth (Colin et al, 2018), microtubule's shape fluctuations (Brangwynne et al, 2006; Katrukha et al, 2017) and centrosome displacement (Piel et al, 2000). Interestingly, by contrast with previous descriptions of physical barriers blocking microtubule growth locally (Katrukha et al, 2017; Colin et al, 2018) our observations show that centrosomal actin filaments, by preventing microtubule growth at the organizing centre, affect the entire microtubule network throughout the cell.
Our results expand the description of cytoskeleton changes during B‐lymphocyte activation (Obino et al, 2016) and show that centrosomal actin filament disassembly promotes the growth of microtubules. Interestingly, the increase in microtubules may contribute to B‐cell polarization, a hallmark of their activation (Yuseff et al, 2011), by promoting centrosome off‐centring. Indeed, a high quantity of microtubules can break network symmetry and force centrosome off‐centring and its displacement to the cell periphery through the reorientation of pushing forces produced at the centrosome by microtubule growth (Letort et al, 2016; Burute et al, 2017; Pitaval et al, 2017). Therefore, centrosomal actin filament disassembly could be involved both in the disengagement of the centrosome from the nucleus (Obino et al, 2016) and in the stimulation and reorganization of microtubule‐based pushing forces to drive centrosome motion towards the cell periphery.
The regulation of microtubule growth at the cell centre complements those mechanisms that regulate microtubule stability at the cell periphery, where microtubule stability is promoted by cell adhesions and their associated actin networks (Akhmanova & Steinmetz, 2015; Byron et al, 2015; Bouchet et al, 2016). Those mechanisms ensure a form of regulation that can bias microtubule network organization locally (Gundersen et al, 2004; Etienne‐Manneville, 2013). At the cell centre, the actin network can adapt the entire microtubule network to cell shape, cell adhesion and cell spreading (Fig 6A). An explanation for this is that cell adhesion and cell spreading trigger the elaboration of actin networks at the cortex, hence reducing the pool of available actin monomers, and potentially sequestering from the centrosome actin filament nucleation and branching factors such as Arp2/3 and WASH (Farina et al, 2016; Obino et al, 2016; Suarez & Kovar, 2016). The reduction in the centrosomal actin network thus allows more microtubules to be nucleated at the centrosome. The interplay at the centrosome between actin filaments and microtubules in response to cell spreading may have important implications for the ability of the cell to sense and adapt to external cues.
Materials and Methods
Cell culture and chemical treatments
Stable Jurkat cell lines expressing centrin1‐GFP (Farina et al, 2016) were cultured in RPMI 1640 (Gibco). Cells were not sorted based on GFP fluorescence. The mouse B lymphoma cell line IIA1.6 (derived from the A20 cell line (American Type Culture Collection #: TIB‐208)) was cultured as reported (Obino et al, 2016) in CLICK medium (RPMI 1640—GlutaMax‐I), supplemented with 0.1% β‐mercaptoethanol and 2% sodium pyruvate. The RPE1 cell line stably expressing centrin1‐GFP (Farina et al, 2016) was cultured in DMEM/F‐12. All media were supplemented with 10% foetal calf serum and penicillin/streptomycin. Cells were cultured at 37°C and 5% CO2. All cell lines were tested monthly for mycoplasma contamination.
Two million IIA1.6 cells were electroporated with 2 μg of EB3‐mCherry plasmid and 2 μg centrin1‐GFP plasmid using the Amaxa Cell Line Nucleofector Kit R (T‐016 programme, Lonza). Cells were incubated in CLICK medium for 8–12 h before analysis.
Cytoskeleton inhibitors (CK666 at 25 μM, SMIFH2 at 25 μM; Latrunculin A at 5 μM; all from Tocris Bioscience) were added in the cell medium for 45 min at 37°C.
