

Swarr et al. identified a regulatory feedback loop between Foxa2 and a downstream lncRNA, Falcor, in the lung.
Keywords: lncRNAs, Foxa2, lung injury, transcription factors, cell adhesion, cell migration
Abstract
Transcription factors (TFs) are dosage-sensitive master regulators of gene expression, with haploinsufficiency frequently leading to life-threatening disease. Numerous mechanisms have evolved to tightly regulate the expression and activity of TFs at the transcriptional, translational, and posttranslational levels. A subset of long noncoding RNAs (lncRNAs) is spatially correlated with transcription factors in the genome, but the regulatory relationship between these lncRNAs and their neighboring TFs is unclear. We identified a regulatory feedback loop between the TF Foxa2 and a downstream lncRNA, Falcor (Foxa2-adjacent long noncoding RNA). Foxa2 directly represses Falcor expression by binding to its promoter, while Falcor functions in cis to positively regulate the expression of Foxa2. In the lung, loss of Falcor is sufficient to lead to chronic inflammatory changes and defective repair after airway epithelial injury. Moreover, disruption of the Falcor–Foxa2 regulatory feedback loop leads to altered cell adhesion and migration, in turn resulting in chronic peribronchial airway inflammation and goblet cell metaplasia. These data reveal that the lncRNA Falcor functions within a regulatory feedback loop to fine-tune the expression of Foxa2, maintain airway epithelial homeostasis, and promote regeneration.
Maintaining precise control over patterns of gene expression throughout development is a critical perquisite to an organism's survival and health. Many transcription factors (TFs) serve as the master regulators of gene expression within broad and complex gene regulatory networks. Each TF can influence the expression of hundreds to thousands of downstream targets within the context of regulatory modules that may establish and maintain lineage identity (Holmberg and Perlmann 2012). Given the central role of TFs in regulating gene expression, it is not surprising that even modest changes in TF dosage are not well tolerated. TFs as a group are significantly overrepresented among the genes for whom haploinsufficiency is known to result in human disease (Dang et al. 2008). Consequently, numerous layers of regulation have evolved to precisely control the expression levels of TFs throughout the life span of an organism (Lee and Young 2013; Voss and Hager 2014).
It is now widely recognized that the majority of the mammalian genome is transcribed despite the fact that <2% of genomic sequence actually encodes for protein (Djebali et al. 2012). Long noncoding RNAs (lncRNAs), defined as RNA transcripts >200 nt in length that do not encode a protein, are a heterogeneous class of transcripts that is increasingly being recognized as critical regulators of gene expression (Ulitsky and Bartel 2013; Jandura and Krause 2017). lncRNAs can act in cis or trans to control gene expression in a precise temporal and spatial manner (Kopp and Mendell 2018). lncRNAs acting in trans may regulate gene expression at the transcriptional level or perform completely independent functions such as acting as a molecular sponge for proteins or miRNAs (Marchese et al. 2017; Noh et al. 2018). lncRNAs acting in cis have been shown to regulate neighboring protein-coding genes (PCGs) by recruiting chromatin regulators or TFs to the locus to modulate gene expression (da Rocha and Heard 2017). In other cases, the act of transcription through the lncRNA locus and/or splicing of the RNA transcript rather than the primary RNA sequence itself is thought to modulate the neighboring PCG's expression (Anderson et al. 2016; Engreitz et al. 2016). Our group demonstrated previously that lncRNAs expressed in the lung, as a class, are statistically more likely to be within close proximity to a TF, including many of the TFs known to direct fundamental aspects of lung development (Herriges et al. 2014).
The TF Foxa2, a member of the forkhead class of DNA-binding proteins, has been shown to act as a “pioneer factor” that engages chromatin to control early development of the node, notochord, and endoderm, followed by subsequent specification and development of endoderm-derived organs, including the liver, pancreas, and lung (Ang and Rossant 1994; Gualdi et al. 1996; Wan et al. 2004a,b, 2005; Lee et al. 2005; Gao et al. 2008). Expression of Foxa2 is first seen in mice at embryonic day 6.5 (E6.5) in the primitive streak and node during gastrulation (Ang and Rossant 1994; Besnard et al. 2004; Uetzmann et al. 2008). Foxa2 plays an essential role in epiblast cell migration and establishment of apical–basal polarity and new intracellular junctions in the early mammalian embryo (Burtscher and Lickert 2009). Data from lung, intestine, and endometrium collectively suggest that Foxa2, either alone or in combination with other TFs such as Nkx2-1 and Cdx2, continues to maintain epithelial identity by promoting cell–cell adhesion (through regulation of genes such as E-cadherin) and suppressing cell migration (Tang et al. 2011; Gosalia et al. 2015; Li et al. 2015; Zhang et al. 2015; Jägle et al. 2017; Lin et al. 2018).
Here we show that the lncRNA Falcor (Foxa2-adjacent long noncoding RNA), which is located downstream from Foxa2, is positionally and functionally conserved between mice and humans and is expressed in a pattern similar to Foxa2 throughout development. Falcor positively regulates the expression of Foxa2 in cis, whereas Foxa2 negatively regulates the expression of Falcor by directly binding to its promoter. While Falcor is not required for normal development, Falcor-null mice exhibit defects in regeneration of the airway epithelium after injury, including incomplete re-epithelialization of the lung airways, goblet cell metaplasia, and chronic peribronchial inflammation. Moreover, specific disruption of the Foxa2–Falcor regulatory feedback loop exacerbates the effects of Foxa2 haploinsufficiency in the airway epithelium, leading to peribronchial inflammation and goblet cell metaplasia. Taken together, these data highlight the importance of the lncRNA Falcor in the maintenance of precise Foxa2 dosage in the setting of tissue homeostasis and regeneration.
Results
Falcor is positionally conserved between mice and humans and is coordinately expressed with Foxa2 throughout development
We first identified Falcor in a screen of lncRNAs expressed in the developing mouse lung as a noncoding transcript ∼2 kb downstream from the 3′ terminus of Foxa2 (Herriges et al. 2014). Initially annotated in mice as RIKEN cDNA clone 9030622O22Rik (ENSMUSG00000086141, Ensembl release 91), Falcor originates from a genomic region 103 kb in length and contains multiple transcript isoforms ranging in length from 2 to 9.2 kb (Fig. 1A). No transcriptional readthrough between the Foxa2 and Falcor locus was detected by either RNA-seq or targeted PCR, verifying it as a unique gene locus. In humans, LINC00261/DEANR1 (ENSG00000259974, Ensembl release 91) is also located ∼2 kb downstream from the 3′ terminus of Foxa2 (Fig. 1A; Jiang et al. 2015). ChIP-seq for the promoter-associated marks Pol2 and H3K4me3 demonstrates discrete peaks surrounding the Falcor transcriptional start site that are distinct from the Foxa2 locus (Fig. 1B). The Falcor promoter region is H3K4me3-high/H3K4me1-low, indicating that this is a bona fide promoter region and not an enhancer element for Foxa2 (Supplemental Fig. 1A). Although these equivalently positioned mouse and human lncRNAs have overall poor primary sequence conservation, the promoter region, transcriptional start site, and first exon exhibit strong conservation across all primates and placental mammals, with conservation extending to Xenopus tropicalis, zebrafish, and tetraodons but not lampreys (Supplemental Fig. 1A).
Figure 1.

