

Abstract
Nonalcoholic fatty liver disease is currently the most common liver disease and is a leading cause of liver‐related morbidity and mortality. However, its pathogenesis remains largely unclear. We previously showed that mice deficient in mitogen‐activated protein kinase (MAPK) phosphatase 5 (MKP5) spontaneously developed insulin resistance and glucose intolerance, which are associated with visceral obesity and adipose tissue inflammation. In this study, we discovered that mice deficient in MKP5 developed more severe hepatic steatosis and steatohepatitis with age or with feeding on a high‐fat diet (HFD) compared to wild‐type (WT) mice, and this was associated with increased expression of proinflammatory cytokines and collagen genes. Increased p38 activation in MKP5 knockout (KO) liver compared to that in WT liver was detected, which contributed to increased expression of lipid droplet‐associated protein cell death‐inducing DFF45‐like effector A (CIDEA) and CIDEC/fat‐specific protein 27 but not CIDEB through activating transcription factor 2 (ATF2). In addition, MKP5 KO liver had higher peroxisome proliferator‐activated receptor gamma (PPARγ) expression compared with WT liver. On the other hand, overexpression of MKP5 or inhibition of p38 activation in hepatocytes resulted in reduced expression of PPARγ. Inhibition of p38 resulted in alleviation of hepatic steatosis in KO liver in response to HFD feeding, and this was associated with reduced expression of CIDEA, CIDEC, and proinflammatory cytokines. Conclusion: MKP5 prevents the development of nonalcoholic steatohepatitis by suppressing p38–ATF2 and p38–PPARγ to reduce hepatic lipid accumulation, inflammation, and fibrosis.
Abbreviations
- ATF2
activating transcription factor 2
- BDL
bile duct ligation
- cDNA
complementary DNA
- CIDE
cell death‐inducing DFF45‐like effector
- Col
collagen
- DAG
diacylglycerol
- DEN
diethylnitrosamine
- ERK
extracellular signal‐regulated kinase
- FSP
fat‐specific protein
- H&E
hematoxylin and eosin
- HCC
hepatocellular carcinoma
- HFD
high‐fat diet
- HSP
heat shock protein
- IL
interleukin
- JNK
c‐Jun N‐terminal kinase
- KO
knockout
- LD
lipid droplet
- MAPK
mitogen‐activated protein kinase
- MCP‐1
monocyte chemoattractant protein 1
- MKP
MAPK phosphatase
- MKP5mut
MKP5 phosphatase‐dead mutant
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NEFA
nonesterified fatty acid
- PA
palmitic acid
- pATF2
phosphorylated activating transcription factor 2
- PBS
phosphate‐buffered saline
- PPARγ
peroxisome proliferator‐activated receptor gamma
- ROS
reactive oxygen species
- RT‐qPCR
real‐time quantitative polymerase chain reaction
- TAA
thioacetamide
- TG
triglyceride
- TNFα
tumor necrosis factor alpha
- WAT
white adipose tissue
- WT
wild type
Nonalcoholic fatty liver disease (NAFLD), ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), is the most common cause of chronic liver disease in developed countries and affects up to one third of the world population.1, 2, 3 Liver steatosis is characterized by accumulation of excessive hepatocellular lipid droplets (LDs) in patients in the absence of other causes of chronic liver diseases, including alcohol, virus, drugs, and autoimmunity. NAFLD can progress from hepatic steatosis to steatohepatitis, cirrhosis, and hepatocellular carcinoma (HCC). The majority of cases of NAFLD are associated with obesity, insulin resistance, and type 2 diabetes; NAFLD in turn increases the risk of type 2 diabetes, cardiovascular and cardiac disease, and chronic kidney disease.1, 3 The primary event of NAFLD is the accumulation of triglycerides (TGs) in hepatocytes in the form of LDs1; this leads to cellular stress and hepatic injury and eventually results in chronic disease. LDs are spherical organelles composed of a core of neutral lipids, mainly TGs and sterol esters, covered by a monolayer of phospholipids, free cholesterol, and specific proteins.2, 4 Accumulating evidence demonstrates that LDs are bioactive organelles with functions beyond mere lipid storage in hepatocytes. There are distinct populations of LDs that differ in their lipid and protein composition and are targeted for lipolysis, secretion in the form of very low‐density lipoprotein, or long‐term storage of lipids.2, 5 The biogenesis and growth, function, and fate of these multifunctional LDs are highly regulated and are integrated within the hepatocellular machinery. Hepatic steatosis is thought to result from dysregulation of the lipid homeostatic process.2
LD‐associated proteins, such as members of the perilipin family of proteins and cell death‐inducing DFF45‐like effectors (CIDEs), play important roles in lipid metabolism and participate in the pathogenic processes of metabolic disorders, including insulin resistance and hepatic steatosis.6 For instance, the expression of perilipin 2 (PLIN2), a member of the perilipin family of proteins, is elevated in human fatty livers, and deficiency of PLIN2 in mice resulted in resistance to diet‐induced fatty liver development, which was associated with reduced hepatic TG accumulation.7, 8 The levels of CIDEA and CIDEC/fat‐specific protein 27 (FSP27) are markedly up‐regulated in steatotic livers and are strongly correlated with the severity of hepatic steatosis in humans.9 In mice, their expression in the liver is correlated with the development of hepatic steatosis. Deficiency of CIDEA or CIDEC/FSP27 in mice resulted in decreased hepatic TG levels and resistance to diet‐induced or genetically mediated hepatic steatosis.9, 10
Mitogen‐activated protein kinase (MAPK) phosphatases (MKPs), also known as dual‐specificity phosphatases (DUSPs), are major negative regulators of MAPKs, including extracellular signal‐regulated kinase (ERK), c‐Jun N‐terminal kinase (JNK), and p38. Accumulating evidence demonstrates that MKPs play important roles in metabolic processes. For instance, it has been shown that mice deficient in MKP1 are resistant to diet‐induced obesity due to increased energy expenditure.11 MKP1 knockout (KO) mice were also resistant to diet‐induced hepatic steatosis, which was associated with increased β‐oxidation of fatty acid in the liver. In addition, it has been observed that the expression of MKP1 is correlated with the development of atherosclerotic lesions and hypercholesterolemia in mice.12 However, its function in atherosclerosis is unknown. MKP3 is another MKP member that plays an important role in metabolism. Interestingly, MKP3 was found to regulate the activity of forkhead box O1 (FOXO1) in hepatic gluconeogenesis.11 It dephosphorylates FOXO1 to promote its nuclear translocation, thereby regulating the expression of gluconeogenic genes. In addition, mice deficient in MKP3 were found to be resistant to diet‐induced obesity and hepatic steatosis, and this was associated with improved systemic insulin sensitivity.13 We recently showed that MKP5 is beneficial in maintaining adipose tissue homeostasis by preventing the development of metabolic disorders.14 Deletion of MKP5 in mice resulted in progressive development of systemic insulin resistance, glucose intolerance, and visceral obesity, which are all associated with the development of adipose tissue inflammation. Here, we report that MKP5 inhibits p38 activation, the expression of CIDEA and CIDEC/FSP27, and the development of inflammation in the liver to prevent development of aging‐associated and diet‐induced NAFLD.
Materials and Methods
Animal Experiments
All animal protocols were approved by the Animal Care and Use Committee of the National University of Singapore. All mice under a C57BL6/J genetic background were housed under a 12‐hour:12‐hour light–dark cycle and an ambient temperature of 22°C and allowed free access to water and food. Mkp5 –/– mice generated previously were crossed with C57BL6/J mice for 12 generations. For diet studies, 7‐ to 9‐week‐old C57BL6/J mice were fed a diet containing 35% lard (TD.03584) purchased from Harlan Laboratories.
Cell Culture
Mouse FL83B hepatocytes (American Type Culture Collection) were cultured in F12K nutrient medium (Invitrogen) supplemented with 10% fetal bovine serum and 1% antibiotics. For p38 inhibitor treatment, cells were pretreated with 20 μM SB203580 for 1 hour, followed by 9 hours of palmitic acid (PA) stimulation.
Histology
Paraffin‐embedded liver sections (5 μm) were deparaffinized and stained with hematoxylin and eosin (H&E) before mounting with mounting medium (Invitrogen). For Oil Red O staining, 6‐μm‐thick frozen sections were cut and stained with Oil Red O (Sigma). Digital images were captured on a light microscope (Olympus). To assess hepatic fibrosis, liver tissue samples were fixed with 2% paraformaldehyde in phosphate‐buffered saline (PBS) with 30% sucrose at 4 °C overnight, washed with PBS for 2 days at 4℃, and then embedded in optimum cutting temperature compound (Sakura FineTek, Singapore). Sections 5 μm thick were then cut on a cryostat (Leica, Singapore) and stained with Masson’s trichrome stains (Sigma‐Aldrich) using the standard protocol. Quantification of Masson’s trichrome staining was done by ImageJ software.
Lipid Test
Serum TGs were measured using the Triglyceride Determination Kit (Sigma) based on the manufacturer’s instructions. Cholesterols were measured using the Cholesterol Determination Kit (Biovision). Nonesterified fatty acids (NEFAs) were measured by the NEFA Determination Kit (Wako Chemicals). Lipids from liver tissues were extracted using the methanol–chloroform method. Snap‐frozen liver fragments of equal weight were homogenized in nine volumes of PBS, and 200 μL of the homogenate was transferred into 1,200 μL of chloroform:methanol (2:1 volume per volume). Then, 100 μL of PBS was added, mixed, and centrifuged at 4,200g for 10 minutes at 4°C; 200 μL of the organic phase was then transferred into a tube and left to dry in the fume hood. The dried lipid residue was resuspended in 1% Triton X‐100 (Sigma) in absolute ethanol. Levels of hepatic TGs, cholesterol, and NEFAs were determined with the respective determination kit.
Chromatin Immunoprecipitation
MKP5‐overexpressed mouse hepatocytes FL83B and control cells were stimulated with PA for 9 hours and crosslinked with formaldehyde at a final concentration of 1% for 10 minutes, followed by quenching with glycine. Cell lysates were fragmented by sonication and precleared with protein G Dynabeads and subsequently precipitated with anti‐phosphor activating transcription factor 2 (pATF2) antibody (Santa Cruz) or normal rabbit immunoglobulin G (Santa Cruz) overnight at 4°C. After washing and elution, crosslink reversal was done by incubating at 65°C for 8 hours. The eluted DNA was purified and analyzed by real‐time polymerase chain reaction (RT‐PCR) with primers specific to Cidea and Fsp27 promoters.
Statistical Analysis
Data are presented as mean ± SEM. Statistical significance of the differences between groups was determined by the two‐tailed unpaired Student t test with GraphPad Prism software. Other materials and methods are described in Supporting Materials and Methods.
Results
Development of Steatohepatitis in MKP5 KO Mice With Aging
We have shown that mice deficient in MKP5 developed insulin resistance at the age of 3 months and glucose intolerance at approximately 5 months; this was associated with increased inflammatory macrophage infiltration in perigonadal adipose tissue and increased adipose tissue inflammation.14 To further understand the role of MKP5 in metabolism, we continuously monitored the metabolic phenotypes of the mice. We observed that livers from MKP5 KO mice were heavier from the age of 11 months and appeared to be paler than livers from wild‐type (WT) mice (Fig. 1A,B). H&E staining of WT and KO liver sections demonstrated the development of microvesicular steatosis in the KO liver but not in the WT liver (Fig. 1C), which is consistent with increased liver TG contents in the KO mice (Fig. 1D). Masson’s trichrome staining of liver sections of WT and KO mice at the age of 14 months showed increased fibrosis in the KO liver (Fig. 1E). In addition, expression of inflammatory genes, including tumor necrosis factor alpha (Tnfα) and monocyte chemoattractant protein 1 (Mcp‐1), and collagen genes, including Col1a1, Col3a1, Col6a1 and Col6a2, were elevated in the liver of KO mice (Fig. 1F). The expression of these genes was not increased in the liver of KO mice compared to that in WT mice at the age of 3 months (Supporting Fig. S1). Together, these results demonstrated the development of steatohepatitis with aging in MKP5 KO mice.
Figure 1.
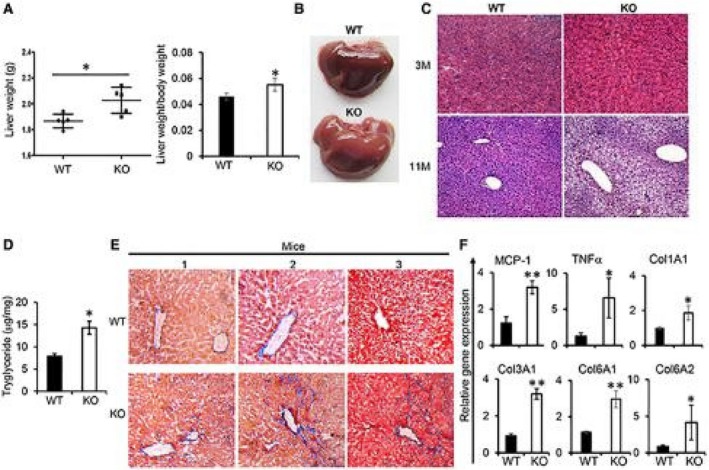
Deficiency of MKP5 in mice resulted in the development of steatohepatitis in aged mice. (A) The weight of the liver from WT (n = 5) and MKP5 KO (n = 5) male mice at the age of 11 months on a chow diet was determined. (B,C) Representative images of WT and KO mice (B) at the age of 11 months and (C) H&E staining of sections of liver at 3 (3M) and 11 (11M) months showed the development of hepatic steatosis in the KO mice. (D) Hepatic TG in WT and MKP5 KO livers was measured using the Triglyceride Determination Kit (Sigma). (E) Representative pictures of Masson’s trichrome staining of liver sections of WT and KO mice at the age of 14 months. (F) RT‐qPCR examination of proinflammatory cytokines and collagen genes in the liver of WT and KO mice at the age of 14 months. The data shown are representative of three to four independent experiments with similar results. (A,D) Data are presented as mean ± SEM. *P < 0.05; **P < 0.01.
MKP5‐Deficient Mice are More Susceptible to Diet‐Induced Hepatic Steatosis
A high‐fat diet (HFD) accelerates the development of obesity and obesity‐associated metabolic disorders, such as hepatic steatosis. To facilitate the study on the role of MKP5 in the pathogenesis of hepatic steatosis, WT and KO mice were fed with an HFD following weaning. Increased susceptibility to HFD‐induced metabolic disorders, including insulin resistance and glucose intolerance, was observed in MKP5 KO mice. Consistent with our previous finding,14 MKP5 KO mice developed insulin resistance but not glucose intolerance at approximately 3 months on a chow diet (Fig. 2A). With HFD feeding for 8 weeks, KO mice developed more severe insulin resistance and glucose intolerance compared to WT mice (Fig. 2A), indicating the requirement of MKP5 for the animals to cope with excessive nutrition.
Figure 2.
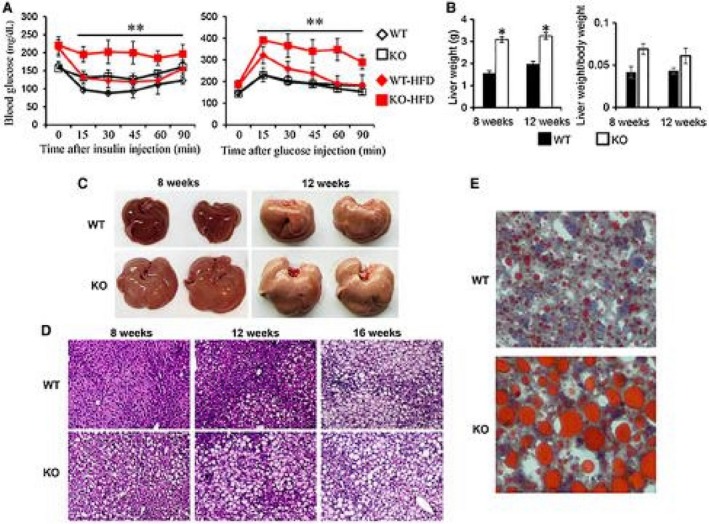
MKP5 KO mice are more susceptible to HFD‐induced hepatic steatosis. WT (n = 5) and MKP5 KO mice were fed with an HFD (TD03584, Harlan) for 8 weeks after weaning. (A) Insulin resistance and glucose tolerance tests were performed. (B) The weight of livers from WT (n = 3‐5) and MKP5 KO (n = 3‐5) mice after 8 or 12 weeks of HFD feeding was measured. (C) Representative images showing livers from WT and MKP5 KO mice after 8 or 12 weeks of HFD feeding. (D) Representative micrographs showing H&E staining on liver sections of WT and MKP5 KO mice receiving the HFD for 8, 12, or 16 weeks. (E) Representative micrographs showing Oil Red O staining on liver sections from WT and MKP5 KO mice receiving 12 weeks of the HFD. Scale bar denotes 100 μM. Data shown are representative of three independent experiments with similar results. (A,B) Data are presented as mean ± SEM. *P < 0.05; **P < 0.01.
In addition, a dramatic increase in liver weight in KO mice compared to WT mice was detected after 8 weeks of HFD feeding (Fig. 2B). In WT mice, gross morphology of the liver appeared to be normal after 8 weeks of HFD feeding (Fig. 2C). H&E staining of WT liver sections showed no development of steatosis at this stage (Fig. 2D). Steatosis was observed in WT mice after 12 weeks of the HFD, and the degree of steatosis appeared more profound at 16 weeks (Fig. 2D,E). In contrast, obvious steatosis in KO mice was observed after 8 weeks of the HFD and was more severe at 12 and 16 weeks compared to WT mice (Fig. 2C,D). Oil Red O staining of liver sections from mice on the HFD for 12 weeks revealed accumulation of small LDs in WT hepatocytes (Fig. 2E). In contrast, accumulation of larger LDs was observed in KO hepatocytes. Collectively, these results demonstrate that MKP5 KO mice are more susceptible to the development of diet‐induced hepatic steatosis than WT mice.
Altered Hepatic Lipid Metabolism in MKP5 KO Mice is Associated With Inflammation in the Liver
To further understand the effect of MKP5 deficiency on lipid metabolism, we measured NEFAs, TGs, and cholesterol in both serum and the liver from WT and MKP5 KO mice after HFD feeding. Serum levels of NEFAs and TGs were comparable between WT and MKP5 KO mice following HFD feeding for 8, 12, or 16 weeks (Fig. 3A). Significantly increased levels of total cholesterol in serum from KO mice were detected compared to serum from WT mice. On the other hand, significantly increased levels of hepatic TGs, total cholesterol, and NEFAs were detected in KO mice compared to levels in WT mice after the HFD for 8, 12, or 16 weeks (Fig. 3B). A comprehensive lipidomic analysis, which included glycerophospholipids, sphingolipids, and neutral lipids, of the extracts from WT and KO livers showed that the difference in lipid content between the two conditions was mainly due to accumulation of TGs, diacylglycerols (DAGs), and cholesterol esters in KO mice (Fig. 3C). The molecular species that are significantly different in each of these lipid classes contain short saturated or monounsaturated acyl chains as well as long polyunsaturated chains (Supporting Fig. S2). Other lipid classes were affected but only in specific molecular species, as shown in the figure. Therefore, the development of hepatic steatosis in MKP5 KO mice is in line with altered hepatic lipid metabolism, which may in turn contribute to the development of hepatic steatosis.
Figure 3.
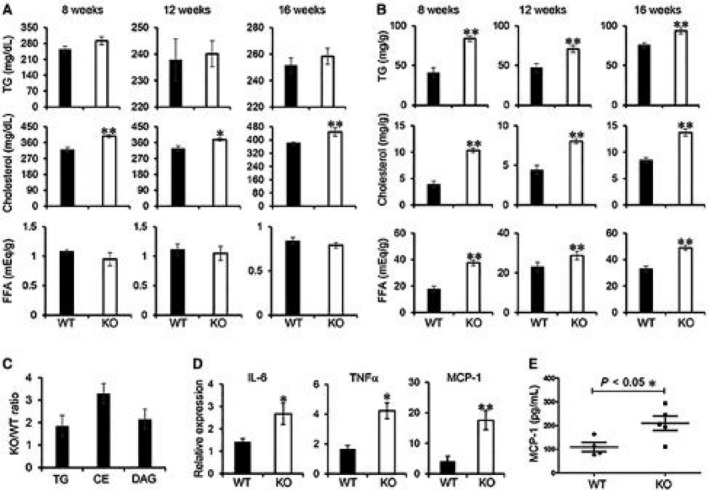
Altered lipid metabolism in MKP5 KO liver is associated with hepatic inflammation. (A,B) Concentrations of TGs, total cholesterol, and FFAs in the (A) serum and (B) liver of WT and MKP5 KO mice fed with the HFD for 8, 12, or 16 weeks were determined using the Triglyceride Determination Kit (Sigma), Cholesterol Determination Kit (Biovision), and NEFA Determination Kit (Wako Chemicals), respectively. (C) Lipidomic analysis of liver extracts in WT (n = 4) and KO (n = 4) mice after 12 weeks of HFD feeding showed accumulation of total TGs, DAGs, and CEs in KO livers. The ratios of TGs, CEs, and DAGs between KO and WT livers are shown. (D) Expression of proinflammatory cytokine genes, including IL‐6, TNFα, and MCP‐1, in the liver from mice fed with the HFD for 12 weeks was determined by qPCR. (E) Serum concentrations of MCP‐1 in WT (n = 4) and MKP5 KO (n = 5) mice after 12 weeks of HFD feeding were determined by enzyme‐linked immunosorbent assay. Data shown are representative of three to four independent experiments with similar results. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01. Abbreviations: CE, cholesterol ester; FFA, free fatty acid.
Next, we tested if enhanced lipid accumulation in MKP5 KO liver leads to the development of NASH. To address this, we examined the expression of proinflammatory cytokines, including interleukin‐6 (IL‐6), TNFα, and MCP‐1, in both WT and KO livers following the HFD for 12 weeks. The expression of all three cytokines was increased in KO livers compared to WT livers (Fig. 3D). Furthermore, serum MCP‐1 levels were significantly higher in KO mice than in WT mice (Fig. 3E). These results demonstrate the development of liver inflammation on top of steatosis in MKP5 KO mice compared to WT mice, suggesting that MKP5 is protective in the pathogenesis of NASH.
MKP5 Inactivates P38 Mapk in the Liver
To evaluate the substrate(s) of MKP5 in the liver, we examined activation of ERK, JNK, and p38 in livers from WT and KO mice. Increased activation of p38 in KO livers compared to WT livers at the age of 11 months on the chow diet was detected (Supporting Fig. S3). Activation of ERK and JNK was comparable between WT and KO mice. Consistently following HFD feeding, significantly increased phosphorylation of p38 but not JNK or ERK was detected in the liver from KO mice compared to WT mice (Fig. 4A). Together, these results demonstrate that p38 is the main target of MKP5 in the liver.
Figure 4.
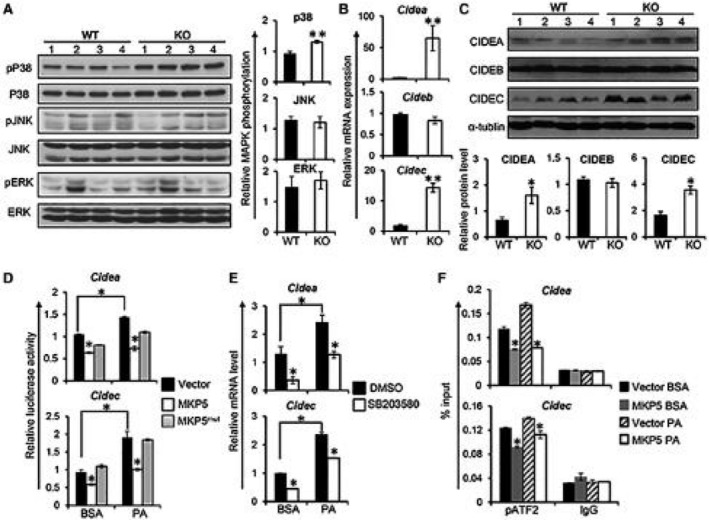
MKP5 regulates CIDEA and CIDEC/FSP27 in the liver through p38. (A) Representative western blots showing the activation of MAPKs, including p38, JNK, and ERK, in the liver from WT and MKP5 KO mice fed with the HFD for 12 weeks. Quantification of immunoblots was performed using ImageJ software. (B) mRNA expression of Cidea, Cideb, and Cidec/Fsp27 in the liver of WT and MKP5 KO mice following HFD feeding was determined by qPCR. (C) Protein levels of CIDEA, CIDEB, and CIDEC/FSP27 in the liver of WT and MKP5 KO mice were examined by western blots. (D) Overexpression of MKP5 inhibits Cidea and Cidec promoter activity. The Cidea or Cidec promoter region was cloned into a pGL3 luciferase vector and was separately cotransfected with MKP5 or MKP5mut together with a pRL reporter vector containing a cDNA encoding Renilla luciferase renilla luciferase‐null vector into FL83B hepatocytes for a dual‐luciferase assay after stimulation with or without PA. (E) p38 inhibition reduced the expression of Cidea or Cidec but not Cideb in mouse hepatocytes. FL83B mouse hepatocytes were pretreated with p38‐specific inhibitor SB203580 (40 μM) for 1 hour, followed by 500 μM of PA stimulation for 9 hours. Cide gene expression was examined by qPCR. (F) MKP5 attenuates pATF2 binding to the promoter of Cidea or Cidec/Fsp27. Mouse hepatocytes with or without MKP5 overexpression were treated with BSA or PA for 9 hours. Cells were harvested for the chromatin immunoprecipitation assay using pATF2 antibody to examine its binding to Cide promoters. The promoter region of Cidea or Cidec/Fsp27 associated with pATF2 was quantified by qPCR. Data shown are representative of three independent experiments with similar results. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01. Abbreviations: BSA, bovine serum albumin; DMSO, dimethyl sulfoxide.
Increased Expression of CIDEA and CIDEC/FSP27 in the Liver From MKP5 KO Mice
To further understand the regulatory role of MKP5 in the liver, we performed a transcriptome analysis of WT and KO livers from mice fed with the HFD for 12 weeks. Pathway analysis showed that p38 pathway genes or genes in ATF2 pathway, which is a major target of p38, were enriched in KO livers (Supporting Fig. S4); this is consistent with the increased activation of p38 in KO livers after HFD feeding compared to WT livers (Fig. 4A). In addition, genes associated with inflammatory cytokine pathways, such as IL‐6 and TNFα, were also found to be enriched in KO livers (Supporting Fig. S4), which correlated with the increased expression of proinflammatory cytokines in the KO liver (Fig. 3C), suggesting the development of liver inflammation in KO mice.
Next, we analyzed the genes that are up‐regulated in the KO liver. The CIDEA gene was identified among the top up‐regulated genes (Supporting Fig. S5). Members of the CIDE protein family, including CIDEA, CIDEB, and CIDEC (also known as FSP27), play important roles in the development of hepatic steatosis.15 Therefore, we examined the expression of Cide genes in both WT and KO livers after HFD feeding. Interestingly, increased expression of CIDEA and CIDEC/FSP27, but not CIDEB, at both messenger RNA (mRNA) and protein levels in the liver of MKP5 KO mice compared with WT mice was detected (Fig. 4B,C), suggesting that MKP5 specifically inhibits the expression of CIDEA and CIDEC/FSP27 during the development of hepatic steatosis.
To further understand the regulation of CIDEA and CIDEC/FSP27 by MKP5, we made luciferase constructs in which the expression of firefly luciferase is controlled by the promoter of Cidea or Cidec/Fsp27. The Cidea or Cidec/Fsp27 luciferase construct was then cotransfected with the full length of MKP5 complementary DNA (cDNA) or MKP5 phosphatase‐dead mutant (MKP5mut) cDNA into FL83B cells, a mouse liver cell line, to perform a dual‐luciferase assay with or without PA stimulation. Overexpression of MKP5 inhibited Cidea and Cidec/Fsp27 promoter activity (Fig. 4D). In contrast, MKP5mut was unable to suppress the promoter activity of Cidea or Cidec/Fsp27 (Fig. 4D). Together, these results demonstrate that MKP5 inhibits Cidea and Cidec/Fsp27 expression, which is dependent on its phosphatase activity.
MKP5 Regulates CIDEA and CIDEC/FSP27 Expression Through P38
Next, we examined if negative regulation of CIDEA and CIDEC/FSP27 by MKP5 is through its regulation of p38. To address this, we first pretreated FL83B cells with p38 inhibitor SB203580, followed by stimulation with PA, to examine the expression of Cide genes. The results showed that SB203580 treatment suppressed the activation of ATF2 (Supporting Fig. S6A), indicating that activation of p38 was inhibited. Expression of both Cidea and Cideac/Fsp27 was increased in hepatocytes in response to PA stimulation, and inhibition of p38 significantly inhibited the expression of both genes with or without PA stimulation (Fig. 4E). We verified this observation with another p38 inhibitor, SB20219. Inhibition of p38 activation with SB20219 also resulted in inhibition of Cidea and Cideac/Fsp27 expression (Supporting Fig. S6B). These results demonstrate that p38 regulates the expression of Cidea and Cideac/Fsp27 in hepatocytes, suggesting that MKP5 regulates the expression of Cidea and Cideac/Fsp27 through p38.
To substantiate these results, we generated MKP5 overexpressing FL83B cells (Supporting Fig. S6C). A chromatin immunoprecipitation assay was carried out to examine the binding of pATF2, a major target of p38, to the promoter of Cidea or Cidec/Fsp27 in FL83B cells with or without MKP5 overexpression. pATF2 binds to the promoter of both Cidea and Cideac/Fsp27 in FL83B cells, and this binding was enhanced by PA stimulation (Fig. 4F). Overexpression of MKP5 significantly reduced the binding of pATF2 to the promoter of Cidea or Cideac/Fsp27 with or without stimulation (Fig. 4F). Together, these data demonstrate that MKP5 inhibits the expression of Cidea and Cidec/Fsp27 through negative regulation of p38 activation.
P38–Peroxisome Proliferator‐Activated Receptor Gamma Axis Contributes to the Regulation of CIDE Genes by MKP5
Studies have revealed that CIDEA and CIDEC/FSP27 are direct mediators of peroxisome proliferator‐activated receptor gamma (PPARγ)‐dependent hepatic steatosis.10, 16 Interestingly, we found that both mRNA and protein expression of PPARγ was increased in KO livers compared to WT livers in response to the HFD (Fig. 5A), suggesting that MKP5 regulates hepatic PPARγ expression. To test the regulation of PPARγ expression by MKP5, we made a PPARγ promoter luciferase construct and cotransfected it with MKP5 cDNA into FL83B hepatocytes for the luciferase assay. MKP5 expression inhibited PA‐induced PPARγ promoter activity, indicating that MKP5 negatively regulates PPARγ expression (Fig. 5B).
Figure 5.
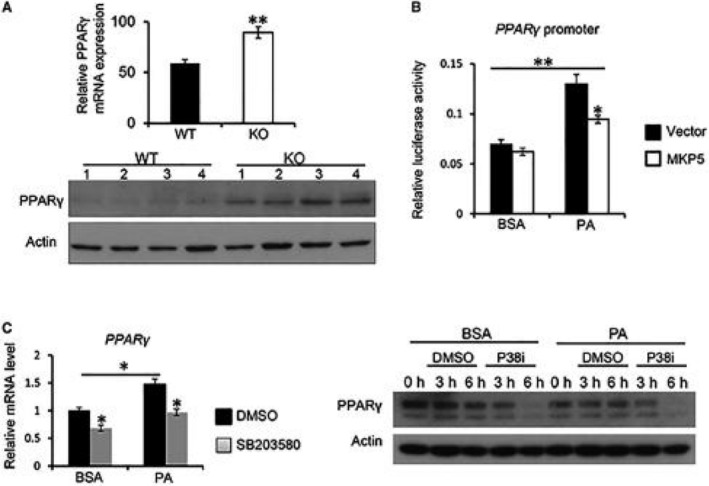
MKP5 negatively regulates PPARγ expression through p38. (A) Increased expression of PPARγ in MKP5 KO livers following HFD feeding. mRNA and protein expression of PPARγ in the liver of WT and MKP5 KO mice following 12 weeks of HFD feeding were determined by qPCR and western blots, respectively. (B) Overexpression of MKP5 inhibits Pparγ promoter activity. The Pparγ promoter region was cloned into a pGL3 luciferase vector and was cotransfected with MKP5 cDNA or control plasmid into FL83B hepatocytes together with a phosphorylated renilla luciferase‐null plasmid for a dual‐luciferase assay with or without PA stimulation for 6 hours. (C) p38 inhibition reduced Pparγ expression in mouse hepatocytes. FL83B mouse hepatocytes were pretreated with p38 inhibitor SB203580 (40 μM) for 1 hour, followed by stimulation with 500 μM of PA for 6 hours. PPARγ expression at both mRNA and protein levels was determined by qPCR and western blots, respectively. Data shown are representative of three independent experiments with similar results. Data are presented as mean ± SEM. *P < 0.05; **P < 0.01. Abbreviations: BSA, bovine serum albumin; DMSO, dimethyl sulfoxide.
It has been shown that p38 regulates PPARγ expression.17 We also found that inhibition of p38 by SB203580 reduced the expression of both mRNA and proteins of PPARγ in FL83B hepatocytes with or without PA stimulation (Fig. 5C). Collectively, these results indicate that MKP5 regulates PPARγ expression through p38, which contributes to its regulation of CIDEA and CIDEC/FSP27 expression.
Inhibition of P38 Alleviates Hepatic Steatosis in MKP5 KO Mice
Our findings suggest that deficiency of MKP5 resulted in increased p38 activation, which promoted the development of steatosis. Inhibition of p38 activation would therefore reduce the severity of steatosis in the KO mice. To address this, we administered SB203580 to WT and KO mice at week 3, after the HFD for 7 weeks in a course of 10‐week HFD feeding. Mice were then killed for examination. Increased liver weight and increased hepatic TG level in KO mice compared to WT mice were detected (Fig. 6A). Inhibition of p38 resulted in reduced liver weight and reduced hepatic TG level in both WT and KO mice. Importantly, TG levels became comparable between WT and KO mice after 7 weeks of p38 inhibition (Fig. 6A). Measurement of MAPK activation demonstrated reduced activation of p38 but not JNK or ERK in the liver of MKP5 KO mice following SB203580 treatment (Supporting Fig. S7). Next, we examined the expression of CIDE family members in the liver from WT and KO mice. We found that mRNA expression of Cidea and Cidec but not Cideb was higher in MKP5 KO mice compared to WT mice in response to the HFD (Fig. 6B). Following 7 weeks of SB203580 administration, the expression of Cidea and Cidec but not Cideb was significantly reduced in the KO liver (Fig. 6B). Reduced protein expression of CIDEA and CIDEC but not CIDEB was also observed in MKP5 KO livers following p38 inhibition (Fig. 6C), suggesting that p38 specifically regulates CIDEA and CIDEC in the liver. Furthermore, the development of hepatic steatosis was obviously alleviated in both WT and KO mice (Fig. 6D). Together, these results demonstrate that activation of p38 in the liver promotes the development of diet‐induced hepatic steatosis, which can be suppressed by MKP5.
Figure 6.
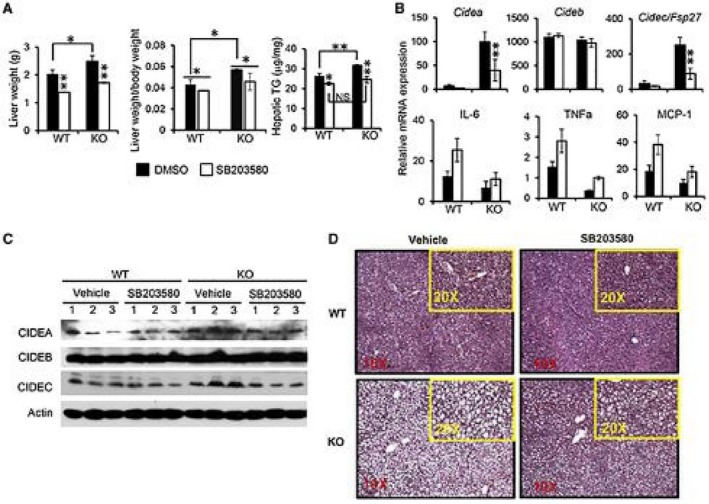
Inhibition of p38 activation alleviates hepatic steatosis in both WT and MKP5 KO mice. WT and MKP5 KO mice were fed with the HFD for 3 weeks after weaning, followed by administration of p38‐specific inhibitor SB203580 by oral gavage with continuation of HFD feeding for 7 weeks. (A) The weight of the liver and hepatic TG levels were determined. (B) mRNA and (C) protein expression of CIDEA, CIDEB, and CIDEC were determined in the liver from WT and KO mice with or without p38 inhibition. (D) H&E staining of liver sections from WT and MKP5 KO mice with or without p38 inhibition. Original magnification ×20. Data shown are representative of two independent experiments with similar results. (A,B) Data are presented as mean ± SEM. *P < 0.05; **P < 0.01. Abbreviation: NS, not significant.
Discussion
We have shown that MKP5 is required for maintenance of white adipose tissue (WAT) homeostasis in aging by preventing the development of adipose tissue inflammation and insulin resistance.14 Mice deficient in this molecule progressively developed insulin resistance, visceral obesity, and glucose intolerance, which are associated with adipose tissue inflammation. Here, we showed that MKP5 plays a protective role in the development of NAFLD, including hepatic steatosis and steatohepatitis. MKP5 KO aged mice developed hepatic steatosis spontaneously, which further progressed to more severe liver inflammation and fibrosis compared to WT mice (Fig. 1). Similarly, more severe steatosis accompanied by liver inflammation in the KO mice compared to WT mice was observed following the HFD for 12 weeks, suggesting the development of steatohepatitis in the KO mice (Figs. 2 and 3).
MKPs are cysteine‐based protein tyrosine phosphatases that are able to dephosphorylate both the tyrosine and threonine residues in the activation loop of MAPKs to inactivate them, and therefore are also known as DUSPs.18 Our previous study showed that deficiency of MKP5 resulted in increased p38 activation in WAT,14 suggesting that p38 is the main substrate of this phosphatase in WAT. In the current study, we detected increased activation of p38 but not JNK or ERK in the liver from MKP5 KO mice compared to WT mice with age or on the HFD (Supporting Fig. S2; Fig. 5A), demonstrating that p38 is also the main target of MKP5 in the liver in response to stimulation that leads to the development of NAFLD.
The function of p38α in liver inflammation and injury has been studied using cell‐/tissue‐specific p38α KO mice. Tormos et al.19 employed bile duct ligation (BDL)‐induced chronic cholestasis to assess the role of p38α in the progression of biliary cirrhosis using liver‐specific p38α KO mice. BDL‐induced chronic cholestasis causes proliferation of hepatocytes and cholangiocytes together with cell death, leukocyte infiltration, oxidative stress, and eventually fibrosis. In this biliary cirrhosis model, it was found that hepatocyte growth was reduced and hepatomegaly was absent in p38α‐deficient mice; this was associated with enhanced expression of inflammatory mediators, including regulated on activation, normal T‐cell expressed, and secreted (also known as CCL5) and intercellular cell adhesion molecule 1, but reduced fibrosis. Reduced survival of the p38α liver‐specific KO mice in response to BDL was observed compared to control mice. In another study using a coenzyme A‐induced hepatitis model where T cells and natural killer T (NKT) cells play critical roles in liver inflammation and damage, Kang et al.20 found that p38α activation in hepatocytes plays a protective anti‐inflammatory role during acute liver injury, whereas its activation in T cells or NKT cells promotes liver inflammation and injury. Interestingly, it was found that deficiency of p38α in hepatocytes resulted in increased JNK activation that contributed to increased chemokine expression for the recruitment of inflammatory cells to the liver, leading to severe inflammation and injury.20 Such a phenomenon was not observed in the liver‐specific p38α KO mice following BDL.19 The roles of p38α in liver injury and HCC has also been investigated using liver‐specific p38α‐deficient mice.21, 22 Sakurai et al.21 used a diethylnitrosamine (DEN)‐induced HCC mouse model and liver‐specific p38α‐deficient mice (p38αΔhep) to examine the role of p38α in liver injury and HCC. It was found that deficiency of p38α in hepatocytes resulted in increased cell death of hepatocytes. This led to an increased release of IL‐1α that stimulates IL‐6 production. IL‐6 stimulates hepatocyte compensatory proliferation, which is important for the development of HCC in response to DEN. In addition, enhanced accumulation of reactive oxygen species (ROS) and reduced expression of heat shock protein (HSP)25, a potent inhibitor of ROS, were observed in p38αΔhep mice. Subsequent administration of chemical antioxidant butylated hydroxyanisole to p38αΔhep mice or overexpression of HSP25 in p38αΔhep mice showed a marked reduction in DEN‐induced liver injury and compensatory proliferation. Interestingly, increased JNK activity was detected in the liver from p38αΔhep mice compared to that in p38α floxed mice. Conversely, administration of a JNK inhibitor inhibited DEN‐induced liver damage and compensatory proliferation. In another study using a thioacetamide (TAA)‐induced HCC mouse model, Sakurai et al.22 showed that p38αΔhep mice had enhanced hepatocarcinogenesis, which was associated with reduced expression of HSP25 and enhanced ROS accumulation in the liver of p38αΔhep mice compared to that in p38αF/F mice in response to TAA treatment. The enhanced ROS accumulation in the liver of p38αΔhep mice was believed to be responsible for the increased liver injury and fibrogenesis. The expression and phosphorylation of MAPK‐activated protein kinase 2 (MAPKAPK2) was down‐regulated in p38αΔhep liver following TAA treatment. Importantly, increased JNK activity was detected in p38αΔhep mouse liver. Therefore, the protective role of p38α showed in these two chemical‐induced HCC mouse models was entangled with JNK. Indeed, it has been shown that p38α negatively regulates cell proliferation and liver cancer development by antagonizing the JNK‐c‐Jun pathway.23 In addition, p38α can regulate the activities of a large number of substrates, including MAPKAPK2, C/EBP homologous protein (CHOP), mitogen‐ and stress‐activated protein kinase (MSK)1/2, and ATF2, which are the source of functional diversity of this pathway.24 In our study, we observed increased activation of p38 but not JNK or ERK (Fig. 4A) in the liver of MKP5‐deficient mice in response to HFD feeding, which led to increased ATF2 activity, increased expression of Cidea and Cidec, increased expression of PPARγ, and increased inflammation for the development of hepatic steatosis and steatohepatitis. Together, these studies suggest that cell‐/tissue‐specific regulation of p38α activation is dependent on the pathologic conditions,20 and this leads to different biological outcomes.
Increased activation of p38 coupled with increased activation of JNK was associated with the development of NAFLD, and inhibiting p38/JNK ameliorates disease severity.25, 26 However, the mechanism of how p38 regulates NAFLD is unclear. In this study, we found that deficiency of MKP5 resulted in enhanced activation of p38 but not ERK or JNK; this was associated with increased hepatic steatosis, inflammation, and fibrosis in response to aging or overnutrition (Figs. 1, 2, 3; Supporting Fig. S3). We further showed that p38 regulates the expression of LD‐associated proteins CIDEA and CIDEC but not CIDEB in the liver; we believe this contributes to the more severe hepatic steatosis observed in the KO mice (Fig. 4B‐F). Mechanistically, we demonstrated that pATF2, a major target of p38, bound to the promoter regions of Cidea and Cidec, thereby enhancing their expression following activation in hepatocytes (Fig. 4D,F). Conversely, inhibition of p38 suppressed the expression of both Cidea and Cidec (Fig. 4E). In addition, expression of PPARγ, a known regulator of Cidea and Cidec/Fsp27, 10 was observed to be enhanced in the liver from KO mice (Fig. 5A). This increased expression of PPARγ was likely due to increased p38 activation caused by the deficiency of MKP5 as we observed that overexpression of MKP5 or inhibition of p38 activation inhibited the expression of PPARγ (Fig. 5B,C), demonstrating that p38 promotes PPARγ expression in the liver. Therefore, it is likely that MKP5 regulates the expression of Cidea and Cidec/Fsp27 through both p38–ATF2 and p38–PPARγ axes, thereby inhibiting the development of hepatic steatosis.
PPARγ, a member of the PPAR subfamily of nuclear hormone receptors, is a ligand‐activated transcriptional factor with a major role in the development of adipocytes and also serves as a receptor for an important class of antidiabetic drugs.27 The expression of PPARγ was found to be elevated in steatotic livers in both animal models and patients with NAFLD,28, 29, 30 suggesting that it plays a role in the pathogenesis of the disease. A study using HFD‐induced hepatic steatosis in low‐density lipoprotein receptor‐deficient mice showed that PPARγ expression induced by its ligand rosiglitazone attenuated the development of NASH.31 However, in leptin‐deficient mice, rosiglitazone treatment resulted in increased oxidative stress and liver steatosis.32 In addition, PPARγ expression in the liver was found to mostly predominate in hepatocytes, and PPARγ deletion in hepatocytes but not in macrophages promoted the development of hepatic steatosis.29, 33 Interestingly, deficiency of PPARγ in hepatocytes or macrophages did not lead to changes in hepatic inflammation in response to HFD feeding. Furthermore, expression of Cidec/Fsp27, a PPARγ‐target gene, promotes hepatic steatosis.10, 34 Genetic differences in the mice used in these studies could be one of the reasons responsible for the observed discrepancy on the role of this molecule in the pathogenesis of NAFLD. We observed the development of both steatosis and hepatic inflammation in MKP5 KO mice, which are associated with increased p38 activation, and inhibition of p38 resulted in reduced expression of Cidea and Cidec/Fsp27 as well as inflammatory cytokines, including IL‐6, TNFα, and MCP‐1 (Fig. 6B). Therefore, p38 activation contributes to both increased accumulation of lipids through both ATF2 and PPARγ and the development of inflammation through ATF2 in the liver (Fig. 7).
Figure 7.
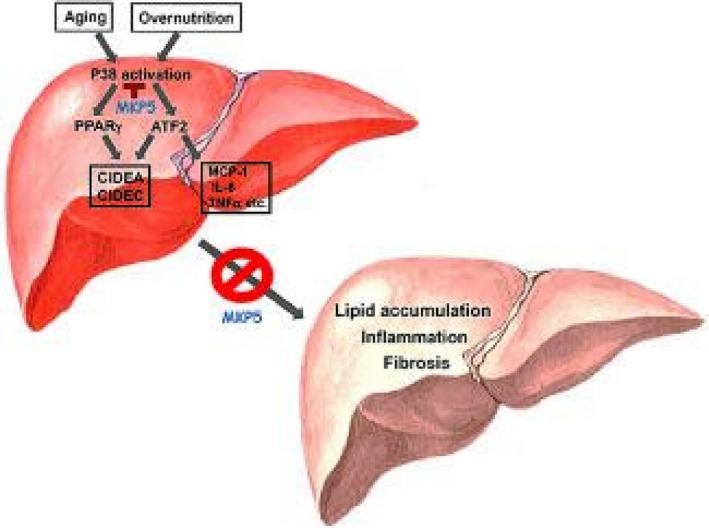
Schematic model for MKP5 in hepatic steatosis and steatohepatitis. Aging or overnutrition increases activation of p38 in the liver. P38 promotes the expression and activity of ATF2 and PPARγ, leading to increased expression of CIDEA, CIDEC/FSP27, and proinflammatory cytokines. Chronic activation of p38–ATF2 and p38–PPARγ axes cooperatively promote the development of steatosis, hepatic inflammation, and fibrosis; this is antagonized by MKP5.
In summary, our study demonstrates that MKP5 plays protective roles in the development of aging‐ and diet‐induced hepatic steatosis and steatohepatitis. It controls the activation of p38–ATF2 and p38–PPARγ axes in the liver, thereby inhibiting excessive accumulation of LDs and the development of inflammation. Our study therefore reveals the mechanism of p38 in the pathogenesis of NAFLD and suggests that MKP5 may be targeted for the development of therapeutics for the prevention and treatment of steatohepatitis.
Supporting information
Supported by the National University Health System of Singapore (T1‐NUHS O‐CRG2016 Oct‐21 to Y.Z.), Singapore National Medical Research Council (NMRC/OFIRG/0059/2017 to Y.Z.), A*STAR‐NHMRC (bilateral grant NHMRC2017‐SG006 to Y.Z.), and National Research Foundation, Prime Minister’s Office, Singapore, under its Campus of Research Excellence and Technological Enterprise program.
Potential conflict of interest: Nothing to report.
[Correction added 2 April, 2019. Author Asim Shabbir was mistakingly omitted from the original publication.]
SEE EDITORIAL ON PAGE https://doi.org/10.1002/hep4.1366
References
- 1. Tamura S, Shimomura I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J Clin Invest 2005;115:1139‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carr RM, Ahima RS. Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res 2016;340:187‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol 2015;62(Suppl.):S47‐S64. [DOI] [PubMed] [Google Scholar]
- 4. Okumura T. Role of lipid droplet proteins in liver steatosis. J Physiol Biochem 2011;67:629‐636. [DOI] [PubMed] [Google Scholar]
- 5. Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: getting to the root of fatty liver. Hepatology 2015;62:964‐967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikura Y, Caldwell SH. Lipid droplet‐associated proteins in alcoholic liver disease: a potential linkage with hepatocellular damage. Int J Clin Exp Pathol 2015;8:8699‐8708. [PMC free article] [PubMed] [Google Scholar]
- 7. Wang C, Zhao Y, Gao X, Li L, Yuan Y, Liu F, et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015;61:870‐882. [DOI] [PubMed] [Google Scholar]
- 8. Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, et al. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation‐related protein. Mol Cell Biol 2006;26:1063‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou L, Xu L, Ye J, Li D, Wang W, Li X, et al. Cidea promotes hepatic steatosis by sensing dietary fatty acids. Hepatology 2012;56:95‐107. [DOI] [PubMed] [Google Scholar]
- 10. Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, et al. Hepatic steatosis in leptin‐deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab 2008;7:302‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee MK, Choi CS, et al. Mice lacking MAP kinase phosphatase‐1 have enhanced MAP kinase activity and resistance to diet‐induced obesity. Cell Metab 2006;4:61‐73. [DOI] [PubMed] [Google Scholar]
- 12. Reddy ST, Nguyen JT, Grijalva V, Hough G, Hama S, Navab M, et al. Potential role for mitogen‐activated protein kinase phosphatase‐1 in the development of atherosclerotic lesions in mouse models. Arterioscler Thromb Vasc Biol 2004;24:1676‐1681. [DOI] [PubMed] [Google Scholar]
- 13. Feng B, Jiao P, Helou Y, Li Y, He Q, Walters MS, et al. Mitogen‐activated protein kinase phosphatase 3 (MKP‐3)‐deficient mice are resistant to diet‐induced obesity. Diabetes 2014;63:2924‐2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Nguyen T, Tang P, Kennedy NJ, Jiao H, Zhang M, et al. Regulation of adipose tissue inflammation and insulin resistance by MAPK phosphatase 5. J Biol Chem 2015;290:14875‐14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu L, Zhou L, Li P. CIDE proteins and lipid metabolism. Arterioscler Thromb Vasc Biol 2012;32:1094‐1098. [DOI] [PubMed] [Google Scholar]
- 16. Viswakarma N, Yu S, Naik S, Kashireddy P, Matsumoto K, Sarkar J, et al. Transcriptional regulation of Cidea, mitochondrial cell death‐inducing DNA fragmentation factor alpha‐like effector A, in mouse liver by peroxisome proliferator‐activated receptor alpha and gamma. J Biol Chem 2007;282:18613‐18624. [DOI] [PubMed] [Google Scholar]
- 17. Xiong Y, Collins QF, An J, Lupo E Jr, Liu HY, Liu D, et al. p38 mitogen‐activated protein kinase plays an inhibitory role in hepatic lipogenesis. J Biol Chem 2007;282:4975‐4982. [DOI] [PubMed] [Google Scholar]
- 18. Zhang Y, Dong C. Regulatory mechanisms of mitogen‐activated kinase signaling. Cell Mol Life Sci 2007;64:2771‐2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tormos AM, Arduini A, Talens‐Visconti R, del Barco Barrantes I, Nebreda AR, Sastre J. Liver‐specific p38alpha deficiency causes reduced cell growth and cytokinesis failure during chronic biliary cirrhosis in mice. Hepatology 2013;57:1950‐1961. [DOI] [PubMed] [Google Scholar]
- 20. Kang YJ, Bang BR, Otsuka M, Otsu K. Tissue‐specific regulation of p38alpha‐mediated inflammation in Con A‐induced acute liver damage. J Immunol 2015;194:4759‐4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, et al. Hepatocyte necrosis induced by oxidative stress and IL‐1 alpha release mediate carcinogen‐induced compensatory proliferation and liver tumorigenesis. Cancer Cell 2008;14:156‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sakurai T, Kudo M, Umemura A, He G, Elsharkawy AM, Seki E, et al. p38alpha inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res 2013;73:215‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, et al. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK‐c‐Jun pathway. Nat Genet 2007;39:741‐749. [DOI] [PubMed] [Google Scholar]
- 24. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J 2010;429:403‐417. [DOI] [PubMed] [Google Scholar]
- 25. Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat‐induced nonalcoholic fatty liver. J Nutr 2011;141:603‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ye P, Xiang M, Liao H, Liu J, Luo H, Wang Y, et al. Dual‐specificity phosphatase 9 protects against non‐alcoholic fatty liver disease in mice via ASK1 suppression. Hepatology 2019;69:76‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem 2008;77:289‐312. [DOI] [PubMed] [Google Scholar]
- 28. Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, et al. Increased expression of PPARgamma in high fat diet‐induced liver steatosis in mice. Biochem Biophys Res Commun 2005;336:215‐222. [DOI] [PubMed] [Google Scholar]
- 29. Moran‐Salvador E, Lopez‐Parra M, Garcia‐Alonso V, Titos E, Martinez‐Clemente M, Gonzalez‐Periz A, et al. Role for PPARgamma in obesity‐induced hepatic steatosis as determined by hepatocyte‐ and macrophage‐specific conditional knockouts. FASEB J 2011;25:2538‐2550. [DOI] [PubMed] [Google Scholar]
- 30. Pettinelli P, Videla LA. Up‐regulation of PPAR‐gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP‐1c induction. J Clin Endocrinol Metab 2011;96:1424‐1430. [DOI] [PubMed] [Google Scholar]
- 31. Gupte AA, Liu JZ, Ren Y, Minze LJ, Wiles JR, Collins AR, et al. Rosiglitazone attenuates age‐ and diet‐associated nonalcoholic steatohepatitis in male low‐density lipoprotein receptor knockout mice. Hepatology 2010;52:2001‐2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garcia‐Ruiz I, Rodriguez‐Juan C, Diaz‐Sanjuan T, Martinez MA, Munoz‐Yague T, Solis‐Herruzo JA. Effects of rosiglitazone on the liver histology and mitochondrial function in ob/ob mice. Hepatology 2007;46:414‐423. [DOI] [PubMed] [Google Scholar]
- 33. Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver‐specific disruption of PPARgamma in leptin‐deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 2003;111:737‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu X, Park JG, So JS, Lee AH. Transcriptional activation of Fsp27 by the liver‐enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology 2015;61:857‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials