

Abstract
Background:
A promising biomarker for axonal damage early in the disease course of multiple sclerosis (MS) is neurofilament light chain (NfL). It is unknown whether NfL has the same predictive value for MS diagnosis in children as in adults.
Objective:
To explore the predictive value of NfL levels in cerebrospinal fluid (CSF) for MS diagnosis in paediatric and adult clinically isolated syndrome (CIS) patients.
Methods:
A total of 88 adult and 65 paediatric patients with a first attack of demyelination were included and followed (mean follow up-time in adults: 62.8 months (standard deviation (SD) ±38.7 months) and 43.8 months (SD ±27.1 months) in children). Thirty control patients were also included. Lumbar puncture was done within 6 months after onset of symptoms. NfL was determined in CSF using enzyme-linked immunosorbent assay (ELISA). COX regression analyses were used to calculate hazard ratios (HR) for clinically definite multiple sclerosis (CDMS) diagnosis.
Results:
After adjustments for age, oligoclonal bands (OCB), and asymptomatic T2 lesions on baseline magnetic resonance imaging (MRI), increased NfL levels in both paediatric and adult CIS patients were associated with a shorter time to CDMS diagnosis (children HR = 3.7; p = 0.007, adults HR = 2.1; p = 0.032). For CIS patients with a future CDMS diagnosis, children showed higher NfL levels than adults (geometric mean 4888 vs 2156 pg/mL; p = 0.007).
Conclusion:
CSF NfL levels are associated with CDMS diagnosis in children and adults with CIS. This makes NfL a promising predictive marker for disease course with potential value in clinical practice.
Keywords: CIS, multiple sclerosis, neurofilament light chain, children, adults
Introduction
Childhood-onset multiple sclerosis (MS) occurs in 3%–5% of all MS patients.1,2 Although children with MS have a more inflammatory disease course with a higher relapse rate than adult patients,3–5 clinical follow-up studies suggest that disability progression is slower in children than in adults.4,6,7 However, impairment of age-expected brain growth was seen early in the disease course of paediatric MS patients.8 This indicates that not only neuroinflammation but also neurodegeneration occurs early in childhood-onset MS.
Axonal damage is considered one of the major causes for persisting neurological disability in MS.9 A promising biomarker for axonal damage is neurofilament light chain (NfL).
NfL is an element of the neuron cytoskeleton and is released in the extracellular space after neuronal cell death.10 In healthy individuals, NfL levels increase with age, which reflects neurodegeneration and is part of the physiological ageing process.11
High NfL levels in cerebrospinal fluid (CSF) in adults with clinically isolated syndrome (CIS) have been reported as an independent risk factor for MS diagnosis.12 Furthermore, NfL levels have been associated with brain volume changes in adult CIS patients.12 However, whether NfL is also increased at disease onset in children is still unknown.
The primary purpose of this prospective study was to investigate whether NfL levels can predict diagnosis of clinically definite multiple sclerosis (CDMS) in children with CIS. Our second aim was to compare NfL levels in CSF at time of a first demyelinating event between children and adults. Finally, we examined the association between NfL and signs of axonal loss on magnetic resonance imaging (MRI).
Methods
Study participants
Children and adults were included in either our prospective cohort of adult patients with CIS (PRedicting the OUtcome of a Demyelinating event, PROUD study) or in our prospective cohort of children with acquired demyelinating syndromes (ADS; PROUD-kids study).13,14 Both studies are ongoing multicentre studies initiated by Erasmus MC in Rotterdam, The Netherlands, which is a tertiary referral centre for adult and paediatric MS patients (MS Centre ErasMS and National Paediatric MS Centre).
All patients were included between February 2002 and December 2015 within 6 months after a first event of demyelination of the central nervous system (CNS). Adult patients were younger than 50 years of age, and paediatric patients were younger than 18 years. No patients had a history of previous neurological symptoms suggestive for CNS demyelination. Patients with alternative diagnoses were excluded from analyses.
The included patients underwent a baseline brain MRI and routine laboratory tests to rule out other possible diagnoses. A lumbar puncture (LP) was performed and extra CSF was collected and stored at −80°C until use.
Patients were assessed at baseline and were reassessed regularly. At baseline, instructions were given to the patients to contact the hospital in case of suspected exacerbation.
CSF of adult control samples (n = 30) was obtained in the Erasmus MC from patients with neurological symptoms but no objective clinical or paraclinical findings to define a specific neurological disease (symptomatic controls).15
Standard protocol approvals and patient consents
The study protocol was approved by the Medical Ethics Committee of Erasmus MC Rotterdam and of the other participating centres. Written informed consent was obtained from all patients and/or their families.
Definitions
CIS was defined as a first attack of demyelination in the CNS without encephalopathy.16 Clinically definite MS (CDMS) was defined by the Poser criteria as two non-encephalopathic attacks with clinical evidence of two separate lesions.17 ADS in children encompass the first attack of demyelination, including CIS and acute disseminated encephalomyelitis (ADEM).14 Patients presenting with other ADS subtypes than CIS or ADEM were excluded from the analyses. Children were diagnosed with CIS, ADEM and CDMS according to the diagnostic criteria proposed by the International Paediatric Multiple Sclerosis Study Group.13 CDMS was used as the primary outcome. Patients who remained CIS during follow-up are referred to as CIS-CIS and patients who were diagnosed with CDMS during follow-up are referred to as CIS-CDMS. In both children and adults, an exacerbation is defined as sub-acute worsening of existing symptoms or new symptoms after at least 30 days of improvement or stable disease. Symptoms should exist for more than 24 hours, not be preceded by fever and not be caused by an alternative diagnosis.18 All exacerbations were confirmed by neurological examination.
Expanded Disability Status Scale (EDSS) was used to assess disability.19 When patients were diagnosed with CDMS, an EDSS was done annually. EDSS scores performed within 3 months after an exacerbation were not considered. Follow-up was calculated by subtracting the date of first symptoms from the last visit date. Baseline MRI scans were performed at 1.5 T scanners and reviewed blindly. Available T1-, axial T2-, axial and/or sagittal fluid attenuated inversion recovery (FLAIR) images were used. The MRIs were scored on ≥9 T2 lesions, dissemination in space and time, and asymptomatic T2 lesions. The presence of T1-hypointense lesions on baseline MRI were assessed in CDMS patients. T1-hypointense lesions were defined as non-enhancing lesions being hypointense relative to cortical grey matter.20 Patients who did not receive gadolinium were excluded for the analysis of T1-hypointense lesions.
CSF sampling and NfL enzyme-linked immunosorbent assay
Routine CSF diagnostics including IgG index, oligoclonal bands (OCB), cell count and total protein were performed. The remaining CSF was immediately centrifuged for 10 minutes at 3000 r/min to separate the supernatant from cells and cellular elements. After centrifugation, samples were aliquoted and stored at −80°C until use.
CSF analyses for OCB were performed in local laboratories using isoelectric focusing.21 OCB status was regarded as positive if there were ⩾2 unique bands in CSF compared to serum. IgG index above 0.66 was considered as elevated.
NfL levels in CSF were measured batch wise in two rounds, according to the manufacturer’s instructions, using a stable commercially available solid-phase sandwich ELISA (UmanDiagnostics, Umea, Sweden).22 NfL concentrations (picogram per millilitre (pg/mL)) were calculated using a standard curve according to manufacturer’s instructions. All samples were tested double blind and measured in duplicate. The detection limit of the ELISA was 150 pg/mL.
Data analysis
We used SPSS software, version 21.0 (SPSS Inc) and GraphPad Prism5 to perform statistical analyses. After log transformation, NfL levels showed a parametric distribution in both children and adult samples. Therefore, we calculated geometric means for NfL levels. Group comparisons for continuous data were performed using two-tailed t-test for normally distributed variables (NfL, age at onset, follow-up time), and Mann–Whitney U test was used for non-parametric data (time between CIS and LP). We used one-way analysis of variance (ANOVA) and post hoc Bonferroni correction for parametric data to analyse differences between multiple groups. Chi-square or Fisher exact test were performed for categorical variables (gender, type of clinical onset, OCB, elevated IgG index, asymptomatic T2 lesions, ≥9 T2 lesions, and disease-modifying therapies (DMTs) after CIS and before CDMS). Spearman rank correlation was used for correlation analyses between non-parametric continuous variables. Time to CDMS diagnosis was determined by subtracting the date of the first symptoms from the date of diagnosis. COX proportional hazard regression analyses were used to calculate univariate and multivariable hazard ratios (HR) for time to CDMS diagnosis. Known predictors for MS diagnosis were used in the multivariable analyses (OCB, asymptomatic T2 lesions). Patients who were not diagnosed with CDMS during follow-up were considered as censored observations. We used the median of NfL levels to establish cut-off values for high and low levels of NfL in children and adults separately. The p values less than 0.05 were considered significant.
Results
Patients characteristics
A total of 65 children with a first demyelinating event of the CNS, 88 adult patients with CIS, and 30 age- and gender-matched adult control individuals were included in this study.
Of the 65 children, 24 presented with ADEM and 41 with CIS; 25 out of 41 (61%) children with CIS were diagnosed with CDMS during a mean follow-up time of 38.4 months (standard deviation (SD): 21.0 months). The mean follow-up time in adult CIS patients was 68.2 months (SD: 39.3 months) in this period, and 43 (49%) patients were diagnosed with CDMS.
The median time from CIS to CDMS in adult patients was 36.4 months (interquartile range (IQR) 14.4–48.9 months) and in children 10.8 months (IQR 5.0–15.7 months).
The time between CIS and LP was not significantly different between children and adult patients. No patients were receiving DMTs at time of LP.
