

In this review, Poillet-Perez et al. discuss how understanding the underlying mechanisms by which autophagy functions in cancer can guide its development as an anticancer approach.
Keywords: autophagy, cancer, metabolism
Abstract
Macroautophagy (referred to here as autophagy) degrades and recycles cytoplasmic constituents to sustain cellular and mammalian metabolism and survival during starvation. Deregulation of autophagy is involved in numerous diseases, such as cancer. Cancers up-regulate autophagy and depend on it for survival, growth, and malignancy in a tumor cell-autonomous fashion. Recently, it has become apparent that autophagy in host tissues as well as the tumor cells themselves contribute to tumor growth. Understanding how autophagy regulates metabolism and tumor growth has revealed new essential tumor nutrients, where they come from, and how they are supplied and used, which can now be targeted for cancer therapy.
Autophagy is a catabolic process that captures and degrades damaged proteins and organelles in lysosomes. Autophagy is regulated by >30 autophagy-related (ATG) proteins (Klionsky et al. 2011). During this multistep process, cytosolic components are sequestered in double-membrane vesicles called autophagosomes, which then fuse with lysosomes to form autolysosomes (Mizushima 2007; Klionsky and Codogno 2013; Feng et al. 2014). The contents of autolysosomes are broken down by the degradative enzymes supplied by lysosomes into products such as amino acids, nucleic acids, sugars, and fatty acids, which are recycled into central carbon metabolism (Mizushima and Klionsky 2007; Rabinowitz and White 2010; Guo et al. 2016; Zong et al. 2016). In normal cells and tissues, autophagy is active at a low basal level to sustain cellular homeostasis and protein and organelle quality control through the elimination of damaged organelles and protein aggregates. Autophagy can also be induced by different stresses such as nutrient starvation, hypoxia, oxidative stress, and infection to allow adaptation and survival (Komatsu et al. 2005). Autophagy can be selective or nonselective (Khaminets et al. 2016; Mancias and Kimmelman 2016; Pickles et al. 2018; Wyant et al. 2018). Selective autophagy identifies and targets specific cargo for degradation. Selective autophagic degradation pathways include mitophagy for mitochondria, xenophagy for bacteria, ribophagy for ribosomes, and ferritinophagy for ferritin, ensuring that the appropriate substrates are degraded and recycled under specific conditions to maintain homeostasis.
Without autophagy, neonates deficient for Atg5 or Atg7 survive for only 12 h due to the neonatal starvation period, which can be extended to 24 h with force feeding. Atg5- or Atg7-deficient neonates also show reduced concentrations of essential amino acids and branched chain amino acids in the circulation. These results indicate the critical importance of autophagy to provide amino acids necessary to survive the neonatal starvation period (Kuma et al. 2004; Komatsu et al. 2005). Autophagy is also required for adult mice to survive fasting (Fig. 1A; Karsli-Uzunbas et al. 2014). Indeed, conditional (acute) systemic (whole-body) Atg7 deficiency in adult mice leads to gradual depletion of dedicated nutrient stores of lipid in white adipose tissue (WAT), glycogen in liver, and protein in muscle (muscle wasting, also known as cachexia). Fasting conditionally autophagy-deficient adult mice causes rapid depletion of these dedicated nutrient stores and failure to maintain circulating glucose levels, which leads to death from hypoglycemia (Fig. 1A,B). Supplementation of Atg7 conditionaly deficient mice with glucose during fasting rescues muscle wasting and mouse survival (Karsli-Uzunbas et al. 2014). Thus, the loss of autophagy creates a systemic metabolic defect in mammals, possibly increasing dependency on and consumption of circulating nutrients, necessitating excessive catabolism of dedicated nutrient stores (lipid, glycogen, and muscle protein) (Karsli-Uzunbas et al. 2014). The metabolic imbalance created by conditional autophagy deficiency in adult mice may reflect a mechanism of cachexia seen in cancer patients that needs to be explored further.
Figure 1.
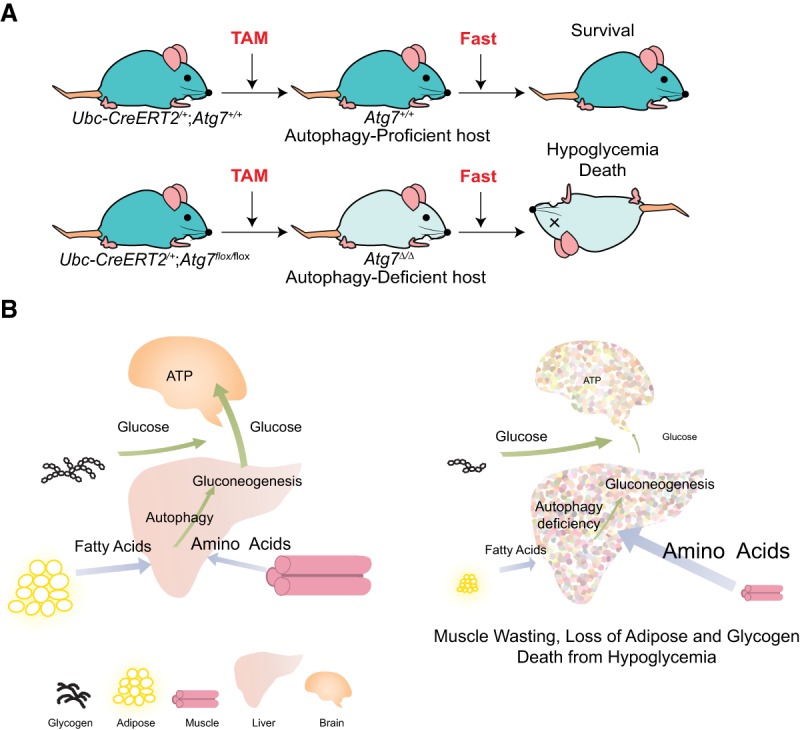
Autophagy is required to sustain circulating nutrients during fasting, critical for the survival of adult mice. (A) Mammalian survival during fasting requires autophagy. Treatment with tamoxifen (TAM) leads to conditional whole-body deletion of Atg7 in adult Ub-CreERT2+/−;Atg7flox/flox mice. While autophagy-proficient hosts can survive during fasting, autophagy-deficient hosts die from hypoglycemia when fasted (Karsli-Uzunbas et al. 2014). (B) Autophagy is essential to sustain circulating nutrients during fasting. Fasting autophagy-deficient hosts leads to the loss of WAT, glycogen in the liver, and proteins in muscle (cachexia) and the failure to maintain circulating glucose.
Autophagy and cancer
Given the role of autophagy in protein and organelle turnover, intracellular trafficking, and mammalian metabolism, it is not surprising that its deregulation has been implicated in numerous diseases such as metabolic diseases, neurodegenerative diseases, infectious diseases, and cancer (Mizushima et al. 2008). For example, conditional whole-body deletion of Atg7 in adult mice increases susceptibility to Streptococcus infection, and most of these mice die at 2–3 mo of age due to neurodegeneration (Karsli-Uzunbas et al. 2014). Mutations of Atg genes have also been identified in various human diseases, such as Parkinson diseases and lysosomal storage disorders (Jiang and Mizushima 2014). It is now clear that autophagy is induced in many cancers and promotes their survival. In doing so, cancers have merely usurped the normal and critical survival mechanism of autophagy to enhance their growth and malignancy; hence, targeting autophagy is an important and novel approach to cancer therapy. Understanding the underlying mechanisms by which autophagy functions in cancer can guide its development as an anticancer approach.
Autophagy in tumor cells promotes cancer
The role of autophagy in cancer has been explored extensively in genetically engineered mouse models (GEMMs). GEMMs for cancer in which autophagy was specifically ablated in tumor cells demonstrated that autophagy loss can promote formation of benign lesions associated with tissue damage and inflammation but that many aggressive cancers require autophagy for growth, survival, and malignancy (White 2012; Kimmelman and White 2017; Onorati et al. 2018). In several of these models, autophagy can act as a tumor-suppressive mechanism during the early stage of tumorigenesis by suppressing reactive oxygen species (ROS), DNA damage, tissue damage, inflammation, and genome instability, which are known inducers of tumor initiation (Karantza-Wadsworth et al. 2007; Komatsu et al. 2007; Mathew et al. 2007, 2009, 2014; Yang et al. 2011; Deretic et al. 2013; Rosenfeldt et al. 2013; Strohecker et al. 2013). In tissues such as the pancreas and liver, where tumor initiation is caused by chronic tissue damage and inflammation, ablation of Atg7 or Atg5 leads to induction of benign pancreatic intraepithelial neoplasia (PANIN) in KrasG12D/+ mice and spontaneous liver adenomas, respectively, that fail to progress to malignancy (Takamura et al. 2011; Rosenfeldt et al. 2013; Yang et al. 2014). As these tumors remain benign, this indicates that even though depletion of autophagy can increase tumor initiation in the pancreases and livers of mice, autophagy is required for tumors to progress to a malignant stage. While these findings from GEMMs are interesting, the general infrequency of mutations in essential autophagy genes in human cancers has indicated that this does not represent a mechanism of cancer causation in humans and that the vast majority of human cancers preserve autophagy function (Laddha et al. 2014; Lebovitz et al. 2015).
Genetic ablation of essential autophagy genes in numerous GEMMs for cancer has revealed an important role for autophagy in promoting tumor growth, survival, and malignancy. In these GEMMs, essential autophagy genes are deleted in tumor cells that arise spontaneously in the context of a normal tumor microenvironment and functional immune system. Deletion of Atg5 or Atg7 in KrasG12D- or BrafV600E-driven lung cancer (Fig. 2A; Guo et al. 2013; Strohecker et al. 2013; Karsli-Uzunbas et al. 2014), BrafV600E;Pten−/−-driven melanoma (Xie et al. 2015), KrasG12D-driven pancreatic ductal adenocarcinoma (PDAC) (Rosenfeldt et al. 2013; Yang et al. 2014), or Pten−/−-driven prostate cancer (Santanam et al. 2016) with or without Trp53 deletion decreases tumor progression. Similar results were obtained with the deletion of Becn1 or FAK family-interacting protein of 200 kDa (Fip200) in Palb2−/− or polyoma middle T (PyMT) oncogene-driven breast cancer GEMMs (Wei et al. 2011; Huo et al. 2013) or the deletion of Atg13 or Unc-51-like autophagy-activating kinase (Ulk1) in a KrasG12D-driven glioblastoma model (Gammoh et al. 2016). Deletion of Atg7 in the intestine epithelium in adenomatous polyposis coli (APC)+/− mice suppresses tumor initiation by increasing the immune response and perhaps also by increasing metabolic stress (Levy et al. 2015). Finally, autophagy is particularly critical for the survival and growth of KrasG12D;Lkb1−/− (liver kinase B1)-driven lung cancer, as these tumors lack LKB1 and the ability to induce AMP kinase and adapt to metabolic stress (Bhatt et al. 2019). Thus, many cancers benefit from and require functional autophagy for their growth and progression.
Figure 2.
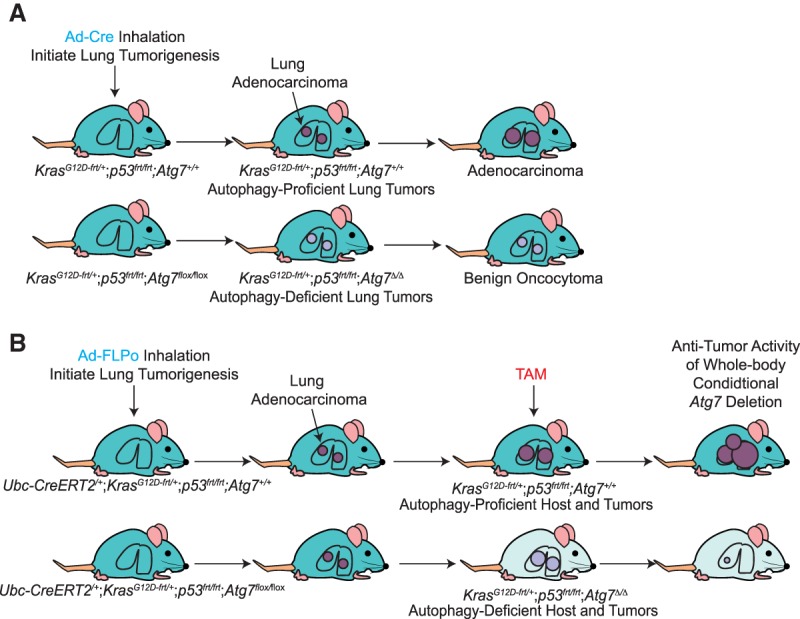
Tumor cell-autonomous autophagy as well as systemic autophagy promote tumor growth. (A) Tumor cell-autonomous autophagy promotes tumor growth. AdenoCre virus inhalation initiates lung tumorigenesis by activation of KrasG12D and deletion of Trp53, and tumor-specific autophagy deficiency is induced by deletion of Atg7. Loss of tumor cell-autonomous autophagy inhibits tumor growth and leads to the development of benign oncocytomas (Guo et al. 2013). (B) Pronounced antitumor activity of systemic autophagy ablation in tumor-bearing mice. AdenoFLPo virus inhalation initiates lung tumorigenesis by activation of KrasG12D and deletion of Trp53. When lung cancer is established, conditional whole-body autophagy deficiency is achieved by tamoxifen (TAM) injection and deletion of Atg7. Loss of host autophagy and tumor cell-autonomous autophagy inhibits tumor growth to a greater extent than tumor cell-autonomous autophagy alone (Karsli-Uzunbas et al. 2014).
There is increasing evidence that selective autophagy, such as mitophagy or ferritinophagy, also has a role in cancer (Chang et al. 2017; Um and Yun 2017; Santana-Codina and Mancias 2018). For mitophagy, several studies have suggested a tumor suppressor role for PARKIN (Fujiwara et al. 2008; Poulogiannis et al. 2010; Zhang et al. 2011), whereas BCL-2-interacting protein 3 (BNIP3) seems to display protumorigenic and antitumorigenic roles (Maes et al. 2014; Chourasia and Macleod 2015; Chourasia et al. 2015). The different isoforms of the selective cargo receptor for ferritinophagy, nuclear receptor coactivator 4 (NCOA4; α and β), may have a differential role by either inhibiting or promoting cancer cell proliferation (Peng et al. 2008; Wu et al. 2011). The exact role of selective autophagy in cancer still needs further investigation, as its involvement seems to be cell type- and context-dependent.
Autophagy sustains tumor cell metabolism
Whereas basal autophagy functions at a low level in normal cells and tissues, numerous cancer cell lines have a high level of basal autophagy, which is necessary to meet elevated metabolic demand and allow cell survival in vitro and tumorigenesis in vivo (Degenhardt et al. 2006; Guo et al. 2011, 2013; Lock et al. 2011; Yang et al. 2011; Viale et al. 2014). Comprehensive metabolic analysis of RAS-driven tumor cells with and without genetic ablation of autophagy revealed that autophagy is required to prevent energy crisis and maintain nucleotide pools during starvation. Autophagy accomplishes this by recycling macromolecules, thereby providing bioenergetic and biosynthetic substrates to the TCA cycle, which maintains energy homeostasis and nucleotide levels (Guo et al. 2011, 2016; Strohecker et al. 2013). In KrasG12D;Lkb1−/−-driven lung cancer models, deficiency in Atg7 reduces the amino acid substrate supply to mitochondria, causing excessive fatty acid oxidation, which depletes lipid stores and promotes energy crisis (Bhatt et al. 2019). Therefore, an important mechanism by which autophagy promotes tumor growth, survival, and malignancy is through its ability to sustain essential metabolic functions of tumor cells. One means by which RAS-driven cancers achieve this is by up-regulating basal autophagy by directly activating the MiT/TFE-regulated transcription program for autophagy and lysosomal biogenesis (Perera et al. 2015).
Autophagy deficiency selectively kills tumor cells, thereby providing a therapeutic window
Knowing that many tumors are autophagy-dependent, as autophagy deficiency compromises growth, survival, and malignancy, raises the question of whether tumors are more sensitive to loss of autophagy than normal tissues. Differential sensitivity between tumor and normal tissues is essential for a therapeutic window in cancer therapy. To test this genetically, conditional whole-body deletion of Atg7 in adult mice with established KRAS-driven lung cancer was performed to simulate the consequences in cancer patients following autophagy inhibitor treatment. Conditional whole-body deletion of Atg7 in mice with lung cancer produced dramatic antitumor activity prior to significant damage to normal tissues (Karsli-Uzunbas et al. 2014). Similar findings were obtained with systemic induction of expression of a dominant-negative ATG4b in mice with KRAS-driven PDAC (Yang et al. 2018). The preferential sensitivity of some tumors compared with normal tissues upon systemic loss of autophagy indicated the existence of a therapeutic window for cancer therapy.
Host autophagy promotes tumor growth
A remarkable finding from the conditional whole-body deficiency in autophagy in mice with cancer was the substantial tumor regression, which was far greater than that which occurred with tumor-specific autophagy ablation. In one example, conditional deletion of Atg7 throughout mice with KrasG12D- and Trp53−/−-driven lung cancers caused greater tumor regression than tumor-specific Atg7 deletion, suggesting that host autophagy as well as tumor cell-autonomous autophagy contributed to tumor growth (Fig. 2A,B; Karsli-Uzunbas et al. 2014). In a second example, conditional induction of systemic expression of a dominant-negative ATG4b in mice with KrasG12D- and Trp53−/+-driven PDAC produced similar results (Yang et al. 2018). In a third example, the important role of host autophagy in tumor growth was demonstrated in a Drosophila melanogaster malignant tumor model in which deletion of Atg13 and Atg14 in different compartments demonstrated that autophagy in the tumor microenvironment and in distant tissues was required for tumor growth (Katheder et al. 2017). To directly test whether host autophagy as well as tumor cell-autonomous autophagy promote tumor growth in mammals, autophagy-competent cancer cell lines were allografted onto autophagy wild-type and autophagy-deficient (conditional whole-body Atg7 deleted) host mice. Remarkably, deletion of host-specific autophagy impaired the growth of multiple cancer cell lines, including melanoma, carcinogen-induced urothelial carcinoma, and nonsmall cell lung cancer (NSCLC) cell lines (Fig. 3A; Poillet-Perez et al. 2018). Similarly, autophagy in stromal cells in the local tumor microenvironment is necessary for the efficient growth of PDAC (Sousa et al. 2016). Thus, autophagy in both the tumor cells themselves and the host promotes tumorigenesis.
Figure 3.
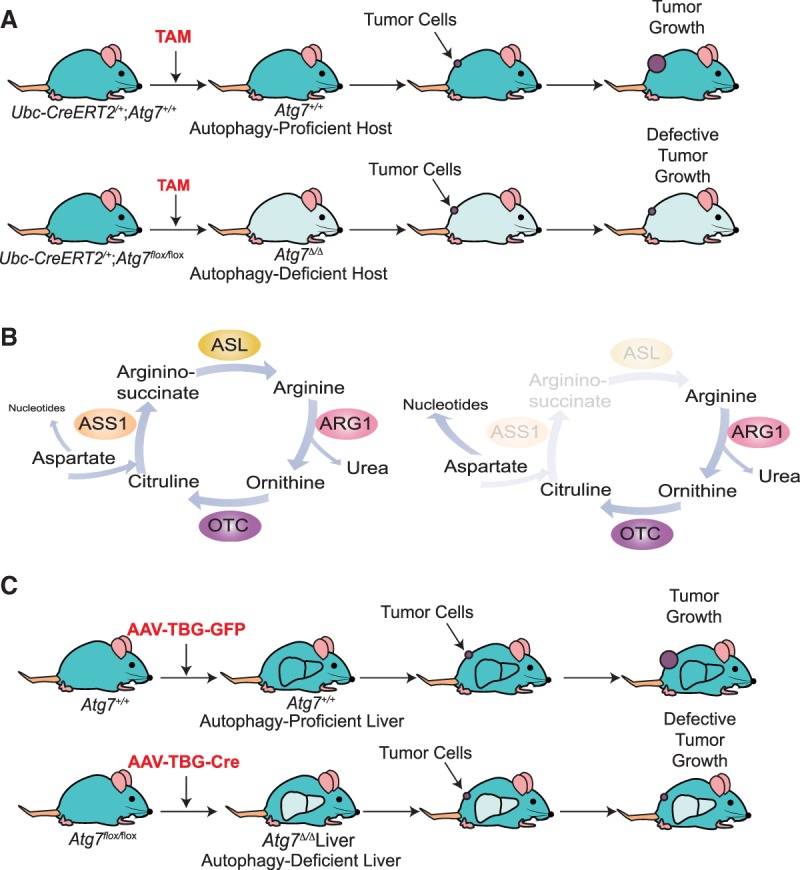
Whole-body autophagy and liver autophagy are essential for the growth of arginine-auxotrophic tumors. (A) Host autophagy promotes tumor growth. Treatment with TAM leads to conditional whole-body deletion of Atg7. Loss of host autophagy dramatically decreases the growth of autophagy-competent tumor cells, demonstrating the role of nontumor cell-autonomous autophagy in tumor growth (Poillet-Perez et al. 2018). (B) Arginine auxotrophy in cancer. Many cancer cells are auxotrophs for arginine, as they do not express argininosuccinate synthase 1 (ASS1) or argininosuccinate lyase (ASL), two enzymes required for de novo arginine biosynthesis. Tumors downregulate expression of these enzymes in order to use aspartate for nucleotide biosynthesis instead of the urea cycle (Rabinovich et al. 2015). (ARG1) Arginase 1; (OTC) ornithine transcarbamylase. (C) Liver autophagy promotes tumor growth. Tail vein injection of AAV-TBG-cre leads to the specific deletion of Atg7 in the liver. Loss of autophagy in the liver mimics the effect of host autophagy loss on tumor growth (Poillet-Perez et al. 2018).
Host autophagy sustains tumor metabolism
As there is clear evidence that host autophagy promotes tumor growth, the next question is as follows: How does host autophagy promote tumor growth? Tumors reprogram their metabolism to support biosynthetic and energetic pathways necessary for growth, proliferation, and survival. Tumors obtain their entire nutrient supply from the host to fuel these pathways (DeBerardinis and Chandel 2016; Pavlova and Thompson 2016; Vander Heiden and DeBerardinis 2017). As autophagy is important for sustaining host metabolism at both the cellular and mammalian levels, particularly during nutrient limitation (Rabinowitz and White 2010; Goldsmith et al. 2014; Karsli-Uzunbas et al. 2014; Kimmelman and White 2017), the loss of host autophagy may create a systemic metabolic defect and unfavorable environment for tumor growth. Two main sources of tumor nutrients are the host circulation and the local tumor microenvironment; as such, they were the first places to look for a role for host autophagy in promoting cancer metabolism.
Using conditional whole-body deletion of Atg7 or Atg5 and autophagy-competent cancer cell lines, it was found that host autophagy maintains tumor growth by sustaining the levels of circulating arginine (Poillet-Perez et al. 2018). These autophagy-deficient mice have altered serum metabolite levels. Among these metabolites, arginine is notably reduced in serum from autophagy-deficient host mice (Karsli-Uzunbas et al. 2014; Poillet-Perez et al. 2018). Arginine is a semiessential amino acid that is derived from three different sources: the diet, de novo synthesis, and protein turnover. Arginine is involved in multiple biological pathways, such as the urea cycle, mechanistic target of rapamycin (mTOR) activation (Chantranupong et al. 2016), and synthesis of nitric oxide, creatine, polyamines, and proteins (Morris 2007). It has long been known that numerous human cancers are arginine auxotrophs due to the silencing of enzymes such as argininosuccinate synthase 1 (ASS1), which is responsible for de novo arginine synthesis (Fig. 3B; Dillon et al. 2004; Delage et al. 2010; Patil et al. 2016). Without expression of the enzymes necessary for de novo arginine biosynthesis, tumor cell growth thereby is completely dependent on a supply of exogenous arginine. Urea cycle dysregulation (UCD) through loss of ASS1 allows cancer cells to use aspartate for pyrimidine synthesis to support proliferation rather than for arginine synthesis and the urea cycle (Fig. 3B; Rabinovich et al. 2015; Nagamani and Erez 2016; Keshet et al. 2018). In a similar way, the mitochondrial electron transport chain can enable aspartate-derived nucleotide synthesis, which is required for tumor growth (Birsoy et al. 2015; Garcia-Bermudez et al. 2018). Moreover, arginine deficiency in arginine-auxotrophic tumors leads to mitochondrial distress and exhausts aspartate by inducing asparagine synthetase, leading to tumor cell death (Cheng et al. 2018). UCD induces nucleotide imbalance that leads to an increase in transversion mutations, worse prognosis, and better response to immune checkpoint blockade (Lee et al. 2018). Given these well-known alterations in de novo arginine biosynthesis, usage, and dependency in cancer, the decreased circulating arginine caused by autophagy deficiency in mice was likely to have significant deleterious consequences for tumor growth. The next questions are as follows: Is this the case, and why does autophagy deficiency in mice cause depletion of circulating arginine?
Unbiased proteomic profiling of serum revealed that in conditional whole-body autophagy-deficient host mice (either Atg7 or Atg5 deleted), low-circulating arginine is associated with increased levels and activity of a major enzyme that degrades arginine: arginase 1 (ARG1) (Poillet-Perez et al. 2018). ARG1 is localized in the liver and is involved in the degradation of arginine to ornithine. ARG1 can be released into the circulation following liver damage (Morris 2012). Indeed, liver-specific deletion as well as whole-body deletion of Atg7 or Atg5 cause the release of ARG1 into the circulation and decrease serum arginine and tumor growth (Fig. 3C). These findings indicate that ARG1 is released from hepatocytes following Atg7 or Atg5 deletion in the liver. Importantly, dietary arginine supplementation partially restores circulating arginine and tumor growth, confirming the importance of arginine for tumor growth (Fig. 4A). Thus, autophagy in the liver prevents ARG1 release from hepatocytes and the degradation of circulating arginine that is important for the growth of arginine-auxotrophic tumors, highlighting a new metabolic vulnerability of cancer (Fig. 4B; Poillet-Perez et al. 2018; Venida and Perera 2019). Similarly, autophagy in pancreatic tumor stromal cells facilitates secretion of alanine, which is taken up by PDAC cells and used to support their metabolism and growth (Fig. 4B; Sousa et al. 2016). Thus, autophagy has an important role in controlling essential tumor nutrients by regulating both the local and systemic amino acid supply. Therapeutic inhibition of autophagy in cancer patients should limit the supply of these essential tumor nutrients, thereby impairing tumor growth in addition to inhibiting tumor promotion through tumor cell-autonomous autophagy.
Figure 4.
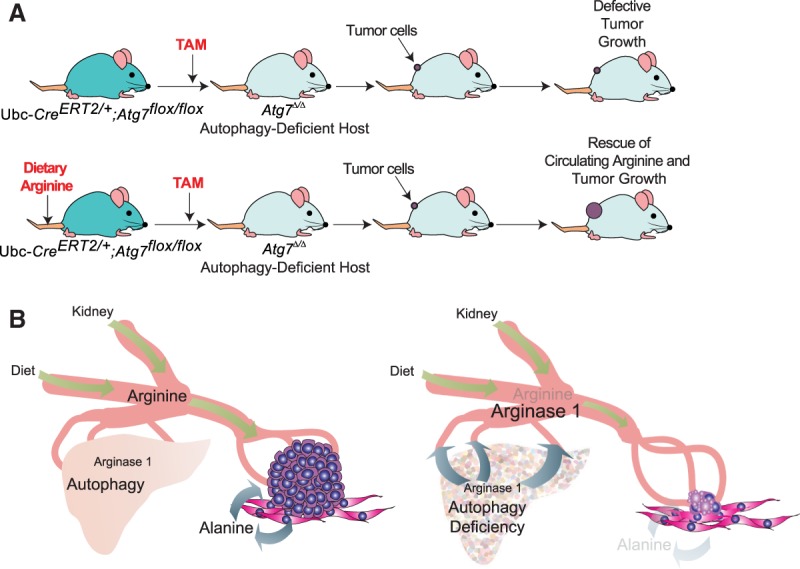
Host autophagy promotes tumor growth through circulating arginine. (A) Dietary arginine supplementation partially rescues tumor growth on autophagy-deficient host mice. Treatment with TAM leads to conditional whole-body deletion of Atg7. Supplementation of the mice with arginine partially rescues tumor growth in autophagy-deficient host mice (Poillet-Perez et al. 2018). (B) Nontumor cell-autonomous autophagy promotes tumor growth by sustaining the supply of amino acids that are essential tumor nutrients. When autophagy is active in the liver, the release of ARG1 from hepatocytes is prevented, thereby maintaining circulating arginine that is important for the growth of arginine-auxotrophic tumors. Loss of autophagy in the liver causes the release of ARG1 from hepatocytes into the circulation, leading to the depletion of circulating arginine and inhibition of the growth of tumors auxotrophic for arginine (Poillet-Perez et al. 2018). Similarly, loss of autophagy in the stroma cells inhibits the secretion of alanine necessary for PDAC growth (Sousa et al. 2016).
Conclusions and future directions
Recent studies have characterized the role of autophagy in tumor metabolism and highlighted the importance of both host and tumor autophagy in promoting metabolism and tumorigenesis. However, many additional questions remain. Although host autophagy sustains circulating arginine, allowing arginine-auxotrophic tumor cells to grow, not all arginine-auxotrophic cancer cell lines are defective for growth on an autophagy-deficient host (Poillet-Perez et al. 2018). This suggests the existence of tumor-specific adaptation mechanisms that need to be further studied.
Liver-specific deletion of Atg7 or Atg5 leads to decreased circulating arginine, but the tumor growth defect is less than that observed with conditional whole-body deletion of Atg7 (Poillet-Perez et al. 2018). These data suggest that in liver-specific deleted hosts, autophagy in the microenvironment may locally feed the tumor with arginine, partially compensating for the loss of circulating arginine. Indeed, autophagy in the tumor microenvironment can provide amino acids as well as extracellular matrix molecules and interleukin-6, which promote PDAC growth and tumors in Drosophila (Sousa et al. 2016; Endo et al. 2017; Katheder et al. 2017). Note that macrophages in the tumor microenvironment can also influence tumor growth through ARG1-dependent depletion of arginine (Zea et al. 2005; Ellyard et al. 2010). While this was not observed in autophagy-deficient hosts, there are likely multiple roles for arginine in tumorigenesis. Moreover, LC3-associated phagocytosis (LAP) in the myeloid compartment, which uses essential autophagy components such as ATG7, also promotes tumor growth by engulfing dying cells and suppressing inflammatory polarization of the tumor-associated macrophages and an antitumor T-cell response (Cunha et al. 2018). Thus, inhibition of autophagy by targeting ATG7 may not only compromise tumor nutrition and metabolism but also induce an antitumor T-cell response through inhibition of LAP or by other mechanisms. Tumors might also be getting arginine through macropinocytosis, a nonselective form of endocytosis that provides amino acid supply (Recouvreux and Commisso 2017; Finicle et al. 2018). In fact, it has been shown that RAS-driven PDAC and PTEN-deficient prostate cancer cells use macropinocytosis to overcome amino acid deprivation and support their growth (Commisso et al. 2013; Kamphorst et al. 2015; Davidson et al. 2017; Kim et al. 2018).
The reason why tumors need arginine for their growth is not totally clear. Arginine could promote tumor growth in different ways: by enabling protein synthesis or by sustaining mTOR activity, nitric oxide, or polyamine synthesis, all of which could be involved in promoting tumor growth (Chantranupong et al. 2016; Morris 2016; Saxton et al. 2016). In autophagy-deficient hosts, circulating arginine is degraded by ARG1 to produce ornithine (Fig. 3B; Poillet-Perez et al. 2018). It will be interesting to use in vivo isotope tracing to determine how arginine is used in tumors grown on wild type and autophagy-deficient hosts.
Circulating nutrients are important for tumor growth, and depleting essential tumor nutrients is an established therapeutic approach for cancer. For example, L-asparaginase is a frontline component in the treatment of acute lymphoblastic leukemia (Koprivnikar et al. 2017). As numerous tumors are auxotrophs for arginine, arginine deprivation therapy for cancer patients using enzymes that catabolize arginine, such as pegylated arginine deiminase (ADI-PEG20) or pegylated ARG1 (PEG-ARG1), are in development (Feun et al. 2015; Patil et al. 2016). Arginine deprivation with ADI-PEG20 in vitro leads to metabolism alteration and up-regulation of glutamine anaplerosis and cell death when used with glutaminase inhibitor (Kremer et al. 2017). The use of ADI-PEG20 alone or in combination with temozolomide in a mouse model of glioblastoma multiforme (GBM) leads to a decrease in tumor growth and extends mouse survival without obvious toxicity (Przystal et al. 2018). The impact of ADI-PEG20 on patients with ASS1-deficient tumors needs to be investigated further, and, encouragingly, a clinical trial demonstrated at least 30% stable disease in numerous cancer types, such as hepatocellular carcinoma (HCC) and melanoma (Patil et al. 2016). However, ADI-PEG20 is isolated from Mycoplasma and is immunogenic in humans; therefore, the use of other arginine-degrading enzymes, such as human PEG-ARG1, might be a more effective alternative. In vitro, PEG-ARG1 induces nonapoptotic cell death in GBM (Khoury et al. 2015). PEG-ARG1 also suppresses the tumor growth of malignant pleural mesothelioma xenografts and inhibits T-cell leukemia cell proliferation by inducing apoptosis through eIF2α phosphorylation (Morrow et al. 2013; Lam et al. 2017), suggesting promising activity of PEG-ARG1 in human cancer. There are only a few clinical trials on PEG-ARG1: A stable disease rate of 26.7% was observed in HCC, and one patient with immunotherapy-resistant melanoma showed a sustained complete remission following treatment with PEG-ARG1 (Yau et al. 2013; De Santo et al. 2018). However, ARG1 is also a mediator of immune suppression. Inhibition of ARG1 with a small molecule inhibitor induced a proinflammatory environment and reduced tumor growth in a preclinical model (Steggerda et al. 2017). The effect of PEG-ARG1 needs to be further investigated in order to better understand the protumorigenic or antitumorigenic role of ARG1. In order to further deprive tumors of arginine, it would be interesting to assess the combination of PEG-ARG1 with autophagy inhibiton. The use of chloroquine (CQ) or LY294002 to inhibit autophagy enhanced the toxicity and apoptosis induced by recombinant human arginase (rhARG) in NSCLC cells in vitro and potentiated the antitumor effect of rhARG in vivo (Shen et al. 2017).
Work on arginine identifies a new metabolic vulnerability of cancer and highlights the importance of better understanding the needs of arginine-auxotrophic tumors in order to best implement this therapy in the right patient population (Savaraj et al. 2010; Shen et al. 2017). In addition to tumors that have silenced enzymes required for de novo arginine biosynthesis, RAS- and BRAF-driven tumors that are highly autophagy-dependent are likely to be responsive to arginine deprivation therapy. Recent insights from understanding the nature of the autophagy addiction of RAS-driven cancers are providing further guidance. For example, coordinate inhibition of CRAF, BRAF, and ATG7 is selectively synthetically lethal in RAS-driven tumor cells in vitro (Lee et al. 2019; White 2019). Moreover, in RAS-driven cancers, inhibition of MAP kinase signaling with MEK or ERK inhibitors further induces autophagy, and their anticancer activity is significantly enhanced by genetic or pharmacologic (CQ) inhibition of autophagy (Bryant et al. 2019; Kinsey et al. 2019). It will be of interest to test whether this autophagy addiction of RAS-driven cancers is all or in part due to the need to maintain circulating arginine essential for tumor growth.
Acknowledgments
This work was supported by National Institutes of Health grant R01CA163591 to E.W. L.P.-P. received support from a postdoctoral fellowship from the New Jersey Commission for Cancer Research (DHFS16PPC034).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.325514.119.
References
- Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, Guo JY. 2019. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev 33: 150–165. 10.1101/gad.320481.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. 2015. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162: 540–551. 10.1016/j.cell.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. 2019. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 10.1038/s41591-019-0368-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Yi HS, Kim HW, Shong M. 2017. Dysregulation of mitophagy in carcinogenesis and tumor progression. Biochim Biophys Acta Bioenerg 1858: 633–640. 10.1016/j.bbabio.2016.12.008 [DOI] [PubMed] [Google Scholar]
- Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP, Sabatini DM. 2016. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165: 153–164. 10.1016/j.cell.2016.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CT, Qi Y, Wang YC, Chi KK, Chung Y, Ouyang C, Chen YR, Oh ME, Sheng X, Tang Y, et al. 2018. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun Biol 1: 178 10.1038/s42003-018-0178-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Macleod KF. 2015. Tumor suppressor functions of BNIP3 and mitophagy. Autophagy 11: 1937–1938. 10.1080/15548627.2015.1085136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, et al. 2015. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163. 10.15252/embr.201540759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. 2013. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497: 633–637. 10.1038/nature12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, et al. 2018. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175: 429–441.e16. 10.1016/j.cell.2018.08.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. 2017. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med 23: 235–241. 10.1038/nm.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Chandel NS. 2016. Fundamentals of cancer metabolism. Sci Adv 2: e1600200 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. 2006. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10: 51–64. 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, Szlosarek PW. 2010. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer 126: 2762–2772. [DOI] [PubMed] [Google Scholar]
- Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13: 722–737. 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santo C, Cheng P, Beggs A, Egan S, Bessudo A, Mussai F. 2018. Metabolic therapy with PEG-arginase induces a sustained complete remission in immunotherapy-resistant melanoma. J Hematol Oncol 11: 68 10.1186/s13045-018-0612-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. 2004. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer 100: 826–833. 10.1002/cncr.20057 [DOI] [PubMed] [Google Scholar]
- Ellyard JI, Quah BJ, Simson L, Parish CR. 2010. Alternatively activated macrophage possess antitumor cytotoxicity that is induced by IL-4 and mediated by arginase-1. J Immunother 33: 443–452. 10.1097/CJI.0b013e3181cd8746 [DOI] [PubMed] [Google Scholar]
- Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, et al. 2017. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology 152: 1492–1506.e24. 10.1053/j.gastro.2017.01.010 [DOI] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. 2014. The machinery of macroautophagy. Cell Res 24: 24–41. 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feun LG, Kuo MT, Savaraj N. 2015. Arginine deprivation in cancer therapy. Curr Opin Clin Nutr Metab Care 18: 78–82. 10.1097/MCO.0000000000000122 [DOI] [PubMed] [Google Scholar]
- Finicle BT, Jayashankar V, Edinger AL. 2018. Nutrient scavenging in cancer. Nat Rev Cancer 18: 619–633. 10.1038/s41568-018-0048-x [DOI] [PubMed] [Google Scholar]
- Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T. 2008. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27: 6002–6011. 10.1038/onc.2008.199 [DOI] [PubMed] [Google Scholar]
- Gammoh N, Fraser J, Puente C, Syred HM, Kang H, Ozawa T, Lam D, Acosta JC, Finch AJ, Holland E, et al. 2016. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 12: 1431–1439. 10.1080/15548627.2016.1190053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, Papagiannakopoulos T, Molina H, Snuderl M, Lewis CA, et al. 2018. Publisher correction: aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol 20: 1228 10.1038/s41556-018-0184-2 [DOI] [PubMed] [Google Scholar]
- Goldsmith J, Levine B, Debnath J. 2014. Autophagy and cancer metabolism. Methods Enzymol 542: 25–57. 10.1016/B978-0-12-416618-9.00002-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. 2011. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470. 10.1101/gad.2016311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. 2013. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27: 1447–1461. 10.1101/gad.219642.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, White E. 2016. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 30: 1704–1717. 10.1101/gad.283416.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S, Barnard N, Ganesan S, Karantza V, White E, et al. 2013. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov 3: 894–907. 10.1158/2159-8290.CD-13-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Mizushima N. 2014. Autophagy and human diseases. Cell Res 24: 69–79. 10.1038/cr.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. 2015. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75: 544–553. 10.1158/0008-5472.CAN-14-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. 2007. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21: 1621–1635. 10.1101/gad.1565707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. 2014. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4: 914–927. 10.1158/2159-8290.CD-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katheder NS, Khezri R, O'Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhasz G, et al. 2017. Microenvironmental autophagy promotes tumour growth. Nature 541: 417–420. 10.1038/nature20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet R, Szlosarek P, Carracedo A, Erez A. 2018. Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18: 634–645. 10.1038/s41568-018-0054-z [DOI] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I. 2016. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16. 10.1016/j.tcb.2015.08.010 [DOI] [PubMed] [Google Scholar]
- Khoury O, Ghazale N, Stone E, El-Sibai M, Frankel AE, Abi-Habib RJ. 2015. Human recombinant arginase I (Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human glioblastoma cells. J Neurooncol 122: 75–85. 10.1007/s11060-014-1698-5 [DOI] [PubMed] [Google Scholar]
- Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, et al. 2018. PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov 8: 866–883. 10.1158/2159-8290.CD-17-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman AC, White E. 2017. Autophagy and tumor metabolism. Cell Metab 25: 1037–1043. 10.1016/j.cmet.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey CG, Camolotto SA, Boespflug AM, Gullien KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, et al. 2019. Protective autophagy elicited by RAF → MEK → ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 10.1038/s41591-019-0367-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Codogno P. 2013. The mechanism and physiological function of macroautophagy. J Innate Immun 5: 427–433. 10.1159/000351979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, Debnath J, Deretic V, Elazar Z, Eskelinen EL, et al. 2011. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7: 1273–1294. 10.4161/auto.7.11.17661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425–434. 10.1083/jcb.200412022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149–1163. 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- Koprivnikar J, McCloskey J, Faderl S. 2017. Safety, efficacy, and clinical utility of asparaginase in the treatment of adult patients with acute lymphoblastic leukemia. Onco Targets Ther 10: 1413–1422. 10.2147/OTT.S106810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JC, Prudner BC, Lange SES, Bean GR, Schultze MB, Brashears CB, Radyk MD, Redlich N, Tzeng SC, Kami K, et al. 2017. Arginine deprivation inhibits the warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS1-deficient cancers. Cell Rep 18: 991–1004. 10.1016/j.celrep.2016.12.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. 2004. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036. 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
- Laddha SV, Ganesan S, Chan CS, White E. 2014. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res 12: 485–490. 10.1158/1541-7786.MCR-13-0614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SK, Li YY, Xu S, Leung LL UKP, Zheng YF, Cheng PN, Ho JC. 2017. Growth suppressive effect of pegylated arginase in malignant pleural mesothelioma xenografts. Respir Res 18: 80 10.1186/s12931-017-0564-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, Gorski SM. 2015. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11: 1668–1687. 10.1080/15548627.2015.1067362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, Auslander N, Keshet R, Stettner N, Silberman A, Agemy L, et al. 2018. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174: 1559–1570.e22. 10.1016/j.cell.2018.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, Caplen NJ, McCormick F, Luo J. 2019. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci 116: 4508–4517. 10.1073/pnas.1817494116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy J, Cacheux W, Bara MA, L'Hermitte A, Lepage P, Fraudeau M, Trentesaux C, Lemarchand J, Durand A, Crain AM, et al. 2015. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat Cell Biol 17: 1062–1073. 10.1038/ncb3206 [DOI] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. 2011. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22: 165–178. 10.1091/mbc.e10-06-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Van Eygen S, Krysko DV, Vandenabeele P, Nys K, Rillaerts K, Garg AD, Verfaillie T, Agostinis P. 2014. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis 5: e1127 10.1038/cddis.2014.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Kimmelman AC. 2016. Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol 428: 1659–1680. 10.1016/j.jmb.2016.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. 2007. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21: 1367–1381. 10.1101/gad.1545107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. 2009. Autophagy suppresses tumorigenesis through elimination of p62. Cell 137: 1062–1075. 10.1016/j.cell.2009.03.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, White E. 2014. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 55: 916–930. 10.1016/j.molcel.2014.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. 2007. Autophagy: process and function. Genes Dev 21: 2861–2873. 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Klionsky DJ. 2007. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 27: 19–40. 10.1146/annurev.nutr.27.061406.093749 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075. 10.1038/nature06639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM Jr. 2007. Arginine metabolism: boundaries of our knowledge. J Nutr 137: 1602S–1609S. 10.1093/jn/137.6.1602S [DOI] [PubMed] [Google Scholar]
- Morris SM Jr. 2012. Arginases and arginine deficiency syndromes. Curr Opin Clin Nutr Metab Care 15: 64–70. 10.1097/MCO.0b013e32834d1a08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SM Jr. 2016. Arginine metabolism revisited. J Nutr 146: 2579S–2586S. 10.3945/jn.115.226621 [DOI] [PubMed] [Google Scholar]
- Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, Wyczechowska D, Reiss K, Rodriguez PC. 2013. Anti-leukemic mechanisms of pegylated arginase I in acute lymphoblastic T-cell leukemia. Leukemia 27: 569–577. 10.1038/leu.2012.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamani SC, Erez A. 2016. A metabolic link between the urea cycle and cancer cell proliferation. Mol Cell Oncol 3: e1127314 10.1080/23723556.2015.1127314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. 2018. Targeting autophagy in cancer. Cancer 124: 3307–3318. 10.1002/cncr.31335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil MD, Bhaumik J, Babykutty S, Banerjee UC, Fukumura D. 2016. Arginine dependence of tumor cells: targeting a chink in cancer's armor. Oncogene 35: 4957–4972. 10.1038/onc.2016.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. 2016. The emerging hallmarks of cancer metabolism. Cell Metab 23: 27–47. 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Li CX, Chen F, Wang Z, Ligr M, Melamed J, Wei J, Gerald W, Pagano M, Garabedian MJ, et al. 2008. Stimulation of prostate cancer cellular proliferation and invasion by the androgen receptor co-activator ARA70β. Am J Pathol 172: 225–235. 10.2353/ajpath.2008.070065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. 2015. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524: 361–365. 10.1038/nature14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigié P, Youle RJ. 2018. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28: R170–R185. 10.1016/j.cub.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, et al. 2018. Autophagy maintains tumour growth through circulating arginine. Nature 563: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, et al. 2010. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci 107: 15145–15150. 10.1073/pnas.1009941107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przystal JM, Hajji N, Khozoie C, Renziehausen A, Zeng Q, Abaitua F, Hajitou A, Suwan K, Want E, Bomalaski J, et al. 2018. Efficacy of arginine depletion by ADI-PEG20 in an intracranial model of GBM. Cell Death Dis 9: 1192 10.1038/s41419-018-1195-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D, et al. 2015. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527: 379–383. 10.1038/nature15529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, White E. 2010. Autophagy and metabolism. Science 330: 1344–1348. 10.1126/science.1193497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recouvreux MV, Commisso C. 2017. Macropinocytosis: a metabolic adaptation to nutrient stress in cancer. Front Endocrinol 8: 261 10.3389/fendo.2017.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. 2013. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504: 296–300. 10.1038/nature12865 [DOI] [PubMed] [Google Scholar]
- Santana-Codina N, Mancias JD. 2018. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 11: E114 10.3390/ph11040114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, DiPaola RS. 2016. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev 30: 399–407. 10.1101/gad.274134.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaraj N, You M, Wu C, Wangpaichitr M, Kuo MT, Feun LG. 2010. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr Mol Med 10: 405–412. 10.2174/156652410791316995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. 2016. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 536: 229–233. 10.1038/nature19079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Zhang X, Fu X, Fan J, Luan J, Cao Z, Yang P, Xu Z, Ju D. 2017. A novel and promising therapeutic approach for NSCLC: recombinant human arginase alone or combined with autophagy inhibitor. Cell Death Dis 8: e2720 10.1038/cddis.2017.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. 2016. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536: 479–483. 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, Li W, MacKinnon AL, Makkouk A, Marguier G, et al. 2017. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer 5: 101 10.1186/s40425-017-0308-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. 2013. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 3: 1272–1285. 10.1158/2159-8290.CD-13-0397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. 2011. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25: 795–800. 10.1101/gad.2016211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JH, Yun J. 2017. Emerging role of mitophagy in human diseases and physiology. BMB Rep 50: 299–307. 10.5483/BMBRep.2017.50.6.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ. 2017. Understanding the intersections between metabolism and cancer biology. Cell 168: 657–669. 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venida A, Perera RM. 2019. Host control of tumor feeding: autophagy holds the key. Cell Metab 29: 236–238. 10.1016/j.cmet.2019.01.009 [DOI] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. 2014. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514: 628–632. 10.1038/nature13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. 2011. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 25: 1510–1527. 10.1101/gad.2051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. 2012. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12: 401–410. 10.1038/nrc3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. 2019. Blockade of RAF and autophagy is the one-two punch to take out Ras. Proc Natl Acad Sci 116: 3965–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Chen F, Sahin A, Albarracin C, Pei Z, Zou X, Singh B, Xu R, Daniels G, Li Y, et al. 2011. Distinct function of androgen receptor coactivator ARA70α and ARA70β in mammary gland development, and in breast cancer. Breast Cancer Res Treat 128: 391–400. 10.1007/s10549-010-1131-5 [DOI] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, Chan SH, Heinze I, Ori A, Sabatini DM. 2018. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360: 751–758. 10.1126/science.aar2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, Mehnert JM. 2015. Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov 5: 410–423. 10.1158/2159-8290.CD-14-1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. 2011. Pancreatic cancers require autophagy for tumor growth. Genes Dev 25: 717–729. 10.1101/gad.2016111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. 2014. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 4: 905–913. 10.1158/2159-8290.CD-14-0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al. 2018. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov 8: 276–287. 10.1158/2159-8290.CD-17-0952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, Pang R, Fan ST, Poon RT. 2013. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs 31: 99–107. 10.1007/s10637-012-9807-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O'Neill A, et al. 2005. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res 65: 3044–3048. 10.1158/0008-5472.CAN-04-4505 [DOI] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. 2011. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci 108: 16259–16264. 10.1073/pnas.1113884108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Rabinowitz JD, White E. 2016. Mitochondria and cancer. Mol Cell 61: 667–676. 10.1016/j.molcel.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]