Falcor and Foxa2 are coordinately expressed throughout development in mice and humans. (A) The lncRNA Falcor is located 2 kb downstream from the TF Foxa2 in both mice and humans. Although overall primary sequence conservation is poor, the promoter and first exon of Falcor and overall positional relationship to Foxa2 are well conserved. Both Falcor and Foxa2 are transcribed from the same strand. (B) ChIP-seq for Pol2 and H3K4me3 from the liver (yellow) and lung (black) demonstrates distinct promoter elements for Falcor and Foxa2. (C–L) Comparison between Falcor and Foxa2 expression in Scgb1a1+ secretory epithelial cells and Tub1a1+ multiciliated epithelial cells. (E, inset) Falcor expression was detected using an RNAscope probe, which suggests nuclear localization of the Falcor transcript. Foxa2, Scgb1a1, and Tub1a1were detected by immunohistochemistry. (M) RNA-seq expression data from 42 different tissue types within the Genotype-Tissue Expression (GTEx) data set reveal coexpression of Falcor with Foxa2 in every tissue in which Foxa2 is expressed. (N) During mouse development, in situ hybridization for Falcor and Foxa2 demonstrates nearly identical patterns, with expression seen in the neural tube (NT) and gut endoderm (GE) at E9.5. (O) In the developing human lung, nearly identical patterns of Falcor and Foxa2 expression are seen in the epithelium by in situ hybridization. (P) The expression of Falcor and Foxa2 is highly correlated within hundreds of human lung tissue samples within the GTEx data set. n = 321. Spearman ρ = 0.808. P < 1 × 10−16. Scale bars, 1000 µm (N), 500 µm (O), 25 µm (C–L).
Falcor and Foxa2 exhibit nearly identical patterns of expression throughout mouse development, with strong expression seen in the neural tube and endoderm-derived organs, including the lung, liver, intestine, and pancreas (Fig. 1C–O). Falcor and Foxa2 are highly expressed in the developing epithelium of the mouse and human lung (Fig. 1O; Herriges et al. 2014). In adult mice, the highest levels of Falcor and Foxa2 are seen in the airway epithelium (Fig. 1C–L). In lung epithelium, subcellular localization of Falcor is predominantly nuclear (Fig. 1E, inset). In humans, Falcor and Foxa2 expression was highly coordinated across 42 tissue types examined (Fig. 1M). Levels of FALCOR and FOXA2 were highly correlated across lung tissue obtained from >300 human samples (n = 321; Spearman ρ = 0.808; P < 1 × 10−16) (Fig. 1P; The GTEx Consortium 2013).
Falcor functions within a regulatory feedback loop to positively regulate the expression of Foxa2
To determine how Falcor may regulate tissue development and regeneration, we used CRISPR–Cas9 technology to ablate Falcor transcription by deleting the Falcor promoter, transcriptional start site, and first exon in mice and used lentiviral shRNA knockdown to deplete the FALCOR RNA transcript in the human airway epithelial cell lines A549 and HBEC-3KT in vitro. We used ChIP-seq data for this region to define the Falcor promoter as H3K4me3-hi/H3K4me1-low and also containing Pol2-binding peaks (Fig. 1B; Supplemental Fig. 1A–D). CRISPR-mediated deletion of the Falcor promoter region resulted in a >90% decrease in Falcor transcript detectable by either RNA-seq or qPCR from the lung epithelium (Fig. 2A; Supplemental Fig. 1E,F). In both mice and humans, loss of Falcor/FALCOR expression resulted in a modest but significant decrease in the expression of Foxa2 (Figs. 2A, 6A–D [below]). In contrast, either global haploinsufficiency or complete deletion of Foxa2 in the lung epithelium using Foxa2flox/flox:Shhcre mice resulted in a marked increase in Falcor expression (Fig. 2B,E). Analysis of the Falcor promoter region demonstrates multiple predicted Foxa1 and Foxa2 TF-binding sites, and FOXA2 ChIP-seq data from human livers demonstrate FOXA2 binding to the FALCOR promoter region, suggesting that Foxa2 may directly regulate Falcor expression (Fig. 2C). Importantly, overexpression of Foxa2 in A549 cells resulted in decreased levels of FALCOR RNA in a direct regulation of Falcor by Foxa2 (Fig. 2D). Taken together, these data suggest a regulatory feedback loop in which Falcor positively regulates the expression of Foxa2, and Foxa2 in turn provides negative feedback to Falcor expression by directly binding to its promoter.
Figure 2.

Falcor and Foxa2 function within a regulatory feedback loop to tune Foxa2 expression. (A) Expression of Falcor and Foxa2 in Falcor−/−, Foxa2+/−, and Falcor+/−;Foxa2+/− mice. Loss of Falcor−/− leads to a 25%–30% reduction in Foxa2 expression, whereas haploinsufficiency for Foxa2 leads to a marked increase in Falcor expression. (B) Conditional deletion of Foxa2 in the lung epithelium in Foxa2flox/flox:Shhcre mutants reveals an increase in Falcor expression by in situ hybridization. The high-magnification inset demonstrates high levels of uniform expression throughout the airway epithelium, with lower levels of increased expression in the distal lung. (C) Foxa2 ChIP-seq data from human livers reveal Foxa2-binding peaks (black and gray bars) within the promoter region of Falcor. (D) Overexpression of Foxa2 in A549 cells in vitro leads to suppression of Falcor expression. (E) qPCR confirms loss of Foxa2 expression and increased Falcor expression in Foxa2flox/flox:Shhcre mutants. Scale bars, 1000 µm. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
The Falcor–Foxa2 regulatory loop is required for maintenance of airway epithelial homeostasis
Falcor−/− mutants were viable with no overt phenotype in adults at homeostasis. To further test the hypothesis that Falcor positively regulates the expression of Foxa2 in cis, we compared Falcor knockout mice (Falcor−/−) with mice haploinsufficient for Foxa2 (Foxa2+/−) and mice haploinsufficient for Falcor and Foxa2 in trans (Falcor+/−;Foxa2+/−) (Fig. 2A; Lakso et al. 1996; Sund et al. 2000). While there was a dosage-dependent decrease in Foxa2 from the Falcor−/− and Foxa2+/− mutants, the Falcor+/−;Foxa2+/− mutants expressed the same levels of Foxa2 as the Foxa2+/− mutants, suggesting either additional levels of compensation to maintain Foxa2 expression levels outside of Falcor control or a failure to reproducibly detect such small gene expression changes (Fig. 2A). Falcor+/−;Foxa2+/− mutants were viable but, by 8−12 wk of age, exhibited areas of peribronchial and perivascular inflammation similar to Foxa2+/− mutants (Fig. 3A–D). This inflammatory infiltrate consisted of predominantly B220+ B cells (Fig. 3E–H). In Falcor+/−;Foxa2+/− mice, the inflammation was most notable in the subepithelial region of airway branch points, reminiscent of inducible bronchus-associated lymphoid tissue (iBALT). At these foci of inflammation, CD3e+ T cells were found interspersed among the B cells (Fig. 3E–H). Goblet cell metaplasia was detectable by both Alcian blue staining and immunohistochemistry for Muc5Ac in Foxa2+/− and Falcor+/−;Foxa2+/− mice, with greater numbers of goblet cells present in the double-heterozygous animals (Fig. 3I–P). However, goblet cell metaplasia was not detected in wild-type or Falcor−/− animals at 8–12 wk of age (Fig. 3I–P). Ciliated and secretory cells were present at roughly normal ratios (Supplemental Fig. 2A–D). Similarly, immunohistochemistry for keratin 8, a cytoskeletal protein ubiquitously expressed in the airway epithelium, did not reveal any gross derangements of airway epithelial cell shape (Supplemental Fig. 2E–H). By 1 yr of age, areas of peribronchial inflammation and goblet cell metaplasia were observed in Falcor−/− mutants, reminiscent of the phenotypic changes seen in Foxa2+/− and Falcor+/−;Foxa2+/− at younger ages (Fig. 3Q–V).
Figure 3.