In all, 16 adult patients (18%) and 14 (34%) children who were not yet diagnosed with CDMS received DMT (glatiramer acetate (n = 11), interferon (n = 19), and natalizumab (n = 1)). The patient characteristics for adults and children are shown in Tables 1 and 2.
Table 1.
Patient characteristics (adults).
| Adults | Controls (n = 30) | CIS patients (n = 88) | CIS-CDMS (n = 43) | CIS-CIS (n = 45) | p value a |
|---|---|---|---|---|---|
| Female sex, no. (%) | 20 (66.7) | 59 (67.0) | 34 (79.1) | 25 (55.6) | 0.02 |
| Ageb, mean (SD), years | 33.4 (±9.5) | 31.2 (±7.2) | 31.9 (±7.1) | 33.6 (±7.3) | 0.28 |
| Follow-up time, mean (SD), months | na | 62.8 (±38.7) | 89.0 (±36.8) | 48.3 (±30.7) | <0.01 |
| Type of clinical onset, no. (%) | |||||
| Optic nerve | na | 41 (46.6) | 21 (48.8) | 20 (44.4) | 0.68 |
| Spinal cord | na | 23 (26.1) | 11 (25.6) | 12 (26.7) | 0.91 |
| Other localization | na | 24 (27.3) | 11 (25.6) | 13 (28.9) | 0.73 |
| OCB (⩾2 bands, %) | na | 63/83 (75.9) | 36/41 (87.8) | 27/42 (64.3) | 0.01 |
| Elevated IgG index (cut-off: 0.66), no. (%) | na | 44/85 (51.8) | 22/41 (53.7) | 22/44 (50.0) | 0.74 |
| Time CIS to LP, median (IQR), weeks | na | 6.1 (2.7–13.2) | 6.0 (2.9–12.6) | 6.7 (2.6–14.1) | 0.97 |
| ⩾9 lesions on T2-weighted images, no. (%) | na | 27 (30.7) | 18 (41.9) | 9 (20.0) | 0.03 |
| Asymptomatic T2 lesions, no. (%) | na | 76 (86.4) | 38 (88.4) | 38 (84.4) | 0.59 |
| MS based on first MRIb, no (%) | na | 16 (18.2) | 11 (25.6) | 5 (11.1) | 0.08 |
| DMT before CDMS diagnosis, no. (%) | na | 16 (18.2) | 12 (27.9) | 4 (8.9) | 0.02 |
| Time between CIS and start DMT, median (IQR), months | na | 27.8 (9.7–43.1) | 29.5 (9.9–46.6) | 14.4 (5.9–29.0) | 0.25 |
CIS: clinically isolated syndrome; CIS-CDMS: patients who are diagnosed with CDMS during follow-up after CIS defined by Poser criteria; CIS-CIS: not diagnosed with CDMS; na: not applicable; DMT: disease-modifying therapy; OCB: oligoclonal bands; Ig: immunoglobulin; LP: lumbar puncture; pg/mL: picogram/millilitre; IQR: interquartile range.
p value calculated between CIS-CDMS and CIS-CIS.
For patients with CIS: age at CIS; for controls: age at lumbar puncture.
Dissemination in space and time at baseline based on McDonald 2010 criteria.
Table 2.
Patient characteristics (children).
| Children | ADS-patients (n = 65) | ADEM (n = 24) | CIS-CDMS (n = 25) | CIS-CIS (n = 16) | p valuea |
|---|---|---|---|---|---|
| Female sex, no. (%) | 38 (58.8) | 16 (66.7) | 16 (64.0) | 6 (37.5) | 0.10 |
| Age, median (IQR), years | 12.5 (5.4–15.5) | 4.1 (2.6–7.2) | 15.0 (13.8–16.0) | 14.2 (9.0–16.4) | 0.52 |
| Follow-up time, mean (SD), months | 43.8 (±27.1) | 53.1 (±33.7) | 44.1 (±22.3) | 29.5 (±15.5) | 0.03 |
| Type of clinical onset, no. (%) | |||||
| Optic nerve | 11 (16.9) | 0 (0.0) | 5 (20.0) | 6 (37.5) | 0.22 |
| Spinal cord | 11 (16.9) | 0 (0.0) | 6 (24.0) | 5 (31.2) | 0.61 |
| Other localization | 7 (10.8) | 0 (0.0) | 4 (16.0) | 3 (18.8) | 1.00 |
| Polyfocal without encephalopathy | 12 (18.4) | 0 (0.0) | 10 (40.0) | 2 (12.5) | 0.08 |
| Polyfocal with encephalopathy | 24 (36.9) | 24 (100.0) | 0 (0.0) | 0 (0.0) | 1.00 |
| OCB (⩾2 bands; %) | 28/54 (51.9) | 0/17 (0.0) | 21/23 (91.3) | 7/14 (50.0) | 0.01 |
| Elevated IgG index (cut-off: 0.66, no. (%)) | 33/50 (66.0) | 4/12 (33.3) | 22/23 (95.7) | 7/15 (46.7) | <0.01 |
| Time first symptoms to LP, median (IQR; weeks) | 2.3 (0.8–7.6) | 1.57 (0.57–2.64) | 6.0 (1.9–12.8) | 1.8 (0.6–7.8) | 0.10 |
| ⩾9 lesions on T2-weighted images, no. (%) | 27 (41.5) | 6 (25.0) | 16 (64.0) | 5 (31.2) | 0.04 |
| Asymptomatic T2 lesions, no. (%) | 55 (8.6) | 22 (91.7) | 24 (96.0) | 9 (56.2) | 0.003 |
| MS based on first MRIb, no. (%) | 10 (15.4) | na | 9 (36.0) | 1 (6.2) | 0.03 |
| DMT before CDMS diagnosis, no. (%) | 14/41 (34.1) | na | 9 (36.0) | 5 (31.3) | 0.75 |
| Time between CIS and start DMT, median (IQR), months | 6.4 (3.3–12.1) | na | 6.3 (1.7–14.8) | 6.5 (3.5–10.4) | 0.72 |
CIS: clinically isolated syndrome; CIS-CDMS: patients who are diagnosed with CDMS during follow-up after CIS defined by Poser criteria; CIS-CIS: not diagnosed with CDMS; na: not applicable; DMT: disease-modifying therapy; OCB: oligoclonal bands; Ig: immunoglobulin; LP: lumbar puncture; pg/mL: picogram/millilitre; IQR: interquartile range.
p value calculated between CIS-CDMS and CIS-CIS.
Dissemination in space and time at baseline based on McDonald 2010 criteria.
NfL levels at time of the first demyelinating event per clinical subgroup
Patients (children and adults) at time of a first attack of demyelination showed higher NfL levels than control individuals; geometric mean 2040 versus 444 pg/mL (p < 0.001). In Figure 1, NfL levels from controls, adult, and paediatric patients are shown.
Figure 1.

CSF Nf levels in controls versus adults versus children. CIS and ADEM patients are included in children. Horizontal lines with error bars indicate geometric mean with 95% CI.
NfL: neurofilament light chain; pg/mL: picogram/millilitre.
In adult patients, NfL levels at time of CIS were higher in the group that was diagnosed with CDMS (CIS-CDMS, n = 43 (49%)) compared to the group that remained CIS during follow-up (CIS-CIS); geometric mean 2156 versus 1342 pg/mL (p = 0.012; Figure 2).
Figure 2.

NfL levels in clinical subgroups of adults and children. Horizontal lines with error bars indicate geometric mean with 95% CI.
NfL: neurofilament light chain; ns: not significant; pg/mL: picogram/millilitre.
In children, we compared NfL levels between CIS-CIS, CIS-CDMS, and ADEM patients. NfL levels at time of CIS in paediatric CIS-CDMS patients (n = 25; 61%) were higher than in paediatric CIS-CIS patients; geometric mean 4888 versus 967 pg/mL (p = 0.01). Children with ADEM did not differ in NfL levels from CIS-CIS and CIS-CDMS children; geometric mean 2683 pg/mL (Figure 2). There was no correlation between time from onset of symptoms to LP and NfL levels in both children and adults.
NfL levels compared between children and adults
Next, we compared NfL levels between children and adult patients at the time of CIS. In the CIS-CDMS group, NfL levels were higher in children compared to adults; geometric mean 4888 versus 2156 pg/mL (p = 0.007). NfL levels were not different between adults and children in the CIS-CIS groups (Figure 2).
Association of NfL levels with time to CDMS diagnosis in children and adults with CIS
To analyse time to CDMS diagnosis, we used median CSF NfL levels in CIS patients as cut-off. This resulted in a cut-off of 1802 pg/mL for adults and 2537 pg/mL for children. These cut-offs were used to divide CIS patients (ADEM excluded) into groups with high and low NfL levels and were subsequently used in the COX regression analysis.
The univariate COX regression analysis showed an HR for CDMS diagnosis of 2.1; p = 0.024 in adults and 3.8 in children with CIS; p = 0.003. Kaplan–Meier curves are shown in Figure 3(a) and (b).
Figure 3.
