

Li et al. show that TWIST2 shapes the epigenetic landscape to drive chromatin opening at oncogenic loci and chromatin closing at myogenic loci. These epigenetic changes redirect MyoD binding from myogenic genes toward oncogenic, metabolic, and growth genes.
Keywords: skeletal muscle, bHLH, chromosome amplification, dedifferentiation, histone modification, rhabdomyosarcoma
Abstract
Rhabdomyosarcoma (RMS) is an aggressive pediatric cancer composed of myoblast-like cells. Recently, we discovered a unique muscle progenitor marked by the expression of the Twist2 transcription factor. Genomic analyses of 258 RMS patient tumors uncovered prevalent copy number amplification events and increased expression of TWIST2 in fusion-negative RMS. Knockdown of TWIST2 in RMS cells results in up-regulation of MYOGENIN and a decrease in proliferation, implicating TWIST2 as an oncogene in RMS. Through an inducible Twist2 expression system, we identified Twist2 as a reversible inhibitor of myogenic differentiation with the remarkable ability to promote myotube dedifferentiation in vitro. Integrated analysis of genome-wide ChIP-seq and RNA-seq data revealed the first dynamic chromatin and transcriptional landscape of Twist2 binding during myogenic differentiation. During differentiation, Twist2 competes with MyoD at shared DNA motifs to direct global gene transcription and repression of the myogenic program. Additionally, Twist2 shapes the epigenetic landscape to drive chromatin opening at oncogenic loci and chromatin closing at myogenic loci. These epigenetic changes redirect MyoD binding from myogenic genes toward oncogenic, metabolic, and growth genes. Our study reveals the dynamic interplay between two opposing transcriptional regulators that control the fate of RMS and provides insight into the molecular etiology of this aggressive form of cancer.
Rhabdomyosarcoma (RMS) is an aggressive pediatric soft tissue tumor expressing hallmarks of the skeletal muscle lineage such as MyoD and Myf5 (Hettmer and Wagers 2010; Saab et al. 2011; Skapek et al. 2019). A key characteristic of RMS is the inability of the tumor cells to undergo myogenic differentiation even in the presence of elevated levels of muscle master regulators such as MyoD and Myf5 (Tapscott and Weintraub 1991; Tapscott et al. 1993; Saab et al. 2011; MacQuarrie et al. 2013; Skapek et al. 2019). Traditionally, RMS was classified into two main histological subtypes: alveolar and embryonal (Skapek et al. 2019). However, recent advances in molecular diagnostics have enabled a more refined classification of RMS into fusion-positive RMS (FPRMS) and fusion-negative RMS (FNRMS), based on the presence or absence of key chromosomal translocations (Tsokos 1994; Skapek et al. 2019). FPRMS is commonly driven by the balanced translocation of chromosomes 2 and 13 to generate an oncogenic PAX3-FOXO1 fusion protein (Saab et al. 2011; Skapek et al. 2019). This fusion protein acts as an overactive transcription factor at PAX3-binding sites colocalized with E-box motifs to organize superenhancers and drive RMS pathogenesis (Cao et al. 2010; Gryder et al. 2017). The mechanism of pathogenesis for FNRMS is less understood. Compared with FPRMS, FNRMS exhibits significantly greater genomic instability (Xu et al. 2018). Traditional oncogenic regulators such as KRAS, NRAS, and TP53 are mutated in only <10% of RMS patient cohorts, respectively, suggesting that other mechanisms are at play (Shern et al. 2014). Bioinformatic analyses of RMS genomic data have revealed copy number alterations of oncogenes and tumor suppressors that play a key role in driving RMS development (Preussner et al. 2018; Xu et al. 2018). While targeting growth is a common strategy for many cancers, targeting myogenic differentiation may represent a unique and exploitable vulnerability in RMS, given that terminally differentiated myofibers are permanently postmitotic (Saab et al. 2011; Tremblay et al. 2014; Xu et al. 2018; Skapek et al. 2019).
Skeletal muscle is a highly regenerative tissue required for animal life. Muscle regeneration occurs through a population of resident stem cells called satellite cells, which are marked by expression of the transcription factor Pax7 (Shi and Garry 2006; Chang and Rudnicki 2014). Upon muscle injury, satellite cells up-regulate the muscle master regulator MyoD to proliferate and repair injured muscle (Shi and Garry 2006; Chang and Rudnicki 2014). Our laboratory recently discovered a unique muscle progenitor through fate mapping of the transcription factor Twist2 (Tw2+ cells) (Liu et al. 2017). These Tw2+ cells are distinct from satellite cells and do not express Pax7 in vivo (Liu et al. 2017). In culture, Tw2+ cells down-regulate Twist2 expression and up-regulate MyoD expression, allowing them to form terminally differentiated myotubes (Liu et al. 2017). We found that overexpression of Twist2 in vitro was sufficient to drive transcriptional and phenotypic repression of the myogenic program (Liu et al. 2017).
Mammalian Twist2 and its paralog, Twist1, differ in their temporal and spatial expression patterns and are important regulators of mesoderm development, epithelial–mesenchymal transition (EMT), and cellular differentiation (Li et al. 1995; Gong and Li 2002; Zhang et al. 2008; Franco et al. 2011; Merindol et al. 2014). Additionally, in vitro studies have also shown that both Twist proteins can inhibit MyoD-induced transdifferentiation of fibroblasts into myoblasts (Spicer et al. 1996; Gong and Li 2002). The functional roles of Twist1 and Twist2 are likely imparted by their DNA-binding activity, which is mediated by the basic helix–loop–helix (bHLH) domain. Like other bHLH transcription factors, both Twist1 and Twist2 are capable of binding canonical CANNTG E-box motifs (Jones 2004; Chang et al. 2015). While Twist2 de novo motif analysis has not been reported previously, de novo motif analysis of Twist1 reveals a preference for CAGATG E-boxes as well as a secondary preference for double E-box motifs (Chang et al. 2015). Other bHLH proteins, such as MyoD and NeuroD, prefer different variations of the middle two nucleotides (Fong et al. 2012, 2015). Along with tissue expression pattern, the exact binding sequence likely defines the target genes and functions of different bHLH transcription factors (Fong et al. 2012, 2015). Indeed, swapping of the bHLH domains between MyoD and NeuroD converts NeuroD into a muscle master regulator and MyoD into a master neurogenic regulator (Fong et al. 2015).
bHLH transcription factors act as direct transcriptional activators of gene expression. The advent of ChIP-seq has revealed that bHLH factors such as N-myc and MyoD are capable of invading enhancer regions throughout the genome to regulate the global chromatin architecture (la Serna et al. 2005; Zeid et al. 2018). These chromatin modifications are often associated with histone acetylation, enabling the activation of tightly regulated transcriptional networks (la Serna et al. 2005; Zeid et al. 2018). In the case of N-myc, epigenetic alterations of occupied enhancers involve coordination and co-occupancy of additional bHLH transcription factors such as TWIST1 to drive activation of the neuroblastoma gene program (Zeid et al. 2018). In the case of muscle differentiation, binding of MyoD to DNA is associated with histone acetylation around myogenic loci to promote muscle gene expression (la Serna et al. 2005; Blum et al. 2012). Recent advances in genome-wide analysis of transcription factor binding and epigenetic remodeling provide a powerful tool for refining existing knowledge of bHLH transcription factor function in a global context. In particular, TWIST2 epigenetic and transcriptional regulation during myogenic differentiation is multifaceted and unexplored.
In this study, we analyzed genomic and transcriptomic sequencing data from RMS tumors of 258 patients and identified frequent copy number amplification of the TWIST2 loci that in turn drives overexpression of TWIST2 in RMS. We show that knockdown of TWIST2 in RMS cell lines results in up-regulation of MYOGENIN, the essential transcriptional switch for myogenic differentiation. Using ChIP-seq and RNA-seq, we uncovered the previously unknown interplay between Twist2 and MyoD during myogenic differentiation and its effect on transcriptional output and the chromatin landscape. Our findings indicate that during differentiation, Twist2 globally shifts MyoD activity from myogenic loci to EMT and tumor growth loci through competition and epigenetic modifications at promoters and enhancers, highlighting the central role of TWIST2 in RMS pathogenesis.
