

Abstract
Epigenetic modifications play a key role in cell development and tumorigenesis. Recent large-scale genomic studies have shown that mutations in the epigenetic machinery and concomitant perturbation of epigenomic patterning are frequent events in tumors. Among all the epigenetic marks, DNA methylation is one of the most studied. Hyper/hypo-methylations on specific regulatory elements are sometimes thought to be correlated with expression of nearby genes. High-throughput bisulfite converted sequencing is currently the technology of choice for studying DNA methylation in base-pair resolution and whole-genome scale. Such broad and high-resolution coverage of the epigenome provide unprecedented opportunities to analyze DNA methylation patterns correlated with tumorigenesis and tumor evolution and progression. However, few pipelines are available to the public to perform systematically DNA methylation analysis. Here, utilizing open source tools, we describe a comprehensive computational methodology to thoroughly analyze DNA methylation patterns during tumor evolution based on bisulfite converted sequencing data, including intra-tumor methylation heterogeneity.
Keywords: DNA methylation, ERRBS, DMRs, Intra-tumor methylation heterogeneity
1. Introduction
Epigenetic modification plays a key role in the regulation of all DNA-based processes including transcription, DNA repair and replication, which are fundamental to tumorigenesis[1]. Recent large-scale genomic studies have shown that mutations in the epigenetic machinery and concomitant perturbation of epigenomic patterning are frequent events in tumors, such as B-cell lymphomas[2]. Among all known epigenetic modifications, DNA methylation is one of the most studied epigenetic markers. DNA methylation consists of the addition of a methyl group to carbon 5 of cytosine within the dinucleotide CpGs. Hyper- or hypo-methylation of genomic regions (promoters or enhancers for example) can lead to repression or activation of nearby gene expression. Several examples of promoter methylation levels inversely correlated with gene expression levels have been identified[3], frequently involving tumor suppressor genes. Thus, identifying differentially methylated cytosines (DMCs) and differentially methylated regions (DMRs), especially for those perturbed in tumors, has become a central point in cancer methylome analysis.
The advent of high-throughput DNA sequencing technologies has provided new opportunities to study DNA methylation, allowing for fast, single base resolution scans in targeted or enriched regions or at the whole-genome scale. Large-scale sequencing projects have generated hundreds of methylation profiles of tumors of different origins and at different stages. In turn, markers based on methylation usually provide important information about cellular phenotypes in normal and disease tissues and in many cases enable improved patient stratification over other approaches based on transcriptomics or mutation profiles. Enhanced reduced representation bisulfite sequencing (ERRBS) is one of such powerful high-throughput sequencing platforms, which can provide high sequencing depth and coverage of millions of CpGs in the human genome [4]. To date, few bisulfite sequencing analysis pipelines exist, even though such data are increasingly widely used. Here, we describe a comprehensive DNA methylation profiling analysis based on ERRBS data. This method is also applicable to reduced representation bisulfite sequencing (RRBS) data or whole-genome bisulfite sequencing (WGBS) data[5]. We focus our analysis on DNA methylation patterns during tumor evolution.
Following generation of an ERRBS data set, a typical analysis workflow consists of first identifying DMCs and DMRs, then correlating those regions to biological relevant genes and pathways. Other analyses relevant to tumor evolution may include quantifying intra-tumor methylation heterogeneity (MH). Indeed, comparing to the genetic code, DNA methylation is more flexible and cells within a cell population may have distinct methylation patterns. Thus a tumor cell population may harbor varying levels intra-tumor MH. Such heterogeneity is emerging as a powerful predictor of tumor evolution, progression and relapse[6]. The analysis of DNA methylation patterns during tumor evolution requires ERRBS samples from different tumor development stages, or at least diagnosis-relapse sample pairs from several patients. Also, ERRBS samples from normal tissues to be used as baseline can further enhance the analysis. The computational analysis scheme is outlined in Figure 1. Sample preparation and high-through sequencing are described there [4]. Sequencing reads from every stage during tumor evolution are mapped independently to a bisulfite converted reference genome, generating separate DNA methylation profile. DNA methylation status for every single CpG is determined in each sample, separately. Next, DMCs and DMRs calling is performed between the normal sample and each tumor stage or between any two tumor stages. Intra-tumor MH analysis is performed as a separate analysis on the same samples. Finally, identified DMCs/DMRs and MH hotspots are analyzed for enriched gene functions, in order to unravel pathways relevant to tumor evolution.
Figure 1.

Schematic of ERRBS data analysis pipelines.
2. Software
ERRBS data quality control: FastQC (available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)[7].
Adaptive quality and adapter trimming: Trim Galore (available at http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)[8].
