

Abstract
Myeloid‐derived suppressor cells (MDSCs) are immature myeloid cells with strong immunosuppressive activity that promote tumor growth. In this study, we describe a mechanism by which cancer cells control MDSCs in human cancers by upregulating TRF2, a protein required for telomere stability. Specifically, we showed that the TRF2 upregulation in cancer cells has extratelomeric roles in activating the expression of a network of genes involved in the biosynthesis of heparan sulfate proteoglycan, leading to profound changes in glycocalyx length and stiffness, as revealed by atomic force microscopy. This TRF2‐dependent regulation facilitated the recruitment of MDSCs, their activation via the TLR2/MyD88/IL‐6/STAT3 pathway leading to the inhibition of natural killer recruitment and cytotoxicity, and ultimately tumor progression and metastasis. The clinical relevance of these findings is supported by our analysis of cancer cohorts, which showed a correlation between high TRF2 expression and MDSC infiltration, which was inversely correlated with overall patient survival.
Keywords: HSPG, immunosurveillance, MDSC, NK cells, TRF2
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Immunology
Introduction
Telomeres are key features of linear chromosomes that preserve genome stability and function. Variations in the telomere state are critical for cell senescence, stem cell biology, and cancer development (Blackburn et al, 2015). Human telomeres are composed of repeated TTAGGG DNA sequences and require the enzyme telomerase for full replication (Gilson & Géli, 2007). To prevent unwanted activation of the DNA Damage Response (DDR), telomeric DNA is associated with shelterin, a protein complex composed of six different subunits (TRF1, TRF2, RAP1, TIN2, TPP1, and POT1 (Giraud‐Panis et al, 2013). During oncogenesis, telomere shortening is associated with apparent antagonistic outcomes; it both favors cancer initiation through mechanisms involving genome instability (Artandi et al, 2000) and prevents cancer progression by activating the DNA Damage Response (DDR) checkpoint to behave as a cell‐intrinsic proliferation barrier (González‐Suárez et al, 2000). Consequently, telomerase reactivation is crucial for cancer progression and nearly 90% of human cancers exhibit upregulation of telomerase (Kim et al, 1994). Therefore, telomeres are considered potential anti‐cancer targets; to date, most studies of telomeres have focused on cell‐intrinsic proliferation barriers initiated by telomerase inhibition (Jafri et al, 2016) and more recently TRF1 inhibition (Bejarano et al, 2017). A disadvantage of these therapeutic approaches is the pro‐aging effects of telomere deprotection in normal tissues.
The expression of the key shelterin subunit TRF2 is regulated by several cancer‐associated signaling molecules including p53 (Fujita et al, 2010), Wnt/β‐catenin (Diala et al, 2013), and WT1 (Wagner et al, 2014) and has been reported to be upregulated in a large panel of human cancers, in association with poor prognosis (Nakanishi et al, 2003; Diehl et al, 2011; Biroccio et al, 2013; Benhamou et al, 2016). In various mouse models, TRF2 inhibition has been shown to impair tumorigenesis independently of its functions in telomere protection and maintenance, but via cell extrinsic effects on immunosurveillance and angiogenesis, suggesting TRF2 to be a potential target in anti‐cancer therapies without pro‐aging telomere deprotective effects (Biroccio et al, 2013; El Maï et al, 2014; Zizza et al, 2019). However, the mechanisms by which TRF2 exerts these multiple oncogenic functions, as well as their impact on overall survival (OS), tumor relapse, metastasis, or treatment response remain largely unknown.
An efficient antitumor immune response is of paramount importance to restrain cancer progression and metastasis, as well as for successful chemotherapy or immunotherapy (Fridman et al, 2017). Because cancer cells possess a variety of sophisticated mechanisms to bypass their recognition and elimination by the immune system (Vesely et al, 2011), targeting these pathways represents an unprecedented opportunity for cancer treatment (Galluzzi et al, 2015; Restifo et al, 2016). Among the immunosuppressive properties of tumors, the recruitment and activation of myeloid‐derived suppressor cells (MDSCs) facilitate cancer progression (Veglia et al, 2018). Although the role of systemic and cancer‐associated inflammation in this process has been established (Ostrand‐Rosenberg & Fenselau, 2018), little is known about the specific pathways by which cancer cells control the immunosuppressive functions of MDSCs and how the cell‐surface molecules of cancer cells can trigger MDSC activation; in particular, the impact of the extracellular matrix and glycocalyx in cancer tissues on MDSC remains elusive.
Previously, we revealed the role of TRF2 in the regulation of the HS3ST4 gene, which encodes an enzyme involved in the sulfation of the heparin sulfate moiety of proteoglycans, preventing the recruitment of natural killer (NK) cells (Biroccio et al, 2013). However, we were unable to decipher the mechanism by which HS3ST4 expression and possibly heparin sulfate proteoglycan (HSPG) biosynthesis keep NK cell activation in check. In this study, we analyzed the tumor immune microenvironment of TRF2 overexpressing tumors in innate immunity competent nude mice xenografted with human transformed fibroblasts (Hahn et al, 1999; Hahn & Weinberg, 2002) and in fully immunocompetent mice grafted with mouse melanoma cells (B16F10) (Cheli et al, 2012). We found that cancer cells recruited and directly activated MDSCs in a TRF2‐dependent manner, dampening NK and CD8+ T cell cytotoxicity. Moreover, we showed that this MDSC function of TRF2 was associated with its ability to control the glycocalyx structure by acting as a master regulator of the transcription of three genes involved in HSPG biosynthesis. Overall, our results reveal an immunosuppressive mechanism linking the telomeric factor TRF2 to the glycocalyx and MDSCs, which suggests that targeting TRF2 can counteract the immune escape of cancer cells without inducing telomere deprotection.
Results
TRF2 triggers an immunosuppressive tumor microenvironment
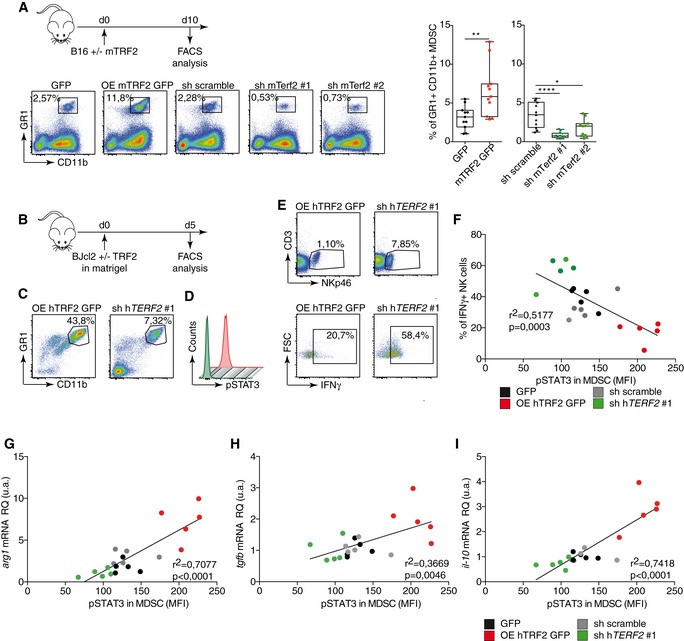
Using the B16F10 melanoma mouse model, we analyzed the effects of TRF2 on intratumoral immune cell infiltration (Fig 1A). Ten days after tumor injection, TRF2 overexpression (conversely, TERF2 knockdown) did not affect global immune cell infiltration (CD45+) or global CD4+, CD3+, or CD8+ T cell infiltration (Fig EV1A). However, intratumoral MDSC infiltration (CD11bHi GR1Hi expressing cells) was strongly dependent on the level of TRF2; its upregulation increased MDSC infiltration by approximately 2.5‐fold, whereas its downregulation decreased infiltration (Fig 1A). Notably, the intratumoral ratio between the two MDSC subpopulations (polymorphonuclear MDSCs [PMN‐MDSCs] and monocytic MDSCs [M‐MDSCs]) was consistent with the findings of a previous report (Fig EV2E and F; Kumar et al, 2016b). Upon TRF2 upregulation, this ratio shifted toward more PMN‐MDSCs (Fig EV2E and F). In addition to MDSC infiltration, TRF2 upregulation (or downregulation) increased (or decreased), respectively, CD25+ Foxp3+ regulatory T cell (Treg) infiltration and decreased (or increased), respectively, activated CD25+ Foxp3–T cell infiltration (Fig EV1B and C) and NK cell recruitment (Fig EV1D). Similarly, xenografted tumors derived from immortalized RasV12 human skin fibroblasts (named BJcl2; Biroccio et al, 2013) injected into nude mice, which are immunodeficient for T cells compartment but still have an active innate immunity including NK cells, exhibited increased or decreased infiltration of MDSCs upon TRF2 overexpression or knockdown, respectively (Fig EV1E).
Figure 1. TRF2 overexpression triggers an immunosuppressive tumor microenvironment.
-
AB16F10 melanoma cells with either mTRF2 overexpression or knockdown were inoculated into the backs of immunocompetent mice. The percentages of intratumoral CD11bHi GR1Hi MDSCs (n = 10 mice), CD25+ Foxp3+ regulatory T cells (Tregs), or CD25+ Foxp3‐activated T cells (n = 5 mice per group) among CD45+ cells were analyzed by fluorescence‐activated cell sorting (FACS) and represented as box plots
-
B–ESchematic representation of the experiment (B). TRF2‐overexpressing or TRF2‐knockdown BJcl2 cells were inoculated into the backs of immunodeficient mice in the presence of Matrigel. Five days later, the percentage of immune cells infiltrating the Matrigel plugs of CD11bHi GR1Hi MDSCs (C), the intranuclear staining of pSTAT3 (D), and CD3− NKp46+ and IFN‐γ+ among CD3− NKp46+ NK cells (E) were determined by FACS (n = 5 mice per group).
-
F–ILinear correlation plot of the percentage of IFN‐ γ+ NK cells, the mean of fluorescence intensity (MFI) of pSTAT3 for MDSCs, and the expression of TRF2 (r 2 = 0.5177; Pearson's correlation, P = 0.003) (F); the mRNA expression level in the Matrigel plugs of arg1, pSTAT3 MFI, and TRF2 expression (r 2 = 0.7077; Pearson's correlation, P < 0.0001) (G); the mRNA expression level in the Matrigel plugs of tgfβ, pSTAT3 MFI, and TRF2 expression (r 2 = 0.3669; Pearson's correlation, P = 0.0046) (H) and the mRNA expression level in the Matrigel plugs of il‐10, pSTAT3 MFI, and TRF2 expression (r 2 = 0.7418; Pearson's correlation, P < 0.0001) (I).
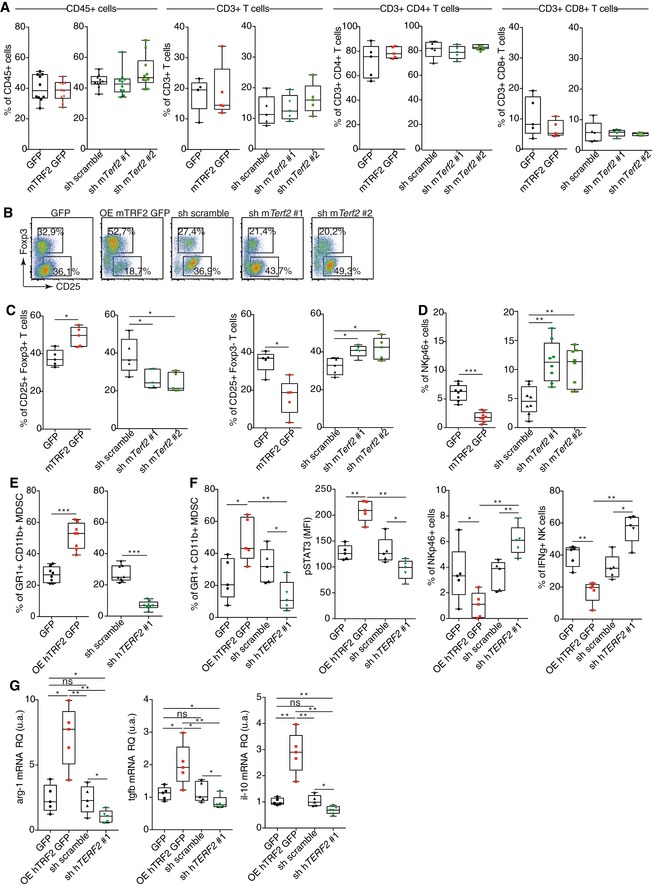
Figure EV1. TRF2 overexpression in cancer cells induces a strong immunosuppressive microenvironment via the recruitment of MDSCs and regulatory T cells.
-
A–DAnalysis of immune cell infiltration in TRF2‐overexpressing or TRF2‐knockdown B16F10 melanoma tumors. Box plots of total immune cell infiltration (CD45+ cells), total T cell infiltration (CD45+ CD3+ T cells), and CD8+ or CD4+ T cells among CD3+ cells infiltrating (A). Representative density plot of regulatory T cells (CD25+ Foxp3+ Treg) and activated T cells (CD25+ Foxp3−) (B). Box plots of infiltrating Treg (CD25+ Foxp3+) and activated T cells (CD25+ Foxp3−) (C) or NK cells (NKp46+ CD3−) (D).
-
EAnalysis of MDSC infiltration in TRF2‐overexpressing or TRF2‐knockdown BJcl2 tumors in Nude mice.
-
F, GData used for the correlation analysis presented in Fig 1F–I. Box plots present the percentages of CD11bHi GR1Hi MDSCs among CD45+ cells infiltrating the Matrigel plug, the mean fluorescence intensity (MFI) of intranuclear pSTAT3 staining in MDSCs, the percentages of intratumoral CD3− NKp46+ NK cells among CD45+ cells, and NKp46+ IFN‐γ+ NK cells (F). The mRNA expression level (relative quantity) in the Matrigel plugs of arg1, tgfβ, and il‐10.