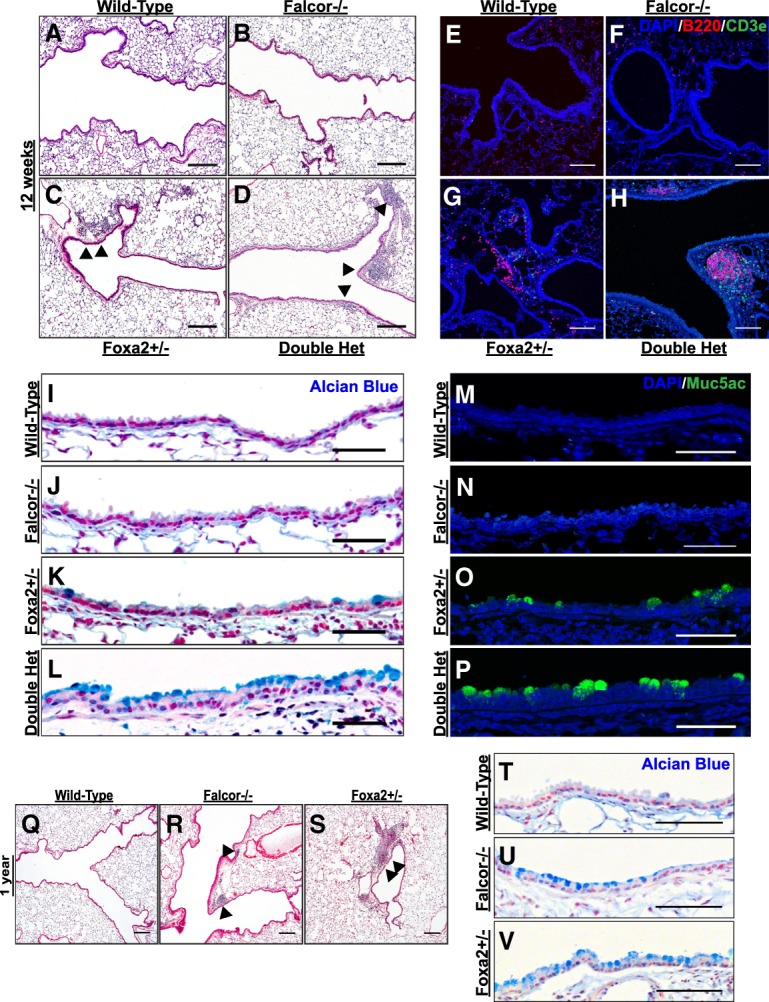
Disruption of the Falcor–Foxa2 feedback loop leads to chronic peribronchial inflammation and goblet cell metaplasia. (A–D) No gross phenotypic changes were observed in the lungs of Falcor−/− or wild-type mice, whereas Foxa2+/− and Falcor+/−;Foxa2+/− exhibited areas of peribronchial and perivascular inflammation (black arrowheads), particularly at airway branch points. (E–H) This inflammatory infiltrate consists predominantly of B220+ B cells intermixed with CD3e+ T cells and was more prominent in Falcor+/−;Foxa2+/− compared with Foxa2+/− mice. (I–L) Alcian blue histochemical staining of lungs from wild-type, Falcor−/−, Foxa2+/−, and Falcor+/−;Foxa2+/− 10- to 12-wk-old mutants. (M–P) Muc5ac immunohistochemical staining of adult lungs from wild-type, Falcor−/−, Foxa2+/−, and Falcor+/−;Foxa2+/− 10- to 12-wk-old mutants. (Q–V) By 1 yr of age, both peribronchial inflammation (Q–S) and goblet cell metaplasia (T–V) were also seen in Falcor−/− mice. Scale bars: 250 µm (A,B,E,F,Q–S), 100 µm (C,D,G–H), 50 µm (T–V), 25 µm (I–P).
To better characterize the transcriptional changes that were occurring due to disruption of Falcor and the Falcor–Foxa2 regulatory loop, RNA-seq was performed on CD326+ (Epcam) lung epithelium isolated from adult wild-type, Falcor−/−, Foxa2+/−, and Falcor+/−;Foxa2+/− mice (Fig. 4A–D; Supplemental Tables 1–3). Principal component analysis (PCA) reveals the distinct nature of the transcriptomic changes in each of these genotypes, with the first dimension of differences driven primarily by Foxa2 dosage (Fig. 4A). Gene set enrichment analysis for the gene expression changes common to Falcor−/− and either Foxa2+/− or Falcor+/−;Foxa2+/− revealed genes related to cell adhesion, cytoskeletal changes, and epithelial development (Fig. 4D). We also compared the transcriptional changes observed in the mouse lung epithelium in Falcor−/− or Foxa2+/− mice with those reported previously in human embryonic stem (ES) cells following knockdown of either FALCOR/DEANR1 or FOXA2 (Jiang et al. 2015). A statistically significant overlap was observed between genes differentially expressed at greater than twofold in Falcor−/− mice compared with FALCOR/DEANR1- or FOXA2-depleted human ES cells (representation factor 2 [P < 0.003] and representation factor 1.6 [P < 0.014], respectively) or when genes differentially expressed in FOXA2+/− mice were compared with FOXA2-depleted human ES cells (representation factor 1.6; P < 1.156 × 10−7) (Supplemental Table 6). Moreover, gene set enrichment analysis of these reported differentially expressed genes from human ES cells was enriched in numerous gene ontology (GO) categories overlapping with those presented in Figure 4, including “cell adhesion molecule binding,” “extracellular matrix binding,” “cell migration,” “regulation of cell motility,” “regulation of cell locomotion,” and “tube morphogenesis” (Supplemental Tables 7, 8). Taken together, these data suggest that loss of Falcor may prime epithelial cells to react poorly to injury or could cause loss of epithelial integrity at homeostasis.
Figure 4.

The Falcor–Foxa2 regulatory loop regulates a transcriptional network involved in maintaining epithelial homeostasis in the adult lung. (A) The transcriptome of lung epithelial cells (Epcam+) was assessed by RNA-seq analysis in wild-type, Falcor−/−, Foxa2+/−, and Falcor+/−;Foxa2+/− adult mice, with each of the four genotypes clustering differently using PCA. (B) Heat maps of the top 100 differentially expressed genes for either Falcor−/− or Foxa2+/− across all four genotypes. (C) Venn diagram of differentially expressed genes (bold numbers) and enriched GO categories (numbers in parentheses) demonstrates greater overlap than expected by chance between the different genotypes, with the greatest number of differentially expressed genes seen in Falcor+/−;Foxa2+/− mice. (D) Significantly enriched GO categories shared across the three genotypes were broadly related to cell adhesion, cell migration, and epithelial and mesenchymal development.
Falcor is necessary for proper repair following airway epithelial injury
Because the Falcor–Foxa2 regulatory loop appears to be involved in maintenance of lung homeostasis through regulation of cell–cell adhesion and cell migration, we hypothesized that loss of Falcor would lead to impaired regeneration after airway epithelial injury. We used the naphthalene model of secretory cell depletion on Falcor−/−, Foxa2+/−, or wild-type control mice. Within 48–72 h, naphthalene is metabolized to a toxic intermediate metabolite within Scgb1a1+ secretory cells, resulting in the loss of the majority of the cells of this lineage. Soon after depletion, the unaffected multiciliated cells flatten and spread out to maintain an intact airway epithelial barrier, and the remaining secretory cells repopulate the airway (Fig. 5A; Stripp et al. 1995). In wild-type mice, 2 wk after injury, regeneration of the secretory cell population is observed, and, by 28 d, the repair process is largely complete (Park et al. 2006; Wang et al. 2013). In both Falcor−/− and Foxa2+/− mice, areas of both peribronchial and perivascular inflammation are evident 1 mo after naphthalene injury (Fig. 5B–G,O). Areas of goblet cell metaplasia were evident in both Falcor−/− and Foxa2+/− mice 1 mo after injury (Fig. 5H–J). Of note, Foxa2 has been shown previously to suppress goblet cell metaplasia by direct negative regulation of Foxa3 and SPDEF expression (Chen et al. 2009, 2010, 2014; Rajavelu et al. 2015). Patches of airway were still devoid of Scgb1a1+ secretory cells 1 mo after injury in Falcor−/− and Foxa2+/− mice (Fig. 5K–N). Changes in gene expression after injury in Falcor−/− lung tissue revealed altered expression of genes related to “apical junction” and “oxidative phosphorylation,” which were also impacted in Foxa2+/− and Falcor−/−;Foxa2+/− mice under homeostasis (Fig. 5P,Q). Other categories were affected in injured Falcor−/− mice that were not affected under homeostasis, including “inflammatory response” and “unfolded protein response” (Fig. 5P,Q). Notably, the phenotype observed in Falcor−/− after injury bears striking similarity to the Falcor+/−;Foxa2+/− mice at homeostasis, with patchy peribronchial inflammation and goblet cell metaplasia (Fig. 3).