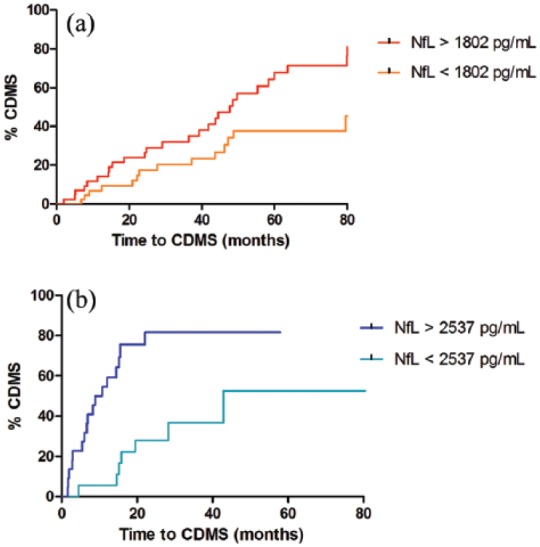
Time from CIS to CDMS in CIS patients with high and low CSF NfL levels. (a) Adults (log-rank test, p = 0.02). (b) Children (log-rank test, p = 0.001). Kaplan–Meier curves showing time to CDMS diagnosis for CIS patients (ADEM excluded) with either high or low CSF NfL levels.
CDMS: clinically definite multiple sclerosis; NfL: neurofilament light chain; pg/mL: picogram/millilitre.
In a multivariable COX regression analysis, we corrected for clinically relevant parameters; the presence of asymptomatic T2 lesions on the baseline MRI and OCB. We also corrected for age at onset, based on correlation with NfL levels in control individuals (Spearman rho = 0.59, p = 0.001). The HR in the multivariable COX regression analysis for high NfL levels was 2.1 in adults (p = 0.032) and 3.7 in children (p = 0.007; Table 3).
Table 3.
Cox regression (univariate and multivariable) hazard ratios for CDMS diagnosis in adults and children with CIS.
| Univariate analysis HR (95% CI) |
p value | Multivariable analysis HR (95% CI) |
p value | |
|---|---|---|---|---|
| Adults | ||||
| Total group (n = 88) | 2.1 (1.1–3.9) | 0.024 | 2.1 (1.1–4.1) | 0.032 |
| After excluding patients with DMT before CDMS diagnosis (n = 72) | 2.2 (1.1–4.7) | 0.035 | 2.2 (1.0–4.9) | 0.061 |
| After excluding CIS-CIS patients with FU < 2 years (n = 78) | 2.0 (1.1–3.8) | 0.027 | 2.1 (1.1–4.1) | 0.034 |
| Children | ||||
| Total group (n = 41) | 3.8 (1.6–9.2) | 0.003 | 3.7 (1.4–9.3) | 0.007 |
| After excluding patients with DMT before CDMS diagnosis (n = 27) | 19.8 (2.5–155.5) | 0.005 | 13.7 (1.6–114.3) | 0.015 |
| After excluding CIS-CIS patients with FU < 2 years (n = 36) | 3.8 (1.6–9.2) | 0.003 | 4.2 (1.6–11.3) | 0.004 |
NfL: neurofilament light chain; HR: hazard ratio; CI: confidence interval; DMT: disease-modifying treatment; CDMS: clinically definite multiple sclerosis; FU: follow-up; CIS: clinically isolated syndrome.
Multivariable analyses: corrected for the presence of asymptomatic T2 lesions on baseline MRI, the presence of OCB and age of onset.
Hazard ratios for CDMS diagnosis in subgroups for adults and children with CIS (ADEM excluded).
A total of 16 (18%) adult CIS patients and 14 (34%) children received DMT before CDMS diagnosis. This could have postponed the second attack. The HR in children increased after excluding patients who received DMT before CDMS diagnosis. In adults, the univariate HR did not change, and the HR in the multivariable analysis showed a trend towards significance.
When we add DMT before CDMS diagnosis into the COX regression model, the HRs did not change; 13 (11%) adults and 9 (22%) children were treated with methylprednisolone within 3 months before LP. When we corrected for this in the COX regression model, the results did not change.
In another subanalysis, we excluded CIS-CIS patients who had less than 2 years of follow-up (children: n = 5, adults: n = 10). After this exclusion, HRs were not altered in adults and increased in children. Table 3 shows the univariate and multivariable HRs including those of the subanalyses.
Association of CSF NfL levels with disability
We did not find a correlation between CSF NfL levels and EDSS scores after CDMS diagnosis. We collected EDSS data from 55/68 (81%) patients who were diagnosed with CDMS. Only 6 patients (4 adults and 2 children) reached an EDSS of 3.0 or more.
Association of CSF NfL levels with signs of axonal damage on MRI
CSF NfL levels were increased in CDMS patients showing T1-hypointense lesions on baseline MRI (adults 20/39, 51%; children 15/23, 65%). We found this in adults (geometric mean 3188 vs 1588 pg/mL; p = 0.001) and in children (geometric mean 8920 vs 1668; p = 0.001; Figure 4(a) and (b)).
Figure 4.

CSF NfL levels in CIS-CDMS adults and children with and without T1-hypointense lesions on baseline MRI. (a) Adults: T1-hypointense lesions versus no T1-hypointense lesions on baseline MRI. (b) Children: T1-hypointense lesions versus no T1-hypointense lesions on baseline MRI. Horizontal lines with error bars indicate geometric mean with 95% CI.