Bisulfite converted sequence reads mapping and cytosine methylation states calling: Bismark (available at http://www.bioinformatics.babraham.ac.uk/projects/bismark/)[9].
DMCs and DMRs calling: RRBSseeqer (available at http://icb.med.cornell.edu/wiki/index.php/Elementolab/)[6].
Regions annotations: ChIPseeqer (available at http://icb.med.cornell.edu/wiki/index.php/Elementolab/)[10].
Pathway analysis: iPAGE (available at http://icb.med.cornell.edu/wiki/index.php/Elementolab/)[11].
ERRBS data analyzing tools: Methylation heterogeneity analysis: errbs-tools including methylCall_from_Bismark.py, regionEpi.R and regionMH.R (available at https://github.com/SpursHeng90/errbs-tools/)[6].
3. Methods
1. Quality control for ERRBS reads
Similar to other high-throughput sequencing technologies, ERRBS is prone to systematic errors and artifacts such as PCR duplicates, GC-content shift and adaptor contamination. In addition to these common problems, ERRBS data can suffer more critical problems such as erroneous methylation status of cytosine introduced by end-repair and low bisulfite conversion rates. Thus, it is always a good idea to perform some simply quality control analysis to avoid any biases that may affect subsequent analyses. ERRBS generate output in FASTQ format, like most high throughput sequencing assays. We have successfully used a publicly available tool--FastQC--to address the quality control problem (Figure 1). Details about how to interpret the FastQC results are available in S. Andrews paper[7]. In general, per base sequence quality should be higher than cutoff (20 in most cases). Also, average per sequence quality should be around 38 (Phred+33, 0–41 scale). The other important feature is there is no overrepresented sequences or adapter contents, which can be potential adapter contamination sources. Ideally, your data set should pass most quality control steps to confirm the data quality for downstream analysis.
General syntax:
$ fastqc [-o output dir] [-f fastq|bam|sam] seqfiles1 … seqfilesN
Example:
$ fastqc -o . -f fastq CLL_S101.RRBS.fq
2. Adapter trimming
Suboptimal adapter contamination may affect read mapping, leading to low mapping efficiencies and even result in incorrect mapping and/or unreliable methylation calling in ERRBS. Even worse, positions filled in during end-repair can introduce artifact methylation status in ERRBS. Also, due to the short size selected fragment size of the restriction endonuclease MspI, ERRBS libraries with longer size have more severe problems from all above sources. To address this problem, we used read trimmers such as trim_galore (Figure 1).
General syntax:
$ trim_galore [options] <filenames>
Example:
$ trim_galore --rrbs –adapter
TGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGC --output_dir . CLL_S101.RRBS.fq
where:
--a/--adapter specifies the adapter sequence.
TGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGC is the standard adapters for Illumina. If no sequence was supplied it will attempt to auto-detect the adapter which has been used (Illumina, Small RNA and Nextera standard adaptors would be used).
--rrbs specifies that input file was an MspI digested RRBS sample.
3. Map ERRBS reads to reference genome
Mapping ERRBS reads to a bisulfite converted genome presents many computational challenges. Alignments should allow for mismatches, especially for potential methylation sites.
Also, alignments should be unique considering the numerous possibilities combining all the methylation status in each read to avoid miscalling of methylation levels. Among all the publicly available mapping tools, we have chosen bismark[9] to map ERRBS reads. Before mapping ERRBS reads to the reference genome, we need prepare the genome to bisulfite converted versions, which can be used by bismark aligner. FASTA/FASTQ format files for genome reference need to be in the genome folder. After genome preparation, we can map ERRBS reads to human genome.
General Syntax:
$ bismark_genome_preparation [options] <path_to_genome_folder>
$ bismark [options] <genome_folder> {−1 <mates1> −2 <mates2> | <singles>}
Example:
$ bismark_genome_preparation <genome_folder>
$ bismark -l 29 --bowtie1 --multicore 6 GRCh37 --output_dir . CLL_S101.RRBS_trimmed.fq
where:
-I specifies the “seed length”, which is the number of bases of the high quality end of the read. Typically, the length of sequencing reads can be used here.
--bowtie1 specifies that bismark will create indexes for bowtie 1[12].
--multicore sets the number of parallel instances of bismark to be run concurrently.
--output_dir specifies that all output files are written into this directory.
4. Cytosine methylation state calling
Once suitable ERRBS alignments are generated, methylation status for individual sites (mostly CpG sites) can be determined. To be consistent with our alignment processes, we utilize a simple script from bismark to achieve this goal.