For the coating of glass coverslips, fibronectin (Sigma‐Aldrich) was used at 10 μg/ml and PLL‐PEG (JenKem Technologies, Texas) at 10 μg/ml in HEPES 10 mM, poly‐L‐Lysine (Invitrogen) was used at 10 μg/ml, and ICAM‐1 (R&D System) was used at 10 μg/ml.
Preparation of BCR‐ligand‐coated beads
Latex NH2‐beads 3 μm in diameter (Polyscience) were activated with 8% glutaraldehyde (Sigma‐Aldrich) for 2 h at room temperature (4 × 107 beads/ml). Beads were washed with PBS and incubated overnight at 4°C with 100 μg/ml of either F(ab′)2 goat anti‐mouse IgG (BCR‐ligand+ beads) or F(ab′)2 goat anti‐mouse IgM (BCR‐ligand− beads; MP Biomedical).
Cell fixation and immuno‐staining
Cells were extracted by incubation for 15 sec with cold cytoskeleton buffer (10 mM MES pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA) supplemented with 0.5% Triton X‐100 and fixed with cytoskeleton buffer supplemented with 0.5% glutaraldehyde for 10 min at room temperature. Glutaraldehyde was reduced with 0.1% sodium borohydride (NaBH4) in 1× PBS for 7 min, and unspecific binding sites were saturated using a solution of 1× PBS supplemented with 2% BSA and 0.1% Triton X‐100 for 10 min. The following primary antibodies were used: monoclonal rat anti‐α‐tubulin (AbD Serotec, Clone YL1/2, 1/1,000) and VHH anti‐green fluorescent protein (GFP)‐human Fc (Recombinant Antibodies Platform (TAb‐IP), Institut Curie, Paris, France, 1/200). The following secondary antibodies were used: AlexaFluor647‐conjugated F(ab′)2 donkey anti‐rat and AlexaFluor488‐conjugated donkey anti‐human (Life Technologies, both 1/200). Actin filaments were stained using AlexaFluor546‐conjugated phalloidin (Life Technologies, #A22283, 1/100).
Isolation of centrosomes
Centrosomes were isolated from Jurkat cells by modifying a previously published protocol (Moudjou & Bornens, 1998; Gogendeau et al, 2015). In brief, cells were treated with nocodazole (0.2 μM) and cytochalasin D (1 μg/ml) followed by hypotonic lysis. Centrosomes were collected by centrifugation onto a 60% sucrose cushion and further purified by centrifugation through a discontinuous (70, 50 and 40%) sucrose gradient. The composition of the sucrose solutions was based on a TicTac buffer (Farina et al, 2016), in which the activity of tubulin, actin and actin‐binding proteins is maintained: 10 mM HEPES, 16 mM Pipes (pH 6.8), 50 mM KCl, 5 mM MgCl2, 1 mM EGTA. The TicTac buffer was supplemented with 0.1% Triton X‐100 and 0.1% β‐mercaptoethanol. After centrifugation on the sucrose gradient, supernatant was removed until only about 5 ml remained in the bottom of the tube. Centrosomes were stored at −80°C after flash freezing in liquid nitrogen.
Protein expression and purification
Tubulin was purified from fresh bovine brain by three cycles of temperature‐dependent assembly/disassembly in Brinkley Buffer 80 (BRB80 buffer: 80 mM Pipes pH 6.8, 1 mM EGTA and 1 mM MgCl2) (Shelanski, 1973). Fluorescently labelled tubulins (ATTO‐488‐ and ATTO‐565‐labelled tubulin) were prepared by following previously published method (Hyman et al, 1991).
Actin was purified from rabbit skeletal‐muscle acetone powder. Monomeric Ca‐ATP‐actin was purified by gel‐filtration chromatography on Sephacryl S‐300 at 4°C in G buffer (2 mM Tris–HCl, pH 8.0, 0.2 mM ATP, 0.1 mM CaCl2, 1 mM NaN3 and 0.5 mM dithiothreitol (DTT)). Actin was labelled on lysines with Alexa‐488 and Alexa‐568. Recombinant human profilin, mouse capping protein, the Arp2/3 complex and GST‐streptavidin‐WA were purified in accordance with previous methods (Michelot et al, 2007; Achard et al, 2010).