NfL: neurofilament light chain; pg/mL: picogram/millilitre; CDMS: clinically definite multiple sclerosis.
Discussion
In this prospective study, we demonstrate that CSF NfL levels in children and adults with a first attack of suspected MS are predictive for CDMS diagnosis. Furthermore, at time of CIS, CSF NfL levels in patients with a future CDMS diagnosis are higher in children than in adult patients. This underlines that not only inflammation is more severe in children3–5 but that children also have more axonal damage early in the disease course of MS than adult patients.8
To our knowledge, we are the first to show that CSF NfL levels are associated with a subsequent diagnosis of CDMS in children with CIS. In addition, the results validate the predictive value of CSF NfL levels for CDMS diagnosis in adult CIS patients.12,23 Both in adults and children, these findings were independent of known predictive factors for CDMS, that is, asymptomatic T2 lesions on baseline MRI and unique OCBs in CSF. Moreover, CSF NfL levels predicted a second attack even better than these currently used markers.
It is essential to improve currently available routes to prediction to prevent unnecessary treatment of patients with low clinical disease activity, especially because these immunomodulatory therapies can have serious side effects. Recently, other potential CSF biomarkers for a future MS diagnosis have been identified.24,25 Our findings draw further attention to the relevance of including CSF analyses as part of routine diagnostics.
Since NfL is considered a biomarker for axonal damage, neurodegeneration, and brain atrophy,26,27 we reasoned that its presence in CSF could be associated with T1-hypointense lesions on MRI, which are signs of axonal loss.28 In children with CIS, these T1-hypointense lesions have been reported to be highly predictive for MS diagnosis.20 Here, we demonstrate that children and adult CIS patients with T1-hypointense lesions on baseline MRI have higher NfL levels than patients without these lesions.
CSF NfL levels in children with ADEM were not significantly different from levels in patients who remained CIS or who were eventually diagnosed with CDMS. Nevertheless, these levels were high in ADEM patients (geometric mean = 2683 pg/mL), indicating considerable axonal damage. Studies have reported cognitive impairment and persistent motor dysfunction in children with ADEM.29,30 Moreover, it has been shown that subsequent white matter maturation and age-expected brain growth are disturbed not only in paediatric MS but also in monophasic ADS, including ADEM.31,32 These findings support the occurrence of damage during the acute phase with a lasting impact, which stresses the importance of adequate follow-up and support after the acute event.
NfL levels have been reported to be also increased in serum of adult CIS and MS patients,33,34 but the predictive value in CIS patients for MS diagnosis of this marker seems limited to CSF.12 As we here aimed to assess and compare prediction in both children and adults with CIS, we restricted in this study to CSF samples.
There are some limitations in this study. First, the range of follow-up is rather wide. We did correct for this in the COX regression analyses, and we also performed a subanalysis after excluding CIS-CIS patients with a follow-up less than 2 years, which did not change our findings. Second, in both the adult and paediatric study, we did not perform a follow-up MRI on a regular basis. Therefore, the Poser criteria were used instead of the McDonald 2010 criteria. In this way, we could show an effect on clinical disease activity (second attack) instead of disease activity measured with MRI. Third, in order to prove an association of CSF NfL levels with EDSS, we will need a longer follow-up period since disability occurs later in the disease course especially in children.6 Fourth, we did not have access to advanced imaging techniques for quantification of neurodegeneration (e.g. T1-hypointense lesion volumes, total brain volume and brain tissue integrity). However, we used the presence of T1-hypointense lesions as an MRI marker for axonal damage,28 because it is easily assessable. Furthermore, we did not include paediatric controls since we did not receive ethical permission to collect paediatric control CSF samples, which made the collection of this rare material not possible. Yet, control groups in other paediatric studies indicate low physiological levels of NfL in the same range as the adult controls in this study.35,36
Finally, although our sample size was relatively small due to limited availability of CSF, we were still able to correct our results for other predictive factors for MS diagnosis. Our findings in children were also compatible to those in adults, further stressing the robustness of the observations.
In conclusion, we show that high levels of CSF NfL are associated with CDMS diagnosis independently of known predictive factors (i.e. asymptomatic T2 lesions and OCB) in both children and adults. CSF NfL levels at time of a first demyelinating event are higher in children than in adults with a future CDMS diagnosis. In addition, this marker for axonal damage is associated with MRI signs of neurodegeneration in both groups. Given that therapeutic interventions might delay disease progression and accumulation of disability,37,38 it is essential to accurately predict MS diagnosis, not the least in the paediatric population. Hence, NfL in CSF is a promising predictive marker for the disease course in both adults and children with a first demyelinating event, with a potential value in future clinical practice.
Acknowledgments
The authors thank the patients of the PROUD and PROUD-kids study and the participating centres, including the members of the Dutch Paediatric MS Study Group.
