

Abstract
Pancreatic ductal adenocarcinoma is one of the deadliest cancers, and its incidence on the rise. The major challenges in overcoming the poor prognosis with this disease include late detection and the aggressive biology of the disease. Intratumoral heterogeneity; presence of a robust, reactive, and desmoplastic stroma; and the crosstalk between the different tumor components require complete understanding of the pancreatic tumor biology to better understand the therapeutic challenges posed by this disease. In this review, we discuss the processes involved during tumorigenesis encompassing the inherent plasticity of the transformed cells, development of tumor stroma crosstalk, and enrichment of cancer stem cell population during tumorigenesis.
Keywords: Pancreatic Cellular Plasticity, Pancreatic Cancer Associated Fibroblasts, Signaling Crosstalk, Cancer Stem Cells
Pancreatic ductal adenocarcinoma [PDA] is one of the deadliest of all human cancers. Despite innovations that have increased survival in other cancers and a surge in research efforts in the past 15 years, the 5-year survival rate for PDA remains <10%, and will soon surpass colon cancer as the second leading cause of cancer-related death in the United States. While early detection will undoubtedly impact PDA survival, it is also imperative that we better understand the biology of the cancer’s progression, so that we know how best to target it upon diagnosis. During its development, all tissues take cues from their macroenvironment as well as their microenvironment. This results in a systematic differentiation pattern. This context- dependent development of tissues in response to their surroundings that is independent of the genetic signature within them is the phenotypic plasticity of any organ or tissue. In this context, the pancreas is a notoriously plastic organ, with its normal epithelia poised to undergo trans-differentiation from a functional glandular epithelium to a proliferative progenitor-like state in cases of tissue damage. Recent studies have suggested that the natural cellular plasticity of pancreatic epithelium may impact not only initial tumorigenesis, but tumor progression, including early-stage invasion and metastasis and metastatic organotropism.1–3 Down-regulation of transcription factors associated with maintaining pancreatic identity is connected to the promotion of both tumor formation4−8 and tumor aggressiveness.9 Here we explore pancreatic cellular plasticity in neoplastic transformation, crosstalk with the tumor microenvironment, and the generation of cancer stem cells (CSCs), in hopes that better understanding these processes can lead to prevention and treatment of pancreatic cancer.
Signal Transduction and Pancreatic Cellular Plasticity
Tumorigenesis in most tissues begins with the mutation of a tumor suppressor gene in a relatively undifferentiated cell type, such as a stem or progenitor cell, locking the cell in a differentiation state compatible with rampant proliferation and inappropriate survival. In contrast, the initiating mutation in >90% of PDA is in the Kras oncogene, with the cell of origin arguably being part of the differentiated parenchyma. In the first PDA model to accurately mimic human pancreatic cancer progression, Hingorani et al10 used a Cre-inducible oncogenic Kras mutant (KrasLSL G12D) knocked into a single allele of the endogenous KRAS2 locus, thus maintaining physiologic levels of oncogene expression upon Cre recombination. When crossed to either a Pdxl-Cre or PtflaCrC/+ driver lines, the resulting KRAS-Crepdxl or “KC” progeny express KrasG12D in pancreatic progenitors, yet show no evident perturbation of organogenesis, in the fully developed organ, which maintains persistent KrasG12D expression in the differentiated parenchymal daughter cells, initial transformation does not become evident until several weeks of age, at which time acinar-to-ductal metaplasia (ADM) and early neoplasia resembling the human disease emerge.10 Thus, not only are developmental progenitor cells resistant to transformation by KrasG1ZD expression, transformation in the fully developed organ is remarkably inefficient. Transformation efficiency is further reduced if mutant Kras expression is initiated only after the animal reaches adulthood, but can be initiated by inducing tissue damage and inflammation with experimental pancreatitis.11
The KC model strongly suggests that the developed parenchyma harbors the true cell of origin for PDA. This naturally leads to questions about the transformation process, for example, what restrains oncogenic Kras from inducing more widespread transformation and, conversely, what signal(s) allow the occasional transformed cell to break through those restraints? Regarding the first question, some studies suggest the existence of a rare facultative stem cell population that has the unique capacity to act as tumor-initiating cells (TICs),12–13 consistent with the stochastic pattern of transformation. However, with several modified KC models showing dramatic increases in both the kinetics and degree of transformation, the adult stem cell model is not a wholly satisfying explanation. Another popular hypothesis is that differentiated epithelia are highly plastic and can be reprogrammed to a transformation-susceptible precursor. This possibility is evident in a very early transgenic mouse model in which constitutive overexpression of the epidermal growth factor receptor (EGFR) ligand transforming growth factor-a induces a prominent ADM response that occasionally leads to neoplasia,14 presumably by constitutively activating the downstream wild-type Kras proteins. A similar process can be induced in vitro by treating primary acinar cells with EGFR ligands,15,16 with lineage tracing demonstrating the resulting metaplastic duct cells being derived from acinar cell transdifferentiation.17 MEK/ERK activity is required for acinar transdifferentiation,18,19 consistent with the process being due to Ras activation. Thus, it was surprising to find that genetic ablation of parenchymal EGFR from the KC mouse model completely abolishes pancreatic transformation in both sporadic and pancreatitis-induced systems.18,20
While it is counterintuitive that a model expressing a constitutively active Kras mutant would require one of its classical upstream activators for transformation, these studies find that EGFR ablation significantly dampens ERK13 and PI3K20 activities compared to control KC mice. This suggests that the protein product of a single mutant allele of Kras is not sufficient to robustly activate its downstream effectors to a level sufficient to initiate transformation. Indeed, in isolated acinar cells, EGFR knockout KC cells show significantly lower levels of active, GTP-loaded Ras,18 correlating to lower levels of active ERK. These data are compatible with the proposed model that mutant Kras itself requires at least initial GTP loading induced by EGFR activity.21 An alternative explanation is that EGFR is required to activate wild-type Ras proteins (NRAS, HRAS, and the protein product of the remaining wild-type Kras allele) to attain the minimal threshold of overall Ras activity required for transformation. This concept is supported by a mouse model where KrasG12D is highly overexpressed in the pancreatic parenchyma, leading to much more rapid and widespread transformation compared to the KC model.22,23 Also, wild-type Ras proteins have been shown to be necessary for Kras-initiated transformation.24,25
