

Abstract
It is unclear what drives the development of fibrosing nonalcoholic steatohepatitis (NASH). We aimed to determine whether cholesterol crystallization within hepatocyte lipid droplets (LDs) distinguishes patients with fibrosing NASH from patients with isolated hepatic steatosis and to study pathways leading to cholesterol accumulation in hepatocyte LDs. Patients with fibrosing NASH (n = 16) were compared to patients with isolated steatosis (n = 14). Almost all patients with fibrosing NASH had free cholesterol staining by filipin (16/16) and cholesterol crystals (15/16) in hepatocyte LDs, mostly in association with the LD membrane, compared to only 3/14 with cholesterol crystals and 3/14 with faint filipin staining in patients with isolated steatosis (P < 0.05). We were unable to identify significant differences in the expression of genes in liver tissue related to cholesterol homeostasis or LD proteins between patients with fibrosing NASH and isolated steatosis. Human hepatoma cell line (HepG2) cells were supplemented with low‐density lipoprotein (LDL)‐cholesterol and oleic acid to develop large LDs, similar to those observed in patients with NASH. Fluorescent markers were used to track the uptake and intracellular trafficking of LDL‐cholesterol. LDL‐cholesterol was taken up by HepG2 cells and transported through the endosomal‐lysosomal compartment directly to LDs, suggesting direct contact sites between late endosomes and LDs. Exposure of HepG2 cells to LDL‐cholesterol resulted in a high concentration of cholesterol and cholesterol crystallization in LDs. Conclusion: Excess cholesterol is stored in the liver primarily within hepatocyte LDs where it can crystallize. Our findings are best explained by direct transport of cholesterol from late endosomes/lysosomes to LDs in hepatocytes. We found a strong association between the presence of LD cholesterol crystals and the development of fibrosing NASH in humans, suggesting a causal relationship.
Abbreviations
- BC
Bodipy‐labeled cholesterol
- BMI
body mass index
- ER
endoplasmic reticulum
- HCV
hepatitis C virus
- HepG2
human hepatoma cell line
- LD
lipid droplet
- LDL
low‐density lipoprotein
- LE/Ly
late endosomes/lysosomes
- MCS
membrane contact site
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OA
oleic acid
Excess dietary cholesterol can lead to the development of experimental nonalcoholic steatohepatitis (NASH) in a variety of animal models.1, 2, 3, 4, 5, 6, 7, 8 In humans, hepatic free cholesterol levels were found to be elevated in NASH9 and dietary cholesterol intake was associated with development of cirrhosis.10 Furthermore, statin use was associated with amelioration of hepatic steatosis, inflammation, and fibrosis,11, 12, 13, 14, 15, 16 albeit mostly in uncontrolled or observational studies or studies using surrogate biochemical or radiographic instead of histologic endpoints. Collectively, these data suggest that hepatic cholesterol is an important etiologic factor that can lead to the progression from isolated steatosis to fibrosing steatohepatitis, both in animal models and in humans.17
The mechanisms by which hepatic cholesterol may promote the development of NASH remain uncertain. We proposed that crystallization of cholesterol within hepatocyte lipid droplets (LDs) represents one such mechanism. We reported that cholesterol crystals were present within the LDs of steatotic hepatocytes in mouse models of NASH induced by a high‐fat high‐cholesterol diet but not in mice with isolated steatosis.4, 5, 18 We found that cholesterol‐lowering drugs that caused dissolution of cholesterol crystals also caused resolution of NASH in a mouse model.5 We also demonstrated that enlarged Kupffer cells surrounded steatotic dead hepatocytes containing cholesterol crystals and appeared to process the remnant LDs within these hepatocytes, forming “crown‐like” structures. Thus, both hepatocytes and Kupffer cells become exposed to cholesterol crystals, which are highly proinflammatory and may contribute to the chronic “sterile inflammation” of NASH.
The extent to which cholesterol crystallization occurs in hepatocyte LDs in human nonalcoholic fatty liver disease (NAFLD) is unclear. More importantly, the extent to which hepatocyte cholesterol crystallization distinguishes the presence of advanced steatohepatitis as opposed to isolated steatosis in humans is unknown. If a strong association exists between the presence of cholesterol crystals in hepatocyte LDs and the presence of fibrosing NASH, it would suggest that these cholesterol crystals have pathogenetic significance in causing NASH rather than simply being innocent bystanders. Furthermore, the presence of hepatocyte cholesterol crystals could be used as a biomarker for the presence or future development of advanced fibrosing NASH. Therefore, we aimed to determine whether the presence of cholesterol crystals in hepatocyte LDs was associated with histologic disease severity in patients with NAFLD.
Mammalian cells acquire cholesterol through receptor‐mediated endocytosis of low‐density lipoprotein (LDL). In most cell types, the major function of cholesterol is to serve as a membrane constituent, especially in the plasma membrane. However, hepatocytes are unique in that they can also store large amounts of cholesterol within their LDs. In fact, we demonstrated that excess body cholesterol in mice is stored primarily in the liver rather than in adipose tissue.18 How cholesterol accumulates and eventually crystallizes in hepatocyte LDs is unclear. Given the potential significance of excess hepatocyte cholesterol in the development of NASH, our second aim was to use a human hepatoma cell line (HepG2) cell culture to study in vitro the route by which endocytosed LDL‐cholesterol accumulates and subsequently crystallizes in the LDs of steatotic hepatocytes.
Patients and Methods
Study Population
Patient data, serum specimens, and liver tissue were derived from two prospective biorepositories at the University of Washington (UW) and the affiliated Veterans Affairs Puget Sound Health Care System (VAPSHCS). These biorepositories prospectively recruited patients undergoing clinically indicated liver biopsies and stored liver tissue that was flash frozen in liquid nitrogen immediately after liver biopsy. We identified patients with NAFLD based on histologic hepatic steatosis on liver biopsy in the absence of hepatitis C virus (HCV; negative serum HCV antibody and HCV RNA), hepatitis B virus (HBV; negative serum HBV surface antigen), excessive alcohol consumption (dedicated alcohol questionnaire administered on the day of the liver biopsy), iron overload (hepatic stain and serum iron markers), or markers of autoimmune liver diseases. Liver slides were prospectively reviewed by a hepatopathologist who scored the grade of steatosis (1‐3), inflammation (0‐3), and ballooning degeneration (0‐2) and the stage of fibrosis (0‐4) according to the system proposed by Kleiner et al.19 From these two biorepositories, we randomly selected patients with NAFLD without prior knowledge of absence/presence of hepatic cholesterol crystals, who had available stored frozen liver tissue, and who either had histologic “isolated steatosis” or “fibrosing NASH” defined as follows: isolated steatosis, also known as nonalcoholic fatty liver20 (n = 14), defined by histologic steatosis grades 1‐3, inflammation grade 0‐1, fibrosis stage 0, and ballooning degeneration grade 0; fibrosing NASH (n = 16), defined by histologic steatosis grades 1‐3, inflammation grade 1‐3, fibrosis stage 1‐3, and ballooning degeneration grade 1‐2. We purposefully selected patients with NASH and fibrosis because of the recent recognition that fibrosis is the histologic feature most strongly associated with adverse long‐term outcomes in patients with NAFLD.21, 22
Of these 30 patients, 7 had been included in a prior publication that described the presence of cholesterol crystals in humans with fatty liver disease but that performed no statistical analyses.4 We compared the two groups with respect to the presence of hepatocyte cholesterol crystals within LDs and the degree of hepatocyte filipin staining for free cholesterol. Relevant clinical characteristics and laboratory tests had been prospectively collected as part of the two biorepositories.