General Syntax:
$ bismark_methylation_extractor [options] <filenames>
Example:
$ bismark_methylation_extractor -s --output . --merge_non_CpG --multicore 6 -- genome_folder <genome_folder> CLL_S101.RRBS_trimmed.fq_bismark.bam
Typically, we need to extract methylation status from those two files:
CpG_OT_CLL_S101.RRBS_trimmed.1bp.fq_bismark.txt and
CpG_OB_CLL_S101.RRBS_trimmed.1bp.fq_bismark.txt. All the reads in files labeled with OT reflect methylation levels of CpGs in forward strand and all the reads in files labeled with OB contain methylation information of CpGs on reverse strand. For the methylation state, “+” and “-” indicate methylated and unmethylated CpGs, respectively. In accordance with this label, “Z” and “z” represent methylation and unmethylated CpGs. Those two labels should agree with each other. After this step, it’s usually important to check the general features of the bismark outputs for all of your samples, such as mapping efficiency, average conversion rate, number of CpGs, average coverage and average CpG methylation levels (Table 1). One of the most important features is average conversion rate, which detects how bisulfite treatment can convert unmethylated Cytosine successfully. This number is usually higher than 99.7% (Table 1). More over, samples should have acceptable mapping efficiency (60% or higher), which varies according to sequencing read length. Also, samples analyzed together should ideally have similar covered CpGs, similar average coverage across the whole data set and similar average CpG methylation levels in each category/group. For example, tumor samples have similar higher average CpG methylation levels versus normal samples (Table 1).
Table 1 –
Bismark output statistics example.
| id | #reads | mapping efficiency | average coversion rate | #CpGs(10X) | average coverage | average CpG methylation levels(%) |
|---|---|---|---|---|---|---|
| 1 | 73293324 | 64.50% | 99.8979 | 2848900 | 51.82 | 34.40% |
| 2 | 76101498 | 66.00% | 99.8947 | 2933770 | 50.14 | 31.50% |
| 3 | 85482964 | 66.50% | 99.8838 | 2822016 | 56.84 | 39.50% |
| 4 | 80272372 | 66.50% | 99.8138 | 2795046 | 56.97 | 35.90% |
| 5 | 64361288 | 66.60% | 99.7396 | 2625874 | 50.8 | 34.50% |
| 6 | 78897537 | 66.90% | 99.8999 | 2782944 | 56.52 | 33.80% |
| 7 | 76009876 | 66.00% | 99.8751 | 2850695 | 52.66 | 38.20% |
| 8 | 75431630 | 65.90% | 99.778 | 2893682 | 52.82 | 38.40% |
| 9 | 76653335 | 65.20% | 99.8868 | 2843240 | 54.8 | 39.00% |
| 10 | 78481481 | 65.00% | 99.8859 | 2808703 | 54.04 | 38.50% |
| 11 | 73287618 | 67.60% | 99.8617 | 2733715 | 52.24 | 37.20% |
| 12 | 73847281 | 67.50% | 99.7764 | 2920328 | 50.99 | 36.50% |
| 13 | 81504963 | 66.30% | 99.8747 | 2822336 | 58.31 | 41.90% |
| 14 | 96892822 | 62.50% | 99.8899 | 2795175 | 59.89 | 48.80% |
| 15 | 43137414 | 64.20% | 99.8866 | 1609370 | 48.55 | 30.80% |
| 16 | 72217478 | 66.50% | 99.7681 | 2741285 | 54.08 | 45.90% |
| 17 | 72922475 | 66.90% | 99.8376 | 2872522 | 53.3 | 38.20% |
| 18 | 75434628 | 66.80% | 99.8349 | 2839250 | 52.93 | 38.30% |
| 19 | 73437411 | 66.30% | 99.8747 | 2767916 | 52.07 | 39.10% |
| 20 | 82936367 | 68.00% | 99.8547 | 2823987 | 52.74 | 36.10% |
5. Data format transformation
This tutorial uses RRBSeeqer to call differentially methylated cytosines and regions. We thus need to convert bismark output files into a format that can be read by RRBSeeqer (Methyl files, Table 2).
Table 2 –
RRBSeeqer input files example.
| chrBase | chr | base | strand | coverage | freqC | freqT |
|---|---|---|---|---|---|---|
| chr1.10542 | chr1 | 10542 | F | 587 | 99.83 | 0.17 |
| chr1.10636 | chr1 | 10636 | F | 57 | 85.96 | 14.04 |
| chr1.10617 | chr1 | 10617 | F | 58 | 100 | 0 |
| chr1.10589 | chr1 | 10589 | F | 58 | 100 | 0 |
| chr1.10631 | chr1 | 10631 | F | 56 | 100 | 0 |
| chr1.10638 | chr1 | 10638 | F | 57 | 85.96 | 14.04 |
| chr1.10609 | chr1 | 10609 | F | 58 | 98.28 | 1.72 |
| chr1.10620 | chr1 | 10620 | F | 59 | 93.22 | 6.78 |
| chr1.10525 | chr1 | 10525 | F | 609 | 95.24 | 4.76 |
| chr1.10497 | chr1 | 10497 | F | 606 | 97.85 | 2.15 |
| chr1.10633 | chr1 | 10633 | F | 58 | 89.66 | 10.34 |
| chr1.133181 | chr1 | 133181 | R | 118 | 61.02 | 38.98 |
| chr1.133218 | chr1 | 133218 | R | 117 | 54.7 | 45.3 |
| chr1.133180 | chr1 | 133180 | F | 131 | 40.46 | 59.54 |
| chr1.133165 | chr1 | 133165 | F | 136 | 88.24 | 11.76 |
| chr1.135028 | chr1 | 135028 | F | 168 | 88.1 | 11.9 |
| chr1.135203 | chr1 | 135203 | R | 77 | 87.01 | 12.99 |
| chr1.135208 | chr1 | 135208 | R | 77 | 90.91 | 9.09 |
| chr1.135173 | chr1 | 135173 | R | 78 | 87.18 | 12.82 |
| chr1.134999 | chr1 | 134999 | F | 170 | 31.18 | 68.82 |
| chr1.135191 | chr1 | 135191 | R | 76 | 94.74 | 5.26 |
| chr1.135179 | chr1 | 135179 | R | 79 | 67.09 | 32.91 |
| chr1.135031 | chr1 | 135031 | F | 168 | 90.48 | 9.52 |
| chr1.135218 | chr1 | 135218 | R | 71 | 78.87 | 21.13 |
| chr1.136911 | chr1 | 136911 | F | 103 | 92.23 | 7.77 |
| chr1.136913 | chr1 | 136913 | F | 101 | 94.06 | 5.94 |
| chr1.136895 | chr1 | 136895 | F | 104 | 67.31 | 32.69 |
| chr1.136876 | chr1 | 136876 | F | 104 | 95.19 | 4.81 |
| chr1.136925 | chr1 | 136925 | F | 103 | 0.97 | 99.03 |
| chr1.137120 | chr1 | 137120 | F | 29 | 96.55 | 3.45 |
| chr1.137157 | chr1 | 137157 | F | 29 | 100 | 0 |
| chr1.137169 | chr1 | 137169 | F | 28 | 0 | 100 |
| chr1.139059 | chr1 | 139059 | F | 13 | 0 | 100 |
| chr1.139029 | chr1 | 139029 | F | 13 | 61.54 | 38.46 |
| chr1.139073 | chr1 | 139073 | F | 13 | 61.54 | 38.46 |
| chr1.237094 | chr1 | 237094 | F | 99 | 7.07 | 92.93 |
| chr1.249382 | chr1 | 249382 | R | 29 | 0 | 100 |
| chr1.249429 | chr1 | 249429 | R | 29 | 93.1 | 6.9 |
| chr1.531247 | chr1 | 531247 | R | 29 | 100 | 0 |
| chr1.531265 | chr1 | 531265 | R | 29 | 100 | 0 |
General Syntax:
$ python methylCall_from_Bismark.py [options] <sample_name> <input_dir> <output_dir>
$ perl epicore2calls.pl <input_file> | gzip > <output_file>
Example:
$ python methylCall_from_Bismark.py -c 10 CLL_S101 bismark_output/ cpg/
$ perl epicore2calls.pl cpg.CLL_S101.mincov10.txt | gzip > cpg.CLL_S101.mincov10.txt.calls.gz
where:
-c specifies the minimum coverage required per CpG site for downstream analysis.
6. DMRs identification patient-by-patient
Identifying DMRs is one of the most important problems in DNA methylation analysis. DMCs and DMRs typically reflect local DNA methylation changes during tumor evolution, such as those occurring in tumors between diagnosis and relapse. Several methods enable discovery of consistently hypo- or hyper-methylated cytosines or regions across several samples. We and other groups have observed significant inter-patient MH in diffuse large b-cell lymphomas (DLBCL), a prevalent non-Hodgkin lymphoma. Hence, we have sought to define DMCs and DMRs on a patient-by-patient basis. We used our in-house tools—RRBSseeqer—to extract DMCs and DMRs within individual patients. In general, we define DMCs using RRBSseeqer_CG followed by a serial of file passing steps (false discovery rate=20%, Chi-square tests). Next, DMRs were defined as regions containing at least five DMCs and whose total methylation difference was more than 10%. The use of five or more DMCs partially overcomes the statistical limitation of individual Chi-square tests based on n=1 patients sample. As the output, we will get DMR files with a single DMR per row including chromosome, start position, end position, size, number of CpGs in DMR and methylation difference.
General Syntax:
$ RRBSseeqer_CG -rrbs1 <control_file> -rrbs2 <experiment_file> | sort_column.pl | sort_column_alpnum.pl > <output_file>
$ Perl RRBSidentifyUpDownDMR.pl --metfile=<input_file> --outfile=<output_file> [options]
Example:
$ RRBSseeqer_CG -rrbs1 cpg.normal.mincov10.txt.calls.gz -rrbs2 cpg.CLL_S101.mincov10.txt.calls.gz -test chi | sort_column.pl 1 | sort_column_alpnum.pl 0 > DMC.CLL_S101.txt
$ perl RRBSidentifyUpDownDMR.pl --metfile=DMC.CLL_S101.txt --dmax=250 -- minmetdx=0.1 --minnumcg=5 –outfile=DMR.CLL_S101.txt
where:
-rrbs1 should be normal samples or earlier tumor stage samples.
-rrbs2 should be tumor samples or later tumor stage samples.
-dmax specifies the largest distance between two DMCs.
-minmetdx specifies the minimum average DNA methylation difference to detect a valid DMR.
-minnumcg specifies the minimum number of DMCs needed to detect a valid DMR.
7. Unsupervised analysis of pathways over-represent in tumor samples
To explore DMRs between Diagnosis and Relapse samples for each patient further, we need to annotate each DMR with the closest gene. We then perform pathway analysis to identify gene sets and pathways over-represented within the DMR associated genes in each patient. Generally, we annotate DMRs to different genomic locations with ChIPseeqerAnnotate from ChIPseeqer. First, we use ChIPseeqerAnnotate to annotate DMRs to genomic locations based on hg19 human genome and RefSeq gene annotations. Then, DMRs overlapped with promoters (defined as ± 2kb windows centered on RefSeq transcription start site) are extracted and converted to iPAGE acceptable file format). The last step is running pathway analysis against known pathways in GO[13]. The outputs list over- or under-representation levels in log10 P values. We then perform unsupervised analysis of genes or pathways can give us a sense how methylation perturbation in each patient compares to each other (Figure 2a).
Figure 2. Examples of DMRs identification and visualization.
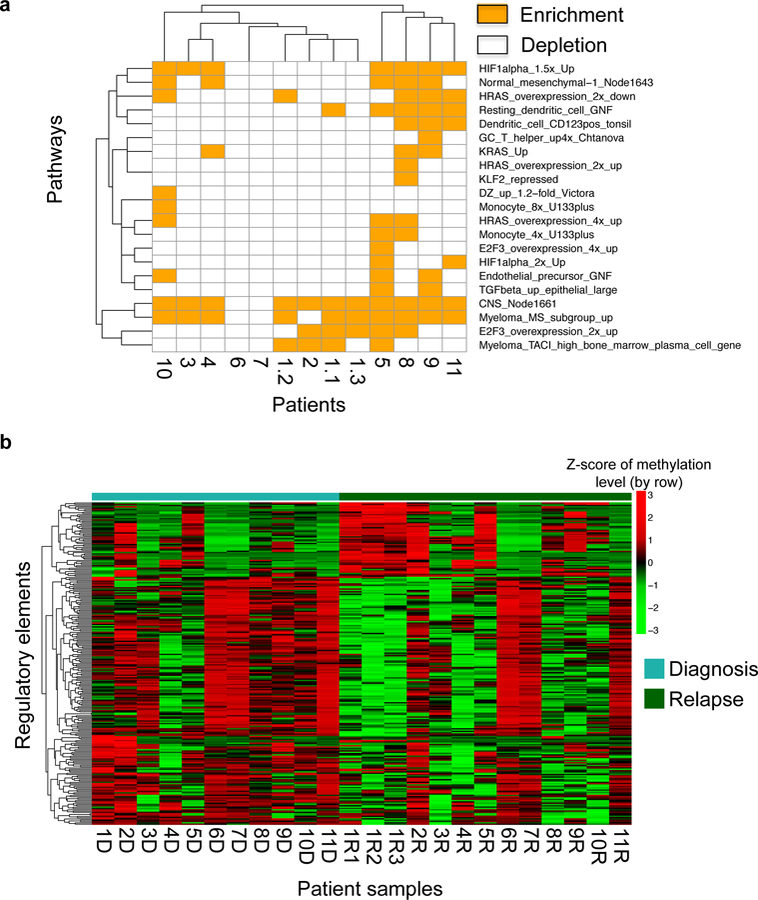
(a) Pathways overexpressed with hypermethylated genes (promoters overlapped with hypermethylation DMRs) of individual patients were illustrated here. Each row represents a single pathway and each column represents a patient pair. (b) Each row represent single differentially methylated regulatory element. Each column represent single diagnosis/relapsed sample from patients. Scale bar represent z-score of methylation level. Values were centered and scaled in row direction.
General Syntax:
$ ChIPseeqerAnnotate --peakfile=<input_file> [options]
$ mergeCSAnnotateGenesColumns.pl --genefile=<input_file> --outfile=<output_file> [options]
$ make_PAGE_input.pl --geneslist=<input_file> [options]
$ page.pl –expfile=<input_file> [options]
Example:
$ ChIPseeqerAnnotate --peakfile=DMR.CLL_S101.txt --genome=hg19 --db=refSeq
$ mergeCSAnnotateGenesColumns.pl -- genefile=DMR.CLL_S101.txt.refSeq.GP.genes.annotated.txt --geneparts=P --showORF=1 -
-outfile=DMR.CLL_S101.pro.txt
$ make_PAGE_input.pl --geneslist=DMR.CLL_S101.pro.txt --refgene=/data/hg19/refSeq
$ page.pl –expfile=DMR.CLL_S101.pro.txt.ORF.txt --pathways=human_go_orf -- cattypes=P,C,F -suffix=GO
where:
--genome specifies using hg19 as reference genome.
--db sepecies using RefSeq gene as gene annotation.
--pathways sets known pathways in GO.
--cattypes specifies using molecular function (F), cellular component (C) and biological process (P) parts of GO.
8. Supervised analysis to call DMRs across all the patients
An alternative way to DMRs patient-by-patient analysis is to directly call DMRs across all the patients. Unlike unbiased DMRs, we selected the region boundaries in this type of analysis, promoter or CpG Islands for example. Enhancers and other methylation sensitivity regulatory elements are also good candidates, such as CTCF binding sites. The methylation level for each region in each sample is calculated by averaging the methylation levels of all CpGs (at least 5 CpGs) inside the corresponding regions.
General Syntax:
$R CMD BATCH --no-save --no-restore [options] regionMethyl.R regionMethyl.log
Example:
$R CMD BATCH --no-save --no-restore ‘--args input_dir=cpg/ output_dir=. regions=promoter22kb.rds regionMethyl.R regionMethyl.log
where:
--no-save specifies that nothing will be saved in the .Rdata file.
--no-restore specifies that R does not read the .Rdata file in the current directory.
--args specifies arguments used from the rscript: input_dir sets the location of CpG Methyl files, all the outputs are written to output_dir, regions are promoter annotations (defined as ± 2kb windows centered on RefSeq transcription start site) in GenomicRanges data types[14], regionMethyl.R is the manuscript and regionMethyl.log is the output record of running this script with R.
This analysis generates a matrix with methylation levels for all the regions across all the samples. The next step is to call DMRs between two groups of samples. While there are several methods for calling DMRs, perhaps one of the simplest methods is to use paired T-test/Wilcox-test between diagnosis and relapse sample pairs followed by correction for multiple testing. A minimum methylation difference is also often used to ensure biological relevance of any significant change. Differentially methylated promoters across all the patients can be visualized using a heatmap (Figure 2b).
9. Intra-tumor MH characterization
It is common to use DMRs or average DNA methylation levels to characterize DNA methylation. However, DNA methylation is measured from cell population, from which methylation patterns are sampled. Bisulfite converted DNA reads may contain multiple CpGs thus enabling a per-read analysis of DNA methylation patterns. In such analyses, loci with identical average DNA methylation levels can have distinct DNA methylation patterns between samples. Such distinct patterns are the result of intra-tumor MH (Figure 3a). Intra-tumor MH has been connected to gene expression levels and to clinical outcome in several types of tumors such as chronic lymphocytic leukemia (CLL) and DLBCL. For example, higher MH are linked to lower gene expression levels in CLL and patients with higher MH levels at diagnosis stage in DLBCL have a higher chance to relapse after chemotherapy[6, 15]. The concept of epipolymorphism has been used to describe and quantify methylation heterogeneity [16]. The epipolymorphism level of a four-CpG locus (reads containing 4 or more contiguous CpGs) was defined as the probability that epialleles randomly sampled from the locus differ from each other. Higher epipolymorphism corresponds to higher intra-tumor methylation heterogeneity and vice-versa. Epipolymorphism can be analyzed at individual loci based on ERRBS data. A global epipolymorphism level can also be calculated. Briefly, epipolymorphism is calculated for each locus in a sample. Epipolymorphism is correlated with methylation level (with epipolymorphism being lowest at highest or lowest methylation levels), therefore global epipolymorphism must be normalized by methylation levels. Our global analysis divides loci into different bins depending on their methylation levels and median epipolymorphism is calculated for each bin. The area under the median line is defined as MH for each patient (Figure 3b–c). With this analysis, we can study the correlation between MH and tumor evolution.
Figure 3. Examples of intra-tumor MH analysis.

(a) Epipolymorphism levels are dependent on DNA methylation levels. All the loci were divided into different groups based on their methylation level and median epipolymorphism of each group is calculated. Genome-wide intra-tumour MH was quantified by area under the median line. (b) Median epipolymorphism lines for diagnosis and relapse tumors from patient 1.1 in our cohort. Intra-tumor MH visibly decreased with tumour evolution. (c) Relapsed samples displayed significant lower intra-tumour MH. All the loci located in gene promoter.
General Syntax:
$R CMD BATCH --no-save --no-restore [options] regionMH.R regionMH.log
Example:
$R CMD BATCH --no-save --no-restore ‘--args sample=CLL_S101 cpg_dir=cpg/ bam_dir=bam/ output_dir=. regions=promoter22kb.rds regionMH.R regionMH.log
where:
--no-save specifies that nothing will be saved in the .Rdata file.
--no-restore specifies that R does not read the .Rdata file in the current directory.
--args specifies arguments used from the rscript: sample sets the sample name, cpg_dir sets the location of CpG Methyl files, bam_dir sets the directory of bam files, all the outputs are written to output_dir, regions are promoter annotations (defined as ± 2kb windows centered on RefSeq transcription start site) in GenomicRanges data types[14], regionMH.R is the manuscript and regionMH.log is the output record of running this script with R.
4. Notes
FastQC performs a series of quality control analyses, such as per base sequence quality, per sequence quality scores, per base sequences content, adapter content and so on. Each test is flagged with a pass (green tick), warning (orange exclamation mark) or fail (red cross), whose status depends on how much a sample deviates from good quality benchmark samples.
When running trim_galore, several parameters need to be considered. First, the default value of-q/--quality <INT> is 20, which means reads with low-quality ends (under 20) are trimmed. This parameter is acceptable for ERRBS analysis but can be less stringent for datasets with low sequencing quality. Ideally, the best way is re-sequenced the data and get enough reads with at least 20 quality score. Also, totally numbers of available reads for downstream analysis should be considered when choose this parameter since there is a trade off between the total number of reads and data quality. Second, the sequencing platform needs to be factored in. ASCII+33 quality scores should be used as phred scores. If not, please remember to use --phred64 option to specify alternative quality scores. Third, when no adapter sequence was provided, it will analyze the first one million sequences of the first specified file and attempt to find the first 12 or 13 bps of the following standard adapters:
Illumina: AGATCGGAAGAGC
Small RNA: TGGAATTCTCGG
Nextera: CTGTCTCTTATA
If using other adapters, it is important to provide correct adapter sequence in this step.
One the most important parameters for trim_galore is -s/--stringency <INT>, which specify the minimum number of required overlap with the adapter sequence. The default value (1) is very stringent since even one overlap with the adaptor sequence would be removed. If less stringent value is used here, there is a higher chance you will include too much adapter contamination into your downstream analysis and distort your results. However, if you use very stringent cutoff (such as the default value), it’s possible that some reads would be mistakenly removed just due to its first base is identical to adapters by chance. In this case, if your data sets don’t have enough sequencing depth, total reads number would be too limited to perform downstream analysis.
When mapping reads to the genome, be aware of your sequencing library context. Directional sequencing library is common. But please add --non_directional parameter if it is not. Also, based on tutorial from bismark[9], use bowtie1 instead of bowtie2 when you try to run alignment faster or the sequencing reads are short.
If your sequencing library is non-directional, it is important to specify this when you are trying to use bismark_methylation_extractor. It is also important to stress that there are four kinds of output files where methylation statuses are stored. They are labeled with OT, OB, CTOT and CTOT. In non-directional model, it is important to extract forward strand CpG methylation information from OT and CTOT files. OB and CTOB files collect reverse strand DNA methylation information. It is also important to stress that extracting methylation information from low coverage CpG is typically considered not reliable due to potential sequencing error for specific base pair in out routinely ERRBS data passing process. It is recommended to remove CpGs covered by less than 10 reads in the original ERRBS method paper[4]. However, considering most people use 3X as the minimum reads in WGBS data, you can choose your own acceptable threshold when you have small library size.
Currently, we can only identify DMCs and DMRs in computational scale. Neither the definition of DMCs/DMRs nor the accuracy of such identifications is confirmed by biological and functional evidences. In summary, there are not widely accepted methods to fulfill this task. Thus, we recommend use different parameters to test the performance of identified DMRs, which can make sure that you identified DMCs and DMRs in proper ways. Also, other tools developed base on different underlying distributions can be tried and used as cross-validation such as methylkit[17].
When comparing two methods calling DMRs, the major advantage to call DMRs across all the patients is that DMRs have identical genomic boundaries. It’s fairly to evaluation methylation levels across all the patients. However, in patient-by-patient method, DMRs are determined automatically per patient, it’s a little tricky to compare DMRs from different positions for specific gene. On the other side, when we test the methylation difference across all the patients versus normal, we might not get significant result when only one or two patients have abnormal methylation levels, but this is also interesting considering dramatic inter-patient MH in tumors. Under this phenomenon, the patient-by-patient methods would work better since it can identify significant DMRs in each patient.
Pathway analysis can only validate the functionality of DMRs and corresponding signature on computational side. We still need other methods to validate those regions. One possible way is to use an orthogonal approach (MassArray) to validate the methylation levels of identified DMRs and confirm the methylation differences between samples. Another way, which maybe more robust, is to change the methylation levels of DMRs and test the following up gene expression level changes, pathway signaling and even proliferation behaviors. Such types of validations need more studies in the future.
Many other analyses can be used to characterize methylation heterogeneity such as M-scores, Eloci and PDR[15, 18, 19]. All these features have positive correlations with MH but they have significant difference in some special cases. For example, M-scores only capture MH in each CpG site, which ignores the heterogeneity relation between adjacent CpGs. Eloci and PDR cannot consider hyper/hypo direction when they estimate MH changes. It is recommended to use different methods to study MH and explore any potential differences between methods.
5. Reference
- 1.Dawson MA, Kouzarides T (2012) Cancer epigenetics: From mechanism to therapy. Cell 150:12–27. [DOI] [PubMed] [Google Scholar]
- 2.Shaknovich R, Melnick A (2011) Epigenetics and B-cell lymphoma. Curr Opin Hematol 18:293–299. doi: 10.1097/MOH.0b013e32834788cf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pike BL, Greiner TC, Wang X, et al. (2008) DNA methylation profiles in diffuse large B-cell lymphoma and their relationship to gene expression status. Leuk Off J Leuk Soc Am Leuk Res Fund UK 22:1035–1043. doi: 10.1038/leu.2008.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akalin A, Garrett-Bakelman FE, Kormaksson M, et al. (2012) Base-Pair Resolution DNA Methylation Sequencing Reveals Profoundly Divergent Epigenetic Landscapes in Acute Myeloid Leukemia. PLoS Genet 8:e1002781. doi: 10.1371/journal.pgen.1002781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meissner A, Gnirke A, Bell GW, et al. (2005) Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33:5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan H, Jiang Y, Boi M, et al. (2015) Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun 6:6921. doi: 10.1038/ncomms7921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrews S (2010) FastQC: A quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/http://www.bioinformatics.babraham.ac.uk/projects/. doi: citeulike-article-id:11583827
- 8.Krueger F (2012) Trim Galore! [http://www.bioinformatics.babraham.ac.uk/projects/trimgalore/]
- 9.Krueger F, Andrews SR (2011) Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giannopoulou EG, Elemento O (2011) An integrated ChIP-seq analysis platform with customizable workflows. BMC Bioinformatics 12:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodarzi H, Elemento O, Tavazoie S (2009) Revealing global regulatory perturbations across human cancers. Mol Cell 36:900–11. doi: 10.1016/j.molcel.2009.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 1–10. doi: gb-2009-10-3-r25 [pii]\r10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed]
- 13.Ashburner M, Ball CA, Blake JA, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aboyoun P, Pages H LM (2010) GenomicRanges: Representation and manipulation of genomic intervals. R Packag version 1:1–5. doi: 10.1007/s13398-014-0173-7.2 [DOI] [Google Scholar]
- 15.Landau D a, Clement K, Ziller MJ, et al. (2014) Locally Disordered Methylation Forms the Basis of Intratumor Methylome Variation in Chronic Lymphocytic Leukemia. Cancer Cell 26:813–825. doi: 10.1016/j.ccell.2014.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landan G, Cohen NM, Mukamel Z, et al. (2012) Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet 44:1207–14. doi: 10.1038/ng.2442 [DOI] [PubMed] [Google Scholar]
- 17.Akalin A, Kormaksson M, Li S, et al. (2012) methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, Garrett-Bakelman F, Perl AE, et al. (2014) Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol 15:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaknovich R, Geng H, Johnson NA, et al. (2010) DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 116:e81–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]