What is it about the pancreas that requires a higher level of Ras activity for transformation than needed in other tissues, such as intestine and lung20? One possibility is the need to overcome acinar cell differentiation in order to generate the metaplastic duct neoplastic progenitor. This acinar cell-centric view of tumorigenesis is supported by the finding that acinar cells are more susceptible to transformation than duct cells,26 and that ADM is blocked by EGFR or MEK inhibition.18,19 That said, recent studies have shown that duct cells can be susceptible to transformation in certain contexts,27–29 and this process is accompanied by a similar change in differentiation status that is simply not as histologically evident as ADM.27 While skepticism remains that acinar cells are the cell of origin of the precancerous pancreatic intraepithelial neoplasms (PanINs) in people, current technology is not sufficient to prove or disprove the possibility. Certainly, within the KC model that was rigorously determined to be the most representative of the human disease, acinar cells are likely the major contributor to these neoplastic lesions. Thus, while the assumption of an acinar cell of origin for PDA provides a convenient context for this discussion, these broad concepts likely apply more universally to parenchymal cell differentiation and transformation in general. With this in mind, one of the earliest events in Kras-initiated transformation of the acinar cell is the down- regulation of several transcription factors important for maintaining acinar cell differentiation (eg, Ptfla,4 Gata6,5 Misti,6 and Nr5a27), while others (eg, Pdxl8) are reassigned from the regulatory regions of acinar differentiation genes to pro-inflammatory and oncogenic pathway genes. Consistent with acinar cell differentiation being the primary barrier to transformation, ablation of each of these acinar identity transcription factors from otherwise wild- type mice leads to different degrees of acinar cell atrophy and, when combined with KrasG12D expression, leads to rapid and widespread transformation.4–8,30 Concomitant with this loss of acinar identity factors is the up- regulation of the duct-specific transcription factor Sox9, the expression of which is required for progression of neoplasia.26
If acinar cell differentiation represents the major barrier to transformation, it stands to reason that physiological Ras signaling is intimately intertwined with the natural plasticity of the pancreatic epithelium. ADM, along with inflammation and fibrosis, is part of the natural regenerative response after pancreatic injury, with ADM proliferating and re-differentiating back to acinar cells after injury resolution.8 Genetic and pharmacologic inhibition of MEK-ERK blocks and reverses ADM formation in experimental pancreatitis,19 demonstrating that the activation of Ras pathway signaling is part of the natural process of pancreatic wound healing. Conversely, cessation of that signal is critical for restoring differentiation and homeostasis.19,31 Besides directly regulating several acinar cell-specific genes, some of the acinar differentiation- related transcription factors also appear to regulate signaling molecules in a manner that broadly serve to dampen RAS pathway signaling.8,32 This suggests that Ras and acinar differentiation may prove to be directly and actively antagonistic in the balance between homeostasis and healing.
Kras itself activates a well-defined oncogenic transcription factor network that includes prominent oncoproteins, such as c-Myc,33 Yap,34 and AP-1 factors,35 among others, many of which have direct effects on acinar differentiation. c-Myc directly represses expression of the acinar cell transcription factor Ptfla,36 leading to a partial loss of acinar cell identity. Activation of YAP signaling by ablation of Mstl/2 kinases is sufficient to induce ADM,37 while ablation of YAP blocks the process in the context of KrasG12D expression.38 in a particularly intriguing demonstration of antagonism between differentiation and Ras signaling, Cobo and colleagues39 found that heterozygous ablation of the acinar differentiation factor Nr5a2 leads to the partial loss of acinar cell identity, accompanied by the cooperative up-regulation of AP-1 transcription factors and several AP-1 target genes, including pro-inflammatory cytokines. Ablation of the AP-1 transcription factor c-Jun from the Nr5a2+/− pancreata is sufficient to restore tissue homeostasis, demonstrating that up-regulation of this oncogenic pathway is required for the progressive loss of tissue identity. Thus, Ras pathway activity can impact acinar cell through many mechanisms, it will be intriguing to fully explore the web of interactions in these and other genetic models that lead to loss of acinar and duct cell identity to determine the extent to which the upstream MAPK pathway and possibly Ras itself contributes to the resulting phenotypes.
Absent from this cell-autonomous view of pancreatic cell plasticity is the common finding that acinar cell atrophy induced by ablation of acinar identity transcription factors is accompanied by an inflammatory response,8,39 as would be expected in conditions of progressive loss of tissue integrity. Even within the limited focus on activation of the Kras pathway, inflammation can induce the activation of EGFR.40 Conversely, parenchymal EGFR is required for the inflammatory and fibrotic responses accompanying pancreatitis and tumorigenesis.41 Understanding this vicious cycle of crosstalk between the parenchyma and the fibro-inflammatory stroma is likely to be a key step in devising avenues for treatment of PDA.
Oncogenic KRAS-Induced Senescence in Pancreatic Ductal Adenocarcinoma
In untransformed and normal cells, activation of oncogenic Ras can induce senescence by up-regulating proteins that are inhibitors of cell proliferation—pl6INK4, pl9ARF, p21CIP, and p53. This prevents expansion of the cells with mutant KRas.42 in pancreatic cancer, oncogenic KRas suppresses premature senescence of pancreatic ductal epithelium. A study by Lee and Bar-Sagi43 in 2010 suggested that this senescence bypass was a potential mechanism that conferred selective advantage to the precursors of the developing PDA. In normal cultured cells, induction of P16INK4A in response to adverse growth conditions is responsible for premature senescence.44 This is similar to that observed in cultured pancreatic epithelial cells. Further, in the presence of oncogenic Kras, loss of Rb from the pancreatic epithelium has been shown to accelerate formation of PanlNs.45 A recent study by Scully et al46 in 2018 showed that basic helix loop helix transcription factor E47 induced growth arrest in PDA by regulating a network of cell-cycle progression genes that induce a senescence like program in pancreatic cancer and restore a degree of acinar cell differentiation.47
Central Role of Inflammation
The interplay of oncogenic Kras and carcinogenesis has been studied extensively in mouse models. As mentioned, KC mice develop lesions with long latency, even as oncogenic Kras is expressed throughout the pancreas, starting during the earliest phases of pancreas development.10,48 Once the lesions do form, they are surrounded by activated fibroblasts and infiltrating immune cells. Supporting evidence on the role of inflammatory cells during pancreatic carcinogenesis derives from the induction of pancreatic inflammation, such as in the commonly used caerulein-driven pancreatitis model, accelerates the onset of pancreatic lesions.11,49–51 While different mechanisms might explain this finding, it is possible that the recruitment of immune cells provides signals that cause the onset of transformation.