All study participants provided informed consent. The study was approved by the Institutional Review Boards of UW and VAPSHCS.
Assessment of Hepatocyte Cholesterol Crystals
Fresh‐frozen liver tissue was embedded in optimal cutting temperature compound and sectioned at a thickness of 10 µm. Sections were allowed to come to room temperature, immediately cover slipped using pure glycerol as the mounting medium without applying any stain, and examined using a Nikon Eclipse microscope with and without a polarizing filter to evaluate for the presence of birefringent crystals typical of cholesterol crystals.4, 5, 18 Cholesterol crystallization within hepatocyte LDs was categorized semiquantitatively and independently by two investigators blinded to liver histology, as follows: 0, no crystals; 1, small birefringent crystals seen in the periphery of LDs; 2, large prominent birefringent crystals in the periphery of LDs. Additionally, ImageJ software (National Institutes of Health, Bethesda, MD) was used to calculate the percentage area that was birefringent as the average of 10 randomly selected magnification ×200 fields.
Assessment of Free Cholesterol Staining With Filipin
Frozen liver sections were stained with filipin, which identifies free cholesterol by interacting with its 3β‐hydroxy group to fluoresce blue,4, 23 and examined using a Nikon Eclipse fluorescence microscope with an excitation 340‐380 nm/emission 435‐485 nm filter in place. Filipin staining of hepatocyte LDs was categorized semiquantitatively and independently by two investigators blinded to liver histology, as follows: 0, no filipin stain; 1, faint filipin staining of some LDs; 2, intense filipin staining of most LDs. ImageJ software was used to calculate the percentage area that was filipin positive as the average of 10 randomly selected magnification ×200 fields.
Human Hepatic Gene Expression Studies
Total RNA was isolated from liver biopsies using RNeasy mini kit (Qiagen, Valencia, CA) and reverse transcribed to complementary DNA. Quantitative real‐time reverse‐transcription polymerase chain reaction was performed in triplicate using TaqMan gene expression assays, and results were expressed as relative gene expression normalized to expression levels of β‐actin as the housekeeping gene. We measured the messenger RNA gene expression of proteins critical for cholesterol synthesis, export, uptake, esterification and conversion to bile acid, proteins related to inflammation, and proteins expressed on LDs (Supporting Table S1).
Cell Culture
Cultured HepG2 cells were supplemented twice/week with LDL‐cholesterol (200 mg/dL) and oleic acid (OA; 200 µM) in order to develop large LDs, as described.18 Following this, Bodipy‐labeled cholesterol (BC) (TOPFLUOR Cholesterol‐810255; Avanti Polar Lipids, Birmingham, AL) was incorporated into LDL24 and introduced into the media of the HepG2 cells, which were studied 2, 4, 6, 24, or 72 hours later to track the uptake and intracellular trafficking of LDL‐cholesterol. BC is a fluorescent cholesterol analogue that has been shown to closely mimic the membrane partitioning and trafficking of cholesterol.24, 25
HepG2 cells were incubated with 1 mg/mL rhodamine B‐dextran (10,000 molecular weight) overnight to label endosomes.25 Additionally, HepG2 cells were incubated with the lysosome marker LysoTracker Red DND‐99 (50 nM) for 2 hours to label lysosomes. Finally, the endoplasmic reticulum (ER) was labeled using CellLight ER‐RFP, BacMam 2.0 (Thermo Fisher Scientific).
HepG2 cells were trypsinized and run on a flow cytometer to measure side scatter. We used the shift in side scatter of the flow cytometry laser beam as a measure of intracellular cholesterol crystallization, as described.26 Additionally, LDs with or without cholesterol crystals were isolated and run on flow cytometry to measure their side scatter and compare it to that induced by pure synthetic cholesterol crystals that we synthesized by cholesterol precipitation.
Results
Patient Characteristics
The majority of patients had diabetes mellitus (53%) and were obese (mean body mass index [BMI], 34 kg/m2), as expected in patients with NAFLD/NASH. There were no substantial differences between patients with NASH versus isolated steatosis in age, race/ethnicity, sex, BMI, prevalence of diabetes, or use of cholesterol‐lowering medications (Table 1). Compared to patients with isolated steatosis, those with NASH had higher mean serum alanine aminotransferase, aspartate aminotransferase, and total cholesterol and triglyceride levels and lower high‐density lipoprotein cholesterol levels, but these differences were not statistically significant (except ALT).
Table 1.