In vitro assays with isolated centrosomes
Experiments were performed in polydimethylsiloxane (PDMS) stencils in order to add/exchange sequentially experimental solutions when needed. PDMS (Sylgard 184 kit, Dow Corning) was mixed with the curing agent (10:1 ratio), degassed, poured into a Petri dish to a thickness of 5 mm and cured for 2 h at 80°C on a hot plate. The PDMS layer was cut to square shape with dimension of 10 × 10 mm and punched using a hole puncher (Ted Pella) with an outer diameter of 6 mm. The PDMS chamber was oxidized in an oxygen plasma cleaner for 40 s at 60W (Femto, Diener Electronic) and brought it into contact with clean coverslip (24 × 30 mm) via a double‐sided tape with 6‐mm hole.
Isolated centrosomes were diluted in TicTac buffer and incubated for 20 min. To remove excess of centrosomes and coating the surface of coverslips, TicTac buffer supplemented with 1% BSA was perfused into the PDMS chamber, which was followed by a second rinsing step with TicTac buffer supplemented with 0.2% BSA and 0.25% w/v methylcellulose. Microtubules and actin assembly at the centrosome were induced using a reaction mixture containing tubulin dimers (labelled with ATTO‐565, 18 μM final) and actin monomers (labelled with Alexa‐488, 0.3–1.0 μM final) in TicTac buffer supplemented with 1 mM GTP and 2.7 mM ATP, 10 mM DTT, 20 μg/ml catalase, 3 mg/ml glucose, 100 μg/ml glucose oxidase and 0.25% w/v methylcellulose. In addition, a threefold molar equivalent of profilin to actin and 60 nM Arp2/3 complex were added in the reaction mixture.
Sequential microtubule and actin filament assembly experiments were carried out based on the aforementioned method. In brief, after assembling microtubules by adding tubulin in the reaction mixture (18 μM final) for 15 min, microtubules were removed by exchanging the reaction mixture with TicTac buffer supplemented with 0.2% BSA and 0.25% w/v methylcellulose. Subsequently, the reaction mixture of actin (1 μM final) with profilin and Arp2/3 was applied to assemble the actin aster. After 15‐min incubation, the tubulin reaction mixture with actin, profilin and Arp2/3 complex was added to assemble both microtubules and actin asters together.
Micropatterning
Micropatterning of microtubules and actin filaments was performed in accordance with previously published methods with modification (Reymann et al, 2010; Portran et al, 2013). In brief, cleaned glass coverslips were oxidized with oxygen plasma (5 min, 60 W, Femto, Diener Electronic) and incubated with polyethyleneglycol silane (5 kDa, PLS‐2011, Creative PEGWorks, 1 mg/ml in ethanol 96.5 and 0.02% of HCl) solution for overnight incubation. PEGylated coverslips were placed on a chromium quartz photomask (Toppan Photomasks, Corbeil, France) using a vacuum holder. The mask‐covered coverslips were then exposed to deep ultraviolet light (180 nm, UVO Cleaner, Jelight Company, Irvine, CA) for 5 min. The PDMS open chamber was assembled as described above. Neutravidin (0.2 mg/ml in 1× HKEM [10 mM HEPES pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM EGTA]) was perfused in PDMS chamber and incubated for 15 min. The biotinylated microtubule seeds, which were prepared with 25% of fluorescent‐dye‐labelled tubulin and 75% biotinylated tubulin in presence of 0.5 mM of GMPCPP as previously described (Portran et al, 2013), were deposited on neutravidin‐coated surface. Subsequently, 1 μM of streptavidin‐WA in 1× HKEM was added into the PDMS chamber. After each step, the excess of unbound proteins was washed away using wash buffer. Microtubules and actin filaments were assembled according to the above protocol (see In vitro assays), except that 120 nM of Arp2/3 complex was used instead of 60 nM. To disassemble actin filaments on the micropatterns, gelsolin (1.6 μM, gift from Robert Robinson laboratory, IMCB, Singapore) was added into the reaction mixture at the last step of the experiment.