In the absence of Kras mutations, myeloid cells promote tissue repair. In a model of experimental ablation of acinar cells, myeloid cells are required for the recovery of the acinar population.52 Ablation of myeloid cells in presence of oncogenic Kras prevents the onset of pancreatic carcinogenesis.53 Ablation of myeloid cells after the formation of low-grade PanlNs allows at least partial re-differentiation of acinar cells.53 Intriguingly, their requirement for myeloid cells during the onset of pancreatic cancer is independent of their immunosuppressive function,53 and might be mediated primarily by their expression of EGFR ligand, a possibility that has yet to be tested experimentally. The observation that myeloid cells are both required for pancreatic repair and for carcinogenesis appears contradictory. A possible explanation is that the myeloid cells present in different settings (repair vs carcinogenesis) are different populations. In support of this notion, inactivation of oncogenic Kras in a mouse model allowing inducible and reversible expression of the oncogene (iKras54) leads to dramatic alteration in myeloid cell gene expression, including down-regulation of immune suppressive factors, such as Arginase 1 and signaling components, such as HBEGF.53 Multiple studies have analyzed the populations of myeloid cells in PaniNs and pancreatic cancer, in general, infiltrating myeloid cells are immunosuppressive and include cellular populations, such as immature myeloid cells [also defined as myeloid-derived suppressor cells) and macrophages.55 Infiltrating myeloid cells are believed to derive, at least in part, from a resident population of pancreatic macrophages that expand during damage. Interestingly, the characteristics of myeloid cells infiltrating the pancreas are regulated, in a non-cell autonomous manner, by oncogenic Kras expressing epithelial cells, in the iKras model, inactivation of oncogenic Kras in the epithelium leads to a drastic reduction of the expression of Arginase 1 and HBEGF in infiltrating myeloid cells. Together, these findings point at a reciprocal loop, whereby oncogenic-Kras expressing epithelial cells induce the accumulation of myeloid cells that, in turn, promote carcinogenesis (Figure 1).
Figure 1.
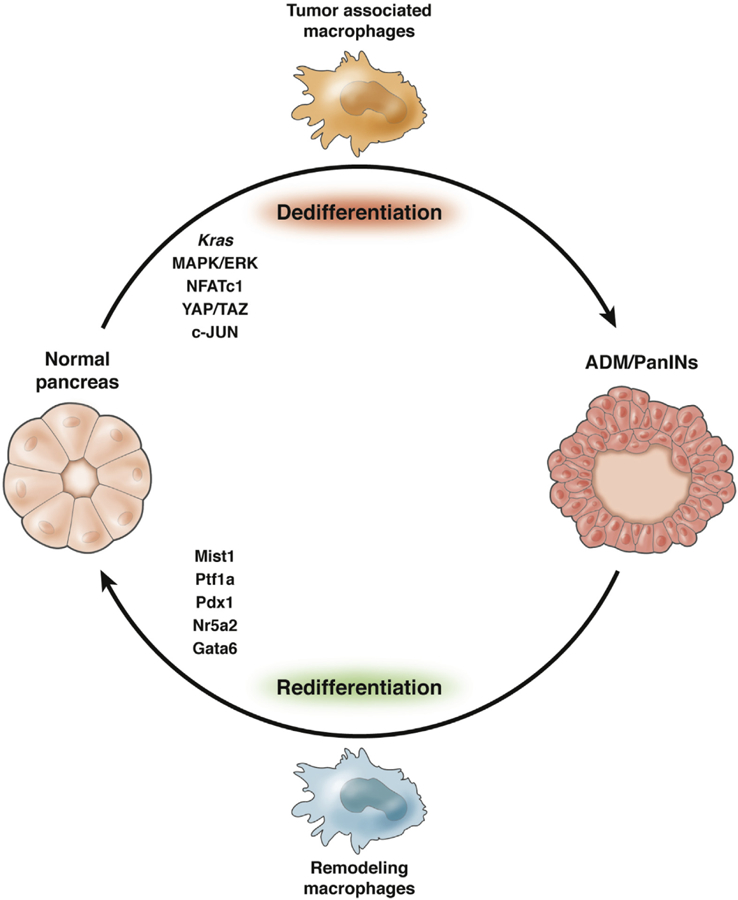
The crosstalk between epithelial cells and components of the microenvironment is regulated by oncogenic Kras. Activation of MAPK/ERK signaling downstream of Kras is required for epithelial cell de-differentiation. In presence of oncogenic Kras, macrophages are polarized to promote tumorigenesis. In contrast, lineage-specific transcription factors and macrophages associated with the tissue repair process antagonize carcinogenesis.
In advanced lesions and invasive cancer, myeloid cells are required to inhibit CD8+ T-cell-mediated anti-tumor immune responses. Experimental depletion of myeloid cells is sufficient to induce anti-tumor immunity and tumor regression.41,56,57 As myeloid cell depletion is not advisable in human patients, several strategies have been employed to target myeloid cells in pancreatic cancer. Reprogramming them to reduce their immune-suppressive function has been explored using a clinically relevant CD40 agonist.58 Inhibition of CXCR2 with a blocking antibody similarly causes changes in myeloid functions and sensitize tumors to immune checkpoint blockade.59 Similarly, targeting CSF1R in macrophages restores T-cell-mediated anti-tumor immune responses.56,60
The crosstalk between cancer cells and myeloid cells is mediated by cytokines, some of which have been studied in detail. Cancer cells secrete granulocyte-macrophage colony- stimulating factor, thus recruiting myeloid cells with immunosuppressive properties.61,62 interleukin (IL) 6 is secreted by multiple cells types within the microenvironment, but the main source is pancreatic fibroblasts.63 1L6, in turn, has pleiotropic effects on different components of the microenvironment, where it activates JAK/STAT3 signaling. Ablation of STAT3 in epithelial cells prevents pancreatic carcinogenesis.64–66 Interestingly, IL6 blockade synergizes with PD1 inhibition to cause tumor regression.66 IL6/Stat3 signaling increases in pancreatic cancer upon inhibition of MEK signaling67; dual targeting with MEK and JAK inhibitors was recently shown to be more effective than either treatment alone, and to enhance synergy with anti-PDl.68 1L6 expression in fibroblasts is heterogeneous, with a population of IL6-high fibroblasts being particularly tumor- promoting, while IL6-low, smooth muscle actin-positive fibroblasts act to restrain tumor growth, at least in 3-dimensional organoids. There is substantial evidence that Hedgehog signaling drives expression of IL6 in fibroblasts, as discussed in the next section.
Fibroblasts in Pancreatic Cancer: A Dual Role?
The accumulation of a fibrotic stroma is a hallmark feature of pancreatic cancer.69 Fibroblast accumulation occurs early during carcinogenesis, and persists in invasive tumors. Current literature points to a “stellate cell” population, characterized by the accumulation of vitamin A rich lipid droplets, as the cell of origin for stromal fibroblasts,70 although lineage-tracing studies are currently missing. Our understanding of stromal fibroblasts has evolved over the past few years, recognizing them as a heterogeneous population with potentially distinct roles.