Comparison of Patients with Nash Versus Isolated Steatosis with Respect to Presence of Hepatocyte Cholesterol Crystals and Other Histologic, Clinical, and Demographic Characteristics
| Histologic Characteristics |
Simple Steatosis n = 14 |
NASH n = 16 |
|---|---|---|
| Hepatocyte cholesterol crystallization, grade,* n (%) | ||
| 0 | 11 (79%) | 1 (6%) |
| 1 | 2 (14%) | 7 (44%) |
| 2 | 1 (7%) | 8 (50%) |
|
Hepatocyte cholesterol crystallization,*
mean grade (SD) |
0.3 (0.6) | 1.4 (0.6) |
|
Cholesterol crystals area*
(% of liver area) (SD) |
0.04% (0.08) |
2.53% (3.8) |
| Hepatic filipin stain grade,* n (%) | ||
| 0 | 11 (79%) | 0 (0%) |
| 1 | 3 (21%) | 6 (38%) |
| 2 | 0 (0%) | 10 (63%) |
| Hepatic filipin stain,* mean grade (SD) | 0.2 (0.4) | 1.6 (0.5) |
|
Filipin stain area*
(% of liver area) (SD) |
0.18% (0.4) |
1.73% (2.7) |
| Hepatic histology,† n (%) | ||
| Steatosis grade | ||
| 0 | 0 (0%) | 0 (0%) |
| 1 | 9 (64%) | 3 (19%) |
| 2 | 5 (36%) | 9 (56%) |
| 3 | 0 (0%) | 4 (25%) |
| Inflammation grade* | ||
| 0 | 0 (0%) | 0 (0%) |
| 1 | 14 (100%) | 8 (50%) |
| 2 | 0 (0%) | 7 (44%) |
| 3 | 0 (0%) | 1 (6%) |
| Ballooning degeneration* | ||
| 0 | 14 (100%) | 0 (0%) |
| 1 | 0 (0%) | 8 (50%) |
| 2 | 0 (0%) | 8 (50%) |
| Fibrosis stage* | ||
| 0 | 14 (100%) | 0 (0%) |
| 1 | 0 (0%) | 10 (63%) |
| 2 | 0 (0%) | 4 (25%) |
| 3 | 0 (0%) | 2 (13%) |
| 4 | 0 (0%) | 0 (0%) |
| Clinical and Demographic Characteristics |
Simple Steatosis n = 14 |
NASH n = 16 |
|---|---|---|
| Age, mean (SD) | 55.4 (10.5) | 53.3 (9.7) |
| Male, n (%) | 10 (71%) | 9 (56%) |
| Race/ethnicity | ||
| Non‐Hispanic white | 13 (93%) | 13 (81%) |
| Non‐Hispanic black | 0 (0%) | 1 (6%) |
| Hispanic | 1 (7%) | 0 (0%) |
| Other | 0 (0%) | 2 (13%) |
| BMI kg/m2 | 34.1 (5.5) | 33.9 (4.8) |
| Diabetes, n (%) | 7 (50%) | 10 (62.5%) |
| Fasting blood levels, mean (SD) | ||
| ALT* (U/L) | 46.9 (28.8) | 76.7 (46.9) |
| AST (U/L) | 33.6 (16.7) | 54.1 (27.4) |
| Total cholesterol | 178 (35.2) | 198.1 (57.1) |
| LDL‐cholesterol | 105.9 (24.7) | 107.6 (41.9) |
| HDL‐cholesterol | 46.6 (22.1) | 34.3 (4.8) |
| Triglyceride | 220.4 (184.7) | 273.3 (149.9) |
| Platelet count | 170.8 (59.6) | 193.6 (76.7) |
| Cholesterol‐lowering medications, n (%) | ||
| Statin | 4 (29%) | 5 (31%) |
| Ezetimibe | 0 (0%) | 1 (6%) |
Characteristics that are significantly (P < 0.05) different between NASH and isolated steatosis.
Scored according to the system proposed by Kleiner et al.19
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; HDL, high‐density lipoprotein.
Patients With NASH Exhibited Excessive Free‐Cholesterol Staining and Cholesterol Crystals Within Hepatocyte LDs, But Patients With Isolated Steatosis Did Not
Patients with NASH had typical histologic features of fibrosing steatohepatitis with hepatic fibrosis and ballooning hepatocytes, while patients with isolated steatosis had no fibrosis or ballooning hepatocytes (Fig. 1; Table 1).
Figure 1.
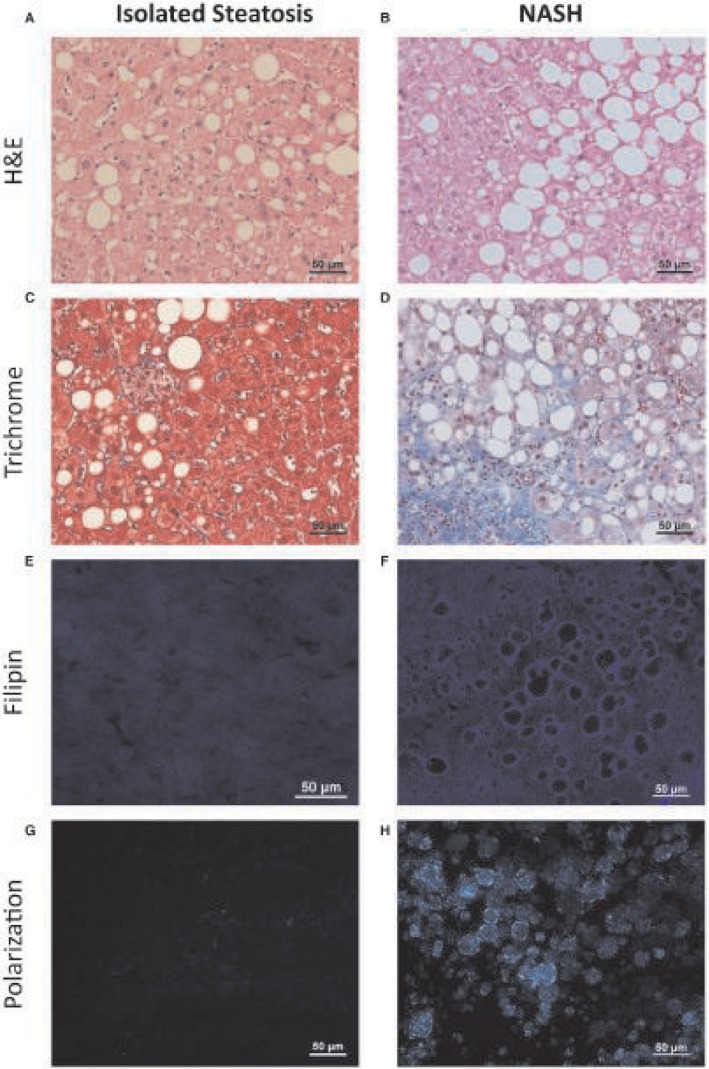
Representative histologic sections from patients with isolated steatosis and patients with NASH. Large LDs were present in (A) patients with isolated steatosis (left column) and (B) patients with NASH (right column). (C,D) Patients with NASH had ballooning hepatocytes and hepatic fibrosis, while patients with isolated steatosis did not. (E‐H) Patients with isolated steatosis had no filipin staining for free cholesterol and no birefringent cholesterol crystals under polarized light within hepatocyte LDs in frozen liver sections. In contrast, the LDs of patients with NASH, exhibited positive filipin staining and birefringent cholesterol crystals. Scale bar, 50 μm. Abbreviation: H&E, hematoxylin and eosin.
Almost all patients with fibrosing NASH had cholesterol crystals under polarized light (15/16; Fig. 1H) and intense or mild free‐cholesterol staining by filipin (16/16; Fig. 1F) within hepatocyte LDs compared to only 3/14 with cholesterol crystals and 3/14 with faint filipin staining in patients with isolated steatosis (P < 0.05). Quantitative assessment of the percentage area of the liver sections taken up by cholesterol crystals or filipin staining confirmed the significant differences between patients with NASH and those with isolated steatosis (Table 1).