Imaging and analysis
Cell imaging was performed on an inverted spinning disc confocal microscope (Nikon) with a EMCCD QuantEM (Photometrics) camera. Z‐stack images (0.5 μm spacing) of fixed cells were acquired with a 60× oil immersion objective (NA 1.4). Live cell images were acquired using ×100 oil immersion objective (NA 1.4) every second at the two planes (centrosome and cortex). Image processing was performed with Fiji (ImageJ) software. Centrosomal actin filaments were quantified as previously described (Obino et al, 2016). Briefly, after selecting manually the centrosome plane, we performed a background subtraction (rolling ball 50 px) on the z‐projection (by calculation of pixel average intensity) of the three planes above and below the centrosome. The total fluorescence of centrosomal actin filaments was measured in a 1.6‐μm‐wide disc centred on the centrosome, and the total fluorescence of microtubules was measured in the entire cell.
The imaging of microtubules, actin filaments and centrosomes in the in vitro experiments was performed with a total internal reflection fluorescence (TIRF) microscope (Roper Scientific) equipped with an iLasPulsed system and an Evolve camera (EMCCD) using 60× Nikon Apo TIRF oil‐immersion objective lens (N.A = 1.49). The microscope stage was maintained at 37°C by means of a temperature controller to obtain an optimal microtubule growth. Multi‐stage time‐lapse movies were acquired using Metamorph software (version 7.7.5, Universal Imaging). Actin‐nucleation activity was quantified by measuring the actin filament fluorescence intensity integrated over a 20 μm diameter at the centre of the actin aster and normalized with respect to initial background intensity. The number of microtubules was manually counted from fluorescence microscopy images. All the measurements were done using Adobe Photoshop CC, and the corresponding graphs were produced using KaleidaGraph 4.0.
Statistics
For the in vitro experiments (Figs 3, 4, 5), statistical differences were identified using the unpaired t‐test with Welch's correction and KaleidaGraph software. For the cellular studies (Figs 1, 2 and 6), statistical differences were computed using GraphPad Prism 7 Software. No statistical method was used to determine sample size. Kolmogorov–Smirnov test was used to assess normality of all data sets. The following tests were used to determine statistical significance: Figs 1B, 2B, F and H, 3D, 4C and 6C (actin and microtubules) and 6E (actin and microtubules): Mann–Whitney test; Figs 3E and F, 4A, 5D and 6C (cell area) and 6E (cell area): unpaired t‐test; Figs 1C and 2C and I: one‐sample t‐test (comparison to a theoretical mean of zero, where zero represents no difference between conditions); Figs 1D, 2D and H, and 5E: Spearman's correlation test. Bar graphs describe the mean ± standard deviation.
Author contributions
DI performed all experiments in reconstituted systems. DO performed all experiments in living cells except those related to EB3 dynamics, which were performed by JP. FF performed preliminary experiments in reconstituted systems. JG and CG were involved in experiments in reconstituted systems. A‐ML‐D, LB and MT designed the project, obtained the funding to support it and supervised the work. MT wrote the manuscript. All authors reviewed/edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Review Process File
Acknowledgements
We acknowledge the Recombinant Antibodies Platform (TAb‐IP) at Institut Curie, Paris, France for providing the VHH anti‐GFP‐human Fc antibody. We thank Pablo Saez (Institut Curie, France) for providing the EB3‐mCherry plasmid and Robert Robinson (IMCB, Singapore) for providing the purified gelsolin. DI was supported by a fellowship from the Uehara Memorial Foundation (Japan). DO was supported by a fellowship from the Fondation pour la Recherche Médicale (FDT20150532056). Funding was obtained from the Association Nationale pour la Recherche (ANR‐PoLyBex‐12‐BSV3‐0014‐001 to A‐ML‐D and ANR‐Mitotube‐12‐BSV5‐0004‐01 to MT) and the European Research Council (ERC‐Strapacemi‐GA 243103 to A‐ML‐D and ERC‐SpiCy 310472 to MT).