Epithelial Signals Promote Expansion of Pancreatic Fibroblasts
Activation of fibroblasts, as measured by expression of smooth muscle actin and by active proliferation, occurs during the earliest stages of pancreatic carcinogenesis, surrounding clusters of ADM.69 Using a model of inducible and reversible oncogenic Kras-driven pancreatic carcinogenesis—the iKras* mouse—our group determined that the activation status of pancreatic fibroblasts is regulated by epithelial signals: inactivation of oncogenic Kras leads to rapid loss of activation of pancreatic fibroblasts and, over time, to their reduction within the pancreas.54
PanIN and pancreatic cancer cells express ligands of the Hedgehog family (prevalently Sonic and Indian Hedgehog).71,72 Although initially believed to function through an autocrine loop. Hedgehog signaling was later determined to act in a paracrine manner.73 Fibroblasts express the cellular machinery to respond to Hedgehog signaling, such as the receptor Patchedl and the co-receptors Gasl, Cdon, and Boc.73,74 In absence of ligand, the transmembrane receptor Patchedl (Ptchl) inhibits Smoothened (Smo), another transmembrane protein. Ligand binding releases the repression and allows activation of downstream signaling, which culminates with the activation of 3 transcription factors in the Gli family (for review see Pasca di Magliano et al75 and Wang et al76). Mammals express 3 Gli factors: Gli1 exists solely as activator; Gli2 can be processed as activator or repressor, but it is most active in the former role, while Gli3 is prevalently a repressor. All 3 Gli genes are expressed in the normal pancreas and in pancreatic cancer (Mathew et al77 and unpublished data). The 3 coreceptors Gasl, Cdon, and Boc bind Hedgehog ligands and are required for signal transduction.78 Initial studies on HH signaling in pancreatic cancer indicated a tumor-promoting role during carcinogenesis.73 Inhibition of HH signaling using the Smo antagonist IPI926 in tumor-bearing KPC mice prolonged survival when combined with gemcita-bine.79 However, IPI926 failed in a clinical trial, with worsened patient outcomes compared to chemotherapy alone, and a different Smo inhibitor, GDC 0449 (Genentech, South San Francisco, CA), provided no benefit.80 Following the disappointing clinical results, a new study in an experimental model showed that KPC mice lacking Shh expression in the epithelium progress to cancer faster than KPC mice expressing Shh.81 IPI926 treatment in KPC mice, this time in the absence of concurrent chemotherapy, shortened survival similarly.81 A possible clue to these contradictory results comes from a study indicating that HH signaling dosage might drive different cellular responses.74 In particular, lowering HH signaling without ablating its activity altogether induces expression of pro-angiogenic factors, such as VEGF and Agptl,81 known Gli targets. Further, ablation of Smo in pancreatic fibroblasts paradoxically results in a compensatory overexpression of Gli2, the main Gli activator.82 Many open questions remain regarding the role of HH signaling in pancreatic cancer. Going forward, it will be of paramount importance to identify the target genes of HH signaling, and gather an understanding of the heterogeneity of fibroblast populations in pancreatic cancer, in fact, while ablation of most fibroblasts in pancreatic cancer resulted in the development of an aggressive, sarcomatoid tumor type, this tumor was, however, sensitive to immune checkpoint inhibition, thus potentially indicating that a targeted combination approach should be developed.83 More recently, the concept of “normalizing” pancreatic fibroblasts has gained traction, with a study showing that high doses of vitamin D might reverse fibroblast activation status.84 Finally, the heterogeneity of fibroblast populations has been described in multiple studies, and subsets that promote or restrain carcinogenesis have been identified.85 Strategies to target fibroblasts are likely to make an impact on pancreatic cancer, considering that fibroblasts are a key mediator of immune suppression in this disease86 and that activation of an immune response represents the best chance at achieving long-term survival.87
Cancer Stem Cells: Cancer Cells With a Survival Advantage
Until now, we have been focused on cellular plasticity as it relates to normal cells in the process of neoplastic transformation. However, the plasticity also pertains to cancer cells, especially in the context of developing effective treatments for pancreatic cancer and overcoming resistance. This is most evident in the evolution of the CSC hypothesis. The concept of CSCs or TICs stems from the notion that a population of tumor cells survived the therapeutic regimen and remained dormant, only to recur as soon as the therapy was withdrawn. Even though the CSCs in several cancers have been studied for decades, their origin has remained an enigma. The earlier studies found that cancer cells within a tumor existed in different phenotypic states that had different functional elements. Among this heterogeneity, the CSCs formed a distinct population of cells that had activated self-renewal pathways, tumor initiation capability, and were responsible for tumor recurrence.88,89 These cells also showed an increased tendency to metastasize and were typically resistant to therapy. Additional studies by Kreso et al90 also indicated that this population of cells were able to reversibly transition between stem and non-stem states as well. These observations—along with the studies that showed that microenvironmental niches like hypoxia, extracellular matrix surrounding the tumor cells, and the inflammatory milieu, can provide cues for the dynamic interconversion between CSC and non-CSC—complicated the understanding of CSCs.
Before the concept of niche influencing the enrichment or origin of CSC population, 2 models determined the origin of CSCs. In the hierarchical model, the CSCs are considered to represent a distinct subset within the tumor that arises when a stem cell escapes regulation and gives rise to an aberrant counterpart with unrestrained self-renewal potential. This population can not only self-renew but also differentiate into a short-lived progeny with restricted proliferative capability.91,92 This indicated that in a clinical setting, eradication of the CSCs would prevent recurrence of the tumor. The stochastic model, however, stated that every cell within a tumor was likely to be a cell of origin that can promote tumor initiation and progression. It also stated that the heterogeneity within the tumor was determined by intrinsic factors like accumulation of genetic mutations.93
These 2 apparently dichotomous models for CSCs can be explained by the CSC plasticity in the presence of the microenvironmental stimuli. This indicated that low oxygen tension, cytotoxic drugs, and oxidative and nutritional stress that the tumor is typically subjected to could influence the interconversion of a non-CSC population to a CSC population. Looking deeper into the concept of plasticity, it becomes evident in the presence of microenvironment- induced stress, a sub-population of cancer cells within the tumor evolves in a Darwinian manner to give rise to CSCs, which are cells with a distinct survival advantage and can tide through all unfavorable conditions that the host presents (Figure 2).
Figure 2.