The degree of free‐cholesterol staining and cholesterol crystallization of hepatocyte LDs was graded semiquantitatively, with representative pictures shown in Fig. 2. Free‐cholesterol staining by filipin (Fig. 2E) corresponded to the birefringent cholesterol crystals seen under polarized light (Fig. 2F) and outlined the membrane of the LDs.
Figure 2.
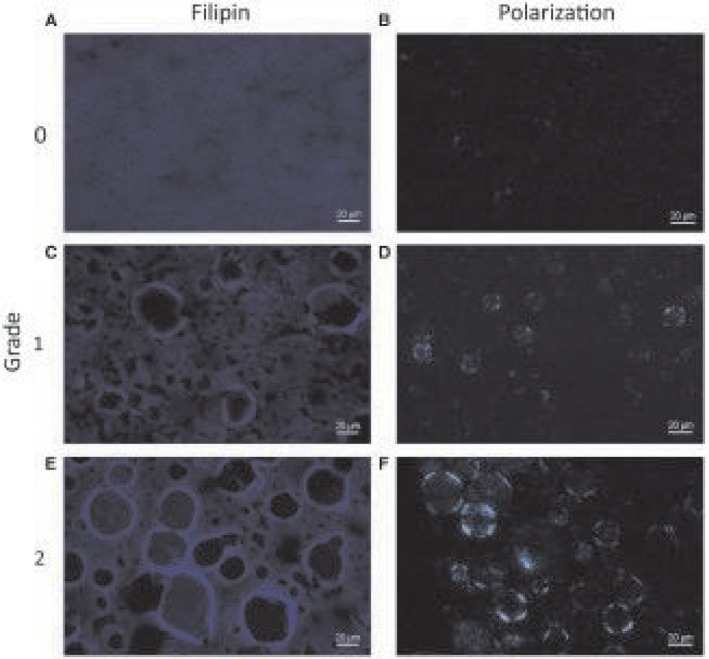
Staining for free cholesterol and birefringent cholesterol crystals. The extent of filipin staining for free cholesterol and birefringent cholesterol crystals under polarized light in human liver biopsies was graded semiquantitatively into (A,B) grade 0 (none), (C,D) grade 1 (faint), and (E,F) grade 2 (intense). Quantitative assessment of the mean percentage area of each liver section staining positive confirmed differences between grade 0 (0.09% for filipin and 0.02% for cholesterol crystals), grade 1 (1.06% for filipin and 0.15% for cholesterol crystals), and grade 2 (2.25% for filipin and 4.18% for cholesterol crystals). Scale bar, 20 μm.
Hepatic gene expression studies, performed in 26 of 30 patients in whom we isolated hepatic RNA, did not reveal any statistically significant differences in the expression of genes related to cholesterol homeostasis, inflammation, or LD proteins between patients with fibrosing NASH and isolated steatosis or between patients with or without cholesterol crystals (Supporting Table S1).
HepG2 Cells Exposed to OA and LDL‐Cholesterol Develop Large LDs With Cholesterol Crystals
HepG2 cells exposed to LDL‐cholesterol and OA for only 3 days developed small LDs without cholesterol crystals (Fig. 3A,C). However, when exposed to LDL‐cholesterol and OA for at least 20 days, they developed large LDs with birefringent cholesterol crystals (Fig. 3B,D), similar in appearance to the ones we noted in humans in vivo. HepG2 cells exposed to both LDL‐cholesterol and OA accumulated greater concentrations of free cholesterol than cells exposed to LDL‐cholesterol alone, OA alone, or control media (Fig. 4A). Only cells exposed to both LDL‐cholesterol and OA developed birefringent cholesterol crystals.
Figure 3.
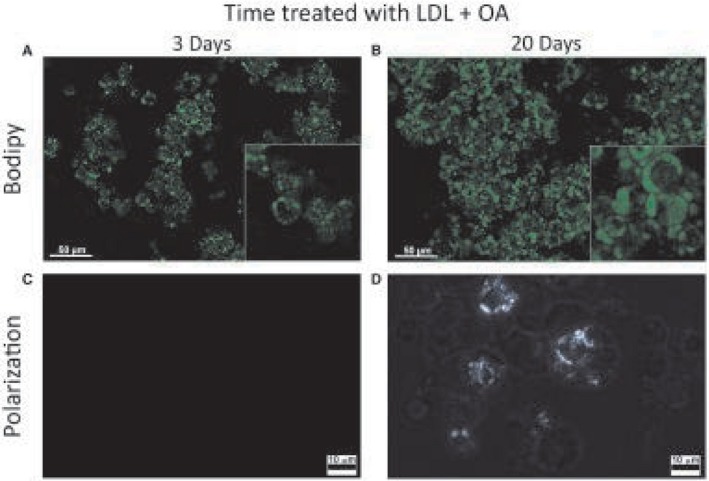
LD development. (A) HepG2 cells exposed to LDL + OA for 3 days only develop small LDs without cholesterol crystals. (B) When exposed to LDL + OA for 20 days, they develop large LDs with cholesterol crystals. (A,B) Scale bar, 50 μm. (C,D) LDL + OA for 3 days and 20 days, respectively, under polarized light. Scale bar, 10 μm.
Figure 4.