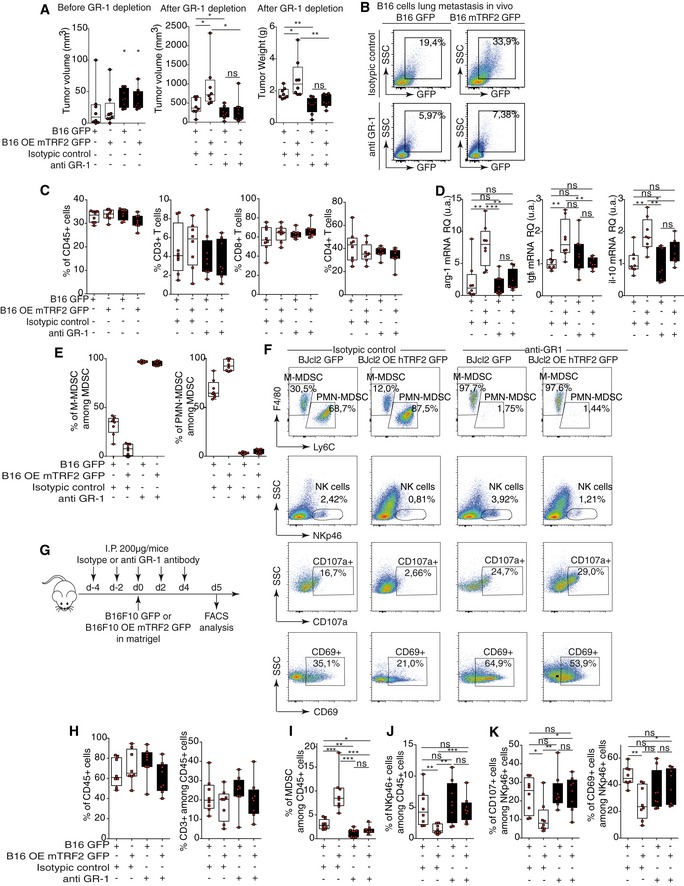
Figure EV2. The TRF2‐dependent immunosuppression is abolished by MDSC depletion.
-
AB16F10 tumor volumes before (left panel) and tumor volume and weight after anti‐GR1 depletion (right panel).
-
BB16F10 GFP+ metastasis infiltrating lung from subcutaneous tumor‐bearing mice (from panel A) after anti‐GR1 depletion.
-
CTotal immune cell infiltration (CD45+ cells), total T cell infiltration (CD45+ CD3+ T cells), and CD8+ or CD4+ T cells among CD3+ cells in TRF2‐overexpressing or TRF2‐knockdown B16F10 tumors treated with or without the anti‐GR1 antibody (from panel A).
-
DThe mRNA expression level (relative quantity) in the tumors of arg1, tgfβ, and il‐10.
-
EPMN‐MDSCs and M‐MDSCs distribution among total intratumoral MDSCs in B16F10 TRF2‐overexpressing tumors after anti‐GR1 depletion.
-
FDensity plot of infiltrating M‐MDSCs, PMN‐MDSCs, total NK cells, and CD107a+ or CD69+ NK cells in control or anti‐GR1‐treated Matrigel plugs generated with BJcl2 cells.
-
G–KMatrigel plug assay using B16F10 cells in anti‐GR1 treated animals (G). Total immune cell infiltration (CD45+ cells) (H), total T cell infiltration (H), MDSCs (I), total NK cells (J), and activated NK cells (K).
We used a Matrigel plug assay to analyze the early immune responses triggered by cancer cells with various TRF2 expression levels (Fig 1B). As observed in tumors, overexpression (knockdown) of TRF2 increased (or decreased) MDSC recruitment, respectively (Figs 1C and EV1F). In addition, using phosphorylated STAT3 (pSTAT3) as a marker for MDSC activation and known regulator of the immunosuppressive functions of MDSC (Marvel & Gabrilovich, 2015), we observed that the TRF2 level was correlated with MDSC activation (Figs 1D and F, and EV1F). The proportion of IFN‐γ+ NK cells was negatively correlated (R 2 = 0.5117, P = 0.0003) with the pSTAT3 level in MDSCs and with TRF2 expression in cancer cells (Fig 1F), indicating that TRF2‐dependent MDSC recruitment in vivo is associated with inhibition of NK cell cytotoxicity. In the same Matrigel plug assay, we observed that the expression of three immunosuppressive molecules, arginase 1 (Arg‐1), IL‐10, and TGF‐β (Ostrand‐Rosenberg & Fenselau, 2018), which are expressed by MDSCs to trigger NK and T cell suppression (Gabrilovich & Nagaraj, 2009; Nagaraj & Gabrilovich, 2012; Sceneay et al, 2012), is upregulated when TRF2 is overexpressed (Fig EV1G) and is correlated with the level of pSTAT3 in infiltrating MDSCs (Fig 1G–I; r 2 = 0.7077, P < 0.0001 for arg‐1; r 2 = 0.3669, P = 0.0046 for tgf‐β; r 2 = 0.7418, P < 0.0001 for il‐10). Overall, these results show, using a tumor growth experiment or Matrigel plug assay with B16F10 mouse melanoma cells implanted into immunocompetent mice and human BJcl2 cells xenografted in Nude mice that a high level of TRF2 in cancer cells induces a strong immunosuppressive tumor microenvironment by promoting MDSC and Treg cell infiltration and inhibiting NK cell recruitment and degranulation.
TRF2 promotes tumorigenesis and metastasis in an MDSC‐dependent manner
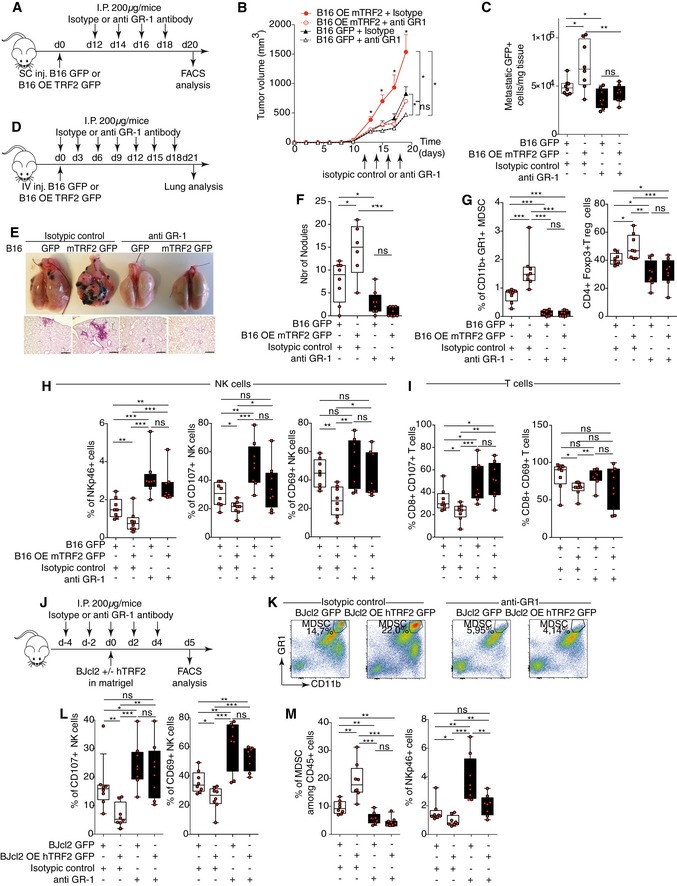
Next, we determined whether the pro‐tumorigenic and immunosuppressive effects of TRF2 overexpression are dependent on MDSCs. MDSCs were selectively depleted in mice by repeated injection of anti‐GR‐1 antibodies (Kumar et al, 2016a; Svoronos et al, 2017). Indeed, such a treatment abolished the intratumoral infiltration of MDSCs, in particular PMN‐MDSCs (Figs 2G, K and M, and EV2E and F). The pro‐tumorigenic and metastatic effects of TRF2 overexpression were obliterated in these MDSC‐depleted mice (Figs 2A–F and EV2A and B). The ability of TRF2‐overexpressing cancer cells to metastasize was strongly reduced when MDSCs were depleted, either when we searched for natural diffusion of cancer cells into the lungs (GFP+ cells infiltrating lungs) from a primitive subcutaneous tumor (Figs 2C and EV2B) or when we used the classical metastasis model by intravenous injection into the tail vein (Fig 2D–F). In both cases, MDSC depletion was sufficient to inhibit the increased number of GFP+ cells infiltrating the lungs, the number of nodules, or the metastasis burden induced by TRF2 overexpression. Although MDSC depletion did not alter the infiltration of CD45, CD3, CD4, or CD8 cells (Fig EV2C), it reversed the immunosuppressive tumor microenvironment created by TRF2 upregulation (i.e., Treg cell infiltration) (Fig 2G), NK cell inhibition (Fig 2H), and CD8+ T cell inhibition (Fig 2I). This rescue by MDSC depletion of the TRF2‐dependent immunosuppressive environment was confirmed by a Matrigel plug assay (Figs 2J–M and EV2F–K). Overall, these results show that TRF2‐mediated regulation of MDSCs plays a central role in the TRF2‐dependent increases in tumorigenicity and metastasis, shaping the tumor immune microenvironment.
Figure 2. TRF2 promotes tumorigenesis and metastasis in an MDSC‐dependent manner.
-
A–CB16F10 melanoma cells with or without mTRF2 overexpression were inoculated into the backs of immunocompetent mice and treated with anti‐GR1 antibody or isotypic control (200 μg/IP). The tumor volume was determined every 2 days using hemi‐ellipsoid formula (B). On day 19 of the tumor growth experiment, the percentages of GFP+ cells infiltrating the lungs were determined by FACS and presented as the number of GFP+ cells/mg lung tissue (C).
-
D–FB16F10 melanoma cells with or without mTRF2 overexpression were inoculated into tail vein of immunocompetent mice and treated with anti‐GR1 antibody or isotypic control (200 μg/IP) every 3 days. On day 21, lung metastasis was analyzed by counting macroscopic metastatic nodules and hematoxylin and eosin (H&E) staining (E and F), scale bars = 500 μm.
-
G–ITumor‐infiltrating immune cells from tumors of the experiment shown in (A) were analyzed by FACS: MDSCs and CD25+ Foxp3+ Tregs (G), NK cells (H), and CD8+ T cells (I).
-
J–MBJcl2 cells with or without hTRF2 overexpression were inoculated into the backs of immunodeficient nude mice and treated with anti‐GR1 antibody or isotypic control (200 μg/IP) as indicated in (J) and NK cells recruitment, degranulation, and MDSC infiltration were monitored by FACS (K‐M). Density plot of CD11bHi GR1Hi MDSC staining in control or anti‐GR1‐treated Matrigel plugs (K). Box plots of the percentages of infiltrating or activated NK cells (CD107a or CD69) (L), of CD11bHi GR1Hi MDSC or NKp46+ NK cells (M) infiltrating the Matrigel plugs assay among the CD45+ cells.
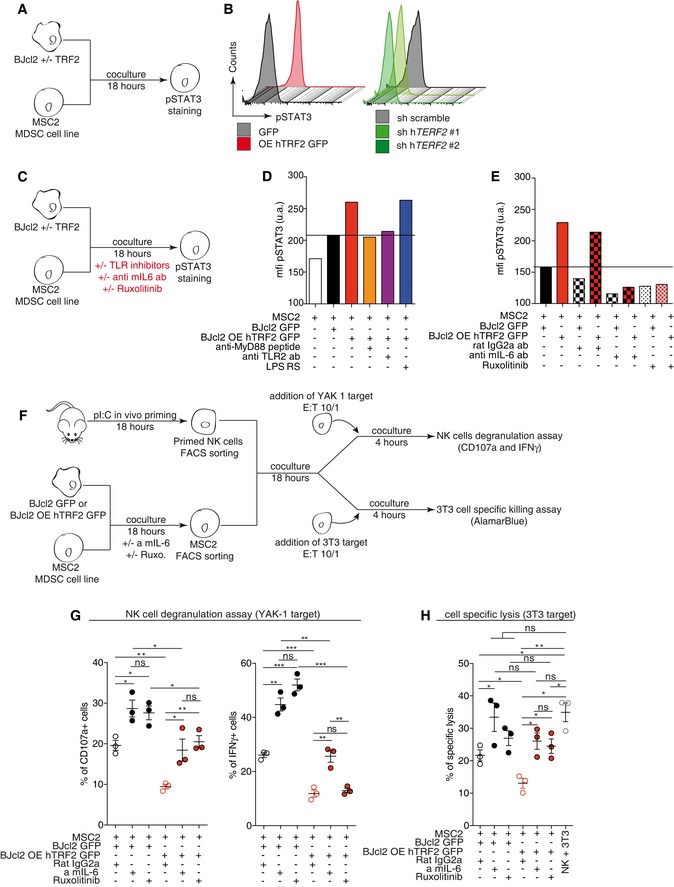
Cancer cells directly activate MDSCs in a TRF2‐dependent manner via the TLR2/MyD88/IL‐6/STAT3 pathway
The above‐mentioned results suggest that the immunosuppressive environment controlled by the level of TRF2 in cancer cells results from MDSC recruitment and/or activation. Therefore, we explored whether MDSCs are directly regulated by cancer cells as a function of the TRF2 level. We co‐cultured BJcl2 or B16F10 cells in the presence or absence of TRF2 overexpression, in a mouse MDSC line (MSC2 cells; Bruchard et al, 2012) and analyzed the pSTAT3 level in MSC2 cells (Figs 3A and B, and EV3A–C). In TRF2‐overexpressing cancer cells, the pSTAT3 level was significantly increased in MSC2 cells, whereas it was decreased after TERF2 knockdown in cancer cells (Figs 3B and EV3C). Interestingly, when the pSTAT3 level was assayed after co‐culture with conditioned medium (Fig EV3D), we detected no differences (Fig EV3E), suggesting that cell contact is required. Next, we investigated whether MDSCs are activated by TRF2‐overexpressing cancer cells via the Toll‐like receptor (TLR)/MyD88 pathway (Fig 3C–E). After determining the optimal concentration of each inhibitor (Fig EV3G and H), we co‐cultured BJcl2 cancer cells in the presence or absence of TRF2 overexpression and MSC2 cells in the presence or absence of a TLR4 antagonist (lipopolysaccharide [LPS‐RS]), an anti‐mouse TLR2‐blocking antibody, or a MyD88‐inhibitory peptide. The blocking of TLR4 by LPS‐RS did not affect the level of pSTAT3 in MSC2 cells; however, treatment with the anti‐TLR2 antibody or anti‐MyD88 peptide was sufficient to inhibit the increase of pSTAT3 in MSC2 cells co‐cultured with TRF2‐overexpressing cancer cells (Figs 3D and EV3F). Since the TLR2/MyD88 pathway does not directly trigger STAT3 phosphorylation, we explored whether activation of the TLR2/MyD88 pathway induces a secondary signal that leads to STAT3 phosphorylation, specifically focusing on IL‐6 (Skabytska et al, 2014). We assessed whether a mouse‐specific anti‐IL‐6 blocking antibody or JAK1/2 inhibitor (Ruxolitinib) can block the increase of STAT3 phosphorylation induced by TRF2 overexpression (Figs 3E and EV3G and H). Adding rat IgG2a isotypic control to the co‐culture did not affect the increased pSTAT3 level; however, inhibition of mouse IL‐6 or JAK1/2 completely inhibited this increase. Taken together, these data show that overexpression of TRF2 triggers the TLR2/MyD88 pathway in MDSCs, which in turn favors STAT3 phosphorylation in an IL‐6/JAK1/2‐dependent manner.
Figure 3. Cancer cells directly activate MDSCs in a TRF2‐dependent manner via the TLR2/MyD88/IL‐6/STAT3 pathway that inhibit NK cell degranulation and cytotoxicity.
-
A, BAnalysis of STAT3 phosphorylation by MSC2 cells after 18 h co‐culture with BJcl2 overexpressing or compromised for TRF2 (A). Histograms of the pSTAT3 mean fluorescence intensity (MFI) (B).
-
C–ESame experiment as in (A, B) ± inhibitors: anti‐MyD88 peptide inhibitor, anti‐mouse TLR2 antibody, or lipopolysaccharide (LPS)‐RS, an antagonist of TLR4 in (D) and anti‐mouse IL‐6 or JAK1/2 inhibitor (Ruxolitinib) in (E).
-
F–HSchematic representation of the MDSC suppression assay (F). TRF2‐overexpressing or TRF2‐knockdown BJcl2 cells were co‐cultured with MSC2 cells for 18 h ± mouse IL‐6 blocking antibody or ruxolitinib. After FACS sorting, MSC2 cells were co‐cultured for 18 h with primed and purified NK cells before a 4 h challenge with YAK‐1. NK cell degranulation and IFN‐γ production were analyzed by FACS (G). Alternatively, a 4 h challenge with NIH3T3 cells was performed and the ability of NK cells to kill NIH3T3 cells were determined by colorimetric analysis (AlamarBlue) (H).
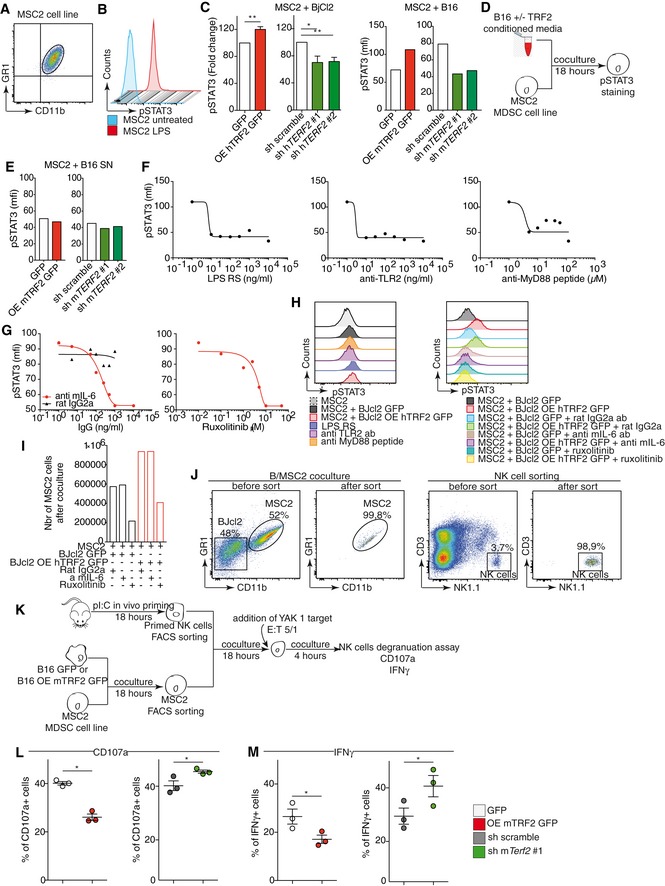
Figure EV3. TRF2‐mediated regulation of MDSCs via TLR2/MyD88 pathway inhibits NK cell functionality.
-
ADensity plot of CD11b GR1 staining in MSC2 cells.
-
BMSC2 cells were activated with 1 μM lipopolysaccharide (LPS), and pSTAT3 levels were determined 18 h after stimulation by FACS.
-
CSTAT3 phosphorylation by MSC2 cells after co‐culture with BJcl2 (left panel) or B16F10 cells (right panel) overexpressing or knockdown for terf2.
-
D, ESTAT3 phosphorylation by MSC2 cells after co‐culture with conditioned media from BJcl2 (left panel) or B16F10 (right panel) overexpressing or knockdown for terf2.
-
F, GDose–response analysis of pSTAT3 levels in MSC2 cells after treatment with various doses of the TLR4 antagonist LPS‐RS, anti‐mouse TLR2 antibody, or anti‐MyD88 peptide (F) or with various doses of anti‐mouse IL‐6 antibody or JAK1/2 inhibitor (G).
-
HHistograms of the pSTAT3 MFIs depending on the concentration of the molecule.
-
IMSC2 cell number after 18 h of co‐culture with BJcl2 cells ± anti‐mouse IL‐6 antibody or JAK1/2 inhibitor.
-
JRepresentative density plot to assessed purity control for MSC2 sorting from the co‐culture or NK cell sorting from splenocytes.
-
K–MSchematic representation of the MDSC suppression assay (K). TRF2‐overexpressing or TRF2‐knockdown B16F10 cells were co‐cultured with MSC2 cells for 18 h. After FACS sorting, MSC2 cells were co‐cultured for 18 h with primed and purified NK cells before a 4 h challenge with YAK‐1 E/T ratio of 5:1. NK cell degranulation (L) and IFN‐γ production (M) were analyzed by FACS.
The TRF2‐mediated activation of MDSCs inhibits NK cell function
We assessed whether the dosage of TRF2 in cancer cells affects the immunosuppressive capacity of MDSCs on NK cell activation using an in vitro suppression assay (Figs 3F–H and EV3J–M). The overexpression or knockdown of TRF2 in BJcl2 cells (Fig 3F–H) or B16F10 cells (Fig EV3J–M) was conducted in co‐culture with MSC2 cells for 18 h; MSC2 cells were then sorted by fluorescence‐activated cell sorting (FACS) (Figs 3F and EV3J and K). Simultaneously, NK cells poly I:C‐primed in vivo for 18 h were sorted by FACS (Figs 3F and EV3J and K). Sorted MSC2 and NK cells were then co‐cultured for 18 h at a 1:1 ratio and finally challenged by adding the target cells (YAK‐1 or 3T3 cells) for 4 h (Figs 3F and EV3K). NK cell degranulation capacity and IFN‐γ production were determined by flow cytometry (Figs 3G and EV3L and M), and the cytotoxicity of NK cells toward the target was assessed using a viability assay (Fig 3H). After co‐culturing MSC2 and cancer cells, we noticed that TRF2 overexpression in cancer cells increased the number of MSC2 cells (Fig EV3I), suggesting that TRF2 enhances MDSC proliferation. Interestingly, this proliferative effect was not altered when IL‐6 was blocked, but was strongly reduced when JAK1/2 was inhibited, suggesting that TRF2 enhances MDSC proliferation in a JAK/STAT‐dependent manner. We also observed that direct co‐culture of TRF2‐overexpressing cancer cells and MSC2 cells, either with BJcl2 (Fig 3G) or with B16F10 cells (Fig EV3L and M), significantly decreased NK cell degranulation and IFN‐γ production. Inversely, TRF2 knockdown in cancer cells led to significant increases in NK cell degranulation capacity and IFN‐γ production (Fig EV3L and M). Overexpression of TRF2 not only inhibited NK cell functionality but also strongly affected NK cell cytotoxicity (Fig 3H). Since we observed that STAT3 phosphorylation was dependent on the IL‐6/JAK1/2 pathway, we explored whether inhibition of JAK1/2 or IL‐6 was sufficient to reverse the inhibitory effect on NK cell functionality. Interestingly, we observed that blocking IL‐6 or JAK1/2 restored NK cell degranulation (Fig 3G) and killing capacity (Fig 3H), which were inhibited by TRF2 overexpression. Taken together, our data show that direct contact between TRF2‐overexpressing cancer cells and MDSCs is sufficient to trigger TLR2 signaling, which enhances the immunosuppressive capacity of MDSCs on NK cells via an IL‐6/JAK1/2‐dependent mechanism.
TRF2 regulates the expression of ITS‐associated genes involved in glycocalyx synthesis
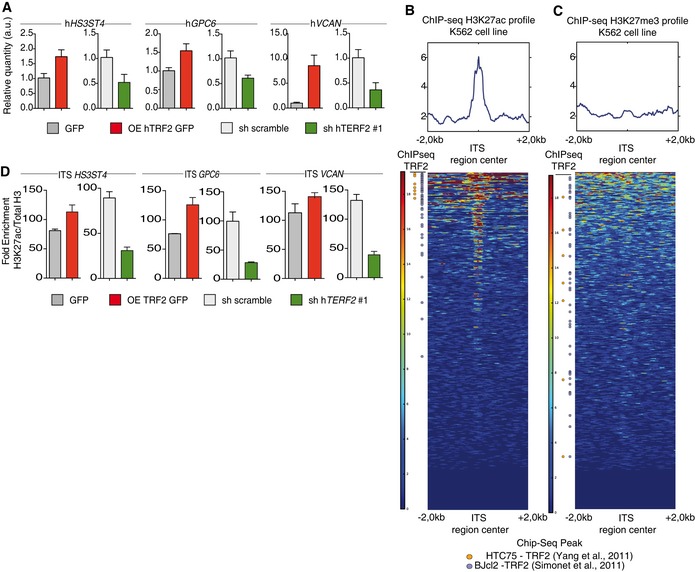
Next, we assessed the mechanism by which TRF2 controls the ability of cancer cells to recruit and activate MDSCs. Because TRF2 binds an extratelomeric range of TTAGGG repeats (ITSs) (Simonet et al, 2011; Yang et al, 2011) to regulate the expression of HS3ST4 (Biroccio et al, 2013), a gene involved in HSPG biosynthesis, and as HSPGs shape the tumor microenvironment and promote tumor growth (Knelson et al, 2014; Hammond et al, 2014), we systematically searched the genome for ITS‐associated genes (Dataset EV1) involved in HSPG biosynthesis using the Reactome online tool (http://reactome.org/PathwayBrowser/) (Dataset EV2). This search revealed two ITS‐associated genes in addition to HS3ST4: VCAN, whose product activates the TLR2 pathway (Kim et al, 2009), and GPC6, which encodes glypican 6 (Dataset EV2). The expression of GPC6 and VCAN was dependent on TRF2 expression, at both the RNA and protein levels (Figs 4A and EV4A–C). These results indicate that TRF2 behaves as a transcriptional regulator of three HSPG‐related genes. Therefore, we examined whether the binding of TRF2 to ITSs induces chromatin changes involved in transcriptional regulation. Using Encode data (Fig 4B and C), we evaluated the specific enrichment of the activating mark H3K27ac in a large set of TRF2‐bound ITSs (Figs 4B and C, and EV4H and Dataset EV4). This specific enrichment of ITS chromatin in H3K27ac is conserved through evolution (Fig EV4D–H and Dataset EV3), suggesting that it plays an important role. Interestingly, almost all the genes for which we previously revealed an ITS by TRF2 ChIP‐seq (Simonet et al, 2011) are also identified in the H3K27ac ChIP‐seq from ENCODE (Figs 4B and EV4H). We then tested whether the binding of TRF2 to ITSs triggers H3K27 acetylation and found that enrichment of H3K27ac in ITSs associated with HS3ST4, VCAN, and GPC6 was decreased (increased) upon TRF2 downregulation (overexpression) (Figs 4D and EV4I). These results suggest that binding of TRF2 to ITSs converts these sequences into positive regulatory elements that enhance the transcription of HSPG genes.
Figure 4. TRF2 regulates the expression of ITS‐associated genes involved in glycocalyx synthesis.
-
AGene expression analysis by quantitative polymerase chain reaction (qPCR) of TRF2 target genes following TRF2 overexpression or knockdown.
-
B, CHeatmap of H3K27ac (B) or H3K23me3 (C) peaks on the 4 kilobase (Kb) region around genomic ITSs (ENCODE data for the K562 cell line). The mean intensity of the region is depicted in the histograms. ITSs are clustered depending of the peak mean intensity obtained from H3K27ac or H3K27me3 chromatin immunoprecipitation sequencing (ChIP‐seq) of the 4 Kb region. ITSs detected by ChIP‐seq using an anti‐TRF2 antibody are shown on the left.
-
DSpecific qPCR for HS3SH4, GPC6, and VCAN ITSs after ChIP using an anti‐H3K27ac antibody in TRF2‐overexpressing or TRF2‐knockdown BJcl2 cells. Data represent fold enrichment of the H3K27ac level compared with the total H3 level.
Figure EV4. TRF2 regulates the expression of a set of ITS‐associated genes involved in HSPG biosynthesis.
-
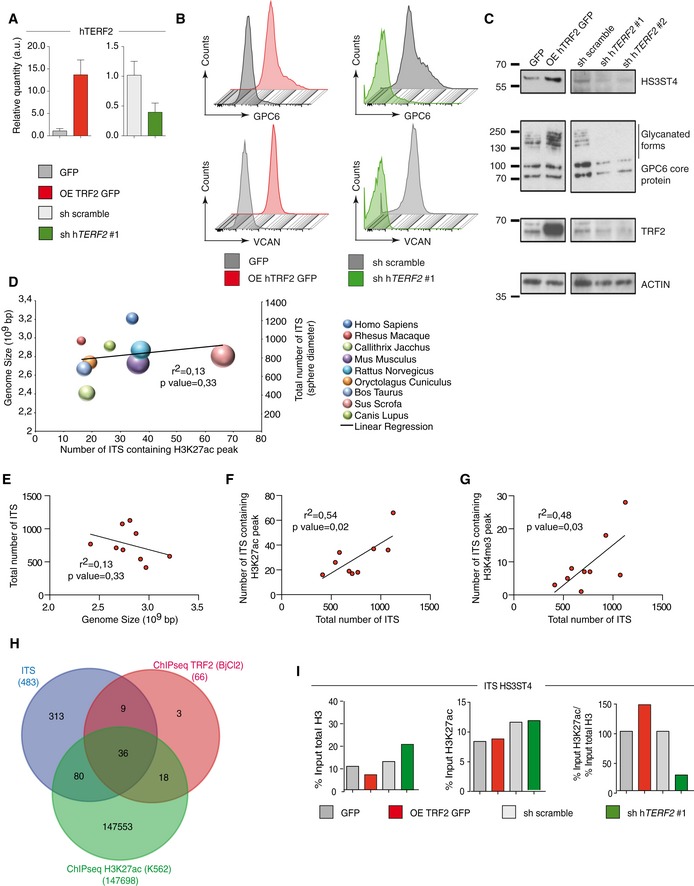
AqPCR analysis of TRF2 expression in BJcl2 cells in which TRF2 target gene expression was determined in Fig 4A and D.
-
BFACS analysis of GPC6 and VCAN expression in BJcl2 cells overexpressing or compromised for TRF2
-
CWestern blot analysis of HS3ST4, GPC6, and VCAN expression in BJcl2 cells overexpressing or compromised for TRF2
-
DAnalysis of the correlation among the genome size (y‐axis), total number of ITSs (z‐axis corresponding to sphere diameter), and number of ITSs containing H3K27ac peaks (x‐axis).
-
E–GCorrelation between genome size and total number of ITSs (D), between total number of ITSs and number of ITSs containing H3K27ac peaks (F), and between total number of ITSs and number of ITSs containing H3K4me3 peaks (G)
-
HVenn diagram of genes identified by TRF2 chromatin immunoprecipitation sequencing (ChIP‐seq; red), genes bearing ITSs (blue), and all H3K27ac peaks (green).
-
IHistograms of the percentage input of total H3 or input of H3K27ac used for immunoprecipitation or the ratio of the percentage input of total H3 to that of H3K27ac used for immunoprecipitation for the HS3ST4 ITS.
HS3ST4, GPC6, and VCAN are effectors of the MDSC‐dependent oncogenic properties of TRF2
Next, we evaluated the roles of HS3ST4, GPC6, and VCAN in TRF2‐mediated immunosuppression (Fig 5A–H and Appendix S1A–C). We analyzed the impact of TRF2 target genes on immune infiltration (Matrigel plug assay) and observed that VCAN downregulation decreased total immune cell infiltration, whereas downregulation of HS3ST4 or GPC6 did not alter total immune cell infiltration (Appendix S1B and C). However, all three genes affected MDSC or NK cell recruitment/activation (Fig 5C and Appendix Fig S1B). Indeed, downregulation of HS3ST4 and GPC6 significantly decreased MDSC recruitment (Fig 5B), whereas only HS3ST4 downregulation increased NK cell recruitment (Fig 5C). In contrast, NK cell degranulation (C107a+ NK cells) was positively affected by downregulation of GPC6 and VCAN but not HS3ST4 (Fig 5D), whereas only GPC6 affected CD69+ NK cell infiltration (Fig 5E). Thus, MDSC recruitment was mediated by HS3ST4 and GPC6, and as expected, NK cell recruitment by HS3ST4 (Biroccio et al, 2013)), whereas NK cell degranulation and activation were dependent on GPC6 and VCAN. The roles of these genes in MDSC activation were confirmed using co‐culture assays, in which HS3ST4, GPC6, or VCAN expression was knocked down in TRF2‐overexpressing or TRF2‐downregulated cancer cells (Fig 5F–H). VCAN knockdown did not affect STAT3 phosphorylation, whereas HS3ST4 or GPC6 knockdown decreased it. In cancer cells overexpressing TRF2, HS3ST4 or GPC6 knockdown significantly reduced the level of pSTAT3 (Fig 5G). In addition, double knockdown of TRF2 with either HS3ST4 or GPC6 (Fig 5H) decreased the pSTAT3 level in a similar manner to that by HS3ST4 knockdown alone, GPC6 knockdown alone, or TRF2 knockdown alone, suggesting epistatic regulation among TRF2, HS3ST4, and GPC6. Thus, the TRF2‐dependent regulation of HS3ST4 and GPC6 is essential for STAT3 phosphorylation in MDSCs. Finally, since we showed that the oncogenic properties of TRF2 were dependent on MDSC recruitment and immunosuppressive functions (Figs 1, 2, 3) and that STAT3 phosphorylation is dependent on HS3ST4 and GPC6, we evaluated the oncogenic properties of HS3ST4 and GPC6 in tumor growth experiments. Their overexpression or downregulation affected neither cell proliferation nor clonogenicity in vitro (Appendix Fig S1D and E). However, in xenograft experiments using BJcl2 cells, downregulation of GPC6 or HS3ST4 decreased tumor weight (Fig 5I), delayed tumor occurrence (Appendix Fig S1F), and reduced tumor growth (Appendix Fig S1G). Conversely, overexpression of GPC6 or HS3ST4 increased cancer tumorigenicity, accelerated tumor occurrence, and increased tumor growth. In summary, TRF2 regulates the expression of three HSPG genes that mediate MDSC recruitment and activation and promote tumorigenicity.
Figure 5. HS3ST4, GPC6, and VCAN are effectors of the MDSC‐dependent oncogenic properties of TRF2.
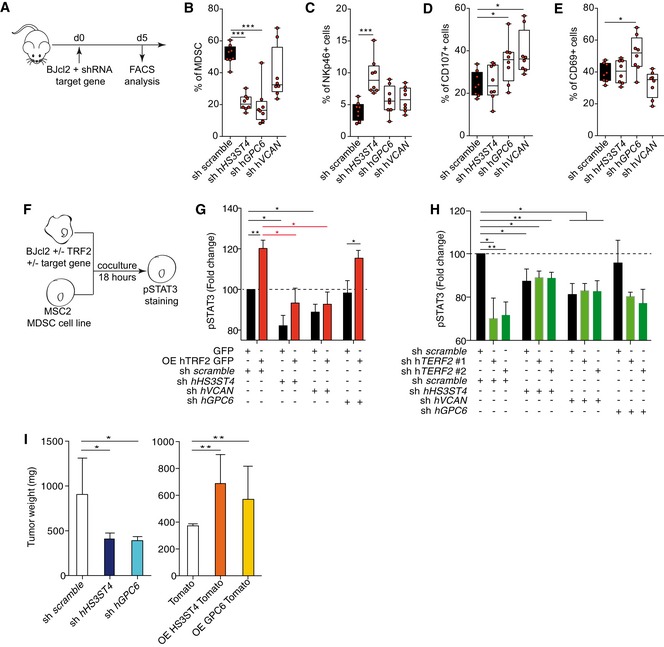
-
ASchematic representation of the Matrigel plug assay with BJcl2 cells knockdown for TRF2 target genes. Immune cell infiltration within the Matrigel plugs was determined by FACS.
-
B–EPercentage of CD11bHi GR1Hi MDSCs (B), CD3‐ NKp46+ NK cells (C), NKp46+ CD107a+ NK cells (D), or CD69+ NK cells (E)
-
F–HAnalysis of STAT3 phosphorylation by MSC2 cells after 18 h co‐culture with BJcl2 overexpressing or compromised for TRF2 and the target genes (F). Histograms of the pSTAT3 mean fluorescence intensity (MFI) of MSC2 cells when TRF2 was overexpressed and the target gene knocked down (G) and when both TRF2 and its target gene were knocked down (H).
-
IHistograms of tumor weights on day 19 after implantation of subcutaneous xenografts containing BJcl2 cells overexpressing or knocked down for HS3ST4 or GPC6.
TRF2 changes glycocalyx structure, which triggers MDSC activation
Because the above‐mentioned results suggest that TRF2 regulates the expression of a network of HSPG genes, we directly evaluated whether TRF2 acts as a general remodeler of glycocalyx structure. First, we analyzed the total heparan sulfate (HS) level by FACS using the 10E4 antibody (van den Born et al, 2005). TRF2 overexpression increased the level of HS staining (Fig 6A), whereas TRF2 knockdown decreased it, indicating that TRF2 enhances HS synthesis. Because HS is an essential component of the glycocalyx, we investigated whether TRF2 modifies the glycocalyx and/or cell stiffness by atomic force microscopy. By analyzing the force curves obtained by cell nano‐indentation, we measured both cell and glycocalyx stiffness simultaneously (Appendix Fig S2A). Interestingly, TRF2 overexpression increased both glycocalyx and cell stiffness (Fig 6B). A visual indication of this TRF2‐dependent cell mechanical property is provided in a heatmap of elastic cell responses to the scanning atomic force microscopy probe (Fig 6C). The modulus heatmap reveals that cells overexpressing TRF2 were stiffer than those expressing GFP. Analysis of the force curves allows measurement of glycocalyx thickness, which was significantly less for TRF2‐overexpressing cells than GFP‐expressing cells (Fig 6B, right panel). To investigate whether these global changes in glycocalyx structure are responsible for the immunosuppressive role of TRF2, we globally inhibited glycocalyx and HS synthesis in cancer cells using sodium chlorate (Kang et al, 2013). As expected, treatment with sodium chlorate was sufficient to inhibit the presence of cell‐surface HS (Appendix Fig S2B and C). We next evaluated its impact on MDSC activation. Although sodium chlorate treatment in MSC2 cells did not alter STAT3 phosphorylation upon LPS stimulation, which ruled out a direct effect of the treatment on MSC2 cells (Appendix Fig S2D), this treatment inhibited TRF2‐dependent STAT3 phosphorylation (Fig 6D and Appendix Fig S2E). We conclude that TRF2‐dependent changes in the glycocalyx lead to MDSC activation.
Figure 6. TRF2 changes glycocalyx structure, which triggers MDSC activation.
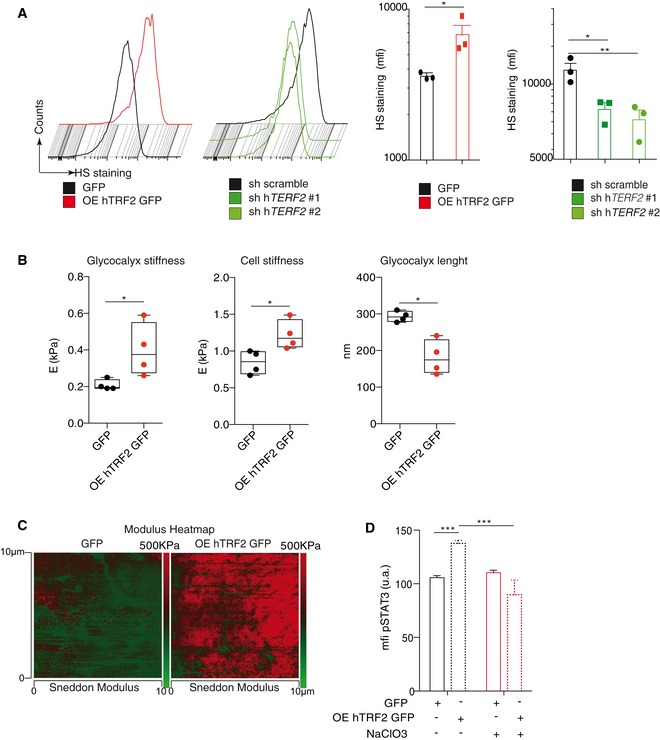
- Analysis of heparan sulfate (HS) expression by FACS using the 10E4 antibody.
- Atomic force microscopic (AFM) analysis of glycocalyx and cell stiffness and glycocalyx length.
- AFM images showing the modulus heatmap (stiffness) in GFP‐expressing or TRF2‐overexpressing cancer cells. At each of the 128 × 128 force curve points, corresponding to an area of 10 μm2, the AFM software automatically displays the Sneddon modulus analysis while scanning the sample. The color bar on the left of the images shows a qualitative representation of the elastic map.
- Analysis of pSTAT3 levels in MDSCs after co‐culture with GFP‐expressing or TRF2‐overexpressing cancer cells treated with or without sodium chlorate (NaClO3).
TRF2 upregulation enhances the response to chemotherapies targeting MDSCs
Because some conventional chemotherapeutic drugs (e.g., 5‐fluorouracil [5‐FU], gemcitabine, and paclitaxel) can target MDSCs (Apetoh & Vegran, 2011; Bruchard et al, 2013; Sevko et al, 2013), TRF2 levels may modulate their efficacy. Therefore, we analyzed the OS of patients according to TRF2 expression level and mode of treatment. The prognoses of patients with high TRF2 expression treated with chemotherapy, radiotherapy, or surgery were worse than those of patients with low TRF2 expression (Figs 7A and EV5B–E, and Dataset EV6), except for gastric cancer patients treated with 5‐FU, ovarian cancer patients treated with gemcitabine, and lung cancer patients treated with cisplatin, paclitaxel, gemcitabine, or 5‐FU (Fig 7A). In contrast, high TRF2 expression seems to be associated with better survival (Figs 7A and EV5B–E). This finding suggests that TRF2 overexpression may favor tumor development in humans but enhance responses to chemotherapies based on 5‐FU or gemcitabine. To test this hypothesis, we implanted B16F10 cells in the presence or absence of TRF2 overexpression in immunocompetent mice treated with or without 5‐FU (Fig 7B). As expected, TRF2 overexpression significantly increased tumor volume (Fig 7C and D) and reduced OS (Fig 7E). After 5‐FU treatment in tumor‐bearing mice, we observed that tumor volume was reduced in both GFP control tumors and TRF2‐overexpressing tumors, with a larger effect on TRF2‐overexpressing tumors (Fig 7D). Following treatment, the volume of TRF2‐overexpressing tumors was comparable with that of control tumors, suggesting that the effect of 5‐FU was enhanced by TRF2 overexpression. Similarly, 5‐FU treatment prolonged mouse OS and prevented the decrease in OS induced by TRF2 overexpression (Fig 7E). Taken together, these data suggest that chemotherapy‐based treatments that kill MDSCs (5‐FU, gemcitabine, and paclitaxel) (Apetoh & Vegran, 2011; Bruchard et al, 2013; Sevko et al, 2013) abolished the pro‐oncogenic effect of TRF2 overexpression.
Figure 7. TRF2 upregulation enhances the response to chemotherapies targeting MDSC.
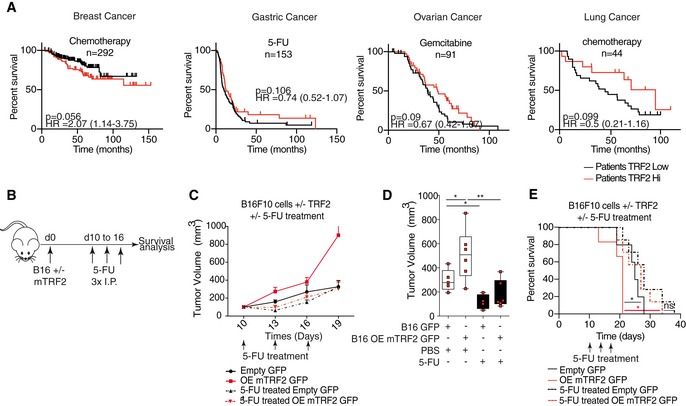
-
AKaplan–Meier curves for overall survival of chemotherapy‐treated patients according to the TRF2 expression level (KM‐plotter, http://kmplot.com/analysis/index.php?p=background).
-
B–ESchematic representation of the mouse experiment. Immunocompetent mice were subcutaneously engrafted with B16F10 cells with or without TRF2 overexpression and treated with three intraperitoneal injections of 50 mg/kg 5‐FU. The tumor volume was determined every 2 days using a caliper, and growth curves over time (C) and tumor volume were determined on day 19 and represented using box plots min to max showing all points with median (D). Overall survival was also determined (E).
Figure EV5. TRF2 upregulation enhances glycocalyx stiffness, which activates MDSCs, enhances the response to chemotherapies targeting MDSC, and decreases overall survival (related to Figs 6, 7, 8).
-
AGraphical model arising from our main results.
-
B–EKaplan–Meier curves for overall survival of chemotherapy‐ or surgery‐treated patients with breast (B), gastric (C), ovarian (D), or lung (E) cancer according to the TRF2 expression level (KM‐plotter, http://kmplot.com/analysis/index.php?p=background).
-
FRNAseq analysis of TERF2 expression of 30 types of malignancies compared with normal tissues. Cases harboring mutations are depicted in the graph.
-
GThe numbers, frequencies, and locations of mutations in the TERF2 gene among 13,490 sequenced cases.
-
H–JThe overall survival of gastric cancer patients (all stages) was analyzed depending on the expression level of HS3ST4 (H) or GPC6 (I) or VCAN (J) compared to TRF2 expression using KM‐plotter.
-
KTable summarizing the median of survival (months), P‐values, and hazard ratio depending on TRF2, HS3ST4, GPC6, or VCAN expression.
-
LVenn diagram associated with Fig 7F showing the intersection between the three categories of patients.
TRF2 upregulation in human malignancies is associated with a poor prognosis, high expression of HSPG genes, and MDSC infiltration
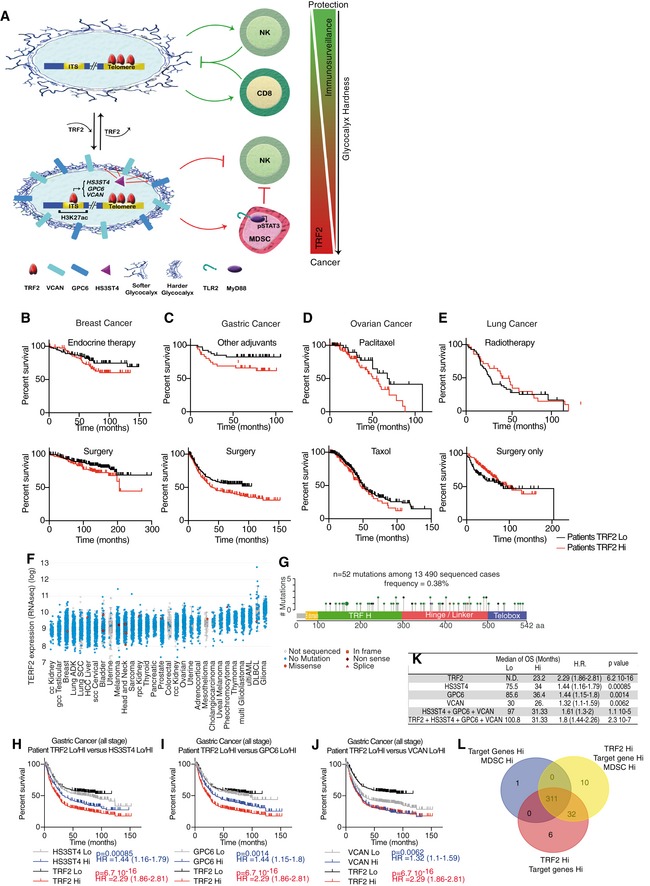
Finally, we investigated whether the relationships among TRF2 upregulation, HSPG gene expression, and MDSCs contribute to patient outcome. Using public data obtained from the Cancer Genome Atlas and KM‐plotter (Szász et al, 2016), we first confirmed that TRF2 is highly expressed in a large panel of human cancers (Fig EV5F). Notably, very few TERF2 mutations were detected in these cancers, suggesting that functional TRF2 is required for tumorigenicity and that the tumorigenic properties of TRF2 depend on its upregulation (Fig EV5G and Dataset EV5). Among all histological types or pathological TNM stages of breast, gastric, ovarian, and lung cancers (Fig 8A), OS was significantly reduced in patients with tumors exhibiting high TRF2 expression. According to hazard ratios (HRs), TRF2 overexpression was significantly associated with an increased rate of mortality (Fig 8A, Appendix Fig S3 and Dataset EV6), by more than twofold, in all stages of gastric cancer (HR = 2.29), stage 3 lung adenocarcinoma (HR = 2.17), and stage 2 lung small cell carcinoma (SCC; HR = 2.03). Strikingly, this increase in mortality rate associated with high TRF2 expression was even greater among stage 1 ovarian and gastric cancers (HR = 6.31 and 4.73, respectively). Taken together, these data suggest that TRF2 is associated with poor prognosis and an increased mortality rate.
Figure 8. TRF2 upregulation in human malignancies is associated with a poor prognosis, high expression of HSPG genes, and MDSC infiltration.
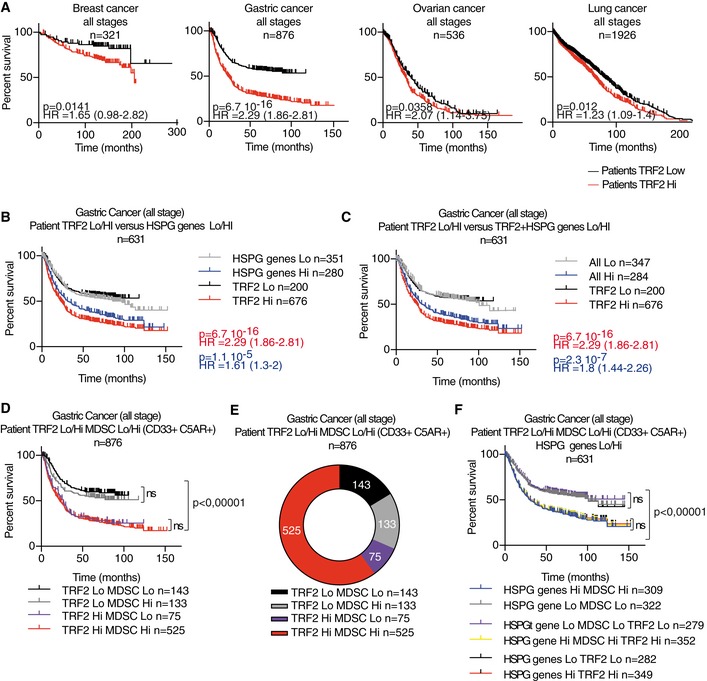
-
AKaplan–Meier curves for overall survival of patients at all stages of cancers (breast, gastric, ovarian, and lung cancer) depending on TRF2 expression level (KMplot, http://kmplot.com/analysis/index.php?p=background).
-
B–DThe overall survival of gastric cancer patients (all stages) was analyzed depending on TRF2 and its target gene expression level (HS3ST4, GPC6, and VCAN collectively named the HSPG genes) or with MDSC level (Lo or Hi), determined as the mean expression level of CD33 and C5AR, using KMplot, http://kmplot.com/analysis/index.php?p=background.
-
ERepartition of number of gastric cancer patients depending on TRF2 and MDSC classification.
-
FOverall survival of gastric cancer patients (all stages) analyzed depending on TRF2, HSPG genes (HS3ST4, GPC6, and VCAN) and MDSC level (Lo or Hi).
We examined OS as a function of the expression levels of TRF2 target genes (HS3ST4, GPC6, and VCAN). Even though some patients are missing in the VCAN gene expression analysis (n = 631 instead of 876), we found that patients with high expression of VCAN, HS3ST4, and GPC6 (collectively, HSPG genes) exhibited OS rates similar to those of patients with high TRF2 expression (Figs 8B and EV5H–J). Interestingly, we noticed similar OS between patients with high expression (Hi) of HSPG and those with TRF2 Hi HSPG Hi (high expression for TRF2, HS3ST4, GPC6, and VCAN), suggesting epistatic relationships among these genes in human gastric tumors (Figs 8C and EV5K). This relationship, together with the fact that the numbers of patients were similar between the TRF2 Hi HSPG Hi (n = 284) and HSPG Hi (n = 280) groups, indicates that most TRF2‐overexpressing tumors also exhibit upregulation of the three HSPG genes and poor prognosis.
Next, we estimated MDSC infiltration by scoring the level of co‐expression of CD33 and C5AR as surrogate markers (Markiewski et al, 2008; Nitta et al, 2013; Janelle et al, 2014). Most patients with high TRF2 expression (gastric cancers) also exhibited marked MDSC infiltration and reduced OS (Fig 8D and E). Consistent with the epistatic relationships detected among high TRF2 levels, high HSPG gene expression, high MDSC infiltration, and reduced OS, TRF2 Hi MDSC Hi patients had OS rates identical to those of TRF2 Hi MDSC Hi HSPG Hi and HSPG Hi MDSC Hi patients (Figs 8F and EV5L). Moreover, TRF2 expression was positively correlated with high HSPG gene expression (multiple regression, P = 6.7 × 10−16; Appendix Fig S4A) and MDSC infiltration (multiple regression, P = 8.4 × 10−25; Appendix Fig S4B). The expression levels of the three HSPG genes were correlated with MDSC infiltration levels (Spearman's correlation, 8.62 × 10−39) (Appendix Fig S4A–H). Finally, we used chi‐square analysis to compare variable independence between TRF2 expression and MDSC infiltration (Fig 8D) or TRF2 expression, MDSC infiltration, and HSPG expression (Fig 8F) and found that TRF2 expression, MDSC infiltration, and expression of HSPG target genes (HS3ST4, GPC6, and VCAN) appear to be interdependent (Appendix Fig S4I and J).
Overall, we conclude that high TRF2 expression in a large cohort of gastric cancer patients was associated with worse prognoses in an epistatic relationship with the expression of the three HSPG genes regulated by TRF2.
Discussion
In this study, we revealed a mechanism by which cancer cells recruit and activate MDSCs via the TLR2/MyD88/IL‐6/JAK1/2 pathway, based on upregulation of the telomere protein TRF2. We also demonstrated that this TRF2‐dependent regulation of MDSCs acts as a general suppressor of the immune response by inhibiting NK and T cell responses, establishing a direct link between cancer‐associated telomere changes and the immunosuppressive tumor microenvironment. Remarkably, the role of TRF2 in this process is not to protect telomeres but to regulate the expression of three genes involved in HSPG biosynthesis (HS3ST4, GPC6, and VCAN). In fact, this role of TRF2 in HSPG gene expression is central to shaping the glycocalyx, because TRF2 overexpression profoundly changes its overall structure, increasing its stiffness and reducing its length. The roles of TRF2 in HSPG biosynthesis and MDSC control may have high clinical relevance because the TRF2 upregulation observed in a large number of human cancer patients was correlated with high HSPG expression, MDSC infiltration, and poor prognosis in an epistatic relationship. Moreover, we provide evidence that the tumoral expression level of TRF2 is associated with an increased mortality rate and decreased OS and could be used as a surrogate marker for the efficacy of chemotherapies targeting MDSCs.
The finding that HS3ST4 and GPC6 are required for MDSC recruitment can be explained by glycocalyx changes that modify micro‐gradients of the cytokines, chemokines, and growth factors involved in immune cell recruitment (Simon Davis and Parish, 2013; Knelson et al, 2014). The glycocalyx changes induced by TRF2 might have other oncogenic consequences, for example, by favoring neoangiogenesis (Zizza et al, 2019) or tissue remodeling. The finding that VCAN upregulation controlled MDSC activation is consistent with a previous report showing that the proteoglycan Versican, encoded by VCAN, acts as a TLR2 ligand (Kim et al, 2009) and that TLR2 signaling activates the immunosuppressive function of MDSCs (Skabytska et al, 2014) via an IL‐6 autocrine pathway (Chalmin et al, 2010).
We showed in a previous study that TRF2 overexpression in cancer cells enhances tumorigenicity and that NK cell inhibition, by depleting NK cells in vivo, was the main target of a TRF2‐dependent increase in tumorigenicity (Biroccio et al, 2013). Since the oncogenic role of TRF2 was detected using both immunocompetent and immunodeficient mouse models, and NK cell recruitment and activation rescue were observed when MDSCs were depleted in both models, NK cells are arguably major target cells of MDSC activation controlled by TRF2 in cancer cells.
The increase of STAT3 phosphorylation and immunosuppressive capacity of MDSCs on NK cell degranulation and cytotoxicity (Fig 3), which were induced by TRF2 overexpression in cancers in a TLR2/MyD88‐dependent manner, required IL‐6 secretion and JAK1/2 activity. This finding suggests that changes in HSPG expression are regulated by TRF2 signaling to induce an immunosuppressive autocrine loop via IL‐6/JAK1/2/STAT3 signaling.
We also explored the mechanism by which TRF2 controls the expression of the three HSPG genes. These genes each contain an ITS that serves as a binding site for TRF2. Upon binding, TRF2 induced a change in chromatin structure at the ITS, as revealed by the enrichment of H3K27ac, an activating histone mark (Villar et al, 2015); this suggests that ITSs behave as TRF2‐dependent enhancer elements that activate the expression of neighboring genes. In agreement with this model, TRF2 interacts with the histone acetyl transferase p300, which is responsible for histone acetylation at enhancers (Her & Chung, 2013).
We provided the following evidence that this TRF2‐dependent mechanism of MDSC regulation by HSPG genes plays an important role in human cancers: (i) immunodepletion of MDSCs by antibodies was sufficient to reverse the oncogenic and pro‐metastatic properties of TRF2 in mouse models of tumor formation (Fig 2); (ii) high TRF2 expression was observed in several human malignancies and found to be associated with poor prognosis (Fig 8); (iii) high expression of TRF2 effector genes (HS3ST4, GPC6, and VCAN) and MDSC infiltration was found to be associated with poor prognosis in an epistatic relationship with high TRF2 expression (Fig 8); (iv) the cell‐surface HS (Qazi et al, 2016) or HSPG expression level (Wang et al, 2009; Knelson et al, 2014; Dinccelik Aslan et al, 2015) was crucial for tumor invasion and metastasis and was associated with poor survival (Gan et al, 2010; Matas‐Rico et al, 2016) (Figs 2 and 8); and (v) TRF2‐dependent glycocalyx changes recruited and activated MDSCs as well as the expression of the immunosuppressive effectors il‐10, tgf‐b, and Arg‐1, molecules responsible for NK and T cell inhibition, in the tumor microenvironment (Fig 2) (Platonova et al, 2011; Kodumudi et al, 2012; Fujimura et al, 2014; Kumar et al, 2016a). Thus, TRF2 emerges as a potential target for innovative anti‐cancer therapies. In particular, specific inhibition of TRF2 may show a synergistic effect with strategies targeting immune checkpoints.
Another clinical implication of our results concerns the efficiency of conventional chemotherapies. The negative effects of TRF2 overexpression on OS can be reduced following 5‐FU treatment in gastric cancer, gemcitabine treatment in ovarian cancer, and paclitaxel‐based chemotherapy in lung cancer, three compounds that target MDSCs (Apetoh & Vegran, 2011; Bruchard et al, 2013; Sevko et al, 2013), which suggests that chemotherapy efficiency is enhanced in patients with high TRF2 expression. Accordingly, the reduction in tumor volume and increase in OS observed in mice bearing TRF2‐overexpressing tumors observed after treatment with 5‐FU suggest that TRF2 overexpression enhances the efficiency of 5‐FU. In line with the MDSC depletion data, the increased response to 5‐FU may be linked to better elimination of MDSCs, in turn allowing reactivation of the immune system. Thus, the level of TRF2 expression might be an interesting surrogate marker for chemotherapy responses.
Taken together, the results of this study identify an immunosuppressive pathway for MDSCs that links the telomeric protein TRF2 to glycocalyx reshuffling, suggesting novel strategies to prevent immunosurveillance escape and enhance the efficacy of cancer therapies (Fig EV5A).
From an evolutionary perspective, there is a striking similarity between (i) our finding that the genes encoding glycocalyx components involved in immunosuppression in cancer cells are regulated by TRF2 and (ii) the antigenic variation employed by various pathogens to bypass the host immune response, involving the regulation of genes encoding plasma membrane proteins by TRF2 homologs (Jehi et al, 2014). This similarity suggests a conserved mechanism that couple telomere status to cell‐surface composition, which is likely to ensure efficient coordination between telomere changes and the interaction of cells with their environment.
Materials and Methods
Cell culture and viral transduction
B16F10 (obtained from the ATCC, Manassas, VA), HEK293T, NIH3T3, or the human fibroblast BJ rendered immortal and tumorigenic by SV40 small t and large T, hTERT overexpression and RasV12 overexpression called Bjcl2 (Biroccio et al, 2013) were grown in DMEM (Invitrogen) supplemented with 10% of FCS and 1% of penicillin–streptomycin at 37°C with 5% CO2. MSC2 (obtained from Pr Ghiringhelli laboratory) or YAK‐1 (obtained from Pr Vivier laboratory) was cultured in RPMI medium (Thermo Fisher Scientific) supplemented with 10% FCS and 1% of penicillin–streptomycin (Invitrogen) at 37°C with 5% CO2. Mycoplasma test was performed every 3 months, and experiments are performed only on mycoplasma negative cells. To create lentiviruses, transient transfection with 293T cells was performed as described previously (Biroccio et al, 2013).
| Target | Species | Number | Sequence |
|---|---|---|---|
| TERF2 | Human | #1 | CATTGGAATGATGACTCTGAA |
| TERF2 | Human | #2 | CCTGGAGAGAAGAATCCCAAA |
| HS3ST4 | Human | #1 | CCCATCAGTTTGATTCAATTA |
| GPC6 | Human | #1 | GCTTCCTCTTTCCTTCAGCTA |
| VCAN | Human | #1 | CCGGGTGAATTTCTCCGCATC |
| Terf2 | Mouse | #1 | CCTTGGAATCAGCTATCAATG |
| Terf2 | Mouse | #2 | CAAGGAGGCTGCTGTCATTAT |
Colony assay
200 transduced cells per well were seeded in 6‐well plate. After 10 days, colonies were washed with PBS, fixed for 30 min with acetic acid/methanol, and revealed by crystal violet staining for 30 min. Colonies were then determined by manual counting.
AlamarBlue
Cells were seeded (5 × 103 cells per well in 200 μl) in a 96‐well flat‐bottom plate. 10 μl of AlamarBlue (Bio‐Rad) was added 24 h later, and the absorbance was measured at 570 and 600 nm for 36 h in a Spectrostar Nano plate reader (BMG Labtech). In the case of NK cell killing assay, the NK cells were added for 4 h before washing of the wells and addition of AlamarBlue. The oxydoreduction of the colorant was determined 8 h after. In both experiments, the oxidation–reduction percentage of AlamarBlue was determined as described by the manufacturer.
Animals
Experiments are performed on 8‐ to 12‐week‐old female C57Bl6J mice and NMRI Nude mice from Janvier Labs (France) or from 6‐ to 12‐week‐old male CD1 nu/nu from Charles River (Italy). All mouse experiments were conducted according to local and international institutional guidelines and were approved by either the Animal Care Committee of the IRCAN and the regional (CIEPAL Cote d'Azur #187 and #188) and national (French Ministry of Research #03482.01/02482.2 and # 02973.01/02973.2) authorities.
Tumor growth, metastasis assay, and Matrigel plugs assay experiments
Cells were injected subcutaneously in the back of mice at 106 cells/mouse (n = 8 mice per group). The tumor appearance was evaluated by palpation, the tumor size was measured every 2 days in three dimensions by caliper and tumor volume calculated using the hemi‐ellipsoid formula: π × (L × l × h)/6, where L corresponds to the length, l to the width, and h to the height the tumor, respectively. Tumor, Matrigel plugs, or lung infiltrating cells were collected after enzymatic dissociation by Dispase (Corning), collagenase A (Roche), and DNAse I (Roche) digestion for 30 min at 37°C. After washes in PBS, cells were fixed in formaldehyde 0.5% and the quantity of GFP+ infiltrating cells (metastatic B16F10 cells) was determined by FACS (ARIA III cytometer, DIVA6 software (BD Biosciences) and FlowJo 10 (LLC)). When indicated, mice were treated with three intraperitoneal injections of 50 mg/kg of 5‐FU (days 10, 13, and 16) or with intraperitoneal injections of 200 μg in 100 μl of rat IgG2b isotopic control or anti‐GR1 antibody (BioXcell).
Cells were injected into the hind leg muscles of mice at 105 cells/mouse or 5 × 105 cells/mouse (n = 6 mice per group). The tumor appearance was evaluated by palpation, the tumor size was measured three times a week in two dimensions by a caliper, and tumor weight was calculated using the following formula: a × b2/2, where a and b are the long and short diameter of the tumor, respectively.
For Matrigel assay (Biroccio et al, 2013), 106 cells/mouse in 100 μl of PBS and complemented with 400 μl of Matrigel growth factor reduced (Corning) were inoculated subcutaneously into the back under isoflurane anesthesia. 5 days after, mice were sacrificed and Matrigel plugs were harvested. Infiltrating cells were collected after enzymatic dissociation as described earlier and stained with directly coupled antibodies for 30 min at 4°C after saturation with Fc‐Block anti‐CD16/CD32 antibodies (clone 2.4G2) for 15 min on ice. After washes in 0.5 mM EDTA 2% FCS PBS and fixation with 0.5% FA, cells were analyzed using an ARIA III cytometer with DIVA6 software (BD Biosciences) and FlowJo 10 (LLC).
For metastasis assay, adult C57Bl6j female mice received a single intravenous tail vein injection of 1 × 105 B16F10 GFP or B16F10 overexpressing TRF2‐GFP cells. The mice were subsequently intraperitoneally injected every 3 days with 200 μg in 100 μl of either isotype (rat IgG2b) or anti‐GR‐1 (RB6‐8C5) antibodies from BioXcell. The mice were euthanized 21 days after intravenous injection, and lungs were inflated and perfused through the trachea with 4% paraformaldehyde (PFA), fixed overnight, transferred to 70% ethanol, and subsequently embedded in paraffin. Metastasis burden was determined for each animal by macroscopic quantification of lung nodules (counting). Tumors histology was examined on 5‐μm tissue section stained with hematoxylin and eosin (H&E).
Table of antibodies
| Specificity | Company | Clone | Species | Isotype | Fluorochrome | Reference |
|---|---|---|---|---|---|---|
| Anti‐Ly6G (Gr1) | eBioscience | RB6‐8C5 | rat | IgG2b, k | PE | 12‐5931‐82 |
| Anti‐CD107a FITC | BD Biosciences | 1D4B | Rat | IgG2a/k | FITC | 553793 |
| Anti‐CD107a FITC | BD Biosciences | 1D4B | Rat | IgG2a/k | FITC | 553793 |
| Anti‐CD107b | BD Biosciences | ABL‐93 | Rat | IgG2a/k | FITC | 558758 |
| Anti‐CD11b | BD Biosciences | M1/70 | Rat | IgG2b | APC‐H7 | 550993 |
| Anti‐CD11c | BD Biosciences | HL3 | Hamster | IgG1 | FITC | 557400 |
| Anti‐CD19 FITC | BD Biosciences | 1D3 | Rat | IgG2a, κ | FITC | 553785 |
| Anti‐CD335 (NKp46) | BD Biosciences | 29A1.4 | Rat | IgG2a | PE | 560757 |
| Anti‐CD45 | BD Biosciences | 30‐F11 | Rat | IgG2b | PerCP | 557235 |
| Anti‐CD69 PE‐Cy7 | BD Biosciences | H1.2F3 | Hamster | IgG1/K | PE‐Cy7 | 552879 |
| Anti‐CD8a | BD Biosciences | 53‐6.7 | Rat | IgG2a,K | BV650 | 563152 |
| Anti‐IFN‐gamma | BD Biosciences | 4S.B3 | Mouse | IgG1/K | PE | 554552 |
| Anti‐IFN‐gamma | BD Biosciences | XMG1.2 | Rat | IgG1/K | PE | 554412 |
| Anti‐NK‐1.1 | BD Biosciences | PK136 | Mouse | IgG2a/k | APC | 550627 |
| Anti‐NK‐1.1 | Biolegend | PK136 | Mouse | IgG2a, κ | APC | 108710 |
| Anti‐NKp46 CD335 | BD Biosciences | 29A1.4 | Rat | IgG2a | Alexa 647 | 560755 |
| Anti‐CD107a | BD Biosciences | 1D4B | Rat SD | IgG2a, κ | V450 | 560648 |
| Anti‐CD11b | Biolegend | M1/70 | Rat | IgG2b, κ | BV605 | 101237 |
| Anti‐CD11c | Biolegend | N418 | Armenian Hamster | IgG | PE/dazzle | 117347 |
| Anti‐CD19 | Biolegend | 6D5 | Rat | IgG2a, κ | BV510 | 115545 |
| Anti‐CD19 | Biolegend | 6D5 | Rat | IgG2a, κ | BV785 | 115543 |
| Anti‐CD25 | eBioscience | PC 61.5 | Hamster | IgG1 lambda | PE‐Cy7 | 25‐0251‐82 |
| Anti‐CD335 (NKp46) | BD HORIZON | 29A1.4 | Rat | IgG2a, κ | BV510 | 563455 |
| Anti‐CD3e | BD Biosciences | 145‐2C11 | Hamster | IgG1, κ | FITC | 553062 |
| Anti‐CD3e | Biolegend | 145‐C11 | Armenian Hamster | IgG | PerCP | 100302 |
| Anti‐CD4 | BD Biosciences | GK1.5 | Rat | IgG2b, κ | PE | 553730 |
| Anti‐CD4 | Life Technologies | Monoclonal | Rat | IgG2a | PE‐AF700 | MCD0424 |
| Anti‐CD45 | BD Biosciences | 30‐F11 | Rat | IgG2b, κ | AF700 | 560510 |
| Anti‐CD8a | BD Biosciences | 53‐6.7 | Rat LOU | IgG2a, κ | PerCP‐Cy5.5 | 551162 |
| Anti‐F4/80 | Biolegend | BM8 | Rat | IgG2a, κ | PerCP‐Cy5.5 | 123128 |
| Anti‐F4/80 | Biolegend | BM8 | Rat | IgG2a, κ | BV510 | 123135 |
| Anti‐Ly6C | eBiosciences | HK1.4 | Rat | IgG2c, κ | APC‐eFluor780 | 47‐5932‐82 |
| Anti‐Ly6C/Ly6G (Gr‐1) | Biolegend | RB6‐8C5 | Rat | IgG2b, κ | PE | 108408 |
| Anti‐Ly6C/Ly6G (Gr‐1) | BD Biosciences | RB6‐8C5 | Rat | IgG2b, κ | PE | 553128 |
| Anti‐Ly6C/Ly6G (Gr‐1) | Biolegend | RB6‐8C5 | Rat | IgG2b, κ | BV421 | 108434 |
| Anti‐Ly6G | BD Biosciences | 1A8 | Rat LEW | IgG2a, κ | PE‐Cy7 | 560601 |
| Anti‐Ly6G | Biolegend | 1A8 | Rat | IgG2a, κ | BV421 | 127627 |
| Anti‐Ly6G (Gr‐1) | BD Biosciences | 1A8 | Rat LW | IgG2a, κ | BV711 | 563979 |
| Anti‐stat3 (pY705) | BD PhosphoFlow | 4/P STAT3 | Mouse | IgG2a, κ | AF 647 | 557815 |
| Anti‐h HS3ST4 | Biotechne | #712010 | Mouse | IgG1 | Uncoupled | MAB6085 |
| Anti‐h GPC6 | Biotechne | #348701 | Mouse | IgG1 | Uncoupled | MAB2845 |
| Anti‐h GPC6 | Abcam | Polyclonal | Rabbit | IgG | Uncoupled | Ab170523 |
| Anti‐h VCAN | Abcam | EPR12277 | Rabbit | IgG | Uncoupled | Ab170480 |
Stiffness determination by atomic force microscopy
The experiments were performed in Leibovitz's medium (Life Technologies) supplemented with heat‐inactivated 10% fetal bovine serum (FBS) on cells grown on 50‐mm Willco glass‐bottom dish. After thermal stabilization, for each sample, a minimum of 20 cells were analyzed using the “Point and Shoot” method, collecting at least 100 force–distance curves at just as many discrete points (on average five points for each cell in the perinuclear area). After determining both the deflection sensitivity of the system in the Leibovitz's medium using a clean Willco glass‐bottom dish and the spring constant of the cantilever by means of the thermal tune method, force–distance curves were collected on samples using a velocity of 1 μm/s, in relative trigger mode and by setting the trigger threshold to 400 pN. The apparent Young's modulus was calculated using the NanoScope Analysis 1.80 software (Bruker Nano Surfaces, Santa Barbara, CA, USA) applying to the force curves, after the baseline correction, the Hertz spherical indentation model using a Poisson's ratio of 0.5. For each force curve, after finding the contact point, the force fit boundaries to perform the fit were chosen between 0% and the percentage corresponding to the point in which a clear change in the slope of the curve was observed (y0 in Fig 5). This first fit was used to obtain the apparent Young's modulus of the glycocalyx. By interpolation, the value |x0|, corresponding to the thickness of the cell brush, was found. The apparent Young's modulus of the cell body was obtained using, on the same force curve, fit boundaries between the percentage corresponding to y0 and 70%. To avoid any tilt effect due to the base correction step, only the force curves having their maximum value at 400 pN were considered for performing the fit, and then, only the apparent Young's modulus values corresponding to a fit with R 2 > 0.75 were considered. The cells mechanical proprieties were obtained by atomic force microscopy using a Bioscope Catalyst (Bruker Nano Surfaces, Santa Barbara, CA, USA), coupled with an inverted optical microscope (Leica DMI6000B, Leica Microsystems Ltd., UK). The force–distance curves needed to calculate the apparent Young's modulus were collected using a Borosilicate Glass spherical tip (5 μm of diameter) mounted on a cantilever with a nominal spring constant of 0.06N/m (Novascan Technologies, Ames, IA USA). The Sneddon modulus heat maps were captured with a SNL‐D probe (Bruker Nano Surfaces, Santa Barbara, CA, USA) in Peak force tapping using a peak force set point of 600 pN and a peak force frequency of 0.25 kHz.
ITS search and mammalian ITS comparison
ITSs motifs were searched on the hg19 genome. We first used BLAST+ (PMID: 20003500) to find all the patterns T2AG3 repeated at least four times and with 1 maximum mismatch per pattern T2AG3. We then used a Python script to merge the positions of distant ITSs closed to < 400 nucleotides and create the final bed file containing a total of 483 ITSs. Mammalian ITSs were identified using the same criteria.
ITSs were annotated using “Bedtools window” as follows: the closest gene (first gene), genes present in a ± 50 Kb interval, and genes present in a ± 200 Kb interval. For each species, the ITS file was compared with the refGene file. For each gene, the transcription start (txStart) and end position (txEnd) were chosen. The position of each ITS was compared with the positions of the genes. Results were summarized using a homemade Python script in files containing the ITS ID, ITS position, closest gene, genes present within the ± 50 Kb interval, and genes present within the ± 200 Kb interval.
Data were compared with published ChIP‐seq data (H3K4me3, H3K27ac) (ENCODE). For each species, overlapping regions were identified between ITS and H3K27ac files, ITS and H3K4me3 files, and ITS and H3K4me3 H3K27ac files thanks to a homemade Python script using the Pybedtools library with “BedTool.intersect()” method. Statistical analysis was performed using Pybedtools.BedTool: “randomstats”. A thousand iterations were chosen, corresponding to a P‐value of 0.001.
TCGA and overall survival analysis
Standardized TCGA data were obtained from Broad Institute TCGA Genome Data Analysis Center (2016, https://doi.org/10.7908/c11g0km9) The expression level and mutations of TERF2 in 30 cancer type were analyzed using the Genomic Data Commons cBioPortal website (http://www.cbioportal.org/). Data were collected to be analyzed using Prism 7.0 (GraphPad).
Overall survival (OS) analysis was performed using Kaplan–Meier plotter (KM‐plotter) website (http://kmplot.com) following recommendations of the authors (Szász et al, 2016) and discussions with Dr Balázs Győrffy. For each cancer type (lung, prostate, gastric, or breast), patient OS was determined based on TERF2 expression level. The optimal cut‐off and the median of survival were determined by KM‐Plotter for each cohort. No dataset was excluded. Down‐staging analysis was performed depending on tumor stage, treatment, or TRF2 target gene as indicated. In all cases, P‐values and hazard ratios (HR) were determined by KM‐plotter and Chi2 analysis and curves were generated using Prism 7.0 (GraphPad).
MDSC co‐culture and NK cell degranulation and killing assay in vitro
Primary NK cells (CD45+, CD3‐NKp46+) were FACS sorted from c57Bl/6j naïve splenocytes previously stimulated in vivo for 18 h by an intraperitoneal injection of 150 μg of Poly I:C) (Invivogen, Toulouse, France). In parallel, a co‐culture was carried out for 18 h with transduced B16F10 or BJcl2 and MSC2 cells as indicated (Figs 3 and EV3). The MSC2 cells are then sorted by FACS (Gr‐1+ CD11b+). Purity controls on the post‐sort fractions are performed (Fig EV3). Finally, sorted NK and MSC2 were co‐cultured for 18 h, in presence or not of mouse IL‐6 blocking antibody or JAK1/2 inhibitor (Ruxolitinib) (see section Inhibitors). Then, the degranulation capacity of NK cells was tested after challenge with YAK‐1 (NK targets) and the killing capacity of NK cells was analyzed using NIH3T3 as target cells. YAK‐1 cells are added to the culture for 4 h in presence of monensin and brefeldin (BD Bioscience) at the effector/target ratio of 5:1 or 10:1. Degranulation activity of the NK cells is then measured by FACS by the anti‐CD107a and IFN‐γ staining. Alternatively, NIH3T3 cells are plated in 96‐well plates and, after adhesion of NIH3T3 cells, the NK/MSC2 co‐culture was added at the effector/target ratio 10:1 for 4 h. Wells were then washed, and the viability of remaining NIH3T3 cells was assessed using AlamarBlue colorimetric method (see section AlamarBlue).
Intranuclear phospho‐STAT3 staining
MSC2 cells or cells from Matrigel plugs or mouse tumors were harvested and fixed with PFA 4% for 30 min at room temperature. Cells were washed in perm/wash buffer (BD Biosciences) and resuspended in buffer III (BD Biosciences) for 30 min on ice. After washes, cells are resuspended in perm/wash buffer and saturated with Fc‐Block. Then, anti‐GR1, anti‐CD11b, and anti‐pSTAT3 or isotypic controls (BD Biosciences) were directly added and incubated for 30 min. After washes, the staining is analyzed on a FACS ARIA III (BD Biosciences). pSTAT3 of MSC‐2 cells was determined after 24 h treatment with 100 ng/ml LPS (Invivogen), with or without NaClO3 (50 mM), after 24 h co‐culture with cancer cell or conditioned media from cancer cell overexpressing or compromised for TRF2 and TRF2 target genes (VCAN, HS3ST4, or GPC6).
Toll‐like receptor pathway and IL‐6/JAK‐STAT pathway inhibitors
Using MSC2 cells pretreated with LPS (100 ng/ml during 24 h), optimal doses for each inhibitor have been determined by analyzing pSTAT3 level by FACS. Then, the optimal doses (LPS‐RS 30 ng/ml, anti‐mTLR2 antibody 30 ng/ml, anti‐MyD88 peptide 10 μM, anti‐mIL‐6 250 ng/ml, or ruxolitinib 10 μM) were applied in MSC2/cancer cell co‐culture assay.
Heparan sulfate analysis by flow cytometry
Cells were harvested with cold PBS–EDTA 3 mM and adjusted at 1 × 106 cells/ml. After Fc‐Block saturation, 10 μg of anti‐heparan sulfate (Clone 10E4 from Amsbio) per million of cells were added for 30 min on ice. After washes and incubation with specific APC coupled anti‐IgM secondary antibody (eBioscience), cells were washed and analyzed with a FACS ARIA III (BD Biosciences). The inhibition of heparan sulfate synthesis by sodium chlorate (NaClO3) was tested by treating the cells with increasing doses of NaClO3 and analyzing the 10E4 staining as described. The optimal doses of 50 mM were determined and used for experiments.
ChIP‐qPCR
1 × 107 transduced BJcl2 cells are fixed with 1% formaldehyde for 10 min at room temperature. The reaction is stopped by 1M glycine. Nuclei are extracted using nuclear extraction buffer (50 mM Tris–Cl pH 8, 10 mM EDTA, 1% SDS, protease inhibitors) and then sonicated using Bioruptor (Diagenode) for 14 cycles alternating 30 s of sonication 30 s without sonication. Chromatin fragmentation, previously washed using the PCR Clean‐Up kit (Promega), was checked (500 bp) by migration on a 1% agarose gel. 30 μl of the sonicated DNA is recovered before IP called “Input.” Chromatin IP is carried out overnight at 4°C with stirring (see antibody list below). DNA is immunoprecipitated using magnetic beads coupled to the G protein (Thermo Fisher Scientific) and washed. DNA–protein interaction was broken by addition of 5M NaCl overnight at 65°C. DNA extraction is carried out by phenol/chloroform method. DNA derived from the ChIP is amplified by qPCR using different pairs of primers specifically targeting the ITS region of the different genes:
HS3ST4 (5′CTGGGAGACTTTGTGCGTCT3′‐3′CTTGAGCTTCCATCCTCTGG5′),
VCAN (5′GCGGATCATGAGGTCAGGAG3′‐3′ACTGGGACTACAGGCTAGGG5′),
GPC6 (5′TCGCTAATCGATGCATAACC3′‐3′TGCTGAAGGCTGTTGAACTT5′).
The enrichment of the ChIP was standardized with respect of histone H3 amount present at the level of the ITS but also in relation to the internal controls (input and GAPDH promoter).
| Specificity | Company | Species | Reference |
|---|---|---|---|
| Anti‐total histone H3 | Abcam | Rabbit polyclonal IgG | ab1791 |
| Anti‐H3K27ac | Abcam | Rabbit polyclonal IgG | ab4729 |
| Anti‐H3K27me3 | Millipore | Rabbit polyclonal IgG | 07‐449 |
RT–qPCR
1 × 106 cells are lysed to extract the RNAs using RNeasy Plus Mini kit (Qiagen). RNAs concentration and quality are determined by Nanodrop 2000/2000c (ThermoFisher). Reverse transcription (RT) from 1 μg of RNA was performed using Applied Biosystems reverse transcription kit in a T100 thermocycler (Bio‐Rad). qPCRs were made on cDNAs obtained using ROCHE's Fast universal Sybr Green master (ROX) on a StepOne plus thermocycler (Applied Biosystems) using primers described below.
| Fwd | Rev | |
|---|---|---|
| h gpc6 | TGGAAGACAAGTTAAGCCAACAA | TTCTTATGCCTGGACACAAAAGT |
| h hs3st4 | AAACCCGAGATCCCCACCTT | CAGCGCATAGATCCCTATTCG |
| h versican | GTAACCCATGCGCTACATAAAGT | GGCAAAGTAGGCATCGTTGAAA |
| h terf2 | GACCTTCCAGCAGAAGATGCT | GTTGGAGGATTCCGTAGCTG |
| m il‐10 | TGTACAGCTGCCGCACACA | TGTACAGCTGCCGCACACA |
| m tgf‐β | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| m arg‐1 | CCAGAGAATGGAAAGTCAGTGT | GCAGATATGCAGGGAGTCACC |
| m terf2 | AAGCCCAGAGCTCTCAACC | TCCACTTGTCTTTGGATGCTT |
Statistical analysis
Reasonable sample size was chosen to ensure adequate reproducibility of results and was based on our previous studies. Mouse experiments are performed on n = 8 individuals (or 5 in some cases) as indicated in Fig legends. Mice were equally divided for treatments and controls. A formal randomization tool was not employed. For survival analysis, patients were separated based on optimal cut‐off of the expression value of the marker determined using KMplot. Animal data are stopped in end point analysis (time point indicated in each Fig legend) before to reach the ethical end point fixed in advance following the international ethical recommendation on mouse care (see details in mouse experiment procedure). No data were excluded. No outliers were excluded. Statistical tests were all performed with GraphPad Prism 7 software including the Student's t‐test, the Mann–Whitney test, the log‐rank test, Pearson correlation, chi‐square analysis, and two‐way ANOVA tests. The corresponding tests and P‐values are mentioned in the legend of each Figure.
Author contributions
JC‐V performed all the experiments with the help of CI, LC, VMR, and RC; SP performed AFM analysis; PZ, CL, and AB helped in in vivo tumorigenicity experiments with GPC6 and HS3ST4 compromised cells; MR‐M helped in virus production; NS and MS helped in B16F10 IV injection and metastasis analysis; OC, NS, EG, BG, and JC‐V performed bioinformatics analysis. FG gave MSC2 cell line and provided advice on MDSC part; EV provided advice, ideas, and technical support for the NK cell part; FA provided precious advices on heparan sulfate biology; EV, FA, and FG were involved in the writing of the manuscript. JC‐V, AB, and EG coordinated the study and JC‐V and EG wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Review Process File
Acknowledgements
We thank the Animal core facility (IRCAN/CAL), CytoMed, GenoMed, PICMI, Histology, and atomic force microscopy facilities of IRCAN which are supported by le FEDER, Ministère de l'Enseignement Supérieur, Région Provence Alpes‐Côte d'Azur, Conseil Départemental 06, ITMO Cancer Aviesan (plan cancer), Cancéropole PACA, the Association pour la Recherche sur le Cancer (ARC), the Infrastructures en Biologie Santé et Agronomie (IBiSA), the University of Nice Sophia‐Antipolis and l'Inserm. We thank the FHU OncoAge. This work was supported by grants from the the Fondation ARC pour la recherche sur le cancer (emergence no. PJA 20151203504 and Labelisation no. PGA20160203873), cancéropole PACA and INCa emergence No. 2016‐04, Fondation de France (postdoc fellowship), Ligue Contre le Cancer (EG Equipe labellisée), and “Investments for the Future” LABEX SIGNALIFE (reference ANR‐11‐LABX‐0028‐01). EV laboratory is supported by funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (TILC, grant agreement No. 694502); the Agence Nationale de la Recherche; Equipe Labellisée “La Ligue,” Ligue Nationale contre le Cancer, MSDAvenir, Innate Pharma and institutional grants to the CIML (INSERM, CNRS, and Aix‐Marseille University) and to Marseille Immunopôle. The work in A.B. laboratory has been financially supported by the Italian Association for Cancer Research (AIRC, #16910 and # 21579) and PZ was recipient of a fellowship from the Umberto Veronesi Foundation (FUV).
The EMBO Journal (2019) 38: e100012
Contributor Information
Annamaria Biroccio, Email: annamaria.biroccio@ifo.gov.it.
Eric Gilson, Email: eric.gilson@unice.fr.
References
- Apetoh L, Vegran F (2011) Restoration of antitumor immunity through selective inhibition of myeloid derived suppressor cells by anticancer therapies. Curr Mol Med 11: 365–372 [DOI] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA (2000) Telomere dysfunction promotes non‐reciprocal translocations and epithelial cancers in mice. Nature 406: 641–645 [DOI] [PubMed] [Google Scholar]
- Bejarano L, Schuhmacher AJ, Méndez M, Megías D, Blanco‐Aparicio C, Martínez S, Pastor J, Squatrito M, Blasco MA (2017) Inhibition of TRF1 telomere protein impairs tumor initiation and progression in glioblastoma mouse models and patient‐derived xenografts. Cancer Cell 32: 590–607.e4 [DOI] [PubMed] [Google Scholar]
- Benhamou Y, Picco V, Raybaud H, Sudaka A, Chamorey E, Brolih S, Monteverde M, Merlano M, Nigro Lo C, Ambrosetti D et al (2016) Telomeric repeat‐binding factor 2: a marker for survival and anti‐EGFR efficacy in oral carcinoma. Oncotarget 7: 44236–44251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biroccio A, Cherfils Vicini J, Augereau A, Pinte S, Bauwens S, Ye J, Simonet T, Horard B, Jamet K, Cervera L et al (2013) TRF2 inhibits a cell‐extrinsic pathway through which natural killer cells eliminate cancer cells. Nat Cell Biol 15: 818–828 [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350: 1193–1198 [DOI] [PubMed] [Google Scholar]
- van den Born J, Salmivirta K, Henttinen T, Östman N, Ishimaru T, Miyaura S, Yoshida K, Salmivirta M (2005) Novel heparan sulfate structures revealed by monoclonal antibodies. J Biol Chem 280: 20516–20523 [DOI] [PubMed] [Google Scholar]
- Bruchard MEL, Mignot GEG, Derangère VDE, Chalmin F, Chevriaux AEL, Végran F, Boireau W, Simon B, Ryffel B, Connat JL et al (2012) Chemotherapy‐triggered cathepsin B release in myeloid‐derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 19: 57–64 [DOI] [PubMed] [Google Scholar]
- Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, Boireau W, Simon B, Ryffel B, Connat JL et al (2013) Chemotherapy‐triggered cathepsin B release in myeloid‐derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 19: 57–64 [DOI] [PubMed] [Google Scholar]
- Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy‐Martin J‐P, Boireau W, Rouleau A, Simon B, Lanneau D et al (2010) Membrane‐associated Hsp72 from tumor‐derived exosomes mediates STAT3‐dependent immunosuppressive function of mouse and human myeloid‐derived suppressor cells. J Clin Invest 120: 457–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Fenouille N, Allegra M, Hofman V, Hofman P, Bahadoran P, Lacour J‐P, Tartare‐Deckert S, Bertolotto C et al (2012) Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 31: 2461–2470 [DOI] [PubMed] [Google Scholar]
- Diala I, Wagner N, Magdinier F, Shkreli M, Sirakov M, Bauwens S, Schluth‐Bolard C, Simonet T, Renault VM, Ye J et al (2013) Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep 14: 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl MC, Idowu MO, Kimmelshue KN, York TP, Jackson‐Cook CK, Turner KC, Holt SE, Elmore LW (2011) Elevated TRF2 in advanced breast cancers with short telomeres. Breast Cancer Res Treat 127: 623–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinccelik Aslan M, Gumus Akay G, Elhan A, Unal E, Tukun A (2015) Diagnostic and prognostic significance of glypican 5 and glypican 6 gene expression levels in gastric adenocarcinoma. Mol Clin Oncol 3: 584–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Maï M, Wagner K‐D, Michiels J‐F, Ambrosetti D, Borderie A, Destree S, Renault V, Djerbi N, Giraud‐Panis M‐J, Gilson E et al (2014) The telomeric protein TRF2 regulates angiogenesis by binding and activating the PDGFRβ promoter. Cell Rep 9: 1047–1060 [DOI] [PubMed] [Google Scholar]
- Fridman WH, Zitvogel L, Sautès‐Fridman C, Kroemer G (2017) The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 14: 717–734 [DOI] [PubMed] [Google Scholar]
- Fujimura T, Kambayashi Y, Aiba S (2014) Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 1: 1433–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Horikawa I, Mondal AM, Jenkins LMM, Appella E, Vojtesek B, Bourdon J‐C, Lane DP, Harris CC (2010) Positive feedback between p53 and TRF2 during telomere‐damage signalling and cellular senescence. Nat Cell Biol 12: 1205–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S (2009) Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9: 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G (2015) Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28: 690–714 [DOI] [PubMed] [Google Scholar]
- Gan HK, Rosenthal MA, Dowling A, Kalnins R (2010) A phase II trial of primary temozolomide in patients with grade III oligodendroglial brain tumors. Neuro Oncol 12: 500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson E, Géli V (2007) How telomeres are replicated. Nat Rev Mol Cell Biol 8: 825–838 [DOI] [PubMed] [Google Scholar]
- Giraud‐Panis M‐J, Pisano S, Benarroch‐Popivker D, Pei B, Le Du M‐H, Gilson E (2013) One identity or more for telomeres? Front Oncol 3: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Suárez E, Samper E, Flores JM, Blasco MA (2000) Telomerase‐deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet 26: 114–117 [DOI] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464–468 [DOI] [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA (2002) Rules for making human tumor cells. N Engl J Med 347: 1593–1603 [DOI] [PubMed] [Google Scholar]
- Hammond E, Khurana A, Shridhar V, Dredge K (2014) The role of heparanase and sulfatases in the modification of heparan sulfate proteoglycans within the tumor microenvironment and opportunities for novel cancer therapeutics. Front Oncol 4: 195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her YR, Chung IK (2013) p300‐mediated acetylation of TRF2 is required for maintaining functional telomeres. Nucleic Acids Res 41: 2267–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri MA, Ansari SA, Alqahtani MH, Shay JW (2016) Roles of telomeres and telomerase in cancer, and advances in telomerase‐targeted therapies. Genome Med 8: 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelle V, Langlois M‐P, Tarrab E, Lapierre P, Poliquin L, Lamarre A (2014) Transient complement inhibition promotes a tumor‐specific immune response through the implication of natural killer cells. Cancer Immunol Res 2: 200–206 [DOI] [PubMed] [Google Scholar]
- Jehi SE, Wu F, Li B (2014) Trypanosoma brucei TIF2 suppresses VSG switching by maintaining subtelomere integrity. Cell Res 24: 870–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Liu M, Fan Y, Deng X (2013) A potential gravity‐sensing role of vascular smooth muscle cell glycocalyx in altered gravitational stimulation. Astrobiology 13: 626–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266: 2011–2015 [DOI] [PubMed] [Google Scholar]
- Kim S, Takahashi H, Lin W‐W, Descargues P, Grivennikov S, Kim Y, Luo J‐L, Karin M (2009) Carcinoma‐produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457: 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knelson EH, Nee JC, Blobe GC (2014) Heparan sulfate signaling in cancer. Trends Biochem Sci 39: 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodumudi KN, Weber A, Sarnaik AA, Pilon‐Thomas S (2012) Blockade of myeloid‐derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol 189: 5147–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E et al (2016a) CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor‐ associated macrophage differentiation. Immunity 44: 303–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Patel S, Tcyganov E, Gabrilovich DI (2016b) The nature of myeloid‐derived suppressor cells in the tumor microenvironment. Trends Immunol 37: 208–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewski MM, DeAngelis RA, Benencia F, Ricklin‐Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD (2008) Modulation of the antitumor immune response by complement. Nat Immunol 9: 1225–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvel D, Gabrilovich DI (2015) Myeloid‐derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 125: 3356–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matas‐Rico E, van Veen M, Leyton‐Puig D, van den Berg J, Koster J, Kedziora KM, Molenaar B, Weerts MJA, de Rink I, Medema RH et al (2016) Glycerophosphodiesterase GDE2 promotes neuroblastoma differentiation through glypican release and is a marker of clinical outcome. Cancer Cell 30: 548–562 [DOI] [PubMed] [Google Scholar]
- Nagaraj S, Gabrilovich DI (2012) Regulation of suppressive function of myeloid‐derived suppressor cells by CD4+ T cells. Semin Cancer Biol 22: 282–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Kawai T, Kumaki F, Hiroi S, Mukai M, Ikeda E, Koering C‐E, Gilson E (2003) Expression of mRNAs for telomeric repeat binding factor (TRF)‐1 and TRF2 in atypical adenomatous hyperplasia and adenocarcinoma of the lung. Clin Cancer Res 9: 1105–1111 [PubMed] [Google Scholar]
- Nitta H, Wada Y, Kawano Y, Murakami Y, Irie A, Taniguchi K, Kikuchi K, Yamada G, Suzuki K, Honda J et al (2013) Enhancement of human cancer cell motility and invasiveness by anaphylatoxin C5a via aberrantly expressed C5a receptor (CD88). Clin Cancer Res 19: 2004–2013 [DOI] [PubMed] [Google Scholar]
- Ostrand‐Rosenberg S, Fenselau C (2018) Myeloid‐derived suppressor cells: immune‐suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 200: 422–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platonova S, Cherfils Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, André P, Dieu‐Nosjean M‐C, Alifano M, Régnard J‐F et al (2011) Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res 71: 5412–5422 [DOI] [PubMed] [Google Scholar]
- Qazi H, Shi Z‐D, Song JW, Cancel LM, Huang P, Zeng Y, Roberge S, Munn LL, Tarbell JM (2016) Heparan sulfate proteoglycans mediate renal carcinoma metastasis. Int J Cancer 139: 2791–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo NP, Smyth MJ, Snyder A (2016) Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 16: 121–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sceneay J, Chow MT, Chen A, Halse HM, Wong CSF, Andrews DM, Sloan EK, Parker BS, Bowtell DD, Smyth MJ et al (2012) Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res 72: 3906–3911 [DOI] [PubMed] [Google Scholar]
- Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, Shurin GV, Shurin MR, Umansky V (2013) Antitumor effect of paclitaxel is mediated by inhibition of myeloid‐derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol 190: 2464–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon Davis DA, Parish CR (2013) Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity. Front Immunol 4: 470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet T, Zaragosi L‐E, Philippe C, Lebrigand K, Schouteden C, Augereau A, Bauwens S, Ye J, Santagostino M, Giulotto E et al (2011) The human TTAGGG repeat factors 1 and 2 bind to a subset of interstitial telomeric sequences and satellite repeats. Cell Res 21: 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skabytska Y, Wölbing F, Günther C, Köberle M, Kaesler S, Chen K‐M, Guenova E, Demircioglu D, Kempf WE, Volz T et al (2014) Cutaneous innate immune sensing of Toll‐like receptor 2‐6 ligands suppresses T cell immunity by inducing myeloid‐derived suppressor cells. Immunity 41: 762–775 [DOI] [PubMed] [Google Scholar]
- Svoronos N, Perales‐Puchalt A, Allegrezza MJ, Rutkowski MR, Payne KK, Tesone AJ, Nguyen JM, Curiel TJ, Cadungog MG, Singhal S et al (2017) Tumor cell‐independent estrogen signaling drives disease progression through mobilization of myeloid‐derived suppressor cells. Cancer Discov 7: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szász AM, Lánczky A, Nagy Á, Förster S, Hark K, Green JE, Boussioutas A, Busuttil R, Szabó A, Győrffy B (2016) Cross‐validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7: 49322–49333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veglia F, Perego M, Gabrilovich D (2018) Myeloid‐derived suppressor cells coming of age. Nat Immunol 19: 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ (2011) Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29: 235–271 [DOI] [PubMed] [Google Scholar]
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ et al (2015) Enhancer evolution across 20 mammalian species. Cell 160: 554–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K‐D, Cherfils Vicini J, Hosen N, Hohenstein P, Gilson E, Hastie ND, Michiels J‐F, Wagner N (2014) The Wilms' tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat Commun 5: 5852 [DOI] [PubMed] [Google Scholar]
- Wang W, Xu G‐L, Jia W‐D, Ma J‐L, Li J‐S, Ge Y‐S, Ren W‐H, Yu J‐H, Liu W‐B (2009) Ligation of TLR2 by versican: a link between inflammation and metastasis. Arch Med Res 40: 321–323 [DOI] [PubMed] [Google Scholar]
- Yang D, Xiong Y, Kim H, He Q, Li Y, Chen R, Songyang Z (2011) Human telomeric proteins occupy selective interstitial sites. Cell Res 21: 1013–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zizza P, Dinami R, Porru M, Cingolani C, Salvati E, Rizzo A, D'Angelo C, Petti E, Amoreo CA, Mottolese M et al (2019) TRF2 positively regulates SULF2 expression increasing VEGF‐A release and activity in tumor microenvironment. Nucleic Acids Res 3: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Review Process File