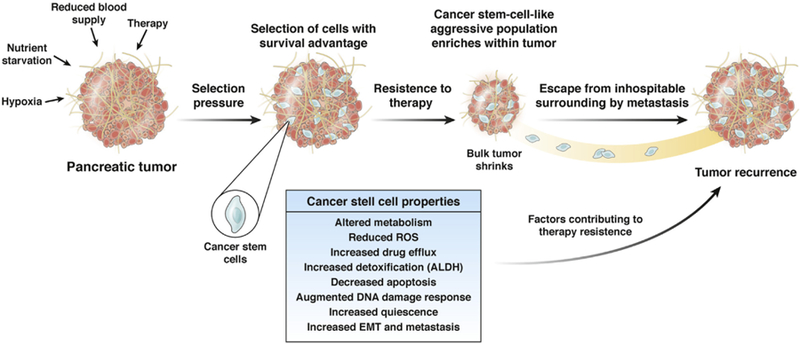
Cancer stem cells. Hypoxia, therapy, and nutritional stress within a pancreatic tumor reprograms a population of cells with increased survival advantage. These are the cells that show classic CSC attributes of therapeutic resistance, decreased apoptosis, and quiescence. This enables them to endure the therapeutic pressure, which shrinks the bulk tumor. The enrichment of the CSC population induces metastatic properties, which enable these cells to leave the primary tumor site and recur at a distant location as a metastatic tumor.
In pancreatic cancer, the CSC was first defined by a CD44+CD24+ESA+ population that was able to form tumors at very low dilutions.94,95 Since then, several markers that can isolate CSC from pancreatic tumors have been reported.23,96–99 The CSC plasticity, with respect to the influence of microenvironment, is relatively less studied in pancreatic cancer. However, studies have shown that extracellular matrix components like collagen can influence clonogenic tumor growth, tumor initiation, and invasion of pancreatic cancer cells via activation of FAK signaling pathway.100,101 Apart from extracellular matrix, hypoxic tumor microenvironment promotes non-CSC to CSC population in pancreatic cancer.102,103 These observations further strengthen the idea that the CSC or TIC populations within the tumor are actually the cells with a distinct survival advantage that enables them to tide over stress conditions, thereby making them therapy-resistant.
Metastasis in Cancer Stem Cell Population: Escaping the Inhospitable Microenvironment
It is becoming increasingly clear that as the local microenvironment is able to select for CSC population, it can also affect its ability to metastasize. Evidence suggests that the metastasis-initiating cells are CSCs that can revert to their functional and proliferative state upon infiltrating a target organ. Their interconversion between the dormant state and subsequent reactivation are governed by intrinsic signaling pathways, which regulate the self-renewal capability of adult stem cells. In addition, metastatic cells undergoing reactivation are nursed by specialized extracellular matrix niches, which support positive signals, such as Wnt and Notch, and attenuate negative signals, such as BMP.104,105 In addition, hypoxic microenvironment is known to promote metastasis along with enriching for CSC population.106 This additionally implies that the plastic nature of CSCs is a part of their survival mechanism that helps them to tide over the unfriendly tumor microenvironment.
Among the many functions of cellular plasticity, one is the ability of cancer cells to dedifferentiate into an embryonic stem-like state. What continues to elude investigators is why a subpopulation of cells characterized by a specific set of surface markers possess these unique properties, while others do not. The existence of specialized cells that can interconvert between stem cell and non-stem cell phenotype is important during embryogenesis, and adult stem cells possess this behavior to some extent. In cancer, reactivation of specialized embryogenic pathways is observed, and has been shown to drive CSC-genesis and tumor formation.107 An extreme case of tumor cell plasticity is observed in vascular mimicry, during which tumor cells transdifferentiate into endothelial-like cells that can form matrix-rich, vascular-like, and perfused channels leading to intravasation and metastasis in breast cancer and melanoma.108–110 Thus, the process of metastasis can be considered as a survival adaptation for a defined population of tumor cells to escape the adverse microenvironment of the primary tumor.
Pancreatic CSC population (whether CD133+ or CD44+/Met+) show an overexpression of genes involved in epithelial-mesenchymal transition (EMT) and increased ability to metastasize and colonize distant tissues. CD44 activated the MT1-MMP-SNAI1 axis to promote metastasis in pancreatic cancer.111 Similarly, CD133 activated ILlB-nuclear factor-κB pathway to promote invasiveness and metastasis in CD133+ TICs.112,113 A recent study by Zhang et al114 showed that the CCR7-CCL21 axis mediated an EMT phenotype in pancreatic CSC via activation of ERK/nuclear factor-κB signaling pathway. Other than nuclear factor-κB and MTl-MMP-SNAi1, the Hh pathway has also been implicated in playing a role in EMT in pancreatic CSCs. Disruption of Hh signaling in pancreatic CSCs resulted in a reduction of self-renewal, EMT, tumorigenesis, invasiveness, chemoresistance, and metastasis.115 Aberrant reactivation of P13K/AKT/mTOR in pancreatic CSC further influenced metastasis in these cells.116
Metabolic Regulation of Reactive Oxygen Species and Oxidative Stress in Cancer Stem Cells
Recent studies that focus not on just the surface markers, but also on the downstream signaling processes in the CSC population, have brought to light the role of survival pathways in pancreatic CSCs. Previously published studies from our laboratory show that CD133+ CSC population arise in the hypoxic niches and keep their mitochondrial activity at the lowest in order to minimize reactive oxygen species (ROS) accumulation and thus turn on anti-apoptotic genes in the presence of chemotherapeutic agents.102 Further, upon inhibition of ROS accumulation by N-acetylcysteine, the treatment-resistant CD133+ cells became susceptible to apoptosis and showed sensitivity to standard chemotherapy.