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Roos M van der Vuurst de Vries, Yu Yi M Wong, Julia Y Mescheriakova, E Daniëlle van Pelt, Tessel F Runia, Theodora AM Siepman, Marie-José Melief, Annet F Wierenga-Wolf, and Marvin M van Luijn reports no competing interests. Naghmeh Jafari received honoraria for serving on advisory boards for Teva, Merck, Roche and Genzyme. Johnny P Samijn received honoraria for serving on advisory boards for Merck Serono and Genzyme. Received travel grants from Merck Serono. He participated in trials with Biogen Idec, Merck Serono, Roche and Genzyme. Rinze F Neuteboom participates in paediatric MS trials sponsored by Sanofi and Novartis. Rogier Q Hintzen received honoraria for serving on advisory boards for Biogen Idec, Roche and Sanofi. He participated in trials with Biogen Idec, Merck Serono, Roche, Genzyme and Novartis.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The study was supported by the Dutch MS Research Foundation.
Contributor Information
Roos M van der Vuurst de Vries, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Yu Yi M Wong, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Julia Y Mescheriakova, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
E Daniëlle van Pelt, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Tessel F Runia, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Naghmeh Jafari, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Theodora AM Siepman, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Marie-José Melief, Department of Immunology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Annet F Wierenga-Wolf, Department of Immunology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Marvin M van Luijn, Department of Immunology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
Johnny P Samijn, Department of Neurology, Maasstad Hospital, Rotterdam, The Netherlands.
Rinze F Neuteboom, Department of Paediatric Neurology, Paediatric MS Centre, Erasmus MC-Sophia, Rotterdam, The Netherlands.
Rogier Q Hintzen, Department of Neurology, MS Centre ErasMS, Erasmus MC, Rotterdam, The Netherlands.
References
- 1. Banwell B, Ghezzi A, Bar-Or A, et al. Multiple sclerosis in children: Clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol 2007; 6: 887–902. [DOI] [PubMed] [Google Scholar]
- 2. Chitnis T, Glanz B, Jaffin S, et al. Demographics of pediatric-onset multiple sclerosis in an MS center population from the Northeastern United States. Mult Scler 2009; 15: 627–631. [DOI] [PubMed] [Google Scholar]
- 3. Van der Vuurst de Vries RM, van Pelt ED, Mescheriakova JY, et al. Disease course after clinically isolated syndrome in children versus adults: A prospective cohort study. Eur J Neurol 2017; 24: 315–321. [DOI] [PubMed] [Google Scholar]
- 4. Boiko A, Vorobeychik G, Paty D, et al. Early onset multiple sclerosis: A longitudinal study. Neurology 2002; 59: 1006–1010. [DOI] [PubMed] [Google Scholar]
- 5. Yeh EA, Weinstock-Guttman B, Ramanathan M, et al. Magnetic resonance imaging characteristics of children and adults with paediatric-onset multiple sclerosis. Brain 2009; 132: 3392–3400. [DOI] [PubMed] [Google Scholar]
- 6. Renoux C, Vukusic S, Mikaeloff Y, et al. Natural history of multiple sclerosis with childhood onset. N Engl J Med 2007; 356: 2603–2613. [DOI] [PubMed] [Google Scholar]
- 7. Simone IL, Carrara D, Tortorella C, et al. Course and prognosis in early-onset MS: Comparison with adult-onset forms. Neurology 2002; 59: 1922–1928. [DOI] [PubMed] [Google Scholar]
- 8. Aubert-Broche B, Fonov V, Narayanan S, et al. Onset of multiple sclerosis before adulthood leads to failure of age-expected brain growth. Neurology 2014; 83: 2140–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Filippi M, Bozzali M, Rovaris M, et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis. Brain 2003; 126: 433–437. [DOI] [PubMed] [Google Scholar]
- 10. Petzold A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J Neurol Sci 2005; 233: 183–198. [DOI] [PubMed] [Google Scholar]
- 11. Vagberg M, Norgren N, Dring A, et al. Levels and age dependency of neurofilament light and glial fibrillary acidic protein in healthy individuals and their relation to the brain parenchymal fraction. PLoS One 2015; 10: e0135886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arrambide G, Espejo C, Eixarch H, et al. Neurofilament light chain level is a weak risk factor for the development of MS. Neurology 2016; 87: 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krupp LB, Tardieu M, Amato MP, et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: Revisions to the 2007 definitions. Mult Scler 2013; 19: 1261–1267. [DOI] [PubMed] [Google Scholar]
- 14. Hintzen RQ, Dale RC, Neuteboom RF, et al. Pediatric acquired CNS demyelinating syndromes: Features associated with multiple sclerosis. Neurology 2016; 87: S67–73. [DOI] [PubMed] [Google Scholar]