The EMBO Journal (2019) 38: e99630
See also: F Farina et al (June 2019)
Contributor Information
Laurent Blanchoin, Email: laurent.blanchoin@cea.fr.
Ana‐Maria Lennon‐Duménil, Email: ana-maria.lennon@curie.fr.
Manuel Théry, Email: manuel.thery@cea.fr.
References
- Achard V, Martiel J‐L, Michelot A, Guérin C, Reymann A‐C, Blanchoin L, Boujemaa‐Paterski R (2010) A “primer”‐based mechanism underlies branched actin filament network formation and motility. Curr Biol 20: 423–428 [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Steinmetz MO (2015) Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol 16: 711–726 [DOI] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426: 570–574 [DOI] [PubMed] [Google Scholar]
- Antoniades I, Stylianou P, Skourides PA (2014) Making the connection: ciliary adhesion complexes anchor basal bodies to the actin cytoskeleton. Dev Cell 28: 70–80 [DOI] [PubMed] [Google Scholar]
- Assis LHP, Silva‐Junior RMP, Dolce LG, Alborghetti MR, Honorato RV, Nascimento AFZ, Melo‐Hanchuk TD, Trindade DM, Tonoli CCC, Santos CT, Oliveira PSL, Larson RE, Kobarg J, Espreafico EM, Giuseppe PO, Murakami MT (2017) The molecular motor Myosin Va interacts with the cilia‐centrosomal protein RPGRIP1L. Sci Rep 7: 43692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au FKC, Jia Y, Jiang K, Grigoriev I, Hau BKT, Shen Y, Du S, Akhmanova A, Qi RZ (2017) GAS2L1 is a centriole‐associated protein required for centrosome dynamics and disjunction. Dev Cell 40: 81–94 [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa‐Paterski R, Sykes C, Plastino J (2014) Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94: 235–263 [DOI] [PubMed] [Google Scholar]
- Bornens M (1977) Is the centriole bound to the nuclear membrane? Nature 270: 80–82 [DOI] [PubMed] [Google Scholar]
- Bornens M, Moudjou M (1999) Studying the composition and function of centrosomes in vertebrates. Methods Cell Biol 61: 13–34 [DOI] [PubMed] [Google Scholar]
- Bouchet BP, Gough RE, Ammon Y‐C, van de Willige D, Post H, Jacquemet G, Altelaar AFM, Heck AJR, Goult BT, Akhmanova A (2016) Talin‐KANK1 interaction controls the recruitment of cortical microtubule stabilizing complexes to focal adhesions. Elife 5: e18124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, MacKintosh FC, Kumar S, Geisse NA, Talbot J, Mahadevan L, Parker KK, Ingber DE, Weitz DA (2006) Microtubules can bear enhanced compressive loads in living cells because of lateral reinforcement. J Cell Biol 173: 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretou M, Kumari A, Malbec O, Moreau HD, Obino D, Pierobon P, Randrian V, Sáez PJ, Lennon‐Duménil AM (2016) Dynamics of the membrane–cytoskeleton interface in MHC class II‐restricted antigen presentation. Immunol Rev 272: 39–51 [DOI] [PubMed] [Google Scholar]
- Burakov AV, Nadezhdina ES (2013) Association of nucleus and centrosome: magnet or velcro?. Cell Biol Int 37: 95–104 [DOI] [PubMed] [Google Scholar]
- Burke TA, Christensen JR, Barone E, Suarez C, Sirotkin V, Kovar DR (2014) Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr Biol 24: 579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette DT, Schaefer AW, Ji L, Danuser G, Forscher P (2007) Filopodial actin bundles are not necessary for microtubule advance into the peripheral domain of Aplysia neuronal growth cones. Nat Cell Biol 9: 1360–1369 [DOI] [PubMed] [Google Scholar]
- Burute M, Prioux M, Blin G, Truchet S, Letort G, Tseng Q, Bessy T, Lowell S, Young J, Filhol O, Théry M (2017) Polarity reversal by centrosome repositioning primes cell scattering during epithelial‐to‐mesenchymal transition. Dev Cell 40: 168–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron A, Askari JA, Humphries JD, Jacquemet G, Koper EJ, Warwood S, Choi CK, Stroud MJ, Chen CS, Knight D, Humphries MJ (2015) A proteomic approach reveals integrin activation state‐dependent control of microtubule cortical targeting. Nat Commun 6: 6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD (2004) LFA‐1/ICAM‐1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity 20: 589–599 [DOI] [PubMed] [Google Scholar]
- Chodagam S, Royou A, Whitfield W, Karess R, Raff JW (2005) The centrosomal protein CP190 regulates myosin function during early Drosophila development. Curr Biol 15: 1308–1313 [DOI] [PubMed] [Google Scholar]
- Coles CH, Bradke F (2015) Coordinating neuronal actin‐microtubule dynamics. Curr Biol 25: R677–R691 [DOI] [PubMed] [Google Scholar]
- Colin A, Singaravelu P, Théry M, Blanchoin L, Gueroui Z (2018) Actin‐network architecture regulates microtubule dynamics. Curr Biol 28: 2647–2655.e4 [DOI] [PubMed] [Google Scholar]
- Derivery E, Sousa C, Gautier JJ, Lombard B, Loew D, Gautreau A (2009) The Arp2/3 activator WASH controls the fission of endosomes through a large multiprotein complex. Dev Cell 17: 712–723 [DOI] [PubMed] [Google Scholar]
- Dogterom M, Koenderink GH (2019) Actin–microtubule crosstalk in cell biology. Nat Rev Mol Cell Biol 20: 38–54 [DOI] [PubMed] [Google Scholar]
- Elie A, Prezel E, Guérin C, Denarier E, Ramirez‐Rios S, Serre L, Andrieux A, Fourest‐Lieuvin A, Blanchoin L, Arnal I (2015) Tau co‐organizes dynamic microtubule and actin networks. Sci Rep 5: 9964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne‐Manneville S (2013) Microtubules in cell migration. Annu Rev Cell Dev Biol 29: 471–499 [DOI] [PubMed] [Google Scholar]
- Fakhri N, Wessel AD, Willms C, Pasquali M, Klopfenstein DR, MacKintosh FC, Schmidt CF (2014) High‐resolution mapping of intracellular fluctuations using carbon nanotubes. Science 344: 1031–1035 [DOI] [PubMed] [Google Scholar]
- Farina F, Gaillard J, Guérin C, Couté Y, Sillibourne J, Blanchoin L, Théry M (2016) The centrosome is an actin‐organizing centre. Nat Cell Biol 18: 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Forges H, Bouissou A, Perez F (2012) Interplay between microtubule dynamics and intracellular organization. Int J Biochem Cell Biol 44: 266–274 [DOI] [PubMed] [Google Scholar]
- Gogendeau D, Guichard P, Tassin AM (2015) Purification of centrosomes from mammalian cell lines. Methods Cell Biol 129: 171–189 [DOI] [PubMed] [Google Scholar]
- Gundersen GG, Gomes ER, Wen Y (2004) Cortical control of microtubule stability and polarization. Curr Opin Cell Biol 16: 106–112 [DOI] [PubMed] [Google Scholar]
- Gupton SL, Salmon WC, Waterman‐Storer CM (2002) Converging populations of f‐actin promote breakage of associated microtubules to spatially regulate microtubule turnover in migrating cells. Curr Biol 12: 1891–1899 [DOI] [PubMed] [Google Scholar]
- Huber F, Boire A, López MP, Koenderink GH (2015) Cytoskeletal crosstalk: when three different personalities team up. Curr Opin Cell Biol 32: 39–47 [DOI] [PubMed] [Google Scholar]
- Hutchins BI, Wray S (2014) Capture of microtubule plus‐ends at the actin cortex promotes axophilic neuronal migration by enhancing microtubule tension in the leading process. Front Cell Neurosci 8: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A, Drechsel D, Kellogg D, Salser S, Sawin K, Steffen P, Wordeman L, Mitchison TJ (1991) Preparation of modified tubulins. Methods Enzymol 196: 478–485 [DOI] [PubMed] [Google Scholar]
- Katrukha EA, Mikhaylova M, van Brakel HX, van Bergen en Henegouwen PM, Akhmanova A, Hoogenraad CC, Kapitein LC (2017) Probing cytoskeletal modulation of passive and active intracellular dynamics using nanobody‐functionalized quantum dots. Nat Commun 8: 14772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina I, Rottner K, Small JV (1998) Targeting, capture, and stabilization of microtubules at early focal adhesions. J Cell Biol 142: 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letort G, Nedelec F, Blanchoin L, Théry M (2016) Centrosome centering and decentering by microtubule network rearrangement. Mol Biol Cell 27: 2833–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López MP, Huber F, Grigoriev I, Steinmetz MO, Akhmanova A, Koenderink GH, Dogterom M (2014) Actin‐microtubule coordination at growing microtubule ends. Nat Commun 5: 4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahuzier A, Shihavuddin A, Fournier C, Lansade P, Faucourt M, Menezes N, Meunier A, Garfa‐Traoré M, Carlier M‐F, Voituriez R, Genovesio A, Spassky N, Delgehyr N (2018) Ependymal cilia beating induces an actin network to protect centrioles against shear stress. Nat Commun 9: 2279–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelot A, Berro J, Guérin C, Boujemaa‐Paterski R, Staiger CJ, Martiel J‐L, Blanchoin L (2007) Actin‐filament stochastic dynamics mediated by ADF/cofilin. Curr Biol 17: 825–833 [DOI] [PubMed] [Google Scholar]
- Mimori‐Kiyosue Y (2011) Shaping microtubules into diverse patterns: molecular connections for setting up both ends. Cytoskeleton 68: 603–618 [DOI] [PubMed] [Google Scholar]
- Mohan R, John A (2015) Microtubule‐associated proteins as direct crosslinkers of actin filaments and microtubules. IUBMB Life 67: 395–403 [DOI] [PubMed] [Google Scholar]
- Moudjou M, Bornens M (1998) Method of centrosome isolation from cultured animal cells In Cell biology: a laboratory handbook, Celis J. (ed.), pp 111–119. San Diego, CA: Academic Press; [Google Scholar]
- Obino D, Farina F, Malbec O, Sàez PJ, Maurin M, Gaillard J, Dingli F, Loew D, Gautreau A, Yuseff M‐I, Blanchoin L, Théry M, Lennon‐Duménil A‐M (2016) Actin nucleation at the centrosome controls lymphocyte polarity. Nat Commun 7: 10969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, You Y, Huang T, Brody SL (2007) RhoA‐mediated apical actin enrichment is required for ciliogenesis and promoted by Foxj1. J Cell Sci 120: 1868–1876 [DOI] [PubMed] [Google Scholar]
- Piel M, Meyer P, Khodjakov A, Rieder CL, Bornens M (2000) The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells 7. Cell 149: 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitaval A, Senger F, Letort G, Gidrol X, Guyon L, Sillibourne J, Thery M (2017) Microtubule stabilization drives 3D centrosome migration to initiate primary ciliogenesis. J Cell Biol 216: 3713–3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portran D, Gaillard J, Vantard M, Théry M (2013) Quantification of MAP and molecular motor activities on geometrically controlled microtubule networks. Cytoskeleton 70: 12–23 [DOI] [PubMed] [Google Scholar]
- Reymann A, Martiel J, Cambier T, Blanchoin L, Boujemaa‐Paterski R, Théry M (2010) Nucleation geometry governs ordered actin networks structures. Nat Mater 9: 827–832 [DOI] [PubMed] [Google Scholar]
- Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, Prosser BL (2016) Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352: aaf0659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez OC, Schaefer AW, Mandato CA, Forscher P, Bement WM, Waterman‐storer CM (2003) Conserved microtubule – actin interactions in cell movement and morphogenesis. Nat Cell Biol 5: 599–609 [DOI] [PubMed] [Google Scholar]
- Roostalu J, Surrey T (2017) Microtubule nucleation: beyond the template. Nat Rev Mol Cell Biol 18: 702–710 [DOI] [PubMed] [Google Scholar]
- Sanchez AD, Feldman JL (2017) Microtubule‐organizing centers: from the centrosome to non‐centrosomal sites. Curr Opin Cell Biol 44: 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelanski M (1973) Chemistry of the filaments and tubules of brain. J Histochem Cytochem 21: 529–539 [DOI] [PubMed] [Google Scholar]
- Stevenson VA, Kramer J, Kuhn J, Theurkauf WE (2001) Centrosomes and the Scrambled protein coordinate microtubule‐independent actin reorganization. Nat Cell Biol 3: 68–75 [DOI] [PubMed] [Google Scholar]
- Suarez C, Carroll RT, Burke TA, Christensen JR, Bestul AJ, Sees JA, James ML, Sirotkin V, Kovar DR (2014) Profilin regulates F‐actin network homeostasis by favoring formin over Arp2/3 complex. Dev Cell 32: 43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C, Kovar DR (2016) Internetwork competition for monomers governs actin cytoskeleton organization. Nat Rev Mol Cell Biol 17: 799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry M, Racine V, Piel M, Pépin A, Dimitrov A, Chen Y, Sibarita J‐B, Bornens M (2006) Anisotropy of cell adhesive microenvironment governs cell internal organization and orientation of polarity. Proc Natl Acad Sci USA 103: 19771–19776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzbekov R, Kireyev I, Prigent C (2002) Centrosome separation: respective role of microtubules and actin filaments. Bilo Cell 94: 275–288 [DOI] [PubMed] [Google Scholar]
- Walentek P, Quigley IK, Sun DI, Sajjan UK, Kintner C, Harland RM (2016) Ciliary transcription factors and miRNAs precisely regulate Cp110 levels required for ciliary adhesions and ciliogenesis. Elife 5: e17557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Chen L, Ding Y, Jin J, Liao K (2008) Centrosome separation driven by actin‐microfilaments during mitosis is mediated by centrosome‐associated tyrosine‐phosphorylated cortactin. J Cell Sci 121: 1334–1343 [DOI] [PubMed] [Google Scholar]
- Wieczorek M, Bechstedt S, Chaaban S, Brouhard GJ (2015) Microtubule‐associated proteins control the kinetics of microtubule nucleation. Nat Cell Biol 17: 907–916 [DOI] [PubMed] [Google Scholar]
- Wu C‐T, Chen H‐Y, Tang TK (2018) Myosin‐Va is required for preciliary vesicle transportation to the mother centriole during ciliogenesis. Nat Cell Biol 20: 175–185 [DOI] [PubMed] [Google Scholar]
- Yuseff MI, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, Randrian V, Larochette N, Vascotto F, Desdouets C, Jauffred B, Bellaïche Y, Gasman S, Darchen F, Desnos C, Lennon‐Duménil AM (2011) Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity 35: 361–374 [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Waterman‐Storer CM, Cohan CS (2002) Focal loss of actin bundles causes microtubule redistribution and growth cone turning. J Cell Biol 157: 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Movie EV1
Review Process File