Results
TWIST2 and TWIST1 genes are highly amplified in fusion-negative RMS
Previously, our laboratory identified a previously unrecognized progenitor through lineage tracing of the transcription factor Twist2 (Liu et al. 2017). We showed that Tw2+ cells down-regulate Twist2 expression in culture and that overexpression of Twist2 was capable of repressing myogenesis (Liu et al. 2017), providing evidence for a physiological role of Twist2 in regulating myoblast differentiation. To explore the potential role of Twist2 in myogenic regulation within pathological contexts, we analyzed SNP array data from 258 RMS patients (Xu et al. 2018) for copy number gain of both TWIST2 and TWIST1 loci (Fig. 1A). TWIST amplification was not significantly associated with FPRMS cases; however, both TWIST2 and TWIST1 were significantly amplified in FNRMS cases. Coordinately, we observed increased expression of TWIST2 and TWIST1 in FNRMS patients but not FPRMS patients, implicating TWIST genes as previously unknown oncogenes for FNRMS patients (Fig. 1B). Strikingly, 72% (114 out of 158 cases) of FNRMS patients contain copy number amplification events on loci of either TWIST2, TWIST1, or both (Fig. 1C). In contrast, somatic mutations in known cancer driver genes such as KRAS, NRAS, and TP53 occur in <10% of FNRMS patient cohorts. We also found that TWIST expression levels decreased during differentiation of human myoblasts (Fig. 1D), suggesting that decreased TWIST2 gene expression may play a role during normal muscle differentiation.
Figure 1.
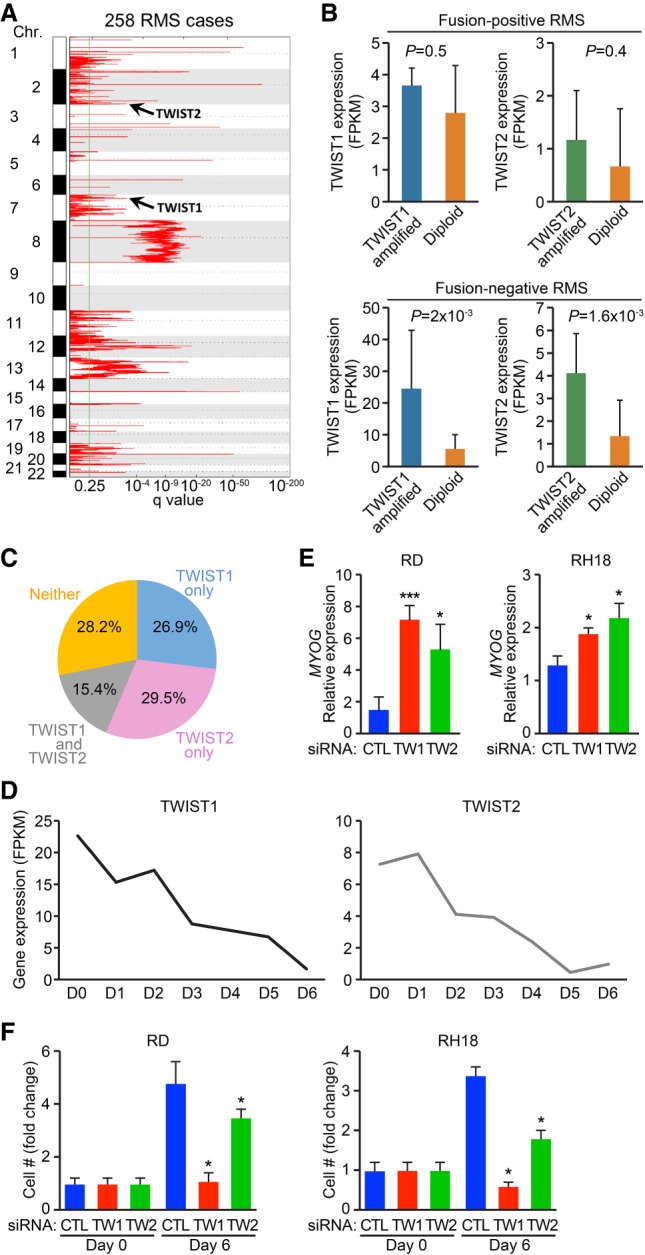
Integrative genomic analysis identifies TWIST genes as potential oncogenes for FNRMS. (A) Charts show genomic regions with statistically significant copy number gains identified by the Genomic Identification of Significant Targets in Cancer 2.0 (GISTIC2) algorithm in 258 RMS cases. Additional amplified loci can be found in Xu et al. (2018). (B) Separate integrated analysis on copy number alterations and gene expression for TWIST1 and TWIST2 genes in fusion-positive (top panel) and fusion-negative (bottom panel) RMS. TWIST expression in patient tumors containing TWIST amplification were compared with those without TWIST amplification (diploid). (C) Venn diagrams display the percentage of RMS cases with copy number amplification events on TWIST1 (blue), TWIST2 (pink), or both (gray). (D) Charts show gene expression for TWIST1 and TWIST2 in human myoblasts cultured in growth medium (D0) or differentiation medium for the indicated number of days (D1–D6). (E,F) Measurement of MYOGENIN expression in RD cells (E, left) or RH18 cells (E, right) or cell proliferation in RD cells (F, left) or RH18 cells (F, right) upon siRNA-mediated knockdown of TWIST2 or TWIST1. Unpaired two-tailed t test: (*) P < 0.05; (***) P < 0.0005.
In order to validate the pathological role of TWIST2 amplification in blocking myogenic differentiation of RMS, we performed siRNA knockdown of TWIST2 and TWIST1. We first screened various RMS cell lines to identify which ones exhibited the highest level of TWIST2 expression. We found that RD and RH18 cells showed higher levels of TWIST2 expression (Supplemental Fig. S1A). We then performed siRNA knockdown of TWIST2 and TWIST1 and validated knockdown by real-time PCR (Supplemental Fig. S1B,C). Loss of either TWIST2 expression or TWIST1 expression resulted in a significant increase in the expression of MYOG, with protein being detectable in RD cells treated with siTWIST1 (Fig. 1E; Supplemental Fig. S1D). Loss of either TWIST2 or TWIST1 also resulted in up-regulation of fast myosin light chain (MYL1) in RD but not RH18 cells, likely due to differing genetic backgrounds (Supplemental Fig. S1E,F). We also found that knockdown of TWIST2 and TWIST1 reduced RD and RH18 cell accumulation (Fig. 1F). In accordance, knockdown of TWIST2 and TWIST1 in RD cells resulted in decreased EdU labeling (Supplemental Fig. S1G). These results suggest that amplification of TWIST in primary RMS tumors may impair differentiation and drive oncogenesis.
Twist2 is a reversible inhibitor of myogenic differentiation
A hallmark of RMS is the inability of tumor cells to undergo normal myogenic differentiation even in the presence of MyoD and Myf5 (Saab et al. 2011). To explore the mechanism by which Twist2 represses myogenic differentiation, we first generated a doxycycline (Dox)-inducible Twist2-IRES-GFP (iTwist2) stable Tw2+ cell line (Fig. 2A) and tested whether Twist2-mediated myogenic repression was a permanent or reversible event. iTwist2 cells were provided 10 µg/mL Dox in growth medium (GM) on day 1 to induce expression of Twist2 and a GFP reporter (Fig. 2B). On the subsequent day, iTwist2 cells were switched to differentiation medium (DM) supplemented with Dox (Fig. 2B). After 3 d in DM, iTwist2 cells were either maintained on Dox or switched to vehicle (Fig. 2B). iTwist2 cells receiving continuous Dox treatment were unable to differentiate, as shown by a lack of myosin heavy chain (Myosin) costaining with GFP (Fig. 2B). However, iTwist2 cells removed from Dox were able to resume myogenic differentiation, as shown by formation of GFP-positive myotubes (Fig. 2B). This result revealed that Twist2 represses myogenesis in a reversible manner.
Figure 2.

Twist2 reversibly inhibits myogenic differentiation and promotes dedifferentiation in vitro. (A, top) Schematic of Twist2 Tet-On vector. (Bottom) Generation of an iTwist2 stable cell line in Tw2+ myoblasts. (B) Experimental scheme of Twist2 reversibly inhibiting differentiation. Dox was added to iTwist2 cells to induce Twist2-IRES-GFP expression. On day 2, GM was replaced with DM. On day 5, Dox was removed to shut off Twist2 expression. Cells were fixed and stained on day 9 for fast myosin (red), GFP (green), and nuclei (blue). Arrows refer to differentiated myotubes that previously expressed Twist2. Scale bar, 100 µm. (C) Schematic of dedifferentiation tracing system using iTwist2 primary myoblasts. (D) Time line of dedifferentiation tracing experiment. Cells were mixed on day 1 and differentiated on day 2. On day 6 of differentiation, Dox was added to induce Twist2 expression. On day 8, cells were switched back to GM to enhance dedifferentiation. Cells were fixed and stained on day 10 for fast myosin (red), GFP (green), and nuclei (blue). Arrows indicate dsRed+ dedifferentiated mononuclear cells. Scale bar, 50 µm. (E) Quantification of myotube dedifferentiation. The number of nuclei within mononuclear dsRed+ cells was quantified as a percentage of total nuclei. For each condition, two to three fields were quantified for each of three biological samples. Unpaired two-tailed t-test: (*) P < 0.05.
We next sought to determine whether Twist2 expression in differentiated myotubes could drive dedifferentiation into mononuclear cells. We first modified our iTwist2 system to enable lineage tracing with a dsRed reporter (iTwist2-tracer), which allowed us to distinguish mononuclear dedifferentiated myoblasts from mononuclear cells that never differentiated (Fig. 2C; Supplemental Fig. S2A,B). Since Tw2+ cells express tdTomato, which is indistinguishable from the dsRed reporter, we generated iTwist2-tracer primary myoblasts for dedifferentiation experiments. Following previously published dedifferentiation schemes (Wang et al. 2015), we allowed iTwist2-tracer cells to differentiate for 4 d before adding Dox (Fig. 2C,D). Cells were kept in DM + Dox for two additional days before being switched to GM + Dox. iTwist2-tracer cells that did not receive Dox maintained their differentiated state, as shown by immunofluorescence and Western blotting for Myosin and GFP (Fig. 2D; Supplemental Fig. S2C). However, iTwist2 tracer cells that received Dox on day 6 were able to undergo dedifferentiation back into mononuclear cells within 2 d of switching to GM, as evidenced by the presence of significantly increased dsRed+ mononuclear cells and down-regulation of Myosin expression (Fig. 2D,E; Supplemental Fig. S2C).
We also used live-cell imaging to capture the dedifferentiation event of a myofiber after induction of Twist2 expression (Supplemental Movies S1, S2). Interestingly, while dedifferentiated cells did not re-enter the cell cycle (data not shown), they were able to redifferentiate when placed back into DM, suggesting that Twist2-mediated dedifferentiation was also reversible (Supplemental Fig. S2D). Taken together, these data suggest that Twist2 is a potent but reversible inhibitor of myogenesis. We believe these results are generalizable to Twist1 as well, since both Twist1 and Twist2 impair myogenic differentiation (Supplemental Fig. S2E). Additionally, both Twist1- and Twist2-overexpressing myoblasts closely resemble the refractile morphology of RD cells (Supplemental Fig. S2F), providing additional in vitro evidence for the role of TWIST proteins in RMS pathogenesis.
Twist2 recognizes a conserved E-box and double-E-box motif
Our previously published data suggested that Twist2 represses the expression of MyoD target genes but not that of MyoD itself (Liu et al. 2017). To explore Twist2-binding dynamics in the context of differentiation, we performed genome-wide ChIP-seq for 3xTy1-Twist2 and MyoD in Tw2+ myoblasts in GM and DM conditions (Fig. 3A). We used a Twist2 overexpression system to model the role of TWIST2 copy number amplification and overexpression in RMS patients. Previously published data suggest that endogenous and exogenous bHLH transcription factors bind similar sites in vitro (Yao et al. 2013). Peak-calling quality was validated by principle component analysis and correlation matrix of samples (Supplemental Fig. 3A,B). Using MEME suite, we performed de novo motif analysis on Twist2-occupied peaks to identify the Twist2 consensus motif. We found that the most enriched and significant primary motif in both GM and DM was a canonical CANNTG E-box motif with a preference for GA or GC as the third and fourth nucleotides (Fig. 3B). We also identified a double-E-box motif as an enriched binding sequence for Twist2 (Fig. 3C). The double-E-box consisted of the primary Twist2-binding motif followed by a secondary E-box 5 bp upstream or downstream (Fig. 3C,D). This 5-bp-separated double-E-box motif has been identified previously in both human and Drosophila cells as an evolutionarily conserved motif recognized by the WR domain of Twist1 (Chang et al. 2015). The Twist2 WR domain is mostly identical to that of Twist1, which may explain why Twist2 also recognizes double E-boxes (Supplemental Fig. S3C). To explore the potential role of double-E-box motifs, we performed gene ontology (GO) analysis on Twist2 target genes with and without 5-bp-separated double-E-box motifs. While single-E-box-containing genes were enriched for developmental pathways, genes associated with double E-boxes were enriched for pathways regulating cellular migration, adhesion, and muscle development (Fig. 3E; Supplemental Fig. S3D).
Figure 3.

Twist2 recognizes a conserved E-box and double-E-box motif. (A) Schematic of ChIP-seq for 3xTy1-Twist2 and MyoD in Tw2+ cells in GM or DM. (B) De novo motif analysis of the primary Twist2-binding motif in GM and DM performed by MEME-ChIP. (C) De novo motif analysis of a secondary double-E-box Twist2-binding motif in GM and DM performed by MEME-ChIP. (D) Relative incidence of Twist2 bound to single versus double E-boxes of various spacing. (E) Panther pathway analysis of Twist2-bound genes associated with 5-bp double-E-box motifs. (F) Twist2-binding distribution across the genome. (TSS) Transcription start site.
Next, we explored where Twist2 binds throughout the genome. The majority of Twist2 peaks in both GM and DM occurred in intronic and intergenic regions, which is reflective of many bHLH transcription factors (Fig. 3F). This is likely due to the prevalence of E-boxes scattered across the genome but could also suggest a role for Twist2 in chromatin remodeling at distal regulatory regions. Additionally, a significant fraction of Twist2 peaks is found at promoter regions, which likely indicates direct Twist2 transcriptional targets.
Twist2 is a direct transcriptional activator of EMT and a direct repressor of myogenesis
Overexpression of Twist2 in myoblasts caused drastic morphological changes in DM but not in GM, suggesting differential Twist2 binding in GM and DM conditions (Supplemental Fig. 2D,E; Liu et al. 2017). Through our ChIP-seq analysis, we identified both up-regulated and down-regulated Twist2 peaks in DM compared with GM (Supplemental Fig. S4A). The up-regulated peaks are associated with genes involved in developmental, metabolic, and muscle pathways as well as extracellular matrix (ECM), cell–cell junction, and cytoskeletal compartments (Supplemental Fig. S4B,C). In the past, Twist2 has been viewed as a transcriptional repressor; however, more recent studies have suggested roles for Twist2 in activating gene transcription, particularly in pathways involving EMT (Franco et al. 2011; Shi et al. 2014).
To determine whether Twist2 functions as an activator or repressor in muscle cells, we intersected our Twist2 ChIP-seq data set with our previously published Twist2 overexpression RNA-seq data set (Supplemental Fig. S4D; Liu et al. 2017). We identified Twist2 promoter peaks from Twist2 ChIP-seq and mapped them to the nearest genes to identify putative Twist2 direct targets (Supplemental Fig. S4D; Supplemental Table 1). We then analyzed our RNA-seq data to determine whether these genes were up-regulated or down-regulated upon Twist2 overexpression (Fig. 4A). We found that Twist2 acts as both a direct activator and repressor (Fig. 4A). Through GO analysis, we found that Twist2 directly activates genes involved in developmental, cellular adhesion, and ECM remodeling pathways while repressing those involved in muscle development and function (Fig. 4B,C).
Figure 4.

Twist2 is a direct transcriptional activator of EMT and a direct repressor of myogenesis. (A) Scatter plot of Twist2 direct target gene expression identified by ChIP-seq and RNA-seq intersection in DM. The cutoff was twofold or greater change and Padj < 0.05. (B) GO enrichment analysis of target genes activated by Twist2, as shown in A. (C) GO enrichment analysis of genes repressed by Twist2, as shown in A. (D, left) Genome browser shot displaying Twist2 binding within the Snai2 locus. (Right) Up-regulation of Snai2 expression induced by Twist2. (E) Transwell migration assay. Ctrl-infected Tw2+ cells and RD cells were used as negative and positive controls, respectively. The number of invading cells was quantified as a relative intensity compared with zero cells 24 h after seeding using Calcein AM. Unpaired two-tailed t-test: (*) P < 0.05; (**) P < 0.005. (F, left) Genome browser shot displaying Twist2 binding within the Myog locus. (Right) Down-regulation of Myog expression induced by Twist2.
In addition to its roles in development, Twist2 is also a master regulator of EMT. Previously, we showed that Twist2 overexpression up-regulated Snai2 (Liu et al. 2017). SNAI2 is a known regulator of RMS pathogenesis (Xu et al. 2018), mediator of TWIST1-induced EMT (Casas et al. 2011; Rinon et al. 2011), and inhibitor of MyoD (Soleimani et al. 2012). We found that the Snai2 locus contains multiple Twist2-binding sites and is highly up-regulated upon Twist2 overexpression (Fig. 4D). A hallmark of EMT is the ability of cells to invade through a basement membrane. Using a transwell migration assay, we found that overexpression of Twist2 was capable of enhancing the cellular invasiveness of myoblasts to levels comparable with RD cells (Fig. 4E). We also examined muscle gene loci, where Twist2 presumably acts to inhibit differentiation. Analysis of the Myogenin locus revealed ample Twist2 binding and subsequent down-regulation of its expression (Fig. 4F). Taken together, these data confirm Twist2 as a direct activator of development and EMT and a repressor of myogenesis.
Twist2 drives global redirection of MyoD DNA binding
Given that Twist2 binds to MyoD target genes and represses their transcription, we investigated whether Twist2 impairs the binding of MyoD to these genomic sites. We first analyzed MyoD peaks in Tw2+ myoblasts by ChIP-seq. Similar to Twist2, we found that MyoD exhibited preferences for intronic and intergenic regions (Supplemental Fig. S5A). De novo motif analysis revealed that MyoD preferred its canonical GC- and CC-containing E-boxes (Fig. 5A; Cao et al. 2010). Unlike Twist2, MyoD peaks did not enrich for any significant secondary motifs, including the double-E-box motif found with Twist1 and Twist2 (Fig. 5A). Protein sequence alignment of MyoD with the WR domains of Twist1 and Twist2 shows little to no identity, indicating that double-E-box binding may be a function unique to the Twist family transcription factors (Supplemental Fig. S3B). Previous studies have suggested that bHLH transcription factors bind both private and shared E-box sequences (Fong et al. 2012, 2015). Comparison of our Twist2 and MyoD motifs reveals that both proteins bind to a common GC E-box (Fig. 5B); however, MyoD also binds to a private CC E-box, while Twist2 binds to a private GA E-box (Fig. 5B). While the GA E-box likely recruits Twist2 to genes involved in EMT and ECM remodeling, the overlap of Twist2 and MyoD at the GC E-box suggests that Twist2 and MyoD may directly compete for binding sites that are important for myogenic regulation (Shield et al. 1996; Cao et al. 2010). Indeed, decreased MyoD binding to GC E-boxes relative to CC E-boxes was observed in RMS cells (MacQuarrie et al. 2013).
Figure 5.

Twist2 drives global redirection of MyoD binding. (A) De novo motif analysis of the MyoD-binding motif in GM and DM performed by MEME-ChIP. MyoD has no preference for secondary motifs. (B) Comparison of Twist2 and MyoD private and shared E-box variants. (C) Scatter plot of differential MyoD peaks in the presence and absence of Twist2 in DM. Differential peak cutoff was defined as signal twofold or greater change and Padj < 0.05. (D, top) Heat map depicting differential binding of MyoD peaks in the presence or absence of Twist2 in DM. (Bottom) ChIP signal distribution plot of differentially bound MyoD peaks in the presence or absence of Twist2. (E) Genome browser shot depicting Twist2 and MyoD competitive binding at the Klhl40 locus. (F) GREAT (Genomic Region Enrichment of Annotations Tool) analysis of MyoD peaks down-regulated in TW2-DM versus GFP-DM. (G) GREAT analysis of MyoD peaks up-regulated in TW2-DM versus GFP-DM.
Next we analyzed our global MyoD peak changes in the presence or absence of Twist2 to determine the impact of Twist2 expression on MyoD binding (Fig. 5C). In both GM and DM, a significant number of MyoD peaks are differentially regulated in the presence of Twist2 overexpression (Fig. 5C,D; Supplemental Fig. S5C). Down-regulated MyoD peaks are co-occupied by Twist2, suggesting that Twist2 directly competes with MyoD (Fig. 5D). Examples of this are shown at the Klhl40 and Mymk loci, where, in the absence of Twist2, MyoD binds strongly to the promoters (Fig. 5E; Supplemental Fig. S5D). However, in the presence of Twist2, MyoD binding is significantly reduced, while Twist2 now occupies the same binding sites (Fig. 5E; Supplemental Fig. S5D). On the global scale, Twist2 displaces MyoD from genes involved in cytoskeletal reorganization and muscle differentiation (Fig. 5F). We also found that Twist2 expression enabled MyoD to bind to a significant number of previously inaccessible sites (Fig. 5C,D). These new sites were associated with genes involved in developmental, metabolic, growth, and cancer pathways (Fig. 5G; Supplemental Table 2). Interestingly, motif analysis of down-regulated MyoD peaks identified the presence of TGIF2/1, MEF2C, and TEAD2 motifs, while motif analysis of up-regulated MyoD peaks revealed the presence of NF1, BATF, RUNX2, and HOXB3 motifs (Supplemental Fig. S5E,F). These coincident motifs are similar to those identified as differentially bound MyoD sites in RD cells versus normal myotubes (MacQuarrie et al. 2013), providing further evidence for Twist2-mediated redirection of MyoD binding.
The bHLH domain of Twist2 is critical for repressing myogenesis
The results so far support the hypothesis that DNA-binding specificity is a primary determinant of Twist2 and MyoD activities (Fong et al. 2015). Since binding sequence preference is largely specified by the bHLH domain (Fong et al. 2015), we swapped the bHLH domains of Twist2 and MyoD to generate chimeric proteins (Fig. 6A,B). We tagged these chimeric constructs with a myc tag and infected Tw2+ primary myoblasts with them to determine how they impacted differentiation (Fig. 6C). After a 4-d course of differentiation, we found that cells infected with the chimera containing the MyoD bHLH region within Twist2, referred to here as Twist2(M), were able to undergo normal differentiation when compared with MyoD- and GFP-infected controls (Fig. 6D,E; Supplemental Fig. S6). However, cells infected with the chimera containing the Twist2 bHLH within MyoD, referred to here as MyoD(T), failed to differentiate (Fig. 6D,E). Additionally, the cells infected with the latter construct resembled the enlarged and fibroblastic morphology of Twist2-infected cells (Fig. 6D), suggesting that the Twist2 bHLH was also sufficient to repress myogenesis and activate an EMT and ECM remodeling profile. These data show that the major determinant of Twist2 function arises from its unique binding signature, which allows it to interfere with MyoD binding at shared GC E-boxes.
Figure 6.

The bHLH domain of Twist2 is critical for repressing myogenesis. (A) Sequence alignment of Twist2 and MyoD bHLH domains. The asterisk represents identical amino acids. (B) Chimeric Twist2 and MyoD bHLH constructs. Numbers represent amino acid number. (C) Experimental scheme to test the effect of Twist2 and MyoD chimeras on differentiation. (D) Tw2+ myoblasts infected with GFP, MyoD, Twist2, Twist2(M), or MyoD(T) were differentiated for 4 d and subsequently fixed and stained for Myosin (My32; green), tdTomato (red), and nuclei (blue). Scale bar, 50 µm. (E) The number of differentiated cells was quantified as a differentiation index based on the percentage of nuclei within My32+ cells compared with total nuclei. Unpaired two-tailed t-test: (****) P < 0.00005.
Twist2 dynamically regulates global chromatin organization during myogenesis
Recent studies have identified a key role for MyoD in driving RMS growth and initiation (Gryder et al. 2017; Tenente et al. 2017). During our analysis, we noticed a significant number of MyoD peaks that were gained only in the presence of Twist2 overexpression (Fig. 5C,D). Additionally, these peaks were enriched for the canonical MyoD GC E-box motif and were associated with genes involved in development, metabolism, growth, and cancer pathways (Fig. 5G; Supplemental Fig. S5F). These observations suggested that Twist2 may play an additional role in chromatin remodeling to alter MyoD accessibility to target genes. To study how Twist2 affects global chromatin changes, we analyzed the active and repressive histone modifications H3K27ac and H3K27me3, respectively, by ChIP-seq in cells maintained in GM and DM as well as in the presence or absence of Twist2 (Supplemental Fig. S7A). We first validated peak-calling quality through principle component analysis and matrix correlation (Supplemental Fig. S7B,C). On a global scale, we observed changes in both H3K27ac and H3K27me3 that coincided with Twist2 binding in GM and DM (Fig. 7A). During differentiation, up-regulated Twist2 peaks were associated with increased deposition of H3K27ac, while down-regulated Twist2 peaks were associated with a loss of H3K27ac (Fig. 7A). Additionally, down-regulated Twist2 peaks were also associated with a slight gain of H3K27me3 marks (Fig. 7A). Interestingly, up-regulated Twist2 peaks during differentiation exhibit both H3K27ac and H3K27me3 marks, suggesting chromatin bivalency (Fig. 7A). This finding may explain the reversibility of myogenic repression imparted by Twist2.
Figure 7.

Twist2 dynamically regulates global chromatin organization during differentiation. (A, left) Heat map depicting differential binding of Twist2 in GM versus DM conditions and the effect on H3K27ac and H3K27me3. (Right) ChIP signal distribution plot of Twist2, H3K27ac, and H3K27me3 peaks associated with up-regulated and down-regulated Twist2 peaks in DM versus GM conditions. (B) Scatter plot depicting differential binding of H3K27ac in the presence and absence of Twist2 in DM. Differential binding cutoff was set at a twofold or greater change and Padj < 0.05. (C, left) Heat map depicting the effect of Twist2 on differential binding of MyoD and H3K27ac. (Right) ChIP signal distribution plot of Twist2, MyoD, and H3K27ac peaks associated with up-regulated and down-regulated MyoD peaks in TW2-DM versus GFP-DM. (D, left) Heat map depicting the effect of Twist2 on differential binding of MyoD and H3K27me3. (Right) ChIP signal distribution plot of TWIST2, MyoD, and H3K27me3 peaks associated with up-regulated and down-regulated MyoD peaks in TW2-DM versus GFP-DM. (E) Genome browser shot depicting H3K27ac and H3K27me3 changes at the Mymk locus during differentiation in the presence and absence of Twist2. (F) GREAT analysis of H3K27ac peaks down-regulated in TW2-DM versus GFP-DM. (G) Genome browser shot depicting gain of H3K27ac and MyoD peaks in the Notch3 locus upon Twist2 binding. (H) GREAT analysis of H3K27ac peaks up-regulated in TW2-DM versus GFP-DM.
We also observed broad changes in chromatin accessibility during differentiation in the presence and absence of Twist2 (Fig. 7B–D). At myogenic loci where Twist2 competes with MyoD, we observed a significant decrease in H3K27ac and a corresponding increase in H3K27me3 (Fig. 7C,D). An example of this occurs at the Mymk locus, where a sharp increase in H3K27ac signal was observed during normal differentiation (Fig. 7E). In the presence of Twist2, this increase was significantly reduced, suggesting that Twist2 prevents the deposition of active H3K27ac marks at myogenic loci (Fig. 7E). Additionally, repressive H3K27me3 marks were normally eliminated at the Mymk locus during differentiation but were maintained in the presence of Twist2 (Fig. 7E). On a genome-wide level, Twist2 impairs H3K27ac deposition primarily at myogenic loci (Fig. 7F).
In addition to competition, Twist2 overexpression also induces new MyoD peaks at metabolic and developmental loci (Fig. 5G). These peaks are associated with Twist2 binding as well as increased H3K27ac deposition, suggesting that Twist2 opens chromatin loci of cancer-related genes to enable MyoD binding (Figs. 5G, 7C). An example of this occurs at the Notch3 locus (Fig. 7G). NOTCH is a regulator of muscle stem cell quiescence (Bjornson et al. 2012; Raimondi et al. 2012), myogenesis (Supplemental Fig. S7D), and RMS differentiation (Kuang et al. 2007). In the absence of Twist2, there is little H3K27ac within the Notch3 locus, corresponding with the absence of MyoD binding (Fig. 7G). Upon Twist2 overexpression, there is a sharp increase in H3K27ac that coincides with Twist2 binding. Additionally, the opening of chromatin now allows MyoD to bind a previously inaccessible site (Fig. 7G). At a genome-wide level, Twist2 induces H3K27ac at EMT and ECM remodeling loci (Fig. 7H). In contrast to H3K27ac, H3K27me3 marks at cancer-related loci are unchanged in the presence or absence of Twist2 (Fig. 7D). This pattern is similar to Twist2-mediated epigenetic regulation during differentiation, where we also observed chromatin bivalency (Fig. 7A).
In order to capture potential transcription factors that might cooperate with Twist2 to remodel the chromatin, we performed motif analysis on Twist2 peaks within up-regulated and down-regulated H3K27ac peaks. Those motifs were then associated with known transcription factors. Unique transcription factors with motifs found only in the lost H3K27ac peaks include MYOG, HAND1, and MEF2, while those found in gained H3K27ac peaks include TWIST1 and NFIA (Supplemental Fig. S7E,F). Shared motifs include those for AP-1, TEAD2, and RUNX (Supplemental Fig. S7E,F). Some of these motifs are also found within differentially regulated MyoD-binding sites in RD cells compared with C2C12 myoblasts (MacQuarrie et al. 2013), further implicating Twist2 as a key mediator of altered MyoD binding in RMS. Our motif analyses also allow us to speculate on the mechanism by which Twist2 induces MyoD binding. Twist2 binding at GC E-boxes is associated with down-regulation of H3K27ac peaks, while Twist2 binding at GA E-boxes is associated with up-regulation of H3K27ac (Supplemental Fig. S7E,F). However, the predominant MyoD motif in both up-regulated and down-regulated regions was a GC E-box (Supplemental Fig. S5E,F), suggesting that Twist2 binding to GA E-boxes results in opening of chromatin, which may uncover nearby GC E-boxes for MyoD to bind. Taken together, these results suggest that in addition to its roles in direct transcriptional regulation, Twist2 functions to block the opening of chromatin at myogenic gene loci while simultaneously promoting opening of chromatin at developmental and EMT loci. Additionally, these epigenetic changes can shape the binding profile of MyoD, allowing it to subvert its normal physiological function in favor of new pathological functions.
Discussion
In the present study, we identified TWIST2 as a key oncogene that is amplified and highly expressed in FNRMS. Additionally, Twist2 acts as a reversible inhibitor of myogenic differentiation with the ability to promote myotube dedifferentiation. To explore the mechanistic basis of Twist2-mediated myogenic repression, we used ChIP-seq and RNA-seq techniques to generate the first comprehensive functional genomics analysis of Twist2 and its interplay with MyoD during differentiation. Our findings demonstrate that Twist2 regulates gene expression through both direct promoter/enhancer binding and histone modification. Additionally, Twist2-mediated epigenetic modifications are sufficient to redirect MyoD binding from myogenic toward oncogenic loci. This function not only is vital for maintaining an undifferentiated state in myogenic progenitors but also plays a significant role in RMS pathogenesis.
We focused our study on Twist2 due to our discovery of a Twist2-expressing lineage of muscle progenitors (Liu et al. 2017). Our iTwist2 system showed that Twist2 is capable of reversibly repressing myogenesis and promoting myotube dedifferentiation. It is interesting to note that previously characterized regulators of myotube dedifferentiation such as Twist1, Msx1, and a variety of small molecules all play roles in cytoskeletal processes (Odelberg et al. 2000; Paliwal and Conboy 2011; Mastroyiannopoulos et al. 2013; Yang et al. 2014; Wang et al. 2015). Given the well-established role of Twist transcription factors in regulating EMT and cellular invasion, it appears that a key aspect of dedifferentiation involves cytoskeletal remodeling that is incompatible with the organization of mature sarcomeres. Interestingly, we observed that iTwist2 dedifferentiated myotubes do not begin proliferating, similar to previous observations (Duckmanton et al. 2005; Wang et al. 2015). Recent studies have shown that part of the dedifferentiation process may be mediated by apoptotic proteins and that generation of full proliferative myoblasts requires impairment of p53 and cell cycle inhibitors (Paliwal and Conboy 2011; Wang et al. 2015).
