

Abstract
Matrix metalloproteinases (MMPs) are transcriptionally regulated proteases that have multiple roles in modifying the extracellular matrix (ECM) and inflammatory response. Our previous work identified Mmp28 as a key regulator of inflammation and macrophage polarization during experimental models of pulmonary infection, fibrosis, and chronic smoke exposure. However, the signaling pathways responsible for regulation of macrophage Mmp28 expression remain undefined. This study utilized murine macrophages obtained from wild type, Tlr2−/−, Tlr4−/−, MyD88−/−, Ticam1Lps2 (Trifmutant), and Ifnar1−/− mice to test the hypothesis that macrophage Mmp28 expression was dependent on TRIF and type I IFN. Our results support the hypothesis, demonstrating that increased macrophage Mmp28 expression was dependent on type I IFN after LPS and poly(I:C) stimulation. To gain further insight into the function of MMP28, we explored the inflammatory response of macrophages derived from wild type or Mmp28−/− mice to stimulation with poly(I:C). Our data support a role for MMP28 in regulating the macrophage inflammatory response to poly(I:C) because expression of Ccl2, Ccl4, Cxcl10, and Il6 were increased in Mmp28−/− macrophages. Together, these data support a model in which macrophages integrate TRIF- and type I IFN-dependent signaling to coordinate regulation of proteins with the capacity to modify the ECM.
Keywords: Macrophage, Mmp28, Mmp10, IFN
Introduction
Macrophages are innate immune cells that play key regulatory roles in host inflammatory responses to infection and injury, controlling the inflammatory process in part through phagocytosis and killing of microbes and clearance of dying cells and debris.1 In addition, macrophages detect changes in the extracellular environment by recognition of pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) to fine-tune the inflammatory response. In the lung, activation of epithelial cell pattern recognition receptors during infection can induce a robust type I IFN response, which is critical for development of a protective innate immune response to infection. Type I IFN also causes functional changes in macrophages, resulting in diverse phenotypes. In many circumstances, type I IFN increases the antimicrobial responses of macrophages.2,3 Macrophages also produce and secrete type I IFN after detection of PAMP to stimulate autocrine and paracrine pro-inflammatory responses. For example, Gram-negative bacterial LPS activates the TLR4 signaling pathway to induce pro-inflammatory and type I IFN responses through a TRIF-dependent pathway,4 and recognition of dsRNA by TLR3 also utilizes a TRIF-dependent pathway to induce a type I IFN response.4,5
Matrix metalloproteinases (MMPs) are proteases that are produced by multiple cell types, including macrophages, and function not only in regulating degradation of the extracellular matrix (ECM), but also in regulating immune responses, including antiviral responses.6,7 Although transcriptional regulation of MMPs is one of the main mechanisms regulating their function,6 the integration of PAMP and DAMP sensing by macrophages to coordinately regulate modifiers of the ECM remains an understudied area of investigation. Although recent studies have demonstrated that macrophage transcriptional regulation of versican is dependent on the type I IFN response,8 the overall contribution of these stimuli to the regulation of macrophage MMPs has not been investigated.
Our previous studies have demonstrated that macrophage expression of Mmp28 is increased upon stimulation with LPS or poly(I:C) and MMP28 regulates inflammation and macrophage polarization during experimental models of pulmonary infection, fibrosis, and chronic smoke exposure.9–11 Depending on the injury model, there are several consequences of MMP28 deletion. For example, during Pseudomonas aeruginosa pneumonia, Mmp28−/− mice have increased macrophage-specific pro-inflammatory responses with enhanced bacterial clearance.9 In contrast, Mmp28−/− mice exposed to chronic cigarette smoke have blunted inflammatory responses and are protected from emphysema, which could be due in part to decreased macrophage M2 polarization.10 Similarly, we have demonstrated that Mmp28−/− macrophages have impaired M2 polarization and are protected from bleomycin-induced fibrosis.11 Because the airway epithelium, in addition to macrophages, expresses Mmp28, it is not surprising that the role of MMP28 in regulating pulmonary inflammation is likely complex and stimulus dependent.
In order to gain a better understanding of how macrophage MMP28 regulates the inflammatory response, we sought to address two unresolved questions. First, we wanted to define the molecular signaling pathway components required for macrophage Mmp28 expression in response to LPS and poly(I:C). Second, we were interested in determining whether the response of Mmp28−/− macrophages to poly(I:C) was altered in order to provide further insight into MMP28 function. We performed our studies in murine bone marrow-derived macrophages to utilize a genetic approach to test the hypothesis that type I IFN, a common feature of both LPS and poly(I:C) stimulation, was responsible for increased macrophage Mmp28 expression.
Materials and methods
Animal ethics
The University of Washington Institutional Animal Care and Use Committee reviewed and approved all animal procedures prior to initiation of these studies. Wild type controls were gender matched for each knock-out mouse during independent experiments.
Cell culture medium and reagents
The macrophage medium used for culture of murine bone marrow-derived macrophages (BMDMs) was RPMI-1640 containing L-Glutamine and supplemented with 20% L929-conditioned medium, 10% heat-inactivated FBS, and 1% penicillin/streptomycin. High-molecular-mass poly(I:C) was from InvivoGen (San Diego, CA) and Escherichia coli LPS 0111:B4 was from Sigma-Aldrich (St. Louis, MO). Carrier-free recombinant mouse IFN-α1 and mouse IFN-β1 were from BioLegend (San Diego, CA). Mouse CCL2 (DY-479-05), CCL4 (DY451), CXCL10 (DY466-05), and IL-6 (DY460-05) DuoSet ELISA assays were from R&D Systems (Minneapolis, MN).
Isolation, culture, and stimulation of BMDMs
Bone marrow was isolated from the femurs and tibias of male and female wild type (C57BL/6) mice and Mmp28−/−, Tlr2−/−, Tlr4−/−, MyD88−/−, Ticam1Lps2 (Trifmutant), and Ifnar1−/− mice 8–12 wk of age as described previously.12 Cells were plated overnight in macrophage medium. Non-adherent cells were collected and cultured for 6 d additional for growth and differentiation into murine BMDM, as described previously.12 BMDM were re-seeded into 24-well tissue culture plates at 200,000 cells/well and allowed to adhere overnight before performing stimulations. Wild type, Tlr2−/− and Tlr4−/− macrophages were stimulated with 100 ng/ml LPS for 24 h. Wild type, MyD88−/−, and Trifmutant macrophages were stimulated with 50 ng/ml LPS or 10 μg/ml poly(I:C) for 4 h. Wild type and Ifnar1−/− macrophages were stimulated with 10 ng/ml LPS or 10 μg/ml poly(I:C) for 4 h. A concentration of 100 U/ml of either murine recombinant IFN-α1 or IFN-β1 were used to stimulate wild type macrophages for 6 h to assess Mmp28 expression. Wild type and Mmp28−/− macrophages were stimulated with 10 μg/ml poly(I:C) or 100 U/ml IFN-α1 or IFN-β1 for 24 h to assess differential inflammatory responses. Non-stimulated cells were treated with equivalent volume of PBS for all experiments (control). For cytokine measurements, cell supernatants were collected after 24 h and centrifuged for 10 min at 14,000 g to remove cellular debris.
Quantitative real-time PCR
RNA was isolated from cell cultures with the NucleoSpin RNA isolation kit (Clontech Laboratories, Mountain View, CA). A NanoDrop 1000 (Thermo Fisher Scientific, Waltham, MA) was used to determine RNA concentrations and equal amounts of RNA were used to synthesize cDNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). This cDNA was used as the template for quantitative real-time PCR (qPCR) reactions that used the SensiMix II Probe Hi-ROX Kit (Bioline, Taunton, MA) and validated TaqMan FAM primer probes for the murine genes Hprt Mm01545399, Mmp2 Mm00439498, Mmp3 Mm00440295, Mmp8 Mm00439509, Mmp9 Mm00600163, Mmp10 Mm01168399, Mmp11 Mm00485048, Mmp12 Mm00500554, Mmp14 Mm00485054, Mmp28 Mm00712992, Tnf Mm00443258, Ccl2 Mm00441242, Ccl4 Mm00443111, Ccl5 Mm01302427, Il1b Mm00434228, Il6 Mm00446190, and Ifit1 Mm00515153 (Life Technologies, Carlsbad, CA). The ABI 7900HT Fast Real-Time PCR System was used to obtain the average cycle threshold (Ct) from duplicate replicates of each sample; ΔCt of each primer probe was calculated relative to Hprt as described previously.12,13
Statistics
Statistical analyses were performed using GraphPad Prism 7 software. One-way ANOVA with Dunnett's multiple-comparisons test or two-way ANOVA with Sidak's or Tukey's multiple-comparisons tests with a confidence of 95% were used as indicated; otherwise, an unpaired Student's t test was used to compare groups.
Results
LPS and poly(I:C) induce macrophage Mmp28 expression by a TRIF-dependent pathway
We previously demonstrated that several inflammatory stimuli, including E. coli LPS, P. aeruginosa, and Chlamydophila pneumoniae, increase murine BMDM expression of Mmp28.9,11 In addition, treatment of murine BMDM with poly(I:C) also increases Mmp28 expression; however, the signaling pathways required for Mmp28 expression in macrophages have not been fully characterized. LPS-induced TLR4 signaling occurs through utilization of both MyD88-TIRAP and TRIF-TRAM adapter proteins,4 whereas utilization of a TRIF-dependent signaling pathway is a shared feature of TLR3 activation by poly(I:C).5 Based on the ability of both LPS and poly(I:C) to increase Mmp28 expression, we hypothesized that stimulation of Mmp28 expression by LPS was primarily due to a TRIF-dependent pathway. To test this hypothesis, we used murine macrophages derived from Tlr2−/−, Tlr4–/–, MyD88−/−, Trifmutant, and wild type mice. Because standard LPS could be contaminated with bacterial lipopeptides that interact with TLR2, we first tested Mmp28 gene expression in LPS-stimulated cells deficient in TLR2 or TLR4. We found that, in response to LPS stimulation, TLR4, but not TLR2, was required for Mmp28 expression (Figure 1a). Next, we utilized murine macrophages deficient in MyD88 or expressing non-functional TRIF. Whereas MyD88−/− macrophages retained Mmp28 expression in response to LPS (Figure 1b), Trifmutant macrophages had significantly reduced Mmp28 expression compared with wild type controls (Figure 1c), thus demonstrating a selective LPS pathway required for Mmp28 expression. Further, we demonstrate that poly(I:C)-stimulated Mmp28 expression was also dependent on TRIF (Figure 1d). Taken together, these data support our initial hypothesis that macrophage Mmp28 expression is TLR4 and TRIF dependent and TLR2 and MyD88 independent.
Figure 1.
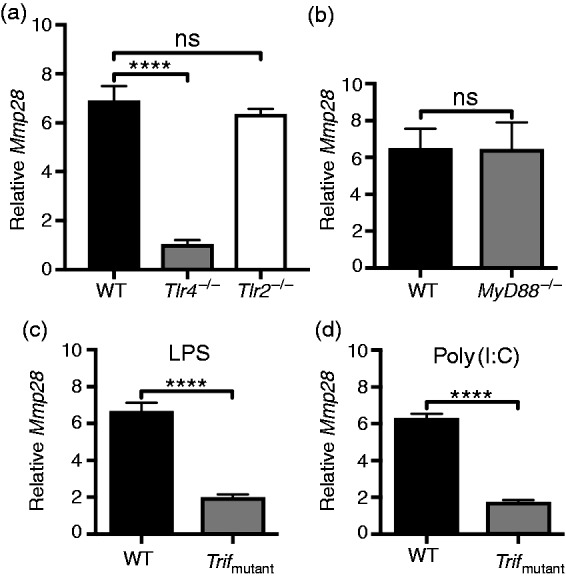
LPS and poly(I:C) increase Mmp28 gene expression by a TRIF-dependent pathway. (a–d) BMDM cells from wild type (WT), Tlr4−/−, Tlr2−/−, and Trifmutant mice were stimulated with LPS for 24 h (a) or LPS (b,c) and poly(I:C) (d) for 4 h and processed to determine mRNA levels of Mmp28 relative to Hprt. Results are shown as the fold change in Mmp28 expression relative to non-stimulated genotype-matched control cells. (a) LPS-induced Mmp28 expression was dependent on TLR4 but not TLR2. (b,c) LPS-induced Mmp28 expression was independent of MyD88 but required TRIF. (d) Poly(I:C)-induced Mmp28 expression was also TRIF dependent. Data represent mean ± SEM from a representative experiment (a) or independent experiments (b–d) with three to four biological replicates per genotype. Statistical analysis was performed using a one-way ANOVA with Dunnett's multiple-comparisons test (a) or Student's unpaired t test (b–d). ****P < 0.0001; ns, not significant (P > 0.05).
Macrophage Mmp28 expression is type I IFN dependent
Because TRIF-dependent pathways activate type I IFN expression and secretion, we investigated whether Mmp28 expression is a type I IFN-dependent gene using two independent approaches. First, we directly tested the ability of type I IFNs, IFN-α or IFN-β, to stimulate macrophage Mmp28 expression. Both IFN-α and IFN-β increased Mmp28 expression, whereas neither recombinant IFN elicited Tnf expression (Figure 2a). To further confirm that type I IFN was responsible for LPS- and poly(I:C)-induced Mmp28 expression, we stimulated macrophages from mice lacking the type I IFN receptor (Ifnar1−/−) compared with wild type cells (Figure 2b). Although Mmp28 expression was induced by both LPS and poly(I:C) in wild type macrophages, it was not increased by these stimuli in Ifnar1−/− macrophages. We also confirmed up-regulation of the type I IFN-responsive transcript Ifit1 in LPS- and poly(I:C)-treated wild type cells that was absent in similarly treated Ifnar1−/− cells. In contrast, LPS-induced increase in Tnf was unaltered in Ifnar1−/− macrophages compared with wild type (Figure 2b). Together, these data support the hypothesis that type I IFN secretion following either LPS or poly(I:C) activation of macrophages results in signaling through IFNAR1 to increase expression of Mmp28.
Figure 2.

Macrophage Mmp28 is regulated by type I IFN. (a) Wild-type (WT) BMDMs were stimulated with PBS (control), LPS (50 ng/ml), IFN-α (100 U/ml), or IFN-β (100 U/ml) for 6 h. The mRNA levels of Mmp28 and Tnf were measured relative to Hprt and normalized to control. All three stimuli, LPS, IFN-α, and IFN-β, increased Mmp28 gene expression. Data are expressed as mean ± SEM (n = 5 independent experiments). Statistical analysis used one-way ANOVA with Dunnett's multiple-comparisons test. (b) WT and Ifnar1−/− BMDMs were stimulated with PBS (control), LPS (10 ng/ml), or poly(I:C) (10 μg/ml) and assessed for Mmp28, Ifit1, and Tnf gene expression at 4 h. Both Mmp28 and Ifit1, but not Tnf, were dependent on IFNAR1 (mean ± SEM; n = 3–5 mice/genotype). Statistical analyses used two-way ANOVA with Sidak's multiple-comparisons test between WT and Ifnar1−/− samples and Tukey's multiple-comparisons test to compare treatment groups within each genotype *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant (P > 0.05).
LPS-induced expression of macrophage Mmp10 is IFNAR1 dependent
Similar to our previous studies with Mmp28, recent data also indicate that Mmp10 expression is increased in macrophages following stimulation with LPS or P. aeruginosa.14 To determine whether macrophage regulation of Mmp10 expression or other Mmps, including Mmp2, Mmp3, Mmp8, Mmp9, Mmp11, Mmp12, and Mmp14, are similar to Mmp28, we measured respective mRNA levels by qPCR in wild type and Ifnar1−/− macrophages stimulated with LPS or poly(I:C). We did not find detectable levels of Mmp2, Mmp3, Mmp11, or Mmp14 in either resting or LPS- or poly(I:C)-activated macrophages (data not shown). Although Mmp8, Mmp9, and Mmp12 were detected, no increase in expression due to LPS or poly(I:C) or dependence on IFNAR1 at this time point was observed (Figure 3). However, consistent with previous findings, LPS increased macrophage Mmp10 expression in wild type cells but was significantly reduced in Ifnar1−/− cells, suggesting that LPS-induced expression of both Mmp10 and Mmp28 is similar and dependent on type I IFN signaling (Figure 3). Although, in contrast to Mmp28, Mmp10 expression was not increased following poly(I:C) stimulation, suggesting that its transcription has additional, as yet undefined, stimulus-specific regulation. Overall, these findings indicate that type I IFN is integral to macrophage regulation of MMPs.
Figure 3.
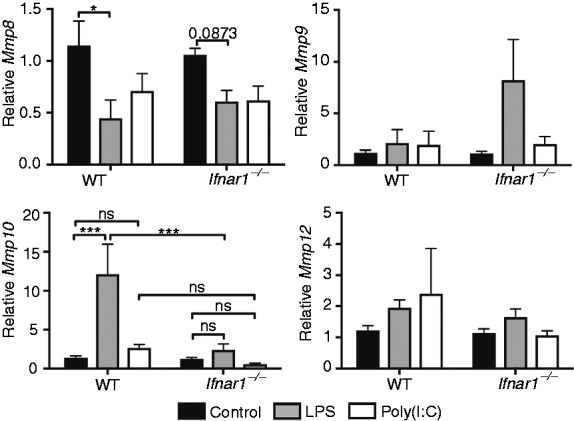
LPS-stimulated increase in macrophage Mmp10 expression is dependent on type I IFN. Wild-type (WT) and Ifnar1−/− BMDMs were stimulated with PBS (control), LPS (10 ng/ml), or poly(I:C) (10 μg/ml) and mRNA levels of Mmp8, Mmp9, Mmp10, and Mmp12 relative to Hprt were measured at 4 h. LPS-induced Mmp10 expression was dependent on IFNAR1; however, poly(I:C) did not increase Mmp10 expression. LPS or poly(I:C) did not increase Mmp8, Mmp9, or Mmp12. Data represent mean ± SEM from five to six mice/genotype. Statistical analyses used two-way ANOVA with Sidak's multiple-comparisons test between WT and Ifnar1−/− samples and Tukey's multiple-comparisons test to compare treatment groups within each genotype. ***P < 0.001; ns, or otherwise not indicated, are not significant (P > 0.05).
MMP28 restrains macrophage pro-inflammatory responses to poly(I:C)
In previous studies, we observed increased pro-inflammatory responses in LPS-stimulated Mmp28−/− macrophages.9,11 However, the role of MMP28 in regulating the inflammatory response to type I IFN or poly(I:C) has not been investigated. To determine whether MMP28 influenced the macrophage response to poly(I:C) stimulation, we measured expression of several inflammatory genes 24 h after PBS or poly(I:C) stimulation of wild type compared with Mmp28−/− murine BMDMs. Expression of Ccl2, Ccl4, Cxcl10, and Il6 was increased in Mmp28−/− macrophages compared with wild type macrophages after stimulation with poly(I:C) (Figure 4a). In contrast, expression of Ccl5, Il1b, and Ifit1 were similar in wild type and Mmp28−/− macrophages (data not shown). We further measured protein levels in cell supernatants to determine whether differences in gene expression between wild type and Mmp28−/− macrophages resulted in changes in chemokine/cytokine secretion. Consistent with the changes in gene expression, there was a significant increase in CCL4, CXCL10, and IL-6 in the supernatants of Mmp28−/− macrophages compared with wild type (Figure 4b). To further evaluate whether loss of MMP28 altered regulation of other MMPs, the mRNA levels of Mmp9, Mmp10, and Mmp12 were measured 24 h after poly(I:C) stimulation. Compared with wild type, Mmp28−/− macrophages had increased Mmp9 and decreased Mmp10 after poly(I:C) (Figure 4c). To determine whether any of the increased inflammatory responses in Mmp28−/− macrophages were the result of differences in the response to type I IFN, we stimulated macrophages with either recombinant IFN-α1 or IFN-β1 and measured inflammatory gene expression. Similar to poly(I:C) treatment, stimulation of Mmp28−/− macrophages with either recombinant IFN-α1 or IFN-β1 resulted in increased Il6 compared with wild type macrophages; however, there was no difference in Ccl2, Ccl4, or Cxcl10 (Figure 4d). These results suggest that, whereas there are some intrinsic differences in the response of Mmp28−/− macrophages to type I IFN, these differences likely do not completely explain the increased pro-inflammatory response to poly(I:C). Potentially, these effects could be due to the altered activation of signaling pathways that are activated by poly(I:C), but not by recombinant type I IFN per se (Figure 5), or that multiple changes due to the loss of MMP28-function combine to change the inflammatory response.2,15,16
Figure 4.

Macrophage MMP28 restrains the pro-inflammatory response to poly(I:C). BMDMs from wild type or Mmp28−/− mice were stimulated with media containing (a,b) poly(I:C) (10 μg/ml) or (c) IFN-α (100 U/ml), IFN-β (100 U/ml), or control media containing PBS for 24 h. The levels of Ccl2, Ccl4, Cxcl10, and Il6 were measured to Hprt and expression was normalized to control treated wild type samples. (a) There was a significant increase in Ccl2, Ccl4, Cxcl10, and Il6 gene expression and (b) in CCL4, CXCL10, and IL-6 protein levels in the cell supernatants in poly(I:C)-treated Mmp28−/− cells compared with similarly treated wild type cells. (c) The levels of Mmp9 and Mmp10 but not Mmp12 were significantly altered relative to Hprt at 24 h in wild type and Mmp28−/− macrophages. (d) In contrast to poly(I:C), only Il6 expression was differentially regulated by MMP28 in response to recombinant type I IFN. Data represent mean ± SEM; n = 3–4 mice per genotype. ND indicates below limit of detection. Statistical analyses used two-way ANOVA with Sidak's multiple-comparisons test. P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Figure 5.

Type I IFN plays an integral role in macrophage regulation of Mmp28 following stimulation with PAMP. Macrophage Mmp28 expression occurs following stimulation with LPS following a TLR4-TRIF-IFNAR1 dependent pathway that occurs independently of TLR2. In addition, poly(I:C) stimulation of macrophage Mmp28 occurs by a TRIF-IFNAR1-dependent pathway. Macrophages derived from Mmp28−/− mice have increased Ccl2, Ccl4, Cxcl10, and Il6 compared with wild type macrophages following poly(I:C) stimulation.
Discussion
Although MMP regulation of ECM has long been recognized, recently described diverse roles of MMPs in regulating other cellular responses have increased the implications for MMPs involved in a number of inflammatory diseases.6 Although prior reports from us9,11 and others17 demonstrated that macrophage Mmp28 expression increases following LPS or poly(I:C) stimulation, the pathways required for this response have not been described previously. In this report, we identified Mmp28 as a type I IFN-responsive gene in murine macrophages (Figure 4). Using a genetic approach, we demonstrated that TRIF and IFNAR1 are required for macrophage expression of Mmp28 during LPS or poly(I:C) stimulation. In addition, stimulation of macrophages with either recombinant IFN-α or IFN-β was sufficient to increase Mmp28 expression. We further discovered that LPS-induced expression of macrophage Mmp10 was dependent on IFNAR1; however, in contrast to Mmp28, Mmp10 expression was not increased in poly(I:C)-treated wild type macrophages, suggesting that the regulation of Mmp10 may be more complex than Mmp28. Although previous examination of the promoter region of Mmp28 from both humans and mice concluded that the DNA sequence does not contain a TATA box or AP-1-binding site close to the transcription start site (classified as a group III MMP promoter region), there are Sp1 and Sp3 sites present that are likely important for transcription.18–22 In addition, there are predicted STAT-binding sites in the promoter region of some MMPs, including MMP-28; however, the role of STATs in regulating the transcription of MMPs has not been well characterized.19,22 A previous study examined transcriptional expression of all MMPs during a murine model of RSV infection and found that expression of several MMPs, including Mmp28, were increased during RSV infection due to a TRIF- and MAVS-dependent response.23 However, this analysis was only performed on whole lung homogenates, whereas our study provides a novel demonstration of macrophage MMP transcriptional activation that is dependent on the type I IFN response. In summary, our data demonstrate that type I IFN is integral to macrophage expression of Mmp10 and Mmp28.
The contribution of type I IFN to regulation of MMPs remains relatively unexplored, with the exception of previous studies that identified that IFNβ-1b reduced MMP9 expression and protein activity in human peripheral blood mononuclear cells24 and human monocyte-derived dendritic cells.25 In contrast, whole-lung homogenate Mmp9 expression was increased during RSV infection by a MAVS-dependent but TRIF-independent mechanism and SAE cells treated with recombinant IFN-β had increased Mmp9 expression.23 Although the underlying mechanism that accounts for differences in these findings are not clear, they highlight the notion that MMPs likely have cell-type-specific functions that will need to be investigated in order to fully understand how MMPs exert their biological functions.6 Our data demonstrating differential regulation of macrophage Mmp10 during stimulation with LPS or poly(I:C) underscore this idea and these differences should be considered when designing murine in vivo models of disease.
In addition to advancing the understanding of macrophage Mmp28 regulation by type I IFN, our work here further investigated whether loss of MMP28 altered macrophage responses to poly(I:C) or recombinant type I IFN. Our results demonstrate that Mmp28−/− macrophages have increased pro-inflammatory responses to poly(I:C) stimulation and at least part of this response, such as Il6, is increased in Mmp28−/− macrophages when stimulated with either recombinant IFN-α or IFN-β. Activation of the NF-κB p65 and ERK1/2 pathways was examined at 5 and 24 h after poly(I:C) stimulation. No statistical differences between wild type and Mmp28−/− macrophages were observed (data not shown). Therefore, the precise underlying mechanism by which MMP28 regulates this inflammatory response remains to be determined. In our previous study, we observed increased Mmp9 mRNA in macrophages obtained from the lungs of cigarette-smoke exposed Mmp28−/− mice compared with wild type.10 Here, we further demonstrate that, after poly(I:C) stimulation, Mmp28−/− macrophages have increased Mmp9 mRNA and decreased Mmp10 mRNA, demonstrating that alterations in MMP28 function likely influence a network of changes that may result in differential regulation of the inflammatory response.
A limitation of our current study is the use of murine cells as a model system compared with human cells. We have found that CD14+ human monocyte-derived macrophages differentiated with M-CSF have very low to undetectable expression of MMP28 (data not shown); therefore, the ability to evaluate whether the regulation of human macrophage MMP28 is similar to the findings we have described here for murine macrophages remains a challenge for the field. However, we have detected MMP28 in alveolar macrophages in human lung samples10 and also in human alveolar macrophage samples from critically ill patients by qPCR (unpublished observations). Multiple studies have described large differences in expression profiles between in vitro cultured human monocyte-derived macrophages, peripheral blood monocytes, and alveolar macrophages;26,27 therefore, future studies examining human alveolar macrophage MMP28 responses to type I IFN pathway activation would be of interest. In summary, our findings suggest a model by which the expression and function of macrophage MMP28 is integrated into to the type I IFN response and future investigation into the function of MMP28 will focus on responses to stimulation of these pathways. In addition, further studies designed to investigate the cell-specific responses of MMPs to determine how they integrate into the overall IFN response will likely yield new insights into the pathogenesis of diseases that have previously been shown to involve the functions of MMPs or type I IFN.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Heart, Lung, and Blood Institute and National Institute of Allergy and Infectious Diseases–National Institutes of Health Grants R01 HL116515 (A.M.M.), T32 HL007828 (M.E.L), R01 AI136468 (C.W.F.), R01 HL122895 (W.A.), and R01 AI104002 (M.G.) and by the University of Washington Cystic Fibrosis Foundation Research Development Program SINGH15R0 (M.E.L.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Cystic Fibrosis Foundation.
References
- 1.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood 2008; 112: 935–945. [DOI] [PubMed] [Google Scholar]
- 2.Ivashkiv LB, Donlin LT. Regulation of type I IFN responses. Nat Rev Immunol 2014; 14: 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I IFNs in infectious disease. Nat Rev Immunol 2015; 15: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. TLR signaling. Cell Death Differ 2006; 13: 816–825. [DOI] [PubMed] [Google Scholar]
- 5.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 2007; 7: 353–364. [DOI] [PubMed] [Google Scholar]
- 6.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004; 4: 617–629. [DOI] [PubMed] [Google Scholar]
- 7.Marchant DJ, Bellac CL, Moraes TJ, et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat Med 2014; 20: 493–502. [DOI] [PubMed] [Google Scholar]
- 8.Chang MY, Kang I, Gale M, Jr, et al. Versican is produced by Trif- and type I IFN-dependent signaling in macrophages and contributes to fine-control of innate immunity in lungs. Am J Physiol Lung Cell Mol Physiol 2017; 313: L1069–L1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manicone AM, Birkland TP, Lin M, et al. Epilysin (MMP-28) restrains early macrophage recruitment in Pseudomonas aeruginosa pneumonia. J Immunol 2009; 182: 3866–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manicone AM, Gharib SA, Gong KQ, et al. Matrix metalloproteinase-28 Is a key contributor to emphysema pathogenesis. Am J Pathol 2017; 187: 1288–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gharib SA, Johnston LK, Huizar I, et al. MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J Leukoc Biol 2014; 95: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long ME, Eddy WE, Gong KQ, et al. MEK1/2 inhibition promotes macrophage reparative properties. J Immunol 2017; 198: 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long ME, Gong KQ, Eddy WE, Liles WC, Manicone AM. Pharmacologic inhibition of MEK1/2 reduces lung inflammation without impairing bacterial clearance in experimental Pseudomonas aeruginosa pneumonia. Pneumonia (Nathan) 2017; 9: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMahan RS, Birkland TP, Smigiel KS, et al. Stromelysin-2 (MMP10) moderates inflammation by controlling macrophage activation. J Immunol 2016; 197: 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chow KT, Gale M., Jr SnapShot: IFN signaling. Cell 2015; 163: 1808–1808.e1. [DOI] [PubMed] [Google Scholar]
- 16.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med 2007; 13: 460–469. [DOI] [PubMed] [Google Scholar]
- 17.Hald A, Rono B, Lund LR, Egerod KL. LPS counter regulates RNA expression of extracellular proteases and their inhibitors in murine macrophages. Mediators Inflamm 2012; 2012: 157894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Illman SA, Lohi J, Keski-Oja J. Epilysin (MMP-28): Structure, expression and potential functions. Exp Dermatol 2008; 17: 897–907. [DOI] [PubMed] [Google Scholar]
- 19.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol 2007; 211: 19–26. [DOI] [PubMed] [Google Scholar]
- 20.Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol 2008; 40: 1362–1378. [DOI] [PubMed] [Google Scholar]
- 21.Rodgers UR, Kevorkian L, Surridge AK, et al. Expression and function of matrix metalloproteinase (MMP)-28. Matrix Biol 2009; 28: 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newby AC. Metalloproteinase production from macrophages: A perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp Physiol 2016; 101: 1327–1337. [DOI] [PubMed] [Google Scholar]
- 23.Foronjy RF, Taggart CC, Dabo AJ, et al. Type-I IFNs induce lung protease responses following respiratory syncytial virus infection via RIG-I-like receptors. Mucosal Immunol 2015; 8: 161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuve O, Chabot S, Jung SS, Williams G, Yong VW. Chemokine-enhanced migration of human peripheral blood mononuclear cells is antagonized by IFN beta-1b through an effect on matrix metalloproteinase-9. J Neuroimmunol 1997; 80: 38–46. [DOI] [PubMed] [Google Scholar]
- 25.Bartholome EJ, Van Aelst I, Koyen E, et al. Human monocyte-derived dendritic cells produce bioactive gelatinase B: Inhibition by IFN-beta. J Interferon Cytokine Res 2001; 21: 495–501. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Pritchard DK, Wang X, et al. cDNA microarray analysis reveals fundamental differences in the expression profiles of primary human monocytes, monocyte-derived macrophages, and alveolar macrophages. J Leukoc Biol 2007; 81: 328–335. [DOI] [PubMed] [Google Scholar]
- 27.Morrell ED, Radella F, 2nd, Manicone AM, et al. Peripheral and alveolar cell transcriptional programs are distinct in acute respiratory distress syndrome. Am J Respir Crit Care Med 2018; 197: 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]