The role of ROS regulation and oxidative stress are not very well studied in pancreatic CSC. However, in embryonic stem cells as well as in other CSCs, they are known to play a significant role in not only chemoresistance, but also in preserving quiescence and protecting against microenvironmental stress.40,117 Studies showed that in breast CSCs isolated from MDA-MB231 and MCF7 mammospheres, there was low ROS accumulation after radiation, and this was further associated with an increase in expression of GCLM, an enzyme that is the rate-limiting step of GSH synthesis.118,119 Decreased ROS also correlated with increased Fox01 levels. The study also showed that inhibition of GSH resulted in decreased “sternness” properties.119 In acute myeloid leukemia, low level of ROS is associated with increased expression of GPX3, further confirming that CSC populations strive to maintain a reduced level of ROS in them in order to maintain their resistant and quiescent phenotype.120 Increased expression of GSH, peroxidasin, as well as thioredoxin, plays a role in maintaining low ROS and thus quiescence in stem cells in a number of other cancers.121,122
Transcriptional Regulation of Therapy Resistance in Cancer Stem Cells
The oxidative stress and thus ROS-mediated regulation of CSC property is tightly regulated at the transcriptional level. The FoxO group of transcription factors have been implicated in playing a major role in maintaining ROS levels in stem cells. These transcription factors play a key role in survival, regulation of proliferation, as well as DNA repair and cell death. These transcription factors further synchronize the oxidative stress response via regulation of the GSH biosynthesis pathway123 and ATM-mediated DNA repair pathways in hematopoietic stem cells.124 ATM proteins are involved in DNA damage repair control, which in turn depends on NADPH production and activity of mTOR pathway.125,126 In pancreatic cancer, ATM and the related protein ATDC play a vital role in tumor initiation as well as in radioresistance.123
Another transcription factor that regulates the oxidative stress in CSCs is NRF2. Interestingly, NRF2-mediated antioxidant response intercalates with both unfolded protein response/endoplasmic reticulum stress-mediated cell survival, as well as via hypoxia-activated stress response. GRP78, the primary regulator of unfolded protein response and thus endoplasmic reticulum stress, is instrumental in maintaining homeostasis in the cells. Perturbation of this pathway leads to aberrant activation of NRF2-mediated signaling, which in turn activates the therapeutic resistance mechanisms in a cancer cell.127 Up-regulated NRF2 activity triggers an increased antioxidant response in the CSCs that can further contribute to the survival advantage in CSC population. Recent studies show that NRF2 regulates drug transporters, as well as the detoxifying enzyme family aldehyde dehydrogenases, to promote therapeutic resistance in the CSC population.128 The role of NRF2 in pancreatic cancer is beginning to be acknowledged.129 However, the intricate signaling pathways that may be instrumental in their regulation of CSC biology is still not well studied.
The role of transcription factors that actively maintain stress response in a cancer cell are becoming clear in the context of therapeutic resistance. Correlative studies from our laboratory as well as others, have shown that increased expression of HSP70 confers resistance to cancer cells to therapy. Similarly, HSF1, the primary transcription factor for HSP70, is also known to drive survival advantage to breast CSCs.130 Interestingly, CD133+ pancreatic CSCs overexpress HSP70 and HSF1.99 However, whether the therapy resistance of this population is because of these proteins has not yet been validated. Hypoxia is known to enrich for sternness. HIF1A, the primary transcription factor that drives adaptations to low oxygen in cancer cells, is also reported to play an active role in driving therapy resistance.131 However, the specific mechanism by which H1F1 may be instrumental in driving therapy resistance in CSCs has not been studied.
The other transcriptional network that is activated in CSCs is the self-renewal regulators: Sox2-Oct4-Nanog. While these transcription factors have been shown to promote self-renewal, whether they actively drive therapy resistance is not clear. Interestingly, while a large body of literature is focused on defining a population of CSCs with specific surface marker and associated properties of increased metastasis and resistance, there does not appear to be a systematic effort in determining what makes these cells activate the survival pathways to promote resistance, activate quiescence, and trigger metastasis.
Conclusions
Pancreatic cancer is a complex disease. The origins of this disease, originating from an organ that, unlike other organs within the gastrointestinal tract, does not have a clear stem cell compartment require reprogramming of adult cells—whether acinar or ductal—to a progenitor-like state that is susceptible to transformation, in turn, this process, initiated by oncogenic Kras, requires repression of the epithelial differentiation program and activation of factors that are normally expressed during the embryonic development of the pancreas. Further, signaling pathways implicated in embryonic development are re-activated and contribute to the carcinogenesis process. The process of de-differentiation of acinar (and presumably ductal) cells relies on non-cell autonomous signals, and is accompanied by the accumulation of a heterogeneous microenvironment. Among the cellular components of the microenvironment, fibroblasts and immune cells are common. The role and nature of fibroblasts is still poorly defined, but current evidence points at different subsets of them with different properties, ranging from tumor restraining to promotion. The infiltrating immune cells are largely suppressive. Among the immune cells, macrophages and immature myeloid cells are prevalent, and collaborate with fibroblasts to generate the profound immune suppression that is characteristic of pancreatic cancer. Finally, a different layer of heterogeneity exists with the tumor cells themselves, with different subpopulations of cells marked by differential expression of certain markers and by different biological characteristics. The composition of the pancreatic cancer microenvironment and of the tumor cells themselves possibly explains the immense challenge of targeting pancreatic cancer therapeutically.
Acknowledgement
Funding
This study was funded by R01 CA198074, R01CA151588, P30CA46592, American Cancer Society Scholar Grant (to MP), U01 CA224145 (to MR and HC), and R01-CA184274 (to SB).
Abbreviations used in this paper:
- ADM
acrnar-to-ductal metaplasia
- CSC
cancer stem cell
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- IL
interleukin
- PanIN
pancreatic intraepithelial neoplasm
- PDA
pancreatic ductal adenocarcinoma
- ROS
reactive oxygen species
- TIC
tumor-initiating cell
Biography
Howard C. Crawford

Marina Pasca di Magliano

Suiagna Banerjee

Footnotes
Conflicts of interest
This author discloses the following: Sulagna Banerjee is a consultant with Minneamrita Therapeutics LLC. The remaining authors disclose no conflicts.