Figure 5.

Falcor is required for lung epithelial repair after airway injury. (A) Scgb1a1+ secretory cells were depleted from the airway by administration of a single intraperitoneal (IP) dose of naphthalene; 48–72 h after naphthalene administration, the vast majority of secretory cells have been cleared from the airway and the remaining ciliated cells have taken on a squamous configuration in order to maintain epithelial barrier integrity. (B,E) In wild-type mice, airway epithelial repair is essentially complete 28 d after injury. In contrast, in both Falcor−/− (C,F) and Foxa2+/− (D,G) mice, areas of peribronchial (single asterisks) and perivascular (dashed outlines) inflammation and an alveolar infiltrate (double asterisks) are seen 1 mo after injury. (H–J) Areas of goblet cell metaplasia are present in both Falcor−/− and Foxa2+/−, but not wild-type, mice 1 mo after injury. (K–N) Quantitative analysis of the ratio of secretory (Scgb1a1+) to multiciliated (Tub1a1+) epithelial cells 1 mo after injury demonstrates incomplete recovery of the secretory cell population in Falcor−/− and Foxa2+/− mice. (O) Analysis of whole-lung isolate by flow cytometry 28 d after injury reveals an increase in B220+ B cells. (P,Q) Transcriptional analysis of Falcor−/− and wild-type whole lung 28 d after injury demonstrates altered expression of genes involved in immune response, oxidative phosphorylation, unfolded protein response, and apical junctions. Scale bars: 250 µm (B–D), 100 µm (E–G), 25 µm (H–M). (*) P < 0.05; (**) P < 0.01.
The Falcor–Foxa2 regulatory loop is required to maintain epithelial cell–cell adhesion and suppress cellular migration
Our transcriptome data during lung homeostasis suggest that Foxa2 and Falcor may regulate epithelial cell migration. While Foxa2 has been implicated in regulation of cell adhesion and cell motility in other organ systems, this has not been well studied in the lung epithelium (Burtscher and Lickert 2009; Gosalia et al. 2015; Lin et al. 2018). To test whether FALCOR regulated epithelial cell adhesion and migration, we used lentiviral shRNA to knock down FALCOR in the A549 and HBEC-3KT cell lines. In both cell lines, knockdown of FALCOR expression also resulted in significant decreases in FOXA2 expression (Fig. 6A–D). Moreover, in both cell lines, cells depleted of FALCOR demonstrated increased migration in a transwell invasion assay (Fig. 6E–J). Similarly, A549 cells depleted of FALCOR migrated faster across a scratch in the epithelial sheet compared with cells transduced with a scrambled RNA (Fig. 6K–Q). Analysis of differentially expressed genes important for cell adhesion and migration in sorted lung epithelium in vivo revealed that genes important for hemidesmosome or focal adhesion assembly (Dst, Hmcn1, and Macf1), cell–cell adhesion (Dchs1), and cell migration (Dst, Hmcn1, Macf1, Itga7, Ros1, and Dlc1) were differentially expressed in the Foxa2/Falcor mutant allelic series (Supplemental Fig. 3A–D) or following FALCOR depletion in A549 and HBECT-3KT cells (Supplemental Fig. 3E–I). Differential expression of targets in Foxa2+/− mice, in human ES cells following shRNA knockdown of Foxa2, and in ChIP-seq data demonstrating Foxa2 binding in promoter and/or enhancer regions of these genes supports the idea that at least a subset of these differentially expressed genes is a direct transcriptional target of Foxa2. (Supplemental Fig. 3J–L; Jiang et al. 2015). Taken together, these data suggest that the Falcor–Foxa2 regulatory feedback loop serves to maintain cell–substrate adhesion and suppress cell migration and that loss of this regulatory loop leads to disrupted airway epithelial barrier integrity, peribronchial inflammation, and incomplete epithelial regeneration in the lung after injury in vivo (Fig. 7).
Figure 6.

Disruption of the FALCOR–FOXA2 regulatory loop leads to alterations in cell migration and cell adhesion. (A–D) shRNAs targeting either FALCOR or a scrambled control were introduced into A549 (A,B) and HBEC-3KT (C,D) cells using lentivirus, resulting in 70%–90% knockdown of FALCOR, with a concomitant reduction in FOXA2 expression. (E–J) Depletion of FALCOR caused increased migration of cells into a transwell membrane in A549 and HBEC-3KT cells. (K–Q) A549 cells depleted of FALCOR migrated faster into the denuded region during “scratch” assays. Data represent the average of four to six assays ±SEM. Scale bars, 250 µm. (*) P < 0.05; (**) P < 0.01.
Figure 7.
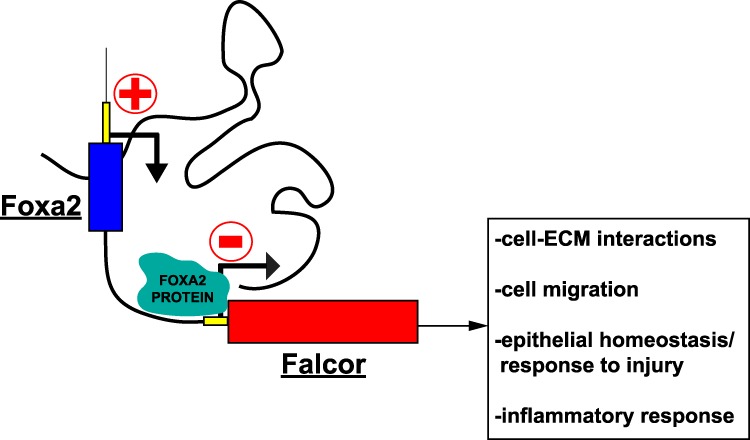
Model for Falcor regulation of Foxa2. The lncRNA Falcor positively regulates the expression of the TF, which in turn provides negative feedback on Falcor expression by direct binding of Foxa2 to the Falcor promoter.
Discussion
Although numerous studies have examined the function and biochemical properties of lncRNAs in vitro, the role of lncRNAs in vivo, including their relevance to human disease, has remained enigmatic (Wapinski and Chang 2011; Li and Chang 2014; Xue et al. 2017). Studies of lncRNAs in vivo have also been complicated by a lack of observed phenotypic effects at homeostasis, leaving it unclear as to whether more subtle phenotypes could be revealed upon stress or injury. Our studies show that the lncRNA Falcor fine-tunes the expression of Foxa2 and that disruption of the Falcor–Foxa2 transcriptional circuit results in subtle defects in lung airway epithelial homeostasis and more dramatic defects in airway regeneration after acute injury. These data reveal that subsets of lncRNAs can tune the expression of neighboring TFs, playing key roles in both tissue homeostasis and regeneration.
The TF Foxa2 plays an essential role in specification of early embryonic endoderm and development of endoderm-derived organs. Almost immediately after establishment of endodermal identity, Foxa2+ endodermal cells acquire features characteristic of epithelium, including apical–basal polarity, proper adhesion to an underlying basement membrane, and establishment of tight intracellular junctions. Loss of Foxa2 during early development impairs the nascent endoderm's ability to establish these core aspects of epithelial identity, ultimately leading to early embryonic lethality (Ang and Rossant 1994; Burtscher and Lickert 2009). Maintenance of epithelial integrity through these processes is critical for the homeostasis of tissues. Our data, along with previous reports, suggest that loss of Foxa2, either directly or by disruption of the Falcor–Foxa2 regulatory loop, is sufficient to alter cell–cell adhesion and migration (Tang et al. 2011; Gosalia et al. 2015; Li et al. 2015; Zhang et al. 2015; Jägle et al. 2017; Lin et al. 2018). Our in vitro studies are also consistent with observations from several other groups (Sha et al. 2017; Wang et al. 2017; Yu et al. 2017; Zhang et al. 2018). Computational analysis of the FOXA2 gene regulatory network in intestinal epithelium has similarly highlighted the central role of this TF in coordinating the expression of genes involved in cell migration, cell motion, and plasma membrane function (Gosalia et al. 2015). Decreased expression of either FALCOR (LINC00261/DEANR1) or FOXA2 has been associated with various forms of malignancy, including lung adenocarcinoma, and has been implicated with both tumor invasion and metastasis (Xu et al. 2001; Basseres et al. 2012; Cao et al. 2013; Smith et al. 2016; Vorvis et al. 2016; Liu et al. 2017; Dhamija et al. 2018; Le Gallo et al. 2018). Decreased expression levels of both FALCOR and FOXA2 has also been reported in ectopic endometrial tissue in endometriosis, a nonmalignant condition that is nonetheless characterized by spread of epithelial cells far beyond their normal place of residence (Sun et al. 2014; Sha et al. 2017; Lin et al. 2018). In the mouse lung, altered Falcor–Foxa2 gene dosage results in chronic inflammatory changes and defective airway epithelial repair after injury. Similarly, loss of Nkx2.1 alone, either directly or by disruption of the NANCI lncRNA–Nkx2.1 feedback loop, leads to impaired cell–cell adhesion, pulmonary inflammation, and loss of lung cell type identity (Snyder et al. 2013; Herriges et al. 2017). Due to redundancy in maintaining core features of epithelial identity, more dramatic effects such as epithelial-to-mesenchymal transition (EMT) or promotion of metastasis in the setting of malignancy likely require alterations in the expression levels of multiple TFs, as was demonstrated recently for lung adenocarcinoma (Li et al. 2015). However, even subtle alterations in epithelial barrier integrity due to modest alterations in expression level of a TF such as Foxa2 or Nkx2.1 could potentially have significant health impacts for a human subjected to recurrent injuries due to environmental exposures and respiratory viral infections (Hammad and Lambrecht 2015; Schleimer and Berdnikovs 2017).
A lncRNA that correlates with the human ortholog of FALCOR was described previously in human ES cells, where it was called DEANR1 and was proposed to be critical for endoderm generation (Jiang et al. 2015). Loss of DEANR1/FALCOR in human ES cells led to severe defects in early endoderm development. In contrast, our study shows that loss of Falcor in mice results in relatively normal development, including in endodermal-derived organs such as the lungs. However, Falcor is required for maintaining normal airway epithelial homeostasis and the response to epithelial injury. A close comparison of the transcriptomic profile in human ES cells depleted of FALCOR/DEANR1 with the mouse data presented here highlights the role of the Falcor–Foxa2 regulatory loop transcriptional regulation of gene targets related to cell adhesion, cell migration, and epithelial development. Moreover, our data are entirely consistent with the cis-regulatory relationship between FALCOR/DEANR1 and FOXA2 proposed by Jiang et al. (2015). Although we were not able to replicate the proposed interaction between FALCOR and SMAD2/3 in lung epithelial cells using targeted RNA immunoprecipitation (RIP) and unbiased proteomics assays, we believe that recruitment of protein complexes augmenting transcription at the FOXA2 locus by FALCOR is a plausible mechanism of action and warrants further investigation. The differences in phenotypic effect size between these two model systems could be due to a more important role for DEANR1/FALCOR in humans or differences between an in vivo animal model and in vitro cell culture systems such as ES cells.
Given the potentially severe consequences for failing to maintain proper expression levels of master TFs such as Foxa2, it is not surprising that multiple layers of regulation have evolved to tightly control their expression at the transcriptional, translational, and posttranslational levels. Our data highlight that although loss of the lncRNA Falcor does not result in an overt developmental phenotype, it nonetheless plays an important role in modulating Foxa2 levels in response to additional stressors such as injury. Even with a single injury to the airway epithelium, loss of this additional layer of regulatory control is sufficient to lead to chronic pulmonary inflammation, impaired epithelial repair, and goblet cell metaplasia. While some lncRNAs may not have an obvious role in regulating gene expression in vivo, our data point to an emerging concept that lncRNAs provide an important tunable mechanism to regulate TF expression. Effects of these lncRNAs may not be readably evident at homeostasis but may be revealed under stress and after injury. This is particularly relevant in the context of disease, as most human diseases do not result from the complete loss of gene function or complete lack of cell lineage specification during development. Instead, subtle derangements in gene expression in the context of environmental exposures or repetitive injuries is the norm, highlighting the importance of future studies aimed at unraveling higher-order gene regulatory mechanisms such as the Falcor–Foxa2 feedback loop to better understand human disease processes.
Materials and methods
Animals
Foxa2 was conditionally deleted from the lung epithelium by crossing Foxa2loxP/loxP mice (Foxa2tm1Khk; JAX, stock no. 022620) with mice expressing Cre recombinase from the Shh locus [Shhtm1(EGFP/cre)Cjt; JAX, stock no. 005622] (Sund et al. 2000; Harfe et al. 2004). Mice globally haploinsufficient for Foxa2 were generated by crossing Foxa2loxP/loxP mice with mice constitutively expressing Cre recombinase driven by EIIA [Tg(EIIa-cre)C5379Lmgd; JAX, stock no. 003724] (Lakso et al. 1996). The Falcor knockout mouse was generated by direct oocyte injection of a 1:1 mixture of two purified pSpCas9(BB)-2A-Puro v2.0 (PX459; Addgene plasmid no. 62988) constructs with sgRNAs flanking the Falcor promoter, transcriptional start site, and first exon (Supplemental Fig. 1B–D; Supplemental Table 4). Founder mice were genotyped for the deletion after confirmation by Sanger sequencing and then crossed to wild-type CD1 mice (Charles River) for more than three generations before studies were performed (Supplemental Fig. 1C,D). Experiments were performed on a mixed C57Bl6 CD1 background. All animal work was performed under the approval and guidance of the University of Pennsylvania and Cincinnati Children's Hospital Institutional Animal Care and Use Committees.
Histology, in situ hybridization, and immunofluorescence
Tissues were fixed in 4% paraformaldehyde, embedded in paraffin wax, and sectioned at 5-µm intervals. Deidentified human fetal lung samples were collected through an Institutional Review Board protocol at Temple University (Philadelphia, PA). Slides were stained with hematoxylin and eosin, Alcian blue, or Weigert's stain for elastin using standard protocols. Complete protocols are available at https://www.pennmedicine.org/departments-and-centers/penn-cardiovascular-institute/core-facilities/histology-and-gene-expression-core#tab-3. Immunofluorescence was performed using the following antibodies: mouse anti-Muc5Ac (1:200; Abcam), rat anti-B220 (1:200; BioLegend), rabbit anti-CD3e (1:200; Invitrogen), rabbit anti-Scgb1a1 (1:500; Seven Hills Bioreagents), mouse anti-Krt8 (1:100, Biolegend), and mouse anti-Tub1a1 (1:1000; Sigma-Aldrich). Slides were mounted with VectaShield mounting medium containing DAPI (Vector Laboratories) and then imaged on either a Nikon Eclipse 90i wide-field or Nikon A1R GaAsP inverted confocal microscope. In situ hybridization was performed as described previously (Zhang et al. 2008; Herriges et al. 2014). In order to study the cell type distribution and subcellular localization of Falcor, RNAScope in situ hybridization was performed per the manufacturer's instructions using a custom-prepared probe (Mm-cust-Falcor; ACD, catalog no. 512881, lot no. 17228B) and the RNAScope 2.5 HD detection reagent-RED kit (Advanced Cell Diagnostics, catalog no. 322360). Probes targeting PPIB (ACD, catalog no. 313911) and the bacterial gene dapB (ACD, catalog no. 310043) were used as positive and negative controls, respectively.
RNA-seq and computational analyses
Lungs from wild-type (n = 5) , Falcor−/− (n = 5), Foxa2+/− (n = 4), and Foxa2+/−;Falcor+/− mice (n = 4) of both genders at 10–12 wk of age were harvested and processed into single-cell suspensions using a dispase (Collaborative Biosciences)/collagase (Life Technologies)/DNase solution. Epithelial cells were enriched using a CD326 (EpCAM) antibody (eBioscience, 17-5791-82) and Dynabeads magnetic sorting (sheep antirat IgG; Invitrogen) according to the manufacturer's instructions. Total RNA was extracted from isolated epithelial cells using Trizol (Invitrogen) and then purified with RNEasy columns (Qiagen) prior to RNA-seq. Library preparation and sequencing for these samples were performed at GeneWiz, LLC. Fastq files were assessed for quality control using the FastQC program. Fastq files were aligned against the mouse reference genome (mm10) using the STAR aligner (Dobin et al. 2013). Duplicate reads were flagged using the MarkDuplicates program from Picard tools. Per-gene read counts for Ensembl (version 67) gene annotations were computed using the Rsubread R package, with duplicate reads removed. Gene counts, represented as counts per million (CPM), were first nominalized using the TMMmethod in the edgeR R package, and genes with 25% of samples with a CPM of >1 were removed. The data were transformed using the VOOM function from the limma R package (Law et al. 2014). Additionally, due to the use of magnetic bead sorting, genes detectable only at low expression levels (VOOM transformed counts per million values [v$E] of <9) were excluded from subsequent analysis to minimize potential effects from nonepithelial cell contamination. Differential gene expression was performed using a linear model with the limma package. Heat maps and PCA plots were generated in R. GO enrichment analysis for the biological process was performed using R package GAGE. ChIP-seq data for H3K4me1, H3K4me3, Pol2, Foxa1, and Foxa2 for the mouse lung and human liver were accessed either directly from the ENCODE Project Web site (http://www.encodeproject.org) or via the University of California at Santa Cruz (UCSC) genome browser (https://genome.ucsc.edu) (ENCODE Project Consortium 2012; Sloan et al. 2016). Human expression data for FALCOR and FOXA2 were obtained for 42 different tissue types from the Genotype-Tissue Expression (GTEx) Project database (The GTEx Consortium 2013). The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the National Cancer Institute; the National Human Genome Research Institute; the National Heart, Lung, and Blood Institute; the National Institute on Drug Abuse; the National Institute of Mental Health; and the National Institute of Neurological Disorders and Stroke. Version 6 of the GTEx data set was obtained directly from the GTEx portal (https://gtexportal.org/home). Expression levels for FALCOR and FOXA2 were plotted across the nine tissue types expressing FALCOR/FOXA2 using the box plot function of ggplot2() in R (Wickham 2016). Expression levels of FALCOR and FOXA2 in individual human lung samples (n = 321) were plotted, and correlation was assessed using a linear regression model with ggplot2 in R. Genomic interval plots were generated using the UCSC genome browser using either the human GRCh37/hg19 or mouse GRCm38/mm10 genome builds. The Gene Expression Omnibus accession number for the RNA-seq is GSE126818.
Quantitative real-time PCR
Total RNA was isolated from the indicated tissues or cell lines using Trizol (Invitrogen) per the manufacturer's protocol. cDNA was synthesized from total RNA by using the SuperScript IV strand synthesis system (Invitrogen). Quantitative real-time PCR was performed using the SYBR Green system (Applied Biosystems) with primers listed in Supplemental Table 5. GAPDH expression values were used to control for RNA quantity. Two-tailed Student's t-test was used for all comparisons involving two groups (e.g., Scrambled vs. Falcor shRNA).
Naphthalene injury
Falcor−/− and Foxa2+/− mice as well as wild-type controls were administered 275 μg/kg naphthalene (Sigma-Aldrich) dissolved in corn oil (MP Biomedicals) via a single intraperitoneal injection (Stripp et al. 1995). Lung tissue was harvested 30 d after injury and processed for histology and immunofluorescence microscopy as described above. The relative numbers of Club to ciliated cell after airway epithelial repair was assess by counting the number of Scgba1a1+ (Club) or Tub1a1+ (ciliated) cells in at least 10 randomly selected airway sections from each of n = 8–10 animals per genotype. To characterize the immune cell infiltrate following injury, the right lungs from wild-type or Falcor−/− animals (n = 4) 28 d after naphthalene injury were minced, incubated for 30 min at 37°C with shaking (150 rpm in digestion buffer: RPMI 1640 with 10% FBS, 15 mM HEPES, 1% [w/v] penicillin/streptomycin, 300 U mL−1 collagenase VIII), and pressed through a 100-µm nylon strainer to obtain single-cell suspension. The cells (1 × 107) were washed and incubated for 10 min at 4°C with antimouse CD16/CD32 and then reincubated for 30 min at 4°C with antimouse CD45 antibody (30-F11), antimouse CD4 antibody (GK1.5), antimouse CD8 antibody (53-5.8), antimouse CD11b antibody (M1/70), antimouse CD11c antibody (N418), antimouse CD19 antibody (6D5), antimouse Ly6G antibody (1A8), and antimouse F4/80 antibody (BM8), all diluted 1:100 (Biolegend). Data were collected with an LSRII flow cytometer (BD Biosciences) and analyzed with the FlowJo analysis software (FlowJo, LLC). For gene expression studies following lung injury, biotinylated cRNA probe libraries were generated from total RNA isolated from homogenized (TissueLyser, Qiagen) whole-lung samples with Trizol (Invitrogen) and then hybridized to Affymetrix human gene 2.0ST arrays. Microarray data were analyzed using the oligo package available at the Bioconductor Web site (http://www.bioconductor.org). The Gene Expression Omnibus accession number for the microarray data is GSE126818.
Cell culture methods and lentiviral shRNA knockdown/Foxa2 overexpression
A549, HEK293T, and HBEC-3KT cell lines obtained from American Type Culture Collection (ATCC) were used for all experiments. HEK293T cells were cultured in DMEM (Life Technologies) plus 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2. A549 cells were cultured in Ham's F12K (Kaighn's) medium (Thermo Fisher Scientific) plus 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2. HBEC-3KT cells were grown in airway epithelial cell basal medium (ATCC, PCS-300-030) supplemental with the bronchial epithelial cell growth kit (ATCC, PCS-300-040). Three unique shRNAs targeting either Falcor or Foxa2 were obtained in the SMARTvector human lentiviral shRNA expression construct (Dharmacon) driven by the mCMV promoter and then screened for the most efficient shRNA, which was used for subsequent experiments (the FALCOR construct used was catalog no. V3SH11246-245241381, clone ID no. V3SH11246-245241381). The SMARTvector nontargeting shRNA was used as a control (catalog no. VSC11708). The pLV.PGK.mFoxa2 construct (Addgene, plasmid no. 33014) was used for Foxa2 overexpression. Lentivirus was produced by transfecting 293T cells with the SMARTvector plasmid or pLV.PGK.mFoxa2, psPAX2 (Addgene, plasmid no. 12259) and pMD2.G (Addgene, plasmid no. 12259) constructs. Virus was harvested 48–72 hours later, purified with FastTrap lentivirus purification kit (Millipore), and applied to target cells with polybrene (final concentration 8 µg/mL; EMD Millipore). Fresh medium was added 12 h after application of virus. Puromycin (3 µg/mL final concentration; Alfa Aesar) was added 24 h later, and all experiments were performed 3–5 d after lentiviral transduction.
Scratch assay
A549 cells were plated to 70% confluency in 12-well plates and then transduced with lenviral shRNA constructs as described above. Over a period of 3 d following transduction, the cells were grown to confluency, and then a vertical and a horizontal “scratch” were made in each well with a 20-µL pipette tip. Cells were photographed at identical points at time 0 and 24 and 48 h following the scratch. Cell migration was assessed by measuring changes in the area devoid of cells per high-powered field over time using ImageJ.
Transwell migration assay
A549 or HBEC-3KT cells were plated to 70% confluency in 12-well plates and then transduced with lentiviral shRNA constructs as described above. Three days after viral transduction, cells were harvested with 0.25% trypsin and reconstituted as a single-cell suspension in serum-free medium, and 5 × 104 cells in 300 µL were applied to the upper well of transwell migration chambers (8 µm pore size; Costar). Standard medium supplemented with 20% FBS was placed into the lower chamber. Sixteen hours later, cells within the transwell membrane were fixed, permeabilized, and stained with Giemsa. Migration was quantified by counting cell number per high-powered field (10× objective) from at least five randomly selected images for n = 4–6 membranes per shRNA construct.
Supplementary Material
Acknowledgments
We thank Hitesh Deshmukh and Jerilyn Gray for their help in characterizing the immune cell population seen after airway injury in transgenic animals. We acknowledge the assistance of the Research Flow Cytometry Core and Confocal Imaging Core at Cincinnati Children's Hospital Medical Center and the Confocal Imaging Core at the University of Pennsylvania. We are thankful for the generous grant support from the following sources: National Institutes of Health grants R01HL132999 (to E.E.M.), R01HL132349 (to E.E.M.), R01HL122993 (to E.E.M.), 4K12HD043245 (to D.T.S.), 7K08HL130666 (to D.T.S.), and R01GM110174 (to B.A.G.) and a Parker B. Francis Fellowship Award (to D.T.S.).
Author contributions: D.T.S., M.H., S.L., A.S., S.F., K.S., and S.Z. performed experiments related to this study. M.M. and D.T.S. performed the bioinformatics analysis. D.T.S., M.M., K.S., and E.E.M. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.320523.118.
References
- Anderson KM, Anderson DM, McAnally JR, Shelton JM, Bassel-Duby R, Olson EN. 2016. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 539: 433–436. 10.1038/nature20128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang SL, Rossant J. 1994. HNF-3β is essential for node and notochord formation in mouse development. Cell 78: 561–574. 10.1016/0092-8674(94)90522-3 [DOI] [PubMed] [Google Scholar]
- Basseres DS, D'Alò F, Yeap BY, Löwenberg EC, Gonzalez DA, Yasuda H, Dayaram T, Kocher ON, Godleski JJ, Richards WG, et al. 2012. Frequent downregulation of the transcription factor Foxa2 in lung cancer through epigenetic silencing. Lung Cancer 77: 31–37. 10.1016/j.lungcan.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard V, Wert SE, Hull WM, Whitsett JA. 2004. Immunohistochemical localization of Foxa1 and Foxa2 in mouse embryos and adult tissues. Gene Expr Patterns 5: 193–208. 10.1016/j.modgep.2004.08.006 [DOI] [PubMed] [Google Scholar]
- Burtscher I, Lickert H. 2009. Foxa2 regulates polarity and epithelialization in the endoderm germ layer of the mouse embryo. Development 136: 1029–1038. 10.1242/dev.028415 [DOI] [PubMed] [Google Scholar]
- Cao WJ, Wu HL, He BS, Zhang YS, Zhang ZY. 2013. Analysis of long non-coding RNA expression profiles in gastric cancer. World J Gastroenterol 19: 3658–3664. 10.3748/wjg.v19.i23.3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, Gregorieff A, Clevers H, Whitsett JA. 2009. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest 119: 2914–2924. 10.1172/JCI35314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wan H, Luo F, Zhang L, Xu Y, Lewkowich I, Wills-Karp M, Whitsett JA. 2010. Foxa2 programs Th2 cell-mediated innate immunity in the developing lung. J Immunol 184: 6133–6141. 10.4049/jimmunol.1000223 [DOI] [PubMed] [Google Scholar]
- Chen G, Korfhagen TR, Karp CL, Impey S, Xu Y, Randell SH, Kitzmiller J, Maeda Y, Haitchi HM, Sridharan A, et al. 2014. Foxa3 induces goblet cell metaplasia and inhibits innate antiviral immunity. Am J Respir Crit Care Med 189: 301–313. 10.1164/rccm.201306-1181OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang VT, Kassahn KS, Marcos AE, Ragan MA. 2008. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet 16: 1350–1357. 10.1038/ejhg.2008.111 [DOI] [PubMed] [Google Scholar]
- da Rocha ST, Heard E. 2017. Novel players in X inactivation: insights into Xist-mediated gene silencing and chromosome conformation. Nat Struct Mol Biol 24: 197–204. 10.1038/nsmb.3370 [DOI] [PubMed] [Google Scholar]
- Dhamija S, Becker AC, Sharma Y, Myacheva K, Seiler J, Diederichs S. 2018. LINC00261 and the adjacent gene FOXA2 are epithelial markers and are suppressed during lung cancer tumorigenesis and progression. Noncoding RNA 5: E2 10.3390/ncrna5010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. 2016. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539: 452–455. 10.1038/nature20149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. 2008. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev 22: 3435–3448. 10.1101/gad.1752608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosalia N, Yang R, Kerschner JL, Harris A. 2015. FOXA2 regulates a network of genes involved in critical functions of human intestinal epithelial cells. Physiol Genomics 47: 290–297. 10.1152/physiolgenomics.00024.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualdi R, Bossard P, Zheng M, Hamada Y, Coleman JR, Zaret KS. 1996. Hepatic specification of the gut endoderm in vitro: cell signaling and transcriptional control. Genes Dev 10: 1670–1682. 10.1101/gad.10.13.1670 [DOI] [PubMed] [Google Scholar]
- Hammad H, Lambrecht BN. 2015. Barrier epithelial cells and the control of type 2 immunity. Immunity 43: 29–40. 10.1016/j.immuni.2015.07.007 [DOI] [PubMed] [Google Scholar]
- Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. 2004. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 118: 517–528. 10.1016/j.cell.2004.07.024 [DOI] [PubMed] [Google Scholar]
- Herriges MJ, Swarr DT, Morley MP, Rathi KS, Peng T, Stewart KM, Morrisey EE. 2014. Long noncoding RNAs are spatially correlated with transcription factors and regulate lung development. Genes Dev 28: 1363–1379. 10.1101/gad.238782.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herriges MJ, Tischfield DJ, Cui Z, Morley MP, Han Y, Babu A, Li S, Lu M, Cendan I, Garcia BA, et al. 2017. The NANCI–Nkx2.1 gene duplex buffers Nkx2.1 expression to maintain lung development and homeostasis. Genes Dev 31: 889–903. 10.1101/gad.298018.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg J, Perlmann T. 2012. Maintaining differentiated cellular identity. Nat Rev Genet 13: 429–439. 10.1038/nrg3209 [DOI] [PubMed] [Google Scholar]
- Jägle S, Busch H, Freihen V, Beyes S, Schrempp M, Boerries M, Hecht A. 2017. SNAIL1-mediated downregulation of FOXA proteins facilitates the inactivation of transcriptional enhancer elements at key epithelial genes in colorectal cancer cells. PLoS Genet 13: e1007109 10.1371/journal.pgen.1007109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandura A, Krause HM. 2017. The new RNA world: growing evidence for long noncoding RNA functionality. Trends Genet 33: 665–676. 10.1016/j.tig.2017.08.002 [DOI] [PubMed] [Google Scholar]
- Jiang W, Liu Y, Liu R, Zhang K, Zhang Y. 2015. The lncRNA DEANR1 facilitates human endoderm differentiation by activating FOXA2 expression. Cell Rep 11: 137–148. 10.1016/j.celrep.2015.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp F, Mendell JT. 2018. Functional classification and experimental dissection of long noncoding RNAs. Cell 172: 393–407. 10.1016/j.cell.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci 93: 5860–5865. 10.1073/pnas.93.12.5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: R29 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gallo M, Rudd ML, Urick ME, Hansen NF; National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program , Merino MJ, Mutch DG, Goodfellow PJ, Mullikin JC, Bell DW. 2018. The FOXA2 transcription factor is frequently somatically mutated in uterine carcinosarcomas and carcinomas. Cancer 124: 65–73. 10.1002/cncr.30971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. 2013. Transcriptional regulation and its misregulation in disease. Cell 152: 1237–1251. 10.1016/j.cell.2013.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Friedman JR, Fulmer JT, Kaestner KH. 2005. The initiation of liver development is dependent on Foxa transcription factors. Nature 435: 944–947. 10.1038/nature03649 [DOI] [PubMed] [Google Scholar]
- Li L, Chang HY. 2014. Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol 24: 594–602. 10.1016/j.tcb.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CM, Gocheva V, Oudin MJ, Bhutkar A, Wang SY, Date SR, Ng SR, Whittaker CA, Bronson RT, Snyder EL, et al. 2015. Foxa2 and Cdx2 cooperate with Nkx2-1 to inhibit lung adenocarcinoma metastasis. Genes Dev 29: 1850–1862. 10.1101/gad.267393.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Yin J, Cheng C, Yang Z, Yang H. 2018. Decreased expression of FOXA2 promotes eutopic endometrial cell proliferation and migration in patients with endometriosis. Reprod Biomed Online 36: 181––187. 10.1016/j.rbmo.2017.11.001 [DOI] [PubMed] [Google Scholar]
- Liu Y, Xiao N, Xu SF. 2017. Decreased expression of long non-coding RNA LINC00261 is a prognostic marker for patients with non-small cell lung cancer: a preliminary study. Eur Rev Med Pharmacol Sci 21: 5691–5695. [DOI] [PubMed] [Google Scholar]
- Marchese FP, Raimondi I, Huarte M. 2017. The multidimensional mechanisms of long noncoding RNA function. Genome Biol 18: 206 10.1186/s13059-017-1348-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh JH, Kim KM, McClusky WG, Abdelmohsen K, Gorospe M. 2018. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip Rev RNA 9: e1471 10.1002/wrna.1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Wells JM, Zorn AM, Wert SE, Laubach VE, Fernandez LG, Whitsett JA. 2006. Transdifferentiation of ciliated cells during repair of the respiratory epithelium. Am J Respir Cell Mol Biol 34: 151–157. 10.1165/rcmb.2005-0332OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajavelu P, Chen G, Xu Y, Kitzmiller JA, Korfhagen TR, Whitsett JA. 2015. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J Clin Invest 125: 2021–2031. 10.1172/JCI79422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleimer RP, Berdnikovs S. 2017. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J Allergy Clin Immunol 139: 1752–1761. 10.1016/j.jaci.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha L, Huang L, Luo X, Bao J, Gao L, Pan Q, Guo M, Zheng F, Wang H. 2017. Long non-coding RNA LINC00261 inhibits cell growth and migration in endometriosis. J Obstet Gynaecol Res 43: 1563–1569. 10.1111/jog.13427 [DOI] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. 2016. ENCODE data at the ENCODE portal. Nucleic Acids Res 44: D726–D732. 10.1093/nar/gkv1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Neff R, Cohn DE, Backes FJ, Suarez AA, Mutch DG, Rush CM, Walker CJ, Goodfellow PJ. 2016. The mutational spectrum of FOXA2 in endometrioid endometrial cancer points to a tumor suppressor role. Gynecol Oncol 143: 398–405. 10.1016/j.ygyno.2016.08.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, Crowley D, Whittaker CA, Meyerson M, Kimura S, et al. 2013. Nkx2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell 50: 185–199. 10.1016/j.molcel.2013.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stripp BR, Maxson K, Mera R, Singh G. 1995. Plasticity of airway cell proliferation and gene expression after acute naphthalene injury. Am J Physiol 269: L791–L799. 10.1152/ajpcell.1995.269.3.C791 [DOI] [PubMed] [Google Scholar]
- Sun PR, Jia SZ, Lin H, Leng JH, Lang JH. 2014. Genome-wide profiling of long noncoding ribonucleic acid expression patterns in ovarian endometriosis by microarray. Fertil Steril 101: 1038–1046 e1037. 10.1016/j.fertnstert.2013.12.035 [DOI] [PubMed] [Google Scholar]
- Sund NJ, Ang SL, Sackett SD, Shen W, Daigle N, Magnuson MA, Kaestner KH. 2000. Hepatocyte nuclear factor 3β (Foxa2) is dispensable for maintaining the differentiated state of the adult hepatocyte. Mol Cell Biol 20: 5175–5183. 10.1128/MCB.20.14.5175-5183.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Shu G, Yuan X, Jing N, Song J. 2011. FOXA2 functions as a suppressor of tumor metastasis by inhibition of epithelial-to-mesenchymal transition in human lung cancers. Cell Res 21: 316–326. 10.1038/cr.2010.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetzmann L, Burtscher I, Lickert H. 2008. A mouse line expressing Foxa2-driven Cre recombinase in node, notochord, floorplate, and endoderm. Genesis 46: 515–522. 10.1002/dvg.20410 [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154: 26–46. 10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorvis C, Hatziapostolou M, Mahurkar-Joshi S, Koutsioumpa M, Williams J, Donahue TR, Poultsides GA, Eibl G, Iliopoulos D. 2016. Transcriptomic and CRISPR/Cas9 technologies reveal FOXA2 as a tumor suppressor gene in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol 310: G1124–G1137. 10.1152/ajpgi.00035.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Hager GL. 2014. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15: 69–81. 10.1038/nrg3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT, Fulkerson PC, Rothenberg ME, Whitsett JA. 2004a. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 131: 953–964. 10.1242/dev.00966 [DOI] [PubMed] [Google Scholar]
- Wan H, Xu Y, Ikegami M, Stahlman MT, Kaestner KH, Ang SL, Whitsett JA. 2004b. Foxa2 is required for transition to air breathing at birth. Proc Natl Acad Sci 101: 14449–14454. 10.1073/pnas.0404424101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, Dingle S, Xu Y, Besnard V, Kaestner KH, Ang SL, Wert S, Stahlman MT, Whitsett JA. 2005. Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. J Biol Chem 280: 13809–13816. 10.1074/jbc.M414122200 [DOI] [PubMed] [Google Scholar]
- Wang Y, Tian Y, Morley MP, Lu MM, Demayo FJ, Olson EN, Morrisey EE. 2013. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2–Bmp4/Rb1 regulatory pathway. Dev Cell 24: 345–358. 10.1016/j.devcel.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xue K, Guan Y, Jin Y, Liu S, Wang Y, Liu S, Wang L, Han L. 2017. Long noncoding RNA LINC00261 suppresses cell proliferation and invasion and promotes cell apoptosis in human choriocarcinoma. Oncol Res 25: 733–742. 10.3727/096504016X14772362173376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O, Chang HY. 2011. Long noncoding RNAs and human disease. Trends Cell Biol 21: 354–361. 10.1016/j.tcb.2011.04.001 [DOI] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York. [Google Scholar]
- Xu XR, Huang J, Xu ZG, Qian BZ, Zhu ZD, Yan Q, Cai T, Zhang X, Xiao HS, Qu J, et al. 2001. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci 98: 15089–15094. 10.1073/pnas.241522398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Zhuo Y, Shan B. 2017. MicroRNAs, long noncoding RNAs, and their functions in human disease. Methods Mol Biol 1617: 1–25. 10.1007/978-1-4939-7046-9_1 [DOI] [PubMed] [Google Scholar]
- Yu Y, Li L, Zheng Z, Chen S, Chen E, Hu Y. 2017. Long non-coding RNA linc00261 suppresses gastric cancer progression via promoting Slug degradation. J Cell Mol Med 21: 955–967. 10.1111/jcmm.13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goss AM, Cohen ED, Kadzik R, Lepore JJ, Muthukumaraswamy K, Yang J, DeMayo FJ, Whitsett JA, Parmacek MS, et al. 2008. A Gata6–Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet 40: 862–870. 10.1038/ng.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yang C, Gao W, Chen T, Qian T, Hu J, Tan Y. 2015. FOXA2 attenuates the epithelial to mesenchymal transition by regulating the transcription of E-cadherin and ZEB2 in human breast cancer. Cancer Lett 361: 240–250. 10.1016/j.canlet.2015.03.008 [DOI] [PubMed] [Google Scholar]
- Zhang HF, Li W, Han YD. 2018. LINC00261 suppresses cell proliferation, invasion and Notch signaling pathway in hepatocellular carcinoma. Cancer Biomark 21: 575–582. 10.3233/CBM-170471 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.