- 15. Teunissen C, Menge T, Altintas A, et al. Consensus definitions and application guidelines for control groups in cerebrospinal fluid biomarker studies in multiple sclerosis. Mult Scler 2013; 19: 1802–1809. [DOI] [PubMed] [Google Scholar]
- 16. Miller DH, Chard DT, Ciccarelli O. Clinically isolated syndromes. Lancet Neurol 2012; 11: 157–169. [DOI] [PubMed] [Google Scholar]
- 17. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann Neurol 1983; 13: 227–231. [DOI] [PubMed] [Google Scholar]
- 18. Schumacher GA, Beebe G, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: Report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci 1965; 122: 552–568. [DOI] [PubMed] [Google Scholar]
- 19. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: An Expanded Disability Status Scale (EDSS). Neurology 1983; 33: 1444–1452. [DOI] [PubMed] [Google Scholar]
- 20. Verhey LH, Branson HM, Shroff MM, et al. MRI parameters for prediction of multiple sclerosis diagnosis in children with acute CNS demyelination: A prospective national cohort study. Lancet Neurol 2011; 10: 1065–1073. [DOI] [PubMed] [Google Scholar]
- 21. Freedman MS, Thompson EJ, Deisenhammer F, et al. Recommended standard of cerebrospinal fluid analysis in the diagnosis of multiple sclerosis: A consensus statement. Arch Neurol 2005; 62: 865–870. [DOI] [PubMed] [Google Scholar]
- 22. Petzold A, Altintas A, Andreoni L, et al. Neurofilament ELISA validation. J Immunol Methods 2010; 352: 23–31. [DOI] [PubMed] [Google Scholar]
- 23. Hakansson I, Tisell A, Cassel P, et al. Neurofilament light chain in cerebrospinal fluid and prediction of disease activity in clinically isolated syndrome and relapsing-remitting multiple sclerosis. Eur J Neurol 2017; 24: 703–712. [DOI] [PubMed] [Google Scholar]
- 24. Komori M, Blake A, Greenwood M, et al. Cerebrospinal fluid markers reveal intrathecal inflammation in progressive multiple sclerosis. Ann Neurol 2015; 78: 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van der Vuurst de Vries RM, Mescheriakova JY, Runia TF, et al. Soluble CD27 levels in cerebrospinal fluid as a prognostic biomarker in clinically isolated syndrome. JAMA Neurol 2017; 74: 286–292. [DOI] [PubMed] [Google Scholar]
- 26. Gaiottino J, Norgren N, Dobson R, et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS One 2013; 8: e75091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kuhle J, Nourbakhsh B, Grant D, et al. Serum neurofilament is associated with progression of brain atrophy and disability in early MS. Neurology 2017; 88: 826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Waesberghe JH, Kamphorst W, De Groot CJ, et al. Axonal loss in multiple sclerosis lesions: Magnetic resonance imaging insights into substrates of disability. Ann Neurol 1999; 46: 747–754. [DOI] [PubMed] [Google Scholar]
- 29. Beatty C, Bowler RA, Farooq O, et al. Long-term neurocognitive, psychosocial, and magnetic resonance imaging outcomes in pediatric-onset acute disseminated encephalomyelitis. Pediatr Neurol 2016; 57: 64–73. [DOI] [PubMed] [Google Scholar]
- 30. Jacobs RK, Anderson VA, Neale JL, et al. Neuropsychological outcome after acute disseminated encephalomyelitis: Impact of age at illness onset. Pediatr Neurol 2004; 31: 191–197. [DOI] [PubMed] [Google Scholar]
- 31. Longoni G, Brown RA, MomayyezSiahkal P, et al. White matter changes in paediatric multiple sclerosis and monophasic demyelinating disorders. Brain 2017; 140: 1300–1350. [DOI] [PubMed] [Google Scholar]
- 32. Aubert-Broche B, Weier K, Longoni G, et al. Monophasic demyelination reduces brain growth in children. Neurology 2017; 88: 1744–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Disanto G, Adiutori R, Dobson R, et al. Serum neurofilament light chain levels are increased in patients with a clinically isolated syndrome. J Neurol Neurosurg Psychiatry 2016; 87: 126–129. [DOI] [PubMed] [Google Scholar]
- 34. Disanto G, Barro C, Benkert P, et al. Serum neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann Neurol 2017; 81: 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shahim P, Darin N, Andreasson U, et al. Cerebrospinal fluid brain injury biomarkers in children: A multicenter study. Pediatr Neurol 2013; 49: 31–39. [DOI] [PubMed] [Google Scholar]
- 36. Pranzatelli MR, Tate ED, McGee NR, et al. CSF neurofilament light chain is elevated in OMS (decreasing with immunotherapy) and other pediatric neuroinflammatory disorders. J Neuroimmunol 2014; 266: 75–81. [DOI] [PubMed] [Google Scholar]
- 37. Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology 2006; 67: 1242–1249. [DOI] [PubMed] [Google Scholar]
- 38. Jokubaitis VG, Spelman T, Kalincik T, et al. Predictors of disability worsening in clinically isolated syndrome. Ann Clin Transl Neurol 2015; 2: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]