References
- 1.Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrell AS, Joly MM, Allen-Petersen BL, et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat Commun 2017; 8:1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reichert M, Bakir B, Moreira L, et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev Cell 2018;45:696–711 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krah NM, De La OJ, Swift GH, et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. Elife 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinelli P, Madriles F, Canamero M, et al. The acinar regulator Gata6 suppresses KrasGI 2V-driven pancreatic tumorigenesis in mice. Gut 2016;65:476–486. [DOI] [PubMed] [Google Scholar]
- 6.Shi G, Zhu L, Sun Y, et al. Loss of the acinar-restricted transcription factor Misti accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology 2009;136:1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Figura G, Morris JP 4th, Wright CV, et al. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut 2014;63:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy N, Takeuchi KK, Ruggeri JM, et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes Dev 2016;30:2669–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 10.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–450. [DOI] [PubMed] [Google Scholar]
- 11.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011;19:728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanger BZ, Stiles B, Lauwers GY, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell 2005;8:185–195. [DOI] [PubMed] [Google Scholar]
- 13.Westphalen CB, Quante M, Wang TC. Functional implication of Dclkl and Dclkl-expressing cells in cancer. Small GTPases 2016:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandgren EP, Luetteke NC, Palmiter RD, et al. Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 1990;61:1121–1135. [DOI] [PubMed] [Google Scholar]
- 15.De Lisle RC, Logsdon CD. Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur J Cell Biol 1990;51:64–75. [PubMed] [Google Scholar]
- 16.Miyamoto Y, Maitra A, Ghosh B, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003; 3:565–576. [DOI] [PubMed] [Google Scholar]
- 17.Means AL, Meszoely IM, Suzuki K, et al. Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 2005;132:3767–3776. [DOI] [PubMed] [Google Scholar]
- 18.Ardito CM, Gruner BM, Takeuchi KK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012;22:304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halbrook CJ, Wen HJ, Ruggeri JM, et al. Mitogen-activated protein kinase kinase activity maintains acinar-to-ductal metaplasia and is required for organ regeneration in pancreatitis. Cell Mol Gastroenterol Hepatol 2017;3:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navas C, Hernandez-Porras I, Schuhmacher AJ, et al. EGF receptor signaling is essential for k-ras oncogene- driven pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H, Daniluk J, Liu Y, et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014; 33:532–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniluk J, Liu Y, Deng D, et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest 2012; 122:1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313–323. [DOI] [PubMed] [Google Scholar]
- 24.Lim K-H, Ancrile BB, Kashatus DF, et al. Tumour maintenance is mediated by eNOS. Nature 2008;452:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grabocka E, Pylayeva-Gupta Y, Jones MJ, et al. Wild- type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell 2014;25:243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopp JL, von Figura G, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Figura G, Fukuda A, Roy N, et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat Cell Biol 2014;16:255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailey JM, Hendley AM, Lafaro KJ, et al. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016; 35:4282–4288. [DOI] [PubMed] [Google Scholar]
- 29.Kopp JL, Dubois CL, Schaeffer DF, et al. Loss of Ren and activation of Kras synergistically induce formation of intraductal papillary mucinous neoplasia from pancreatic ductal cells in mice. Gastroenterology 2018; 154:1509–1523 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flandez M, Cendrowski J, Canamero M, et al. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut 2014;63:647–655. [DOI] [PubMed] [Google Scholar]
- 31.Collins MA, Yan W, Sebolt-Leopold JS, et al. Mapk signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 2014;146:822–834.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoang CQ, Hale MA, Azevedo-Pouly AC, et al. Transcriptional maintenance of pancreatic acinar identity, differentiation, and homeostasis by PTF1A. Mol Cell Biol 2016;36:3033–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh E, Cunningham M, Arnold H, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004;6:308–318. [DOI] [PubMed] [Google Scholar]
- 34.Zhang W, Nandakumar N, Shi Y, et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal 2014;7:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao HF, Wang J, Tony To SS. The phosphatidylinositol 3-kinase/Akt and c-Jun N-terminal kinase signaling in cancer: alliance or contradiction? (Review). Int J Oncol 2015;47:429–436. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Arevalo Lobo VJ, Fernandez LC, Carrillo-de- Santa-Pau E, et al. c-Myc downregulation is required for preacinar to acinar maturation and pancreatic homeostasis. Gut 2018;67:707–718. [DOI] [PubMed] [Google Scholar]
- 37.Gao T, Zhou D, Yang C, et al. Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology 2013;144:1543–1553, 1553 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruber R, Panayiotou R, Nye E, et al. YAP1 and TAZ control pancreatic cancer initiation in mice by direct up-requlation of JAK-STAT3 siqnalinq. Gastroenteroloqy 2016;151:526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cobo I, Martinelli P, Flandez M, et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 2018;554:533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development 2014;141:4206–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057– 5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004;432:307–315. [DOI] [PubMed] [Google Scholar]
- 43.Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell 2010;18:448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. J Clin Invest 2004;113:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carriere C, Gore AJ, Norris AM, et al. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterology 2011. ;141:1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scully KM, Lahmy R, Signaevskaia L, et al. E47 governs the MYC-CDKN1B/p27(KIP1)-RB network to growth arrest PDA cells independent of CDKN2A/p16(INK4A) and wild-type p53. Cell Mol Gastroenterol Hepatol 2018; 6:181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim S, Lahmy R, Riha C, et al. The basic helix-loop-helix transcription factor E47 reprograms human pancreatic cancer cells to a quiescent acinar state with reduced tumorigenic potential. Pancreas 2015;44:718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17:3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morris JP 4th, Cano DA, Sekine S, et al. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 2010;120:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carriere C, Young AL, Gunn JR, et al. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun 2009;382:561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007;11:291–302. [DOI] [PubMed] [Google Scholar]
- 52.Criscimanna A, Coudriet GM, Gittes GK, et al. Activated macrophages create lineage-specific microenvironments for pancreatic acinar-and beta-cell regeneration in mice. Gastroenterology 2014;147:1106–1118 e11. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Yan W, Mathew E, et al. Epithelial-myeloid cell crosstalk regulates acinar cell plasticity and pancreatic remodeling in mice. Elite 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012; 122:639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67:9518–9527. [DOI] [PubMed] [Google Scholar]
- 56.Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013; 73:1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331:1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2( ) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018;67:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Candido S, Abrams SL, Steelman L, et al. Metformin influences drug sensitivity in pancreatic cancer cells. Adv Biol Regul 2018;68:13–30. [DOI] [PubMed] [Google Scholar]
- 61.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012;21:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pylayeva-Gupta Y, Lee KE, Hajdu CH, et al. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012; 21:836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Yan W, Collins MA, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res 2013;73:6359–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fukuda A, Wang SC, Morris JP 4th, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011;19:441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011. ;19:456–469. [DOI] [PubMed] [Google Scholar]
- 66.Mace TA, Shakya R, Pitarresi JR, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018;67:320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Velez-Delgado A, Mathew E, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017;66:124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nagathihalli NS, Castellanos J, Lamichhane P, et al. Inverse correlation of STAT3 and MEK signaling mediates resistance to RAS pathway inhibition in pancreatic cancer. Cancer Res 2018;78:6235–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hezel AF, Kimmelman AC, Stanger BZ, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006;20:1218–1249. [DOI] [PubMed] [Google Scholar]
- 70.Apte MV, Park S, Phillips PA, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 2004;29:179–187. [DOI] [PubMed] [Google Scholar]
- 71.Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003;425:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003;425:846–851. [DOI] [PubMed] [Google Scholar]
- 73.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008;455:406–410. [DOI] [PubMed] [Google Scholar]
- 74.Mathew E, Zhang Y, Holtz AM, et al. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep 2014;9:484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 2003;3:903–911. [DOI] [PubMed] [Google Scholar]
- 76.Wang Y, McMahon AP, Allen BL. Shifting paradigms in Hedgehog signaling. Curr Opin Cell Biol 2007; 19:159–165. [DOI] [PubMed] [Google Scholar]
- 77.Mathew E, Collins MA, Fernandez-Barrena MG, et al. The transcription factor GLI1 modulates the inflammatory response during pancreatic tissue remodeling. J Biol Chem 2014;289:27727–27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev 2007;21:1244–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324:1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim EJ, Sahai V, Abel EV, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res 2014; 20:5937–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu X, Pitarresi JR, Cuitino MC, et al. Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev 2016;30:1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014;159:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017; 214:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res 2014; 2:187–193. [DOI] [PubMed] [Google Scholar]
- 87.Balachandran VP, Luksza M, Zhao JN, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006;66:9339–9344. [DOI] [PubMed] [Google Scholar]
- 89.Nguyen LV, Vanner R, Dirks P, et al. Cancer stem cells: an evolving concept. Nat Rev Cancer 2012;12:133–143. [DOI] [PubMed] [Google Scholar]
- 90.Kreso A, O’Brien CA, van Galen P, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013;339: 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell 2014;14:275–291. [DOI] [PubMed] [Google Scholar]
- 92.Greaves M Cancer stem cells as “units of selection”. Evol Appl 2013;6:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quail DF, Taylor MJ, Postovit LM. Microenvironmental regulation of cancer stem cell phenotypes. Curr Stem Cell Res Ther 2012;7:197–216. [DOI] [PubMed] [Google Scholar]
- 94.Simeone DM. Pancreatic cancer stem cells: implications I for the treatment of pancreatic cancer. Clin Cancer Res 2008;14:5646–5648. [DOI] [PubMed] [Google Scholar]
- 95.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res 2007; 67:1030–1037. [DOI] [PubMed] [Google Scholar]
- 96.Kim MP, Fleming JB, Wang H, et al. ALDH activity selectively defines an enhanced tumor-initiating cell I population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One 2011. ;6:e20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kahlert C, Bergmann F, Beck J, et al. Low expression of aldehyde dehydrogenase 1A1 (ALDH1A1) is a prognostic marker for poor survival in pancreatic cancer. BMC Cancer 2011;11:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herreros-Villanueva M, Zubia-Olascoaga A, Bujanda L. c-Met in pancreatic cancer stem cells: therapeutic implications. World J Gastroenterol 2012;18:5321–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Banerjee S, Nomura A, Sangwan V, et al. CD133+ tumor initiating cells (TIC) in a syngenic murine model of pancreatic cancer respond to Minnelide. Clin Cancer Res 2014;20:2388–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Begum A, Ewachiw T, Jung C, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS One 2017;12:e0180181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Biondani G, Zeeberg K, Greco MR, et al. Extracellular matrix composition modulates PDAC parenchymal and stem cell plasticity and behavior through the secretome. FEBS J 2018;285:2104–2124. [DOI] [PubMed] [Google Scholar]
- 102.Nomura A, Dauer P, Gupta V, et al. Microenvironment mediated alterations to metabolic pathways confer increased chemo-resistance in CD133+ tumor initiating cells. Oncotarget 2016;7:56324–56337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maeda K, Ding Q, Yoshimitsu M, et al. CD133 Modulate HIF-1 alpha expression under hypoxia in EMT phenotype pancreatic cancer stem-like cells. Int J Mol Sci 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Giancotti FG. Mechanisms governing metastatic dormancy and reactivation. Cell 2013;155:750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014;14:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8:851–864. [DOI] [PubMed] [Google Scholar]
- 107.Lonardo E, Hermann PC, Mueller MT, et al. Nodal/activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011;9:433–446. [DOI] [PubMed] [Google Scholar]
- 108.Seftor RE, Hess AR, Seftor EA, et al. Tumor cell vascu-logenic mimicry: from controversy to therapeutic promise. Am J Pathol 2012;181:1115–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wagenblast E, Soto M, Gutierrez-Angel S, et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015;520:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ruf W, Seftor EA, Petrovan RJ, et al. Differential role of tissue factor pathway inhibitors 1 and 2 in melanoma vasculogenic mimicry. Cancer Res 2003;63:5381–5389. [PubMed] [Google Scholar]
- 111.Jiang W, Zhang Y, Kane KT, et al. CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol Cancer Res 2015;13:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nomura A, Gupta VK, Dauer P, et al. NFkappaB-medi- ated Invasiveness in CD133(+) pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol Cancer Res 2018;16:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nomura A, Banerjee S, Chugh R, et al. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015;6:8313–8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang L, Wang D, Li Y, et al. CCL21/CCR7 axis contributed to CD133+ pancreatic cancer stem-like cell metastasis via EMT and Erk/NF-kappaB pathway. PLoS One 2016;11:e0158529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang F, Ma L, Zhang Z, et al. Hedgehog signaling regulates epithelial-mesenchymal transition in pancreatic cancer stem-like cells. J Cancer 2016;7:408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sharma N, Nanta R, Sharma J, et al. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015;6:32039–32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chaudhari P, Ye Z, Jang YY. Roles of reactive oxygen species in the fate of stem cells. Antioxid Redox Signal 2014;20:1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44 breast cancer-initiating cells to radiation. J Natl Cancer Inst 2006;98:1777–1785. [DOI] [PubMed] [Google Scholar]
- 119.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009;458:780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Herault O, Hope KJ, Deneault E, et al. A role for GPx3 in activity of normal and leukemia stem cells. J Exp Med 2012;209:895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim SH, Kwon CH, Nakano I. Detoxification of oxidative stress in glioma stem cells: mechanism, clinical relevance, and therapeutic development. J Neurosci Res 2014;92:1419–1424. [DOI] [PubMed] [Google Scholar]
- 122.Saretzki G, Armstrong L, Leake A, et al. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004; 22:962–971. [DOI] [PubMed] [Google Scholar]
- 123.Wang L, Yang H, Palrrbos PL, et al. ATDC/TRIM29 phosphorylation by ATM/MAPKAP kinase 2 mediates radioresistance in pancreatic cancer cells. Cancer Res 2014;74:1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yalcin S, Zhang X, Luciano JP, et al. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem 2008;283:25692–25705. [DOI] [PubMed] [Google Scholar]
- 125.Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORCI in response to ROS. Proc Natl Acad Sci U S A 2010;107:4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J 2011;30:546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dauer P, Sharma NS, Gupta VK, et al. GRP78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Mol Oncol 2018;12:1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Duong HQ, You KS, Oh S, et al. Silencing of NRF2 reduces the expression of ALDH1A1 and ALDH3A1 and sensitizes to 5-FU in pancreatic cancer cells. Antioxidants (Basel) 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal 2018;29:1727–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang B, Lee CW, Witt A, et al. Heat shock factor 1 induces cancer stem cell phenotype in breast cancer cell lines. Breast Cancer Res Treat 2015;153:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arnason T, Harkness T Development, maintenance, and reversal of multiple drug resistance: at the crossroads of TFPI1, ABC transporters, and HIF1. Cancers (Basel) 2015;7:2063–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]