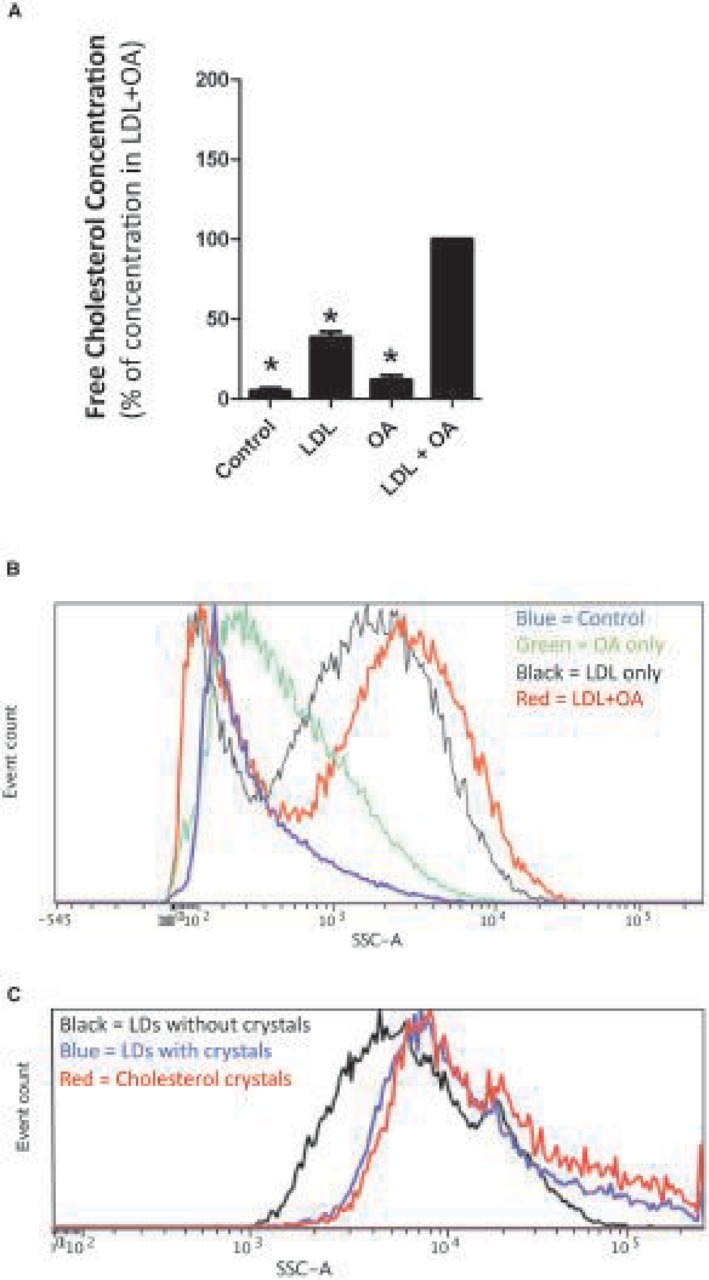
HepG2 cells exposed to control media, OA only, LDL‐cholesterol only (LDL), or both LDL + OA for 20 days. (A) HepG2 cells exposed to LDL + OA accumulate a greater concentration of free cholesterol than cells exposed to control media or LDL alone or OA alone. Data represent mean ± SD; *P ≤ 0.05. (B) Exposure to HepG2 cells to OA + LDL (red line) results in a substantial increase in side scatter (plotted on the x axis) of the flow cytometry laser beam. The side scatter of the laser beam is increased by cholesterol crystals within the LDs of HepG2 cells. (C) Isolated LDs with cholesterol crystals (blue line) produce almost identical side scatter of the flow cytometry laser beam as synthetic cholesterol crystals (red line), while lipid droplets without cholesterol crystals (black line) do not. This provides further evidence of the presence of cholesterol crystals within the LDs. Abbreviation: SSC‐A, side scatter.
HepG2 cells exposed to both OA and LDL‐cholesterol caused a substantial increase in the side scatter of the flow cytometry laser beam (Fig. 4B), suggesting that they contained cholesterol crystals. In order to further demonstrate that the side scatter of the laser beam was caused by cholesterol crystals within the LDs of HepG2 cells, we isolated LDs from cells with and without cholesterol crystals. Isolated LDs with cholesterol crystals produced almost identical side scatter of the flow cytometry laser beam as pure synthetic cholesterol crystals, while LDs without cholesterol crystals did not (Fig. 4C). This provides further evidence of the presence of cholesterol crystals within the LDs.
LDL‐Cholesterol Taken Up By HepG2 Cells is Transported through the Endosomal‐Lysosomal Compartment and Stored Directly in the LDs
HepG2 cells were either pretreated for 2 weeks with LDL + OA to develop large LDs and then exposed to LDL with BC or were directly exposed to LDL with BC without pretreatment. The cells were imaged 2, 4, 6, 24, or 72 hours after exposure to LDL with BC. BC begins to be clearly evident within HepG2 cells at 6 hours and continues to accumulate at 24 and 72 hours. BC accumulated almost exclusively within the LDs (Supporting Fig. S1). The pretreated cells had much larger initial LDs in which the BC was incorporated; otherwise the process was similar in the pretreated and non‐pretreated cells.
In colabeling studies, we found extensive colocalization of BC with lysosomes or BC with endosomes (Fig. 5). This demonstrates that the LDL‐cholesterol taken up by receptor‐mediated endocytosis passes through the endosomal‐lysosomal compartment before accumulating in the LDs, as expected. However, we did not find any significant colocalization of BC with the ER compartment, demonstrating that LDL cholesterol does not pass through the ER before accumulating in the LDs. This suggests that direct membrane contact sites between the endosomal‐lysosomal compartment and the LDs facilitate cholesterol transport between the two organelles.
Figure 5.
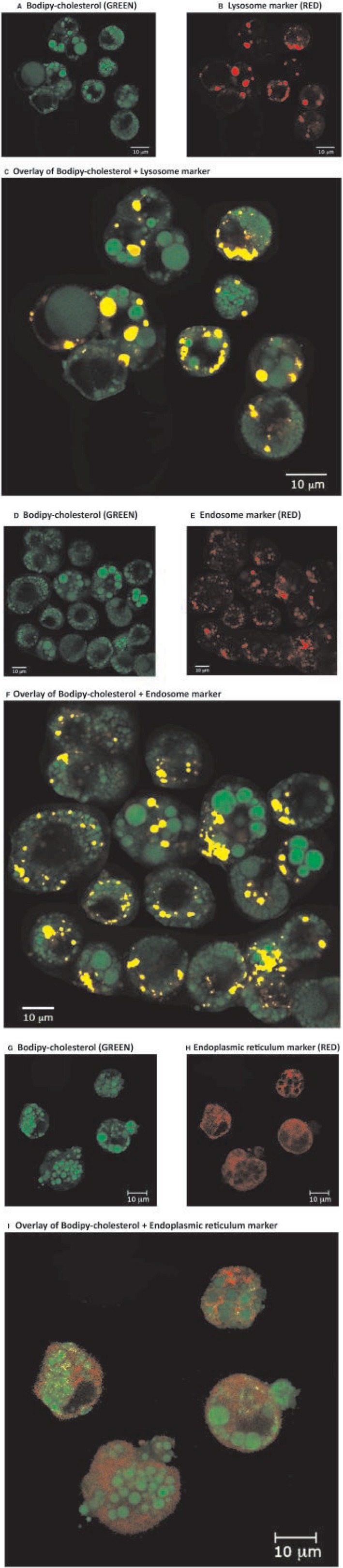
Confocal microscopy of trypsinized HepG2 cells. HepG2 cells were pretreated for 3 weeks with LDL + OA in order to develop large LDs and then exposed to LDL with BC for 1 day (labeled GREEN) to track the uptake and intracellular trafficking of cholesterol. The cells are also labeled with either (A‐C) a lysosome marker (RED, lysosome tracker), (D‐F) an endosome marker (RED, rhodamine dextran), or (G‐I) an ER marker (RED, CellLight ER‐RFP). When RED and GREEN stains are overlaid (i.e., localized in the same compartment), they appear YELLOW. BC (GREEN) accumulates in the LDs. Overlays of different markers show that most of the endosomes and lysosomes contain cholesterol (i.e., they costain with BC and appear YELLOW [C,F]). However, the ER only stains RED and shows little or no costain with BC (i.e., there is very little YELLOW costain [I]). This suggests that LDL‐cholesterol taken up by receptor‐mediated endocytosis goes directly from the endosomal‐lysosomal compartment to the LDs without passing through the ER. Abbreviation: RFP, red fluorescent protein. Scale bar, 10 μm.
