

Abstract
The main focus of cardio-oncology has been the prevention and treatment of the cardiac toxicity of chemotherapy and radiotherapy. Furthermore, several targeted therapies have been associated with unexpected cardiotoxic side-effects. Recently, epidemiological studies reported a higher incidence of cancer in patients with heart failure (HF) compared with individuals without HF. On this basis, it has been proposed that HF might represent an oncogenic condition. This hypothesis is supported by preclinical studies demonstrating that hyperactivation of the sympathetic nervous system and renin–angiotensin–aldosterone system, which is a hallmark of HF, promotes cancer growth and dissemination. Another intriguing possibility is that the co-occurrence of HF and cancer is promoted by a common pathological milieu characterised by a state of chronic low-grade inflammation, which predisposes to both diseases. In this review, we provide an overview of the mechanisms underlying the bidirectional relationship between HF and cancer.
Keywords: Cancer, heart failure, chemotherapy, neurohormonal activation, inflammation, amyloidosis, carcinoid heart disease
Heart failure (HF) and cancer represent two major causes of morbidity and mortality in developed countries.[1,2] The prevalence of these conditions is growing as the age of the population and the burden of shared risk factors, such as diabetes and obesity, are constantly increasing. In past decades, the field of cardio-oncology has predominantly focused on prevention and treatment of cardiovascular disease in cancer survivors, who are particularly prone to developing HF as a result of the cardiotoxicity of many antineoplastic agents and the clustering of cardiovascular risk factors in oncological patients.[3]
The co-occurrence of cancer and HF represents a major clinical problem, because each disease impinges on the treatment of the other disease, and consequently, has a detrimental impact on quality of life and survival.[4,5] In this scenario, the interaction between cardiologists and oncologists is indispensable to ensure optimal management of patients affected by both conditions.[4] In recent years, a previously unappreciated connection between cancer and cardiovascular disease emerged from epidemiological studies reporting an increased risk of incident cancer in HF patients.[6–9] Although the cause of this association is not yet resolved, it has been proposed that HF might represent a cancer-predisposing condition.[9–11] Another intriguing possibility is that the co-occurrence of HF and cancer is promoted by a common pathological milieu characterised by a state of chronic low-grade inflammation, which predisposes to both diseases.[10]
In this review, we provide an overview of the mechanisms underlying the bidirectional relationship between HF and cancer (Figure 1). Whereas pathways driving the increased risk of cardiovascular disease in cancer patients have been the subject of intense investigation, mechanistic links driving the increased risk of malignancy in HF patients have not been elucidated so far. In this respect, we outline below two non-mutually exclusive hypotheses that should be addressed by future preclinical and clinical studies.
Figure 1: Mechanisms Underlying the Bidirectional Relationship Between Heart Failure and Cancer.
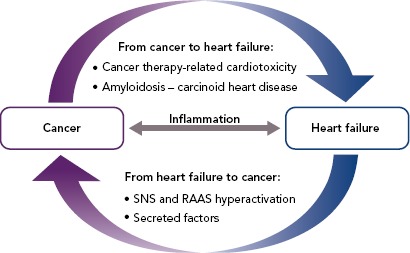
RAAS = renin–angiotensin–aldosterone system; SNS = sympathetic nervous system.
Incident Heart Failure in Cancer
Advances in the treatment of cancer have reduced the morbidity and mortality associated with many types of neoplasms. However, oncological therapies, including chemotherapy, radiotherapy, and newer-generation targeted therapies, may have toxic effects on the heart (Figure 2), up to causing HF either acutely, e.g. by causing acute coronary syndromes or myocarditis-like syndromes, or chronically, by directly impacting on cardiac myocyte function.[12] Because of the substantial improvements in the management of most types of cancer, these complications may have a major impact on the prognosis of patients with malignancy; in fact, they may become the primary clinical problem when cancer is stably controlled or cured.[13]
Figure 2: Mechanisms Underlying Incident HF in Cancer.
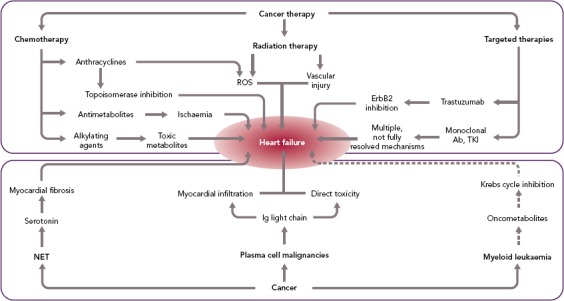
Ab = antibody; Ig = immunoglobulin; NET = neuroendocrine tumours; ROS = reactive oxygen species; TKI = tyrosine kinase inhibitor.
A far less common cause of HF in cancer patients is the secretion of cardiotoxic substances, such as light-chain immunoglobulins or vasoactive mediators associated with monoclonal B-cell proliferation and neuroendocrine tumours (NETs), respectively.
Chemotherapy- and Radiotherapy-induced Heart Failure
Anthracyclines, a class of chemotherapeutic agents commonly used for the treatment of solid and haematologic malignancies, were the first antineoplastic drugs for which a cardiotoxic effect was recognised.[14] Anthracycline cardiotoxicity may manifest as HF with acute or subacute onset, but may also lead to subclinical left ventricular dysfunction insidiously progressing to HF over the course of several years after exposure to the drug.[15] The incidence of anthracycline-related cardiac dysfunction is dose-dependent, and ranges from 5% at a cumulative dose of 400 mg/m2 to 26% for 550 mg/m2.[16] However, a subclinical decrease in systolic function has also been reported for lower doses in survivors of acute lymphoblastic leukaemia.[17]
The antiproliferative effect of anthracyclines stems from their ability to intercalate into nuclear DNA and block topoisomerase 2 activity, consequently inhibiting DNA replication and transcription. Furthermore, these agents cause damage to cellular components by forming complexes with iron and thereby inducing production of reactive oxygen species (ROS). Preclinical studies indicated that oxidative stress might represent the dominant driver of anthracyclines cardiotoxicity, but ROS scavengers failed to prevent doxorubicin-induced cardiomyopathy in humans.[12,18] Moreover, iron chelating agents did not show any cardioprotective effect in a rat model of anthracycline toxicity.[19] A recent experimental study demonstrated that cardiac topoisomerase is a key mediator of doxorubicin-induced cardiotoxicity, possibly accounting for the lack of efficacy of antioxidant agents in this setting. In fact, doxorubicin triggers apoptosis and transcriptomic remodelling in a topoisomerase-dependent manner, ultimately impacting on oxidative phosphorylation and mitochondrial biogenesis.[20]
Overall, in spite of a large body of preclinical research addressing the mechanisms of anthracyclines cardiotoxicity, pharmacological approaches aimed at alleviating this important side-effect are limited to a single agent, dexrazoxane, which achieves its cardioprotective activity via topoisomerase inhibition.[21] Recently, inhibition of the multifunctional kinase, phosphoinositide 3-kinase gamma, was shown to enhance removal of damaged mitochondria in a mouse model of doxorubicin-induced HF, pinpointing a potential therapeutic strategy to protect the heart against anthracyclines toxicity.[22]
Since the discovery of anthracyclines-related cardiotoxicity, many other chemotherapeutic agents have been associated with the development of cardiomyopathy. Alkylating agents, such as cyclophosphamide and ifosfamide, inhibit cell proliferation by inducing DNA damage. Cardiotoxicity associated with these drugs manifests predominantly as conduction disorders and pericarditis, and high-dose regimens can lead to myocarditis and HF.[23,24] Experimental evidence suggests that alkylating agents cause endothelial and myocyte damage secondary to the accumulation of toxic metabolites.[25] Antimetabolites, such as 5-fluorouracil and its pro-drug capecitabine, achieve their cardiotoxic effects mainly by triggering coronary artery vasospasm,[12] but preclinical studies indicate that these agents might also be directly toxic to endothelial cells and cardiac myocytes by triggering ROS production and inducing mitochondrial dysfunction.[26,27]
Radiation therapy represents a standard approach for breast cancer treatment, and often involves the exposure of the heart to high radiation doses. Pericardial fibrosis is the most common radiotherapy-related lesion, but radiation therapy also damages the myocardium.[28] Indeed, although cardiac myocytes are non-proliferating cells, and thus relatively resistant to radiation damage, emerging evidence indicates that chest irradiation can also lead to cardiomyopathy, and preclinical studies pinpointed ROS production with subsequent activation of Ca2+/calmodulin-dependent protein kinase II as a key mediator of radiation damage to cardiac myocytes.[29]
In contrast to cardiac myocytes, endothelial cells are continuously proliferating, and thereby more susceptible to radiation damage. Preclinical studies indicate that the primary lesion associated with radiation therapy is endothelial apoptosis, which might account for the clinical observation of accelerated atherosclerosis in patients receiving radiation therapy to the chest.[30,31] In fact, cardiac radiation dose correlates with the subsequent risk of ischaemic heart disease in breast cancer patients, and even low levels of exposure heighten the risk of coronary events.[32] Furthermore, a recent study observed that radiation therapy in breast cancer patients leads to a dose-dependent increase in the relative risk of HF with preserved ejection fraction (HFpEF).[33] The latter observation corroborates the model according to which microvascular endothelial dysfunction represents a key factor in the pathogenesis of HFpEF.[34]
Antineoplastic Targeted Therapy-induced Heart Failure
Trastuzumab is a humanised monoclonal antibody targeting the human epidermal growth factor receptor (HER)2, a member of the ErbB family of receptors, which is overexpressed in a subset of breast cancer patients.[35] Trastuzumab was initially approved as a first-line treatment for metastatic breast cancer, and is currently indicated for the treatment of HER2-positive breast and gastric cancer. The addition of trastuzumab to adjuvant therapy for breast cancer resulted in asymptomatic left ventricular dysfunction or overt HF in up to 18% and 4% of treated patients, respectively.[36,37] The observation of trastuzumab cardiotoxicity led scientists to interrogate the function of HER2 signalling in preclinical models of cardiac disease, revealing that this pathway plays an important homeostatic role,[38] and is activated in response to cardiac injury; for example, following ischaemia, pressure overload, and anthracycline toxicity (reviewed by De Keulenaer et al.[39]). Therefore, HER2 inhibition does not cause myocardial damage directly, but rather, blocks an important adaptive signalling pathway and thereby renders the heart more susceptible to pathological stressors. Accordingly, trastuzumab-induced cardiac dysfunction is usually completely reversible 4–6 weeks after discontinuation of the drug. However, for reasons yet to be fully elucidated, cardiac function is irreversibly compromised in a minority of patients treated with trastuzumab.[36]
Vascular endothelial growth factor (VEGF) is a key regulator of angiogenesis, the process of new blood vessel formation that sustains tumour growth when its enlargement precludes diffusion of nutrients and oxygen from pre-existing vessels.[40] VEGF signalling has become the target of several antineoplastic agents, such as the humanised antibody bevacizumab and the tyrosine kinase inhibitors (TKI) sunitinib and sorafenib. Drugs targeting VEGF signalling have been linked to a wide spectrum of cardiovascular side-effects, such as hypertension, thromboembolism, and cardiomyopathy.[41,42] In patients treated with anthracyclines for breast cancer, concurrent treatment with bevacizumab increased HF incidence from 4% to 14%.[43] Experimental evidence indicates that the effects of agents inhibiting VEGF signalling on blood pressure and thromboembolic risk might be mediated by decreased production of two vasodilators, nitric oxide and prostacyclin, and increased production of the potent vasoconstrictor, endothelin-1, whose circulating levels were found elevated in patients treated with sunitinib.[44–46] Sunitinib and sorafenib are only two examples of small-molecule TKI for which cardiotoxic effects were recognised. The number of TKI approved for cancer treatment is steadily growing, and cardiovascular side-effects have been reported for many of these drugs, such as the ABL inhibitors dasatinib and nilotinib or the multi-kinase inhibitor regorafenib.[47] Although the incidence of cardiovascular side-effects with these drugs is relatively low, the underlying mechanisms need to be further clarified to improve the safety of TKI currently under development.
Finally, cardiotoxic effects have also been associated with proteasome inhibitors, a class of antineoplastic agents used in the treatment of multiple myeloma and other hematologic malignancies. The first approved agent of this class, bortezomib, might cause HF in up to 4% of treated patients, and the second-generation proteasome inhibitor, carfilzomib, is associated with an even higher cardiotoxicity, with an incidence of cardiovascular adverse events of 18% according to a recent meta-analysis.[48,49] The ubiquitin–proteasome system plays an important adaptive role in the myocardium, and its inhibition is sufficient to cause cardiac dysfunction in pigs.[50] Therefore, cardiotoxicity associated with proteasome inhibitors is likely directly related to their mechanism of action.
Cancer-related Heart Failure
HF can also be the consequence of two rare cardiomyopathies, i.e. light-chain amyloidosis and carcinoid heart disease (Figure 2), although this happens more rarely than following oncological treatments.
Amyloidosis is a disorder characterised by extracellular deposition of a proteinaceous material, coined as amyloid, derived from misfolding of a variety of precursor proteins.[51] Amyloidosis is a systemic disorder and can affect several organs, but amyloid involvement of the heart portends by far the worst prognosis of any other type of organ involvement. Cardiac amyloidosis involves both myocardium and cardiac valves, and manifests as restrictive cardiomyopathy inexorably progressing to overt HF (reviewed by Falk et al.[52] and Gertz et al.[53]). Amyloid light-chain (AL) amyloidosis, which is secondary to overproduction of immunoglobulin light chain by plasma cell malignancies, is the most severe form of the disease, with a median survival of 6 months from HF onset if the underlying dyscrasia is left untreated.[54] Cardiac AL amyloidosis leads to a more severe form of HF despite a lower degree of cardiac hypertrophy, suggesting that AL amyloid protein might have direct toxic effects on cardiac myocytes.[55] Preclinical studies indicate that oxidative stress might represent a dominant driver of AL amyloid cardiotoxic activity.[56] Treatment of cardiac AL amyloidosis is currently limited to optimal management of HF and the underlying amyloidogenic malignancy, whereas therapeutic approaches directly targeting AL deposition in the myocardium are not currently available in the clinical setting.[52]
A rare form of cancer-related cardiac involvement is carcinoid heart disease, which is caused by NETs releasing vasoactive mediators, such as serotonin, bradykinin, and histamine. NETs are rare neoplasms arising from enterochromaffin cells of the gastrointestinal or respiratory tract. Because the mediators released by NETs are efficiently inactivated in the liver and the pulmonary vasculature, carcinoid heart disease usually arises from gastrointestinal NET upon their metastasisation to the liver and predominantly affects the right ventricle, whereas left ventricular involvement is observed in 5–10% of cases and is usually associated with bronchial carcinoids.[57,58] The typical feature of carcinoid heart disease is the formation of endomyocardial fibrotic plaques, ultimately leading to right-sided HF. Furthermore, fibrotic remodelling often also involves the tricuspid valve, causing valvular regurgitation, which contributes to right ventricular decompensation. Medical therapy for carcinoid syndrome is limited to symptomatic relief with somatostatin analogues, which are ineffective toward myocardial and valvular involvement.[59]
While AL amyloidosis and carcinoid heart disease are the only forms of cancer-elicited HF observed in the clinical arena so far, experimental work suggests that other malignancies might affect cardiac function via the release of cardiotoxic oncometabolites. Mutations of the Krebs cycle enzyme, isocitrate dehydrogenase, have been identified in a subset of patients with myeloid leukaemia. In rats, this mutation leads to accumulation and release of D-2-hydroxyglutarate from malignant cells, and this oncometabolite impairs cardiac Krebs cycle activity and contractile function.[60]
Incident Cancer in Heart Failure
Recent epidemiological studies revealed that HF patients carry a higher risk of incident cancer compared with individuals without HF, drawing attention toward another potential link between HF and cancer. This finding was first reported in a community-based case–control study, and subsequently confirmed in a large prospective study based on the Danish national registries.[6,7] Furthermore, a prospective cohort study demonstrated that patients developing HF following acute MI have an increased risk of incident cancer compared with those who do not develop HF after MI.[8] This association might be accounted for by a detection bias due to intensified medical observation following HF diagnosis. However, the increased risk of incident cancer was observed after the second year after HF diagnosis, and the association persisted after excluding cancer diagnoses made in the first years of follow-up.
Furthermore, cancer and HF share several risk factors, such as diabetes and obesity, which might partly explain the association between HF and increased risk of malignancy. In the above-mentioned studies, however, the likelihood of receiving a diagnosis of cancer remained higher in HF patients after adjusting for shared risk factor. Another possibility is that the increased risk of incident cancer is driven by a pro-oncogenic effect of HF medications, but recent meta-analyses addressing this issue do not support this concept.[61] On these grounds, the present authors and other authors have put forward two non-mutually exclusive hypotheses on how HF might lead to an increased risk of cancer, which are discussed in detail below.[9,10]
A third potential mechanism was recently elucidated in a preclinical study that demonstrated that ischemic HF enhances tumour growth via release of mitogenic factors by the failing myocardium.[11] In that study, MI was induced in mice via coronary artery ligation, and infarcted hearts were transplanted in the cervical region of APCmin mice, which are genetically predisposed to develop colorectal neoplasms. Intriguingly, mice transplanted with an infarcted heart developed a higher tumour burden compared with mice receiving a sham-operated heart. Because recipient mice retained their native healthy hearts, the increase in tumour load could not be attributed to haemodynamic impairment related to HF, but was shown to depend on the mitogenic protein, serpinA3, secreted by the failing myocardium. The translational relevance of this mechanism is underscored by the observation that circulating levels of serpinA3 are increased in chronic HF patients.[11] Overall, the results of that study strongly support the concept that a diagnosis of HF represents a risk factor for incident cancer.
The Neurohormonal Hypothesis
Hyperactivation of the sympathetic nervous system (SNS) and renin–angiotensin–aldosterone system (RAAS) is a hallmark of HF with reduced ejection fraction (HFrEF), and substantially contributes to episodes of decompensation as well as to cardiac death. Indeed, medical therapy of HFrEF currently relies on neurohormonal inhibitors: blockers of the beta-adrenergic receptors (AR), through which the catecholamines epinephrine and norepinephrine transmit SNS signals; inhibitors of the angiotensin-converting enzyme that synthesises angiotensin II (AngII); and antagonists of the AngII or aldosterone receptor.[62]
The hypothesis that neurohormonal activation may also account for the increased risk of cancer observed in HF finds its background in a large body of experimental data demonstrating that SNS and RAAS activation promote cancer progression and dissemination via multiple mechanisms. The pro-oncogenic effects of the SNS are predominantly mediated by beta-AR expressed by both cancer cells and, more importantly, non-malignant cells constituting the tumour microenvironment. Specifically, beta-AR signalling was demonstrated to favour tumour growth, induce formation of blood and lymphatic vessels, and promote remodelling of the extracellular matrix, ultimately leading to tissue invasion and metastatic dissemination in vivo.[63] Similarly, AngII promotes tumour vascularisation and invasiveness via type 1 AngII receptors.[64]
An important caveat is that, although SNS and RAAS activation has also been described in HFpEF patients, the latter do not benefit from treatment with beta-AR blockers and RAAS inhibitors, indicating that the role of neurohormonal activation in the progression of HFpEF is not as relevant as in HFrEF.[65,66] Two of the epidemiological studies discussed above included a substantial proportion of HFpEF patients, and cancer incidence was independent of left ventricular ejection fraction.[6,9] Because neurohormonal hyperactivation might not account for the higher incidence of cancer observed in HFpEF, we hypothesise that other factors are involved in this subset of patients.
The ‘Inflammatory Milieu’ Hypothesis
Independent of its aetiology, HF is associated with an increase in circulating and intramyocardial levels of pro-inflammatory cytokines, such as tumour necrosis factor-alpha, interleukin-1, and interleukin-6.[67–70] Inflammation is pivotal to the pathogenesis of atherosclerosis, which underlies the development of ischemic heart disease, the most common cause of HF.[71] In turn, myocardial injury triggers immune system activation, inducing cytokine release, and thereby fostering a vicious cycle of self-sustained inflammation. Furthermore, it has been hypothesised that microvascular endothelial inflammation might decrease myocardial nitric oxide release, thereby inducing cardiac myocyte hypertrophy and impairing relaxation, which is a hallmark of HFpEF.[34]
Indeed, HFpEF patients display elevated concentrations of galectin-3, an inflammatory mediator associated with myocardial fibrosis, and pentraxin 3, an inflammatory marker that was observed to correlate with left ventricular diastolic dysfunction.[72,73] Furthermore, circulating levels of inflammatory markers (tumour necrosis factor-alpha, transforming growth factor-beta, C-reactive protein, procollagen type 1 carboxy-terminal propeptide) were found to be elevated, and correlated with asymptomatic diastolic dysfunction in patients with metabolic syndrome and hypertension.[74] Altogether, a wealth of clinical studies indicate that HFrEF and HFpEF are associated with a state of mild chronic systemic inflammation, but it is currently unresolved whether the latter is a cause or consequence of cardiac dysfunction.
In contrast, chronic inflammation is considered carcinogenic and capable of boosting the transition from early-stage tumours to overt malignancies.[75] In principle, therefore, inflammation might mediate the association of both HFrEF and HFpEF with incident cancer. Although preclinical studies addressing this hypothesis are lacking, this model is corroborated by the results of the Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) trial.[76] In that study, the interleukin-1beta-targeting antibody, canakinumab, reduced the rate of recurrent cardiovascular events in patients with previous MI. Intriguingly, additional analyses revealed that treatment with canakinumab was associated with a dose-dependent trend toward reduction of hospitalisation for HF, which was independent of prior HF history, and a lower risk of incident lung cancer.[77,78] Altogether, the results of the CANTOS trial strongly support the concept that chronic low-grade inflammation represents a fertile substrate for the progression of HF and cancer.
The striking results of the CANTOS trial stand to some degree at odds with studies using broad-spectrum anti-inflammatory agents, namely the tumour necrosis factor-alpha inhibitors, etanercept and infliximab, and the immune-system suppressant, methotrexate, which did not detect any effect of these drugs on cardiovascular events.[77,79,80] An important difference between these studies and the CANTOS trial is that only the latter enrolled patients with modestly elevated C-reactive protein levels, reflecting a state of mild systemic inflammation. Furthermore, although excess inflammation is undoubtedly detrimental, cytokine signalling also mediates adaptive responses in the heart, and future studies should be aimed to more precisely identify signalling pathways associated with maladaptive processes driving the progression of HF.[81,82]
Finally, HF-related inflammation might foster cancer in an indirect way. For instance, a decline in the number of naïve T cells, and a marked increase in highly differentiated effector and memory T cells was recently observed in patients with HF, and is related to elevated levels of interleukin-6.[83] These features are consistent with immunosenescence, which consists of the deterioration of both adaptive and innate immunity, and, given the role played by the immune system in malignant cell elimination, may partly account for the increase in cancer incidence in HF, as it has been postulated for ageing.[84]
Conclusion
Until now, the main focus of cardio-oncology has been the prevention and treatment of cardiotoxic effects of chemotherapeutic agents. In this context, elucidation of the underlying mechanisms is instrumental to the development of strategies to prevent chemotherapy-related cardiomyopathy. While this avenue of research is far from being exhausted as a result of the staggering growth of novel anticancer targeted therapies, a new exciting area of cardio-oncology opens up in front of us, inspired by several lines of evidence linking the pathophysiology of HF to the development and progression of malignancy.
References
- 1.Christ M, Stork S, Dorr M et al. Heart failure epidemiology 2000-2013: insights from the German Federal Health Monitoring System. Eur J Heart Fail. 2016;18:1009–18. doi: 10.1002/ejhf.567. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Armenian SH, Xu L, Ky B et al. Cardiovascular disease among survivors of adult-onset cancer: a community-based retrospective cohort study. J Clin Oncol. 2016;34:1122–30. doi: 10.1200/JCO.2015.64.0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ameri P, Canepa M, Anker MS et al. Cancer diagnosis in patients with heart failure: epidemiology, clinical implications and gaps in knowledge. Eur J Heart Fail. 2018;20:879–87. doi: 10.1002/ejhf.1165. [DOI] [PubMed] [Google Scholar]
- 5.Rohrmann S, Witassek F, Erne P et al. Treatment of patients with myocardial infarction depends on history of cancer. Eur Heart J Acute Cardiovasc Care. 2018;7:639–45. doi: 10.1177/2048872617729636. [DOI] [PubMed] [Google Scholar]
- 6.Banke A, Schou M, Videbaek L et al. Incidence of cancer in patients with chronic heart failure: a long-term follow-up study. Eur J Heart Fail. 2016;18:260–6. doi: 10.1002/ejhf.472. [DOI] [PubMed] [Google Scholar]
- 7.Hasin T, Gerber Y, McNallan SM et al. Patients with heart failure have an increased risk of incident cancer. J Am Coll Cardiol. 2013;62:881–6. doi: 10.1016/j.jacc.2013.04.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hasin T, Gerber Y, Weston SA et al. Heart failure after myocardial infarction is associated with increased risk of cancer. J Am Coll Cardiol. 2016;68:265–71. doi: 10.1016/j.jacc.2016.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto M, Hasegawa T, Asakura M et al. Does the pathophysiology of heart failure prime the incidence of cancer? Hypertens Res. 2017;40:831–6. doi: 10.1038/hr.2017.45. [DOI] [PubMed] [Google Scholar]
- 10.Bertero E, Canepa M, Maack C, Ameri P. Linking heart failure to cancer. Circulation. 2018;138:735–42. doi: 10.1161/CIRCULATIONAHA.118.033603. [DOI] [PubMed] [Google Scholar]
- 11.Meijers WC, Maglione M, Bakker SJL et al. Heart failure stimulates tumor growth by circulating factors. Circulation. 2018;138:678–91. doi: 10.1161/CIRCULATIONAHA.117.030816. [DOI] [PubMed] [Google Scholar]
- 12.Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53:2231–47. doi: 10.1016/j.jacc.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 13.Banke A, Fosbol EL, Moller JE et al. Long-term effect of epirubicin on incidence of heart failure in women with breast cancer: insight from a randomized clinical trial. Eur J Heart Fail. 2018;20:1447–53. doi: 10.1002/ejhf.1168. [DOI] [PubMed] [Google Scholar]
- 14.Tan C, Tasaka H, Yu KP et al. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Cancer. 1967;20:333–53. doi: 10.1002/1097-0142(1967)20:3<333::AID-CNCR2820200302>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 15.van Nimwegen FA, Schaapveld M, Janus CP et al. Cardiovascular disease after Hodgkin lymphoma treatment: 40-year disease risk. JAMA Intern Med. 2015;175:1007–17. doi: 10.1001/jamainternmed.2015.1180. [DOI] [PubMed] [Google Scholar]
- 16.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–79. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 17.Vandecruys E, Mondelaers V, De Wolf D et al. Late cardiotoxicity after low dose of anthracycline therapy for acute lymphoblastic leukemia in childhood. J Cancer Surviv. 2012;6:95–101. doi: 10.1007/s11764-011-0186-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myers C, Bonow R, Palmeri S et al. A randomized controlled trial assessing the prevention of doxorubicin cardiomyopathy by N-acetylcysteine. Semin Oncol. 1983;10(Suppl 1):53–5. [PubMed] [Google Scholar]
- 19.Martin E, Thougaard AV, Grauslund M et al. Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology. 2009;255:72–9. doi: 10.1016/j.tox.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhang S, Liu X, Bawa-Khalfe T et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–42. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 21.Lyu YL, Kerrigan JE, Lin CP et al. Topoisomerase IIbeta-mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–46. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 22.Li M, Sala V, De Santis MC et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. 2018;138:696–711. doi: 10.1161/CIRCULATIONAHA.117.030352. [DOI] [PubMed] [Google Scholar]
- 23.Braverman AC, Antin JH, Plappert MT et al. Cyclophosphamide cardiotoxicity in bone marrow transplantation: a prospective evaluation of new dosing regimens. J Clin Oncol. 1991;9:1215–23. doi: 10.1200/JCO.1991.9.7.1215. [DOI] [PubMed] [Google Scholar]
- 24.Quezado ZM, Wilson WH, Cunnion RE et al. High-dose ifosfamide is associated with severe, reversible cardiac dysfunction. Ann Intern Med. 1993;118:31–6. doi: 10.7326/0003-4819-118-1-199301010-00006. [DOI] [PubMed] [Google Scholar]
- 25.Kurauchi K, Nishikawa T, Miyahara E et al. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res Notes. 2017;10:406. doi: 10.1186/s13104-017-2726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Focaccetti C, Bruno A, Magnani E et al. Effects of 5-fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PloS One. 2015;10:e0115686. doi: 10.1371/journal.pone.0115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Layoun ME, Wickramasinghe CD, Peralta MV, Yang EH. Fluoropyrimidine-induced cardiotoxicity: manifestations, mechanisms, and management. Curr Oncol Rep. 2016;18:35. doi: 10.1007/s11912-016-0521-1. [DOI] [PubMed] [Google Scholar]
- 28.Veinot JP, Edwards WD. Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum Pathol. 1996;27:766–73. doi: 10.1016/S0046-8177(96)90447-5. [DOI] [PubMed] [Google Scholar]
- 29.Sag CM, Wolff HA, Neumann K et al. Ionizing radiation regulates cardiac Ca handling via increased ROS and activated CaMKII. Basic Res Cardiol. 2013;108:385. doi: 10.1007/s00395-013-0385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paris F, Fuks Z, Kang A et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 31.Orzan F, Brusca A, Conte MR et al. Severe coronary artery disease after radiation therapy of the chest and mediastinum: clinical presentation and treatment. Br Heart J. 1993;69:496–500. doi: 10.1136/hrt.69.6.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darby SC, Ewertz M, McGale P et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med. 2013;368:987–98. doi: 10.1056/NEJMoa1209825. [DOI] [PubMed] [Google Scholar]
- 33.Saiki H, Petersen IA, Scott CG et al. Risk of Heart Failure With Preserved Ejection Fraction in Older Women After Contemporary Radiotherapy for Breast Cancer. Circulation. 2017;135:1388–96. doi: 10.1161/CIRCULATIONAHA.116.025434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–71. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 35.Slamon DJ, Leyland-Jones B, Shak S et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 36.Suter TM, Procter M, van Veldhuisen DJ et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J Clin Oncol. 2007;25:3859–65. doi: 10.1200/JCO.2006.09.1611. [DOI] [PubMed] [Google Scholar]
- 37.Slamon D, Eiermann W, Robert N et al. Adjuvant trastuzumab in HER2-positive breast cancer. N EngL J Med. 2011;365:1273–83. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crone SA, Zhao YY, Fan L et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–65. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 39.De Keulenaer GW, Doggen K, Lemmens K. The vulnerability of the heart as a pluricellular paracrine organ: lessons from unexpected triggers of heart failure in targeted ErbB2 anticancer therapy. Circ Res. 2010;106:35–46. doi: 10.1161/CIRCRESAHA.109.205906. [DOI] [PubMed] [Google Scholar]
- 40.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–10. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 41.Elice F, Jacoub J, Rickles FR et al. Hemostatic complications of angiogenesis inhibitors in cancer patients. Am J Hematol. 2008;83:862–70. doi: 10.1002/ajh.21277. [DOI] [PubMed] [Google Scholar]
- 42.Schmidinger M, Zielinski CC, Vogl UM et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26:5204–12. doi: 10.1200/JCO.2007.15.6331. [DOI] [PubMed] [Google Scholar]
- 43.Cobleigh MA, Langmuir VK, Sledge GW et al. A phase I/II dose-escalation trial of bevacizumab in previously treated metastatic breast cancer. Semin Oncol. 2003;30(Suppl 16):117–24. doi: 10.1053/j.seminoncol.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 44.Kappers MH, van Esch JH, Sluiter W et al. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56:675–81. doi: 10.1161/HYPERTENSIONAHA.109.149690. [DOI] [PubMed] [Google Scholar]
- 45.Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol. 1998;274(Pt 2):H1054–8. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- 46.Li W, Croce K, Steensma DP et al. Vascular and metabolic implications of novel targeted cancer therapies: focus on kinase inhibitors. J Am Coll Cardiol. 2015;66:1160–78. doi: 10.1016/j.jacc.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 47.Maurea N, Coppola C, Piscopo G et al. Pathophysiology of cardiotoxicity from target therapy and angiogenesis inhibitors. J Cardiovasc Med (Hagerstown) 2016;17(Suppl 1):S19–26. doi: 10.2459/JCM.0000000000000377. [DOI] [PubMed] [Google Scholar]
- 48.Richardson PG, Sonneveld P, Schuster MW et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N EngL J Med. 2005;352:2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 49.Waxman AJ, Clasen S, Hwang WT et al. Carfilzomib-associated cardiovascular adverse events: a systematic review and meta-analysis. JAMA Oncol. 2018;4:e174519. doi: 10.1001/jamaoncol.2017.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herrmann J, Wohlert C, Saguner AM et al. Primary proteasome inhibition results in cardiac dysfunction. Eur J Heart Fail. 2013;15:614–23. doi: 10.1093/eurjhf/hft034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–96. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 52.Falk RH, Alexander KM, Liao R, Dorbala S. AL (light-chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68:1323–41. doi: 10.1016/j.jacc.2016.06.053. [DOI] [PubMed] [Google Scholar]
- 53.Gertz MA, Benson MD, Dyck PJ et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66:2451–66. doi: 10.1016/j.jacc.2015.09.075. [DOI] [PubMed] [Google Scholar]
- 54.Kyle RA, Linos A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817–22. [PubMed] [Google Scholar]
- 55.Quarta CC, Solomon SD, Uraizee I et al. Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation. 2014;129:1840–9. doi: 10.1161/CIRCULATIONAHA.113.006242. [DOI] [PubMed] [Google Scholar]
- 56.Brenner DA, Jain M, Pimentel DR et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94:1008–10. doi: 10.1161/01.RES.0000126569.75419.74. [DOI] [PubMed] [Google Scholar]
- 57.Lundin L, Norheim I, Landelius J et al. Carcinoid heart disease: relationship of circulating vasoactive substances to ultrasound-detectable cardiac abnormalities. Circulation. 1988;77:264–9. doi: 10.1161/01.CIR.77.2.264. [DOI] [PubMed] [Google Scholar]
- 58.Modlin IM, Sandor A. An analysis of 8305 cases of carcinoid tumors. Cancer. 1997;79:813–29. doi: 10.1002/(SICI)1097-0142(19970215)79:4<813::AID-CNCR19>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 59.Hassan SA, Banchs J, Iliescu C et al. Carcinoid heart disease. Heart. 2017;103:1488–95. doi: 10.1136/heartjnl-2017-311261. [DOI] [PubMed] [Google Scholar]
- 60.Karlstaedt A, Zhang X, Vitrac H et al. Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci U S A. 2016;113:10436–41. doi: 10.1073/pnas.1601650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sipahi I, Debanne SM, Rowland DY et al. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol. 2010;11:627–36. doi: 10.1016/S1470-2045(10)70106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ponikowski P, Voors AA, Anker SD et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18:891–975. doi: 10.1002/ejhf.592. [DOI] [PubMed] [Google Scholar]
- 63.Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15:563–572. doi: 10.1038/nrc3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer. 2010;10:745–59. doi: 10.1038/nrc2945. [DOI] [PubMed] [Google Scholar]
- 65.Kasama S, Toyama T, Kumakura H et al. Effects of candesartan on cardiac sympathetic nerve activity in patients with congestive heart failure and preserved left ventricular ejection fraction. J Am Coll Cardiol. 2005;45((5)):661–7. doi: 10.1016/j.jacc.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 66.Grassi G, Seravalle G, Quarti-Trevano F et al. Sympathetic and baroreflex cardiovascular control in hypertension-related left ventricular dysfunction. Hypertension. 2009;53:205–9. doi: 10.1161/HYPERTENSIONAHA.108.121467. [DOI] [PubMed] [Google Scholar]
- 67.Levine B, Kalman J, Mayer L et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 68.Testa M, Yeh M, Lee P et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J Am Coll Cardiol. 1996;28:964–71. doi: 10.1016/S0735-1097(96)00268-9. [DOI] [PubMed] [Google Scholar]
- 69.Torre-Amione G, Kapadia S, Lee J et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93:704–11. doi: 10.1161/01.CIR.93.4.704. [DOI] [PubMed] [Google Scholar]
- 70.Damas JK, Eiken HG, Oie E et al. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc Res. 2000;47:778–87. doi: 10.1016/S0008-6363(00)00142-5. [DOI] [PubMed] [Google Scholar]
- 71.Libby P, Ridker PM, Hansson GK. Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–38. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Edelmann F, Holzendorf V, Wachter R et al. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail. 2015;17:214–23. doi: 10.1002/ejhf.203. [DOI] [PubMed] [Google Scholar]
- 73.Matsubara J, Sugiyama S, Nozaki T et al. Pentraxin 3 is a new inflammatory marker correlated with left ventricular diastolic dysfunction and heart failure with normal ejection fraction. J Am Coll Cardiol. 2011;57:861–9. doi: 10.1016/j.jacc.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 74.Sciarretta S, Ferrucci A, Ciavarella GM et al. Markers of inflammation and fibrosis are related to cardiovascular damage in hypertensive patients with metabolic syndrome. Am J Hypertens. 2007;20:784–91. doi: 10.1016/j.amjhyper.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 75.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 76.Ridker PM, Everett BM, Thuren T et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 77.Everett BM, Cornel J, Lainscak M Anti-Inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2018. epub ahead of press. [DOI] [PubMed]
- 78.Ridker PM, MacFadyen JG, Thuren T et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–42. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 79.Mann DL, McMurray JJ, Packer M et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 80.Chung ES, Packer M, Lo KH et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 81.Hirota H, Chen J, Betz UA et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–98. doi: 10.1016/S0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 82.Gullestad L, Aukrust P. Review of trials in chronic heart failure showing broad-spectrum anti-inflammatory approaches. Am J Cardiol. 2005;95 doi: 10.1016/j.amjcard.2005.03.008. 17C23C; Discussion 38C–40C. [DOI] [PubMed] [Google Scholar]
- 83.Moro-Garcia MA, Echeverria A, Galan-Artimez MC et al. Immunosenescence and inflammation characterize chronic heart failure patients with more advanced disease. Int J Cardiol. 2014;174:590–9. doi: 10.1016/j.ijcard.2014.04.128. [DOI] [PubMed] [Google Scholar]
- 84.Pawelec G, Derhovanessian E, Larbi A. Immunosenescence and cancer. Crit Rev Oncol Hematol. 2010;75:165–72. doi: 10.1016/j.critrevonc.2010.06.012. [DOI] [PubMed] [Google Scholar]