While the ability of Twist transcription factors to block MyoD-mediated differentiation has been shown previously using reporter constructs and in vitro assays (Spicer et al. 1996; Gong and Li 2002), the exact mechanism has not been explored. In particular, the increasingly evident role of epigenetics in transcription factor biology has not been investigated in the context of Twist-mediated repression of myogenesis. Our study used ChIP-seq and RNA-seq to generate a comprehensive genomics profile of Twist2 function on MyoD binding and chromatin remodeling during myogenic differentiation. Our results suggest that Twist2-mediated myogenic repression occurs at multiple levels across the genome. First, we identified direct competition between Twist2 and MyoD at shared GC E-boxes, which are associated with myogenic genes (MacQuarrie et al. 2013). This interaction results in loss of MyoD and subsequent replacement by Twist2. Second, we also identified Twist2 as a regulator of chromatin accessibility. Twist2 binding at myogenic loci was associated with loss of H3K27ac and gain of H3K27me3. Despite these global changes, Twist2-mediated myogenic repression is reversible. A likely explanation is the presence of bivalent chromatin in regions with strong Twist2 binding during differentiation. Chromatin bivalency typically enables cellular plasticity and is often associated with developmental genes (Bernstein et al. 2006). The reversibility of Twist2 is of key importance in biological contexts. In Drosophila adult muscle precursors and mammalian Tw2+ cells, initial Twist expression is important for maintaining an undifferentiated state; however, subsequent down-regulation of Twist is necessary prior to myogenic differentiation (Gildor et al. 2012; Liu et al. 2017).
Twist2 is not only a repressor of myogenesis but an activator of EMT, cellular invasion, development, and other oncogenic pathways (Yang et al. 2004, 2006; Eckert et al. 2011; Franco et al. 2011; Li et al. 2012; Jung and Yang 2015). Our study corroborates this knowledge by identifying Twist2 as a direct and epigenetic regulator of these pathways through binding of its private GA E-box and double-E-box motifs. We also identified a potential role of Twist proteins in mediating RMS etiology. A prior study found that eight out of 15 RMS samples had high-level expression of TWIST1 (Maestro et al. 1999). Through comprehensive genomic analysis, we discovered that TWIST transcription factors are highly amplified and overexpressed in the majority of FNRMS patients. Additionally, knockdown of TWIST2 and TWIST1 results in significant up-regulation of MYOGENIN expression and a decrease in proliferation. While most cancers are conventionally targeted through exploitation of pathways regulating cell proliferation, RMS is a unique case in which tumor differentiation exists as a possible treatment strategy (Saab et al. 2011). The reversibility of Twist2-mediated myogenic repression suggests that targeting of Twist2 and other amplified oncogenes may enable RMS cells to resume normal differentiation (Xu et al. 2018). Given the difficult nature of targeting transcription factors for therapeutic treatment, additional efforts to generate comprehensive upstream and downstream TWIST2 regulatory networks may reveal additional druggable targets. Since FNRMS patients may experience copy number amplifications of multiple drivers (Xu et al. 2018), the regulatory networks of these drivers could be intersected to identify unique targetable nodes (Gong et al. 2015).
Twist2-induced changes in chromatin structure prevent MyoD from accessing its physiologic target genes but also enable MyoD to bind new target genes. Recent studies have suggested that MyoD and Myf5 adopt key roles in regulating RMS growth and initiation (Olguín et al. 2011; Tenente et al. 2017). Through ChIP-seq of ERMS cell lines, Tenente et al. (2017) observed that both factors regulate genes involved in muscle development, growth factor signaling, smooth muscle and endothelial cell biology, and TGF-β signaling. Knockdown of either MyoD or Myf5 resulted in a near-complete loss of RMS proliferative potential (Tenente et al. 2017). These novel findings suggest that in ERMS, MyoD and Myf5 shift their functions from muscle differentiation toward cancer growth. In support of these findings, we found that Twist2 overexpression remodels the chromatin to shift MyoD binding toward genes involved in EMT, metabolic, smooth muscle, FGFR signaling, and growth pathways. Previous attempts to differentiate RMS by overexpressing MyoD have been unsuccessful (Tapscott et al. 1993). Our data suggest that additional MyoD is likely redirected toward oncogenic genes in the context of RMS. MEF2C-associated sites were found previously to have poor MyoD binding in RD cells (MacQuarrie et al. 2013). Interestingly, Twist2-repressed H3K27ac and MyoD peaks were enriched for MEF2 family motifs. We also noted that many of the Twist2-induced H3K27ac and MyoD peaks contain binding motifs for developmental and cancer regulators such as RUNX, AP-1, and NFI. These motifs were also associated with differentially bound MyoD sites in RD cells relative to normal myotubes (MacQuarrie et al. 2013). These findings implicate TWIST2 as a potential driver of epigenetic changes seen in FNRMS tumorigenesis. In FPRMS, PAX3-FOXO1 and PAX7-FOXO1 may redirect myogenic transcription factors to organize autoregulatory superenhancers that contribute to its unique gene signature (Olguín et al. 2011; Gryder et al. 2017). The similarity of TWIST2 function in this context suggests that oncogene amplification and fusion protein generation represent two parallel pathways in RMS etiology.
This study provides intriguing insights regarding the function of MyoD in RMS. It is tantalizing to hypothesize that MyoD and other transcription factors may adopt novel nonphysiologic roles in the context of RMS through epigenetic remodeling by oncogenic drivers such as TWIST2 and PAX3-FOXO1. How these novel functions contribute to RMS pathogenesis and whether they expose new vulnerabilities for therapeutic treatment will be exciting topics for future studies. Finally, it is becoming increasingly apparent that cancer progression can occur through lineage plasticity and subversion of tissue-specific developmental pathways (Mu et al. 2017). RMS appears to be such a case in which epigenetic remodeling by developmental transcription factors is vital for its formation (MacQuarrie et al. 2013; Gryder et al. 2017; Tenente et al. 2017). Given the depth of knowledge of myogenic regulatory mechanisms and the central role of Twist in these processes, targeting of Twist-regulated pathways may be a key aspect of future RMS therapy.
Materials and methods
Genomic analysis of RMS patient cohort
Genomic data from 258 specimens, collected from 258 patients and deidentified before use, were from sources that were detailed in a previous study from our laboratory (Xu et al. 2018). Genomic analysis of archived patient samples was approved by the University of Texas Southwestern Medical Center Institutional Review Board, and genomic data used in this study have been deposited to the database of Genotypes and Phenotypes (dbGaP) under accession number phs000720. Detailed methods are in the Supplemental Material.
Generation of a Dox-inducible Twist2 stable cell line
Twist2-IRES-GFP was Gateway-cloned into pCW57.1 (a gift from David Root; Addgene, plasmid 41393). For lentiviral production, iTwist2 plasmid was transfected using FuGENE 6 (Roche) into Lenti-X 293T cells (Clontech). Forty-eight hours after transfection, medium was harvested and filtered through a 0.45-µm cellulose filter and then concentrated using Lenti-X concentrator (Clontech). The lentivirus was then resuspended in 10 mL of GM comprised of Ham's F10, 20% fetal bovine serum, 0.2% Primocin (Invivogen), and 2.5 ng µL−1 basic FGF (Gibco). Tw2+ cells were infected with resuspended lentivirus containing 6 µg mL−1 polybrene 24 h after seeding. Twenty-four hours after infection, the viral medium was removed and replaced with GM for an additional 24 h. To generate a stable iTwist2 cell line, 2 µg mL−1 puromycin was added to GM daily on infected Tw2+ cells for 5 d.
Reversible differentiation and dedifferentiation of iTwist2 cells
iTwist2 cells were cultured in GM containing 10 µg mL−1 Dox (GM + Dox) for 24 h. GM + Dox was then replaced with DM comprised of DMEM, 2% horse serum, and 0.2% Primocin and containing 10 µg mL−1 Dox (DM + Dox). After 3 d of DM + Dox treatment, half of the iTwist2 cells continued DM + Dox treatment, and the other half of the iTwist2 cells were changed to DM only.
For dedifferentiation tracing experiments, LSLdsRed and pMSCVhygro-Cre primary myoblasts were cocultured in GM at a 1:1 ratio on day 0. On day 2, GM was replaced with DM to begin differentiation. On day 6, cells were either kept in DM or switched to DM + Dox to begin dedifferentiation. On day 8, DM was replaced with GM, while DM + Dox was replaced with GM + Dox to enhance dedifferentiation as described previously (Paliwal and Conboy 2011; Wang et al. 2015). Cells were then fixed and stained on day 10 or harvested for Western blot.
Immunostaining of cultured cells
Immunostaining of cultured cells was performed as described previously (Liu et al. 2017). The primary antibodies used included fast myosin (1:250; Sigma-Aldrich, My32). Cells were counterstained with Hoechst (1:1000; Thermo Fisher, Hoechst 3342). Alexa fluor secondary antibodies were used according to the manufacturer's instructions. The differentiation index was quantified as the percentage of Myosin+ nuclei over total nuclei. Dedifferentiation was calculated as the ratio of the number of nuclei within dsRed+GFP+ monuclear cells to total nuclei at day 10. Multiple representative fields were counted from three biological replicates.
Transwell migration assay
Transwell migration assay was performed according to the manufacturer's protocol (Corning). First, Ctrl-Tw2+, Twist2-Tw2+, and RD cells were serum-starved for 24 h before the assay. The number of cells migrated was measured using Calcein AM (Thermo Fisher) and a plate reader.
Generating Twist2 and MyoD bHLH domain chimeras
Gene block fragments (IDT) were synthesized with the swapped bHLH domains and Infusion-cloned (Clontech) into pBabe-puro (Addgene, 1764) for retroviral transduction. Retrovirus packaging for GFP, myc-Twist2, myc-MyoD, myc-Twist2(M), and myc-MyoD(T) constructs was performed as described previously (Liu et al. 2017). Infected Tw2+ cells were cultured for 24 h in GM and then differentiated in DM for 4 d. Cells were then fixed and stained for myosin.
ChIP-seq for transcription factors and histone marks
ChIP was performed as described in the ChIP-IT high-sensitivity kit (Active Motif). In brief, Tw2+ cells were infected with 3xTy1-Twist2 or a GFP control and kept in GM or switched to DM 24 h after infection. After 3 d in GM or 4 d in DM, we harvested cells and performed ChIP using antibodies recognizing Ty1 (Diagenode), MyoD (Santa Cruz Biotechnology, sc-32758), H3K27ac (Diagenode, C15410196), and H3K27me3 (Diagenode, C15410195). Sequencing was performed in the McDermott Sequencing Core at University of Texas Southwestern. Data are available in the Gene Expression Omnibus under accession number GSE127998. Detailed ChIP-seq data analysis is in the Supplemental Material.
Statistical analysis
All quantitative data are displayed as mean ± SEM. Unpaired two-tailed t-test was performed for all analyses: P < 0.05 (*), P < 0.005 (**), P < 0.0005 (***), and P < 0.00005 (****).
Supplementary Material
Acknowledgments
We are grateful to the University of Texas Southwestern Live-Cell Imaging Core and McDermott Sequencing Core for technical help and service. We thank Jose Cabrera for graphics assistance, and Zhaoning Wang for helpful discussion with bioinformatics. This work was supported by grants from the Nationals Institutes of Health (NIH; AR-067294, HL-130253, HL-138426, and HD-087351) and the Robert A. Welch Foundation (grant 1-0025 to E.N.O.). S.L. was supported by an NIH F30 fellowship (F30AG056075). N.L. was supported by a grant from the NIH (R01AR071980-01A1). Y. Zheng, L.X., and S.X.S. were supported in part by RP120685-P2 from Cancer Prevention and Research Institute of Texas (CPRIT). S.D.B. and V.S.M. were supported by RP150596 from CPRIT.
Author contributions: S.L. and N.L. designed the study. S.L., Y. Zhang, P.J., Y. Zheng, and M.H. performed experiments. S.L. Y. Zhang, Y. Zheng, S.X.S, N.L., R.B.-D., and E.N.O. interpreted the data. K.C., S.D.B., V.S.M., and L.X. conducted bioinformatic analyses. N.L. and E.N.O. supervised the study. S.L. wrote the manuscript. S.X.S, L.X., N.L., R.B.-D., and E.N.O. edited the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.324467.119.
References
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Bjornson CRR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. 2012. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30: 232–242. 10.1002/stem.773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R, Vethantham V, Bowman C, Rudnicki M, Dynlacht BD. 2012. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev 26: 2763–2779. 10.1101/gad.200113.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. 2010. Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 18: 662–674. 10.1016/j.devcel.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. 2011. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 71: 245–254. 10.1158/0008-5472.CAN-10-2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang NC, Rudnicki MA. 2014. Satellite cells: the architects of skeletal muscle. Curr Top Dev Biol 107: 161–181. 10.1016/B978-0-12-416022-4.00006-8 [DOI] [PubMed] [Google Scholar]
- Chang AT, Liu Y, Ayyanathan K, Benner C, Jiang Y, Prokop JW, Paz H, Wang D, Li H-R, Fu X-D, et al. 2015. An evolutionarily conserved DNA architecture determines target specificity of the TWIST family bHLH transcription factors. Genes Dev 29: 603–616. 10.1101/gad.242842.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckmanton A, Kumar A, Chang Y-T, Brockes JP. 2005. A single-cell analysis of myogenic dedifferentiation induced by small molecules. Chem Biol 12: 1117–1126. 10.1016/j.chembiol.2005.07.011 [DOI] [PubMed] [Google Scholar]
- Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. 2011. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19: 372–386. 10.1016/j.ccr.2011.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong AP, Yao Z, Zhong JW, Cao Y, Ruzzo WL, Gentleman RC, Tapscott SJ. 2012. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev Cell 22: 721–735. 10.1016/j.devcel.2012.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong AP, Yao Z, Zhong JW, Johnson NM, Farr GH, Maves L, Tapscott SJ. 2015. Conversion of MyoD to a neurogenic factor: binding site specificity determines lineage. Cell Rep 10: 1937–1946. 10.1016/j.celrep.2015.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco HL, Casasnovas J, Rodríguez-Medina JR, Cadilla CL. 2011. Redundant or separate entities?—roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res 39: 1177–1186. 10.1093/nar/gkq890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildor B, Schejter ED, Shilo B-Z. 2012. Bidirectional Notch activation represses fusion competence in swarming adult Drosophila myoblasts. Development 139: 4040–4050. 10.1242/dev.077495 [DOI] [PubMed] [Google Scholar]
- Gong XQ, Li L. 2002. Dermo-1, a multifunctional basic helix–loop–helix protein, represses MyoD transactivation via the HLH domain, MEF2 interaction, and chromatin deacetylation. J Biol Chem 277: 12310–12317. 10.1074/jbc.M110228200 [DOI] [PubMed] [Google Scholar]
- Gong W, Koyano-Nakagawa N, Li T, Garry DJ. 2015. Inferring dynamic gene regulatory networks in cardiac differentiation through the integration of multi-dimensional data. BMC Bioinformatics 16: 74 10.1186/s12859-015-0460-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryder BE, Yohe ME, Chou H-C, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, et al. 2017. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov 7: 884–899. 10.1158/2159-8290.CD-16-1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettmer S, Wagers AJ. 2010. Muscling in: uncovering the origins of rhabdomyosarcoma. Nat Med 16: 171–173. 10.1038/nm0210-171 [DOI] [PubMed] [Google Scholar]
- Jones S. 2004. An overview of the basic helix–loop–helix proteins. Genome Biol 5: 226 10.1186/gb-2004-5-6-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H-Y, Yang J. 2015. Unraveling the TWIST between EMT and cancer stemness. Cell Stem Cell 16: 1–2. 10.1016/j.stem.2014.12.005 [DOI] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. 2007. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129: 999–1010. 10.1016/j.cell.2007.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Serna de IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwag CS, Tapscott SJ, Imbalzano AN. 2005. MyoD targets chromatin remodeling complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol 25: 3997–4009. 10.1128/MCB.25.10.3997-4009.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Cserjesi P, Olson EN. 1995. Dermo-1: a novel twist-related bHLH protein expressed in the developing dermis. Dev Biol 172: 280–292. 10.1006/dbio.1995.0023 [DOI] [PubMed] [Google Scholar]
- Li Y, Wang W, Wang W, Yang R, Wang T, Su T, Weng D, Tao T, Li W, Ma D, et al. 2012. Correlation of TWIST2 up-regulation and epithelial–mesenchymal transition during tumorigenesis and progression of cervical carcinoma. Gynecol Oncol 124: 112–118. 10.1016/j.ygyno.2011.09.003 [DOI] [PubMed] [Google Scholar]
- Liu N, Garry GA, Li S, Bezprozvannaya S, Sanchez-Ortiz E, Chen B, Shelton JM, Jaichander P, Bassel-Duby R, Olson EN. 2017. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat Cell Biol 19: 202–213. 10.1038/ncb3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQuarrie KL, Yao Z, Fong AP, Diede SJ, Rudzinski ER, Hawkins DS, Tapscott SJ. 2013. Comparison of genome-wide binding of MyoD in normal human myogenic cells and rhabdomyosarcomas identifies regional and local suppression of promyogenic transcription factors. Mol Cell Biol 33: 773–784. 10.1128/MCB.00916-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH, Hannon GJ. 1999. Twist is a potential oncogene that inhibits apoptosis. Genes Dev 13: 2207–2217. 10.1101/gad.13.17.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroyiannopoulos NP, Antoniou AA, Koutsoulidou A, Uney JB, Phylactou LA. 2013. Twist reverses muscle cell differentiation through transcriptional down-regulation of myogenin. Biosci Rep 33: 903–911. 10.1042/BSR20130068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merindol N, Riquet A, Szablewski V, Eliaou J-F, Puisieux A, Bonnefoy N. 2014. The emerging role of Twist proteins in hematopoietic cells and hematological malignancies. Blood Cancer J 4: e206 10.1038/bcj.2014.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen C-C, Wongvipat J, Ku S-Y, Gao D, Cao Z, et al. 2017. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355: 84–88. 10.1126/science.aah4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odelberg SJ, Kollhoff A, Keating MT. 2000. Dedifferentiation of mammalian myotubes induced by msx1. Cell 103: 1099–1109. 10.1016/S0092-8674(00)00212-9 [DOI] [PubMed] [Google Scholar]
- Olguín HC, Patzlaff NE, Olwin BB. 2011. Pax7-FKHR transcriptional activity is enhanced by transcriptionally repressed MyoD. J Cell Biochem 112: 1410–1417. 10.1002/jcb.23057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal P, Conboy IM. 2011. Inhibitors of tyrosine phosphatases and apoptosis reprogram lineage-marked differentiated muscle to myogenic progenitor cells. Chem Biol 18: 1153–1166. 10.1016/j.chembiol.2011.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preussner J, Zhong J, Sreenivasan K, Günther S, Engleitner T, Künne C, Glatzel M, Rad R, Looso M, Braun T, et al. 2018. Oncogenic amplification of zygotic dux factors in regenerating p53-deficient muscle stem cells defines a molecular cancer subtype. Cell Stem Cell 23: 794–805.e4. 10.1016/j.stem.2018.10.011 [DOI] [PubMed] [Google Scholar]
- Raimondi L, Ciarapica R, De Salvo M, Verginelli F, Gueguen M, Martini C, De Sio L, Cortese G, Locatelli M, Dang TP, et al. 2012. Inhibition of Notch3 signalling induces rhabdomyosarcoma cell differentiation promoting p38 phosphorylation and p21(Cip1) expression and hampers tumour cell growth in vitro and in vivo. Cell Death Differ 19: 871–881. 10.1038/cdd.2011.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinon A, Molchadsky A, Nathan E, Yovel G, Rotter V, Sarig R, Tzahor E. 2011. p53 coordinates cranial neural crest cell growth and epithelial–mesenchymal transition/delamination processes. Development 138: 1827–1838. 10.1242/dev.053645 [DOI] [PubMed] [Google Scholar]
- Saab R, Spunt SL, Skapek SX. 2011. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr Top Dev Biol 94: 197–234. 10.1016/B978-0-12-380916-2.00007-3 [DOI] [PubMed] [Google Scholar]
- Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. 2014. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov 4: 216–231. 10.1158/2159-8290.CD-13-0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Garry DJ. 2006. Muscle stem cells in development, regeneration, and disease. Genes Dev 20: 1692–1708. 10.1101/gad.1419406 [DOI] [PubMed] [Google Scholar]
- Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, et al. 2014. Disrupting the interaction of BRD4 with diacetylated twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25: 210–225. 10.1016/j.ccr.2014.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shield MA, Haugen HS, Clegg CH, Hauschka SD. 1996. E-box sites and a proximal regulatory region of the muscle creatine kinase gene differentially regulate expression in diverse skeletal muscles and cardiac muscle of transgenic mice. Mol Cell Biol 16: 5058–5068. 10.1128/MCB.16.9.5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, Barr FG, Hawkins DS. 2019. Rhabdomyosarcoma. Nat Rev Dis Primers 5: 1 10.1038/s41572-018-0051-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soleimani VD, Yin H, Jahani-Asl A, Ming H, Kockx CEM, van Ijcken WFJ, Grosveld F, Rudnicki MA. 2012. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol Cell 47: 457–468. 10.1016/j.molcel.2012.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer DB, Rhee J, Cheung WL, Lassar AB. 1996. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science 272: 1476–1480. 10.1126/science.272.5267.1476 [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, Weintraub H. 1991. MyoD and the regulation of myogenesis by helix–loop–helix proteins. J Clin Invest 87: 1133–1138. 10.1172/JCI115109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, Thayer MJ, Weintraub H. 1993. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 259: 1450–1453. 10.1126/science.8383879 [DOI] [PubMed] [Google Scholar]
- Tenente IM, Hayes MN, Ignatius MS, McCarthy K, Yohe M, Sindiri S, Gryder B, Oliveira ML, Ramakrishnan A, Tang Q, et al. 2017. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 6: 35 10.7554/eLife.19214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, Judson RN, Thway K, Nadal G, Selfe JL, et al. 2014. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 26: 273–287. 10.1016/j.ccr.2014.05.029 [DOI] [PubMed] [Google Scholar]
- Tsokos M. 1994. The diagnosis and classification of childhood rhabdomyosarcoma. Semin Diagn Pathol 11: 26–38. [PubMed] [Google Scholar]
- Wang H, Lööf S, Borg P, Nader GA, Blau HM, Simon A. 2015. Turning terminally differentiated skeletal muscle cells into regenerative progenitors. Nat Commun 6: 858 10.1038/ncomms8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Zheng Y, Liu J, Rakheja D, Singleterry S, Laetsch TW, Shern JF, Khan J, Triche TJ, Hawkins DS, et al. 2018. Integrative bayesian analysis identifies rhabdomyosarcoma disease genes. Cell Rep 24: 238–251. 10.1016/j.celrep.2018.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939. 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- Yang J, Mani SA, Weinberg RA. 2006. Exploring a new twist on tumor metastasis. Cancer Res 66: 4549–4552. 10.1158/0008-5472.CAN-05-3850 [DOI] [PubMed] [Google Scholar]
- Yang Z, Liu Q, Mannix RJ, Xu X, Li H, Ma Z, Ingber DE, Allen PD, Wang Y. 2014. Mononuclear cells from dedifferentiation of mouse myotubes display remarkable regenerative capability. Stem Cells 32: 2492–2501. 10.1002/stem.1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fong AP, Cao Y, Ruzzo WL, Gentleman RC, Tapscott SJ. 2013. Comparison of endogenous and overexpressed MyoD shows enhanced binding of physiologically bound sites. Skelet Muscle 3: 8 10.1186/2044-5040-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, Scott TG, Nabet B, Erb MA, Winter GE, et al. 2018. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet 50: 515–523. 10.1038/s41588-018-0044-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hassan MQ, Li Z-Y, Stein JL, Lian JB, van Wijnen AJ, Stein GS. 2008. Intricate gene regulatory networks of helix–loop–helix (HLH) proteins support regulation of bone-tissue related genes during osteoblast differentiation. J Cell Biochem 105: 487–496. 10.1002/jcb.21844 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.