Taken together, our results suggest that cholesterol taken up by hepatocytes in the form of LDL is transported through the endosomal‐lysosomal compartment directly onto the LD membrane, where it may crystallize if it exceeds the concentration that can be “solubilized” by the phospholipids in the membrane (Fig. 6).
Figure 6.
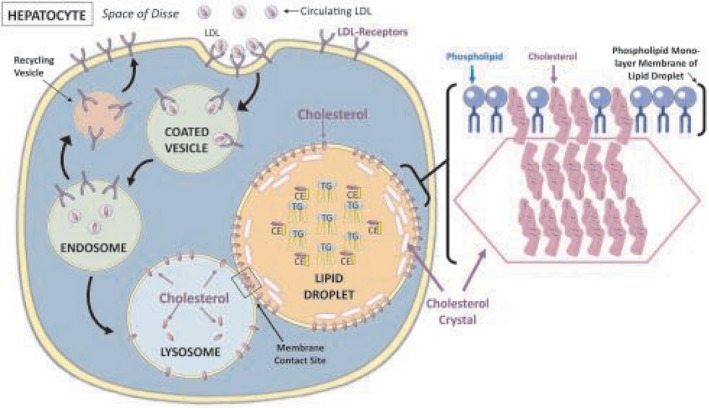
Schematic representation of the cholesterol crystallization process. Circulating LDL‐cholesterol is taken up by hepatocytes by receptor‐mediated endocytosis. The receptor is internalized in clathrin‐coated vesicles. Within the endosomal‐lysosomal compartment, LDL particles are released and cholesterol ester is hydrolyzed to free cholesterol by lysosomal acid lipase and transferred sequentially from NPC2 to NPC1 and onto the membrane of the lysosome. Our results suggest that direct MCSs between lysosomes and LDs facilitate the transport of free cholesterol from the lysosomal membrane to the LD membrane. As the cholesterol concentration in the LD membrane increases, it eventually exceeds the ability of phospholipid headgroups to cover all the cholesterol molecules and excess molecules precipitate adjacent to the membrane, forming cholesterol monohydrate crystals. Abbreviations: CE, cholesterol ester; NPC2/1, Niemann‐Pick disease, type C, intracellular cholesterol transporter 2/1; TG, triglyceride.
Discussion
We demonstrated a very strong association between the presence of cholesterol crystals within hepatocyte LDs and the development of fibrosing NASH in humans. Almost all patients with fibrosing NASH in our study had evidence of cholesterol crystallization in the periphery of hepatocyte LDs in association with the LD membrane. We also showed in vitro that LDL‐cholesterol taken up by receptor‐mediated endocytosis is transported directly from the endosomal‐lysosomal compartment into the LDs of hepatocytes and accumulates almost exclusively within LDs where it crystallizes (Fig. 6). This suggests the presence of direct membrane contact sites between endosomes/lysosomes and LDs that facilitate cholesterol transfer. Collectively, our results suggest that excess hepatic cholesterol preferentially accumulates in hepatocyte LDs, where it may crystallize if it exceeds a threshold concentration, and promotes the development of NASH.
Despite many decades of research, it remains unclear what the “alcohol” equivalent of “nonalcoholic” steatohepatitis is, that is, the primary factor or factors that causatively drive the progressive inflammation and fibrosis of NASH. Our results suggest that crystallization of cholesterol in hepatocyte LDs may be such a factor. Although our study only showed that cholesterol crystals are present in NASH and did not identify specific pathogenetic mechanisms, cholesterol crystals are known to induce cellular toxicity and to activate many proinflammatory pathways, such as the nucleotide‐binding oligomerization domain, leucine rich repeat, and pyrin domain containing 3 (NLRP3) inflammasome,27, 28 that are thought to play a role in NASH.29, 30, 31, 32 In addition, the high concentration and eventual crystallization of cholesterol on the LD membrane should be expected to increase the rigidity of the LD membrane and disrupt the function of proteins residing on the LD membrane, hundreds of which have been described.33, 34 Such effects of membrane cholesterol concentration have been well described in the mitochondrial membrane (where cholesterol affects the function of the 2‐oxoglutarate carrier35) and on the ER membrane (where cholesterol affects the function of sarco(endo)plasmic reticulum Ca2+–adenosine triphosphatase).36, 37 Future work is needed to identify how the cholesterol crystallization in the LD membrane that we described influences or disrupts the function of specific proteins in the LD membrane and of the entire organelle.
It is frequently quoted that adipose tissue is the main site of cholesterol storage in humans.38 However, this is unlikely to be true because adipocyte LDs contain almost no cholesterol esters, the storage form of cholesterol39, 40, 41; instead, they are composed almost entirely of triglycerides. Adipocytes contain small amounts of free cholesterol in association with the membrane of their LD as well as their plasma membrane and other cellular membranes. In contrast, the LDs of hepatocytes can contain very large amounts of cholesterol esters18, 42 (as well as triglycerides). Free cholesterol that enters the hepatocyte LD through its membrane can be esterified and stored in the LD. In previous work, we showed that when mice are fed a diet progressively high in cholesterol, the excess cholesterol accumulates in the liver not in adipose tissue.18 Here, we demonstrate that cholesterol taken up by hepatocytes accumulates in their enlarged LDs. Collectively, these results suggest that the hepatocyte LD can be considered the main storage site of excess body cholesterol.
It is unclear what factors caused increased hepatocyte cholesterol concentration and hence cholesterol crystallization in the patients with NASH. Hepatocyte cholesterol concentration is the sum of the pathways by which hepatocytes acquire cholesterol (endogenous synthesis and uptake of cholesterol‐containing circulating lipoproteins) minus the pathways by which cholesterol is removed from hepatocytes (excretion into bile, conversion to bile acids and excretion into bile, secretion into circulating lipoproteins).17 In addition, esterification of free cholesterol into hydrophobic cholesterol esters stored within the LDs reduces free cholesterol concentration without eliminating the cholesterol from the cell. Extensive dysregulation of hepatic cholesterol homeostasis has been documented in NAFLD, leading to increased hepatic cholesterol levels.43, 44 This dysregulation likely occurs at multiple levels, including increased hydrolysis of cholesterol ester to free cholesterol by hepatic neutral cholesterol ester hydrolase44; increased hepatic cholesterol synthesis related to increased expression and activity (dephosphorylation) of 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase44, 45; increased hepatic levels of active sterol regulatory element‐binding protein 246; increased uptake of cholesterol‐rich lipoproteins44; and decreased cholesterol excretion in bile either as cholesterol or as bile acids.43, 44 A critical limitation is that our gene expression studies failed to identify any key pathways that might be responsible for the cholesterol loading and eventual crystallization in the hepatocytes of patients with NASH. This may be related to the small sample size and intrahepatic variability in gene expression exacerbated by the relatively small pieces of frozen liver tissue available for gene expression studies. Future studies will need to evaluate which ones of the many pathways involved in cholesterol synthesis, uptake, export, and intracellular trafficking are primarily responsible for cholesterol crystallization in patients with NASH.17 Irrespective of what precise mechanisms lead to hepatic cholesterol loading and crystallization, it is likely that currently available pharmacological cholesterol deloading strategies, such as statins, would be effective at reducing hepatic cholesterol concentration and reversing cholesterol crystallization, as we have shown in animal models.5 Although large, well‐designed, observational studies suggested great benefits of statins in ameliorating histologic NASH,15 randomized controlled trials will ultimately be necessary to prove this. Unfortunately, the design of such trials is hampered by the ethics of randomizing patients with NASH to placebo, who most likely will have indications for a statin based on their plasma cholesterol levels.
Great progress has been achieved lately in understanding intracellular cholesterol trafficking, especially through membrane contact sites (MCSs) between organelles that allow cholesterol to transfer from the membrane of one organelle to that of the other. MCSs are facilitated by a protein “tether” (e.g., Rab18 and seipin; different Rab species in the LD regulate interactions with different membrane systems) that connects the membranes of the two interacting organelles and/or by direct lipid bridges formed by continuities of the LD phospholipid monolayer and the outer leaflet of the phospholipid bilayer bordering the partnering organelle.47 LDs have been shown to express MCSs with multiple other organelles, including the ER, mitochondria, and peroxisomes. MCSs have also been described between endosomes and LDs facilitated by Rab5.48 In late endosomes/lysosomes (LE/Ly), CE are hydrolyzed by acid lipase and free cholesterol is delivered by Niemann‐Pick disease, type C, intracellular cholesterol transporter 2 (NPC2) to NPC1, which binds and inserts cholesterol onto the LE/Ly membrane for transport.49 It is likely that cholesterol is transferred from the membrane of the LE/Ly to the membrane of the LD through such direct MCSs between the two organelles.
It is useful to consider why cholesterol crystallizes in the LD membrane rather than in another organellular membrane or subcellular compartment. Our in vitro cell culture model demonstrates that cholesterol taken up by hepatocytes accumulates almost exclusively in the LD, providing an explanation for the high concentration of cholesterol in LDs. Cholesterol is transferred to the LD membrane from the membranes of other organelles by direct MCSs. Although free cholesterol transferred to the LD membrane can be esterified subsequently to cholesterol ester for storage, a high free‐cholesterol concentration might be reached at the LD membrane during this process. Cholesterol is solubilized by the phospholipids in the membrane; these phospholipids have large polar headgroups that act like umbrellas to shield the nonpolar part of cholesterol molecules from water. As the cholesterol concentration in the membrane increases, it eventually exceeds the ability of the phospholipid headgroups to cover all the cholesterol molecules, and excess molecules would be predicted to precipitate adjacent to the membrane, forming cholesterol monohydrate crystals.50 Although theoretical and artificial membrane models predict such cholesterol crystal precipitation, to our knowledge, we describe the first such demonstration of cholesterol crystallization adjacent to phospholipid membranes. Furthermore, the LD membrane is the only cellular membrane that consists of a phospholipid monolayer rather than a bilayer. It is tempting to speculate that cholesterol in the phospholipid monolayer may be more likely to crystallize than in the bilayer.
In summary, we identified a strong association between the presence of cholesterol crystals in hepatocyte LDs and the development of human NASH. Hepatocyte LDs are major sites for the storage of excess body cholesterol. Although most of this cholesterol becomes esterified, free cholesterol can crystallize out of the LD membrane if present in excess. These findings suggest that some cholesterol‐lowering medications may have therapeutic implications for NASH that have not yet been fully explored.
Supporting information
Supported by the U.S. Department of Veterans Affairs, Biomedical and Laboratory Research and Development (I01 BX002910 to G.N.I. and C.S.) and the Diabetes Research Center, University of Washington (P30 DK017047 to C.S.L.).
The content does not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Potential conflict of interest: Dr. Landis received grants from Gilead, Conatus, and Genfit. The other authors have nothing to report.
References
- 1. Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013;57:81‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Subramanian S, Goodspeed L, Wang SA, Kim J, Zeng L, Ioannou GN, et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor‐deficient mice. J Lipid Res 2011;52:1626‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van Rooyen DM, Larter CZ, Haigh WG, Yeh MM, Ioannou G, Kuver R, et al. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011;141:1393‐1403, 1403.e1‐e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown‐like structures distinguish NASH from simple steatosis. J Lipid Res 2013;54:1326‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ioannou GN, Van Rooyen DM, Savard C, Haigh WG, Yeh MM, Teoh NC, et al. Cholesterol‐lowering drugs cause dissolution of cholesterol crystals and disperse Kupffer cell crown‐like structures during resolution of NASH. J Lipid Res 2015;56:277‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng S, Hoos L, Cook J, Tetzloff G, Davis H Jr, van Heek M, et al. Ezetimibe improves high fat and cholesterol diet‐induced non‐alcoholic fatty liver disease in mice. Eur J Pharmacol 2008;584:118‐124. [DOI] [PubMed] [Google Scholar]
- 7. Wouters K, van Bilsen M, van Gorp PJ, Bieghs V, Lütjohann D, Kerksiek A, et al. Intrahepatic cholesterol influences progression, inhibition and reversal of non‐alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett 2010;584:1001‐1005. [DOI] [PubMed] [Google Scholar]
- 8. Yoneda M, Fujita K, Nozaki Y, Endo H, Takahashi H, Hosono K, et al. Efficacy of ezetimibe for the treatment of non‐alcoholic steatohepatitis: an open‐label, pilot study. Hepatol Res 2010;40:566‐573. [DOI] [PubMed] [Google Scholar]
- 9. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081‐1090. [DOI] [PubMed] [Google Scholar]
- 10. Ioannou GN, Morrow OB, Connole ML, Lee SP, et al. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 2009;50:175‐184. [DOI] [PubMed] [Google Scholar]
- 11. Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, et al.; GREACE Study Collaborative Group . Safety and efficacy of long‐term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post‐hoc analysis. Lancet 2010;376:1916‐1922. [DOI] [PubMed] [Google Scholar]
- 12. Athyros VG, Mikhailidis DP, Didangelos TP, Giouleme OI, Liberopoulos EN, Karagiannis A, et al. Effect of multifactorial treatment on non‐alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin 2006;22:873‐883. [DOI] [PubMed] [Google Scholar]
- 13. Foster T, Budoff MJ, Saab S, Ahmadi N, Gordon C, Guerci AD. Atorvastatin and antioxidants for the treatment of nonalcoholic fatty liver disease: the St Francis Heart Study randomized clinical trial. Am J Gastroenterol 2011;106:71‐77. [DOI] [PubMed] [Google Scholar]
- 14. Nelson A, Torres DM, Morgan AE, Fincke C, Harrison SA. A pilot study using simvastatin in the treatment of nonalcoholic steatohepatitis: a randomized placebo‐controlled trial. J Clin Gastroenterol 2009;43:990‐994. [DOI] [PubMed] [Google Scholar]
- 15. Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, et al. Statin use and non‐alcoholic steatohepatitis in at risk individuals. J Hepatol 2015;63:705‐712. [DOI] [PubMed] [Google Scholar]
- 16. Kargiotis K, Athyros VG, Giouleme O, Katsiki N, Katsiki E, Anagnostis P, et al. Resolution of non‐alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World J Gastroenterol 2015;21:7860‐7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab 2016;27:84‐95. [DOI] [PubMed] [Google Scholar]
- 18. Ioannou GN, Subramanian S, Chait A, Haigh WG, Yeh MM, Farrell GC, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. J Lipid Res 2017;58:1067‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 20. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 21. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stål P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 23. Rudolf M, Curcio CA. Esterified cholesterol is highly localized to Bruch’s membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J Histochem Cytochem 2009;57:731‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanerva K, Uronen RL, Blom T, Li S, Bittman R, Lappalainen P, et al. LDL cholesterol recycles to the plasma membrane via a Rab8a‐Myosin5b‐actin‐dependent membrane transport route. Dev Cell 2013;27:249‐262. [DOI] [PubMed] [Google Scholar]
- 25. Holtta‐Vuori M, Sezgin E, Eggeling C, Ikonen E. Use of BODIPY‐cholesterol (TF‐Chol) for visualizing lysosomal cholesterol accumulation. Traffic 2016;17:1054‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, et al. Cholesterol crystals induce complement‐dependent inflammasome activation and cytokine release. J Immunol 2014;192:2837‐2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rajamaki K, Lappalainen J, Oorni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase‐1‐mediated regulation of fibrogenesis in diet‐induced steatohepatitis. Lab Invest 2012;92:713‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dixon LJ, Flask CA, Papouchado BG, Feldstein AE, Nagy LE. Caspase‐1 as a central regulator of high fat diet‐induced non‐alcoholic steatohepatitis. PLoS One 2013;8:e56100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014;59:898‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szabo G, Iracheta‐Vellve A. Inflammasome activation in the liver: focus on alcoholic and non‐alcoholic steatohepatitis. Clin Res Hepatol Gastroenterol 2015;39(Suppl. 1):S18‐S23. [DOI] [PubMed] [Google Scholar]
- 33. Hodges BD, Wu CC. Proteomic insights into an expanded cellular role for cytoplasmic lipid droplets. J Lipid Res 2010;51:262‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su W, Wang Y, Jia X, Wu W, Li L, Tian X, et al. Comparative proteomic study reveals 17beta‐HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2014;111:11437‐11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF‐ and Fas‐mediated steatohepatitis. Cell Metab 2006;4:185‐198. [DOI] [PubMed] [Google Scholar]
- 36. Li L, Hossain MA, Sadat S, Hager L, Liu L, Tam L, et al. Lecithin cholesterol acyltransferase null mice are protected from diet‐induced obesity and insulin resistance in a gender‐specific manner through multiple pathways. J Biol Chem 2011;286:17809‐17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hager L, Li L, Pun H, Liu L, Hossain MA, Maguire GF, et al. Lecithin:cholesterol acyltransferase deficiency protects against cholesterol‐induced hepatic endoplasmic reticulum stress in mice. J Biol Chem 2012;287:20755‐20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krause BR, Hartman AD. Adipose tissue and cholesterol metabolism. J Lipid Res 1984;25:97‐110. [PubMed] [Google Scholar]
- 39. Chung S, Cuffe H, Marshall SM, McDaniel AL, Ha JH, Kavanagh K, et al. Dietary cholesterol promotes adipocyte hypertrophy and adipose tissue inflammation in visceral, but not in subcutaneous, fat in monkeys. Arterioscler Thromb Vasc Biol 2014;34:1880‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wiggers KD, Richard MJ, Stewart JW, Jacobson NL, Berger PJ. Type and amount of dietary fat affect relative concentration of cholesterol in blood and other tissues of rats. Atherosclerosis 1977;27:27‐34. [DOI] [PubMed] [Google Scholar]
- 41. Schreibman PH, Dell RB. Human adipocyte cholesterol. Concentration, localization, synthesis, and turnover. J Clin Invest 1975;55:986‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, et al. Lipid‐induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007;46:1392‐1403. [DOI] [PubMed] [Google Scholar]
- 43. Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non‐alcoholic steatohepatitis. Prog Lipid Res 2013;52:175‐191. [DOI] [PubMed] [Google Scholar]
- 44. Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 2012;15:665‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simonen P, Kotronen A, Hallikainen M, Sevastianova K, Makkonen J, Hakkarainen A, et al. Cholesterol synthesis is increased and absorption decreased in non‐alcoholic fatty liver disease independent of obesity. J Hepatol 2011;54:153‐159. [DOI] [PubMed] [Google Scholar]
- 46. Caballero F, Fernandez A, De Lacy AM, Fernández‐Checa JC, Caballería J, García‐Ruiz C. Enhanced free cholesterol, SREBP‐2 and StAR expression in human NASH. J Hepatol 2009;50:789‐796. [DOI] [PubMed] [Google Scholar]
- 47. Schuldiner M, Bohnert M. A different kind of love ‐ lipid droplet contact sites. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:1188‐1196. [DOI] [PubMed] [Google Scholar]
- 48. Liu P, Bartz R, Zehmer JK, Ying YS, Zhu M, Serrero G, et al. Rab‐regulated interaction of early endosomes with lipid droplets. Biochim Biophys Acta 2007;1773:784‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Du X, Brown AJ, Yang H. Novel mechanisms of intracellular cholesterol transport: oxysterol‐binding proteins and membrane contact sites. Curr Opin Cell Biol 2015;35:37‐42. [DOI] [PubMed] [Google Scholar]
- 50. Huang J, Feigenson GW. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys J 1999;76:2142‐2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials