

Abstract
Nitric oxide (NO) synthesized by eNOS plays a key role in regulation of endothelial barrier integrity but underlying cell signaling pathway is not fully understood at present. Here, we report opposing roles of two different redox-dependent NO metabolites; peroxynitrite (ONOO−) vs. S-nitrosoglutathione (GSNO), in cell signaling pathways for endothelial barrier disruption. In cultured human brain microvessel endothelial cells (hBMVECs), thrombin induced F-actin stress fiber formation causes barrier disruption via activating eNOS. Thrombin induced eNOS activity participated in cell signaling (e.g. RhoA and calcium influx mediated phosphorylation of myosin light chain) for F-actin stress fiber formation by increasing ONOO− levels. On the other hand, thrombin had no effect on intracellular levels of S-nitrosoglutathione (GSNO), another cellular NO metabolite. However, exogenous GSNO treatment attenuated the thrombin-induced cell signaling pathways for endothelial barrier disruption, thus suggesting the role of a shift of NO metabolism (GSNO vs. ONOO−) toward ONOO− synthesis in cell signaling for endothelial barrier disruption. Consistent with these in vitro studies, in animal models of traumatic brain injury and experimental autoimmune encephalomyelitis (EAE), ONOO− scavenger treatment as well as GSNO treatment were effective for attenuation of BBB leakage, edema formation, and CNS infiltration of mononuclear cells. Taken together, these data document that eNOS-mediated NO production and following redox-dependent NO metabolites (ONOO− vs. GSNO) are potential therapeutic target for CNS microvascular disease (traumatic and inflammatory) pathologies.
Keywords: brain endothelial barrier, endothelial nitric oxide synthase (eNOS), actin stress fiber, peroxynitrite (ONOO−), S-nitrosoglutathione (GSNO), nitric oxide
Graphical abstract
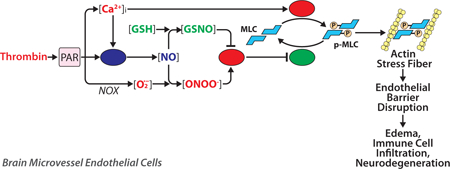
1. Introduction
The blood–brain barrier (BBB) is a specialized microvascular endothelial structure in the central nervous system (CNS). There is growing body of evidence that BBB disruption is associated with multiple CNS diseases including ischemic and hemorrhagic stroke, traumatic brain injury (TBI), multiple sclerosis (MS), and Alzheimer’s disease [1]. In these disease conditions, the BBB disruption contributes to extravasation of peripheral immune/inflammatory cells and blood-borne toxic molecules as well as edema formation leading to CNS inflammation and neurodegeneration [2]. BBB disruption is a complex and multistep process involving several interrelated factors, such as hemostasis (e.g. thrombin), oxidative stress, angiogenesis, and inflammation [2]. Recent studies report that the early events of BBB disruption are partially reversible [3; 4; 5], thus regarded as a potential target for therapeutic interventions [5]. At present the precise mechanism of BBB disruption is not fully understood. However, intracellular Ca2+ ([Ca2+]I) influx and RhoA activation induced sustained phosphorylation of endothelial myosin light chain (MLC) and assembly of F-actin stress fiber leading to deformation of endothelial cells and disassembly of tight junctional complex have been proposed as early events of BBB disruption [6].
Nitric oxide (NO) produced by endothelial nitric oxide synthase (eNOS) is known to play a key role in vascular/endothelial functions [7]. NO is known to exert its biological effect through cGMP-dependent mechanisms [8]. Alternatively, NO also exerts its action via formation of secondary redox derivatives, such as peroxynitrite (ONOO−) and S-nitrosoglutathione (GSNO) [9]. ONOO−, the most powerful oxidative/nitrosative agent, is generated by reaction between NO and superoxide anion (O2•− ) under oxidative stress conditions and has been implicated in various pathological events via irreversible modification of protein tyrosine (3-nitrotyrosine) [10]. On the other hand, GSNO, the most abundant low-molecular-weight S-nitrosothiol synthesized by reaction between NO and glutathione (GSH), participates in various physiological cellular processes via reversible modification of protein thiols, a process termed S-nitrosylation [11; 12]. GSNO is known to be implicated in cardiovascular hemodynamics [13], inhibition of platelet activation [14], and modulation of inflammatory processes [15; 16].
In endothelial cells, eNOS derived NO signaling is known to induce RhoA-mediated cell signaling for endothelial barrier dysfunction via nitration of RhoA (Tyr34) [17; 18]. On the other hand, NO is also reported to inhibit RhoA activity and protects endothelial barrier function by S-nitrosylation of RhoA (Cys16, Cys20, and Cys159) [19]. These studies suggest that NO may play double-edged roles (protective or deleterious) in regulation of endothelial barrier integrity via its conversion to different redox metabolites (GSNO vs. ONOO−) and thus different type of protein modifications (S-nitrosylation vs. tyrosine-nitration). However, mechanisms underlying redox dependent NO signaling (ONOO− vs. GSNO) for endothelial barrier regulation are not well understood at present. Therefore, the aim of this study is to understand mechanisms underlying GSNO vs ONOO− mediated regulation of cell signaling pathways for early event of endothelial barrier disruption (e.g. RhoA/Ca2+ influx dependent MLC phosphorylation) using in vitro cell culture model of thrombin-induced endothelial hyper-permeability.
Thrombin participates in key processes of vascular hemostatic process for control of blood loss [20]. In addition, thrombin also induces non-hemostatic pathological processes via activation of protease-activated receptors (e.g. PAR1, PAR3, and PAR4) [21; 22; 23]. In endothelial cells, thrombin activates PAR1 by cleaving and unmasking of tethered ligand for self-activation [24; 25]. Activated PAR1 induces MLC kinase (MLCK) activation via inducing inositol-1,4,5-trisphosphate (IP3) mediated [Ca2+]i influx [26]. The PAR1 activation also induces RhoA/ROCK activation via regulation of G-proteins [27; 28] leading to inhibition of MLC phosphatase (MLCP). Consequently, activation of MLCK and inhibition of MLCP lead to increased phosphorylation of Ser19 of MLC for assembly of stress fibers and focal adhesions [29]. Because of the similarity in these cell signaling pathways, we studied thrombin-induced hyper-permeability of human brain microvessel endothelial cell (hBMVEC) in culture as an in vitro model for early events in BBB disruption. Using this in vitro model, here, we report that thrombin-induced endothelial eNOS activation for NO synthesis and its conversion to ONOO− increases thrombin-induced [Ca2+]i influx, RhoA activation, and thus MLC phosphorylation for endothelial stress fiber formation associated with endothelial barrier disruption. On the other hand, GSNO inhibits thrombin-induced cell signaling for MLC phosphorylation and endothelial stress fiber formation thus barrier disruption. These observations underscore a role for balance between eNOS derived NO metabolites (ONOO− vs. GSNO) in cell signaling for endothelial barrier integrity.
Secondly, this study also evaluated the potential efficacies of exogenous GSNO and ONOO− scavenging compound FeTPPS on BBB disruption induced by TBI and experimental autoimmune encephalomyelitis (EAE), an animal model for MS. TBI is the leading cause of long‐term neurobehavioral dysfunctions in young as well as in adults. TBI is caused by mechanical forces on brain that results in primary injury, such as shearing injuries, contusions, and hematomas, as well as vascular and parenchymal damage leading to BBB disruption that results in secondary injury, such as edema, inflammation, and hyper-excitability [30]. MS is a debilitating autoimmune inflammatory CNS disorder. The disease is induced by activation of myelin specific autoreactive lymphocytes and their CNS infiltration resulting in encephalitogenic inflammatory disease [31]. Dysregulation of the BBB and transendothelial migration of activated leukocytes are among the earliest cerebrovascular abnormalities seen in MS brains [32]. Therefore, protection of early event of BBB disruption is important for preventing the progression of secondary brain injury in TBI [33] as well as the infiltration of peripheral immune and inflammatory cells under CNS inflammatory disease conditions [32]. In this study, we also reports that BBB disruption in animal model of TBI and EAE are inhibited by ONOO− scavenger (FeTPPS) or exogenous GSNO treatment and thus identifying redox-dependent NO metabolites (ONOO− vs. GSNO) as a potential therapeutic targets for neurovascular integrity in neurological disorders.
2. Material and Methods
2. 1. Reagents
Thrombin was purchase from Sigma-Aldrich (Cat#: T4393, St. Louis, MO). L-NIO [N5-(1-Iminoethyl)-L-ornithine dihydrochloride], SIN-1 (3-morpholinosydnonimine chloride), and BAPTA [1,2-Bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid] were purchase from Tocris (Cat#: 0546, 0756, and 2786, respectively, Minneapolis, MN). RhoA inhibitor I (highly purified C3 transferase) was purchased from Cytoskeleton Inc. (Cat# CT04). FeTPPS [5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrinato Iron (III), Cl] and DETA-NO (diethylamine NONOate/AM) was purchase from Millipore-Calbiochem (Cat#: 341492 and 292505, respectively, Billerica, MA). DAF-FM diacetate (4-amino-5-methylamino-2’,7’-difluorofluorescein diacetate) was purchased from Thermofisher Scientific (Cat# D-23842, Waltham, MA) S-nitrosoglutathione (GSNO) was purchase from World precision instruments (Cat#: GSNO-100, Sarasota, FL). The effective concentration of the GSNO was calculated from the optical absorbance at 338 nm and the reported molar extinction coefficients as described previously [34].
2.2. Cell culture
Primary human brain microvascular endothelial cells (hBMVECs) were purchased from Angio-Proteomie (Cat#: cAP-0002, Atlanta, GA). The cells were cultured in cell culture flasks or plates precoated with Quick Coating Solution (Angio-Proteomie; Cat#: cAP-01) and maintained in Endothelial Growth Medium (Angio-Proteomie; Cat#: cAP-02) at 37°C under 5% CO2/95% air. When the cells were almost confluent, the medium was replaced with endothelial basal medium (Angio-Proteomie; Cat#: cAP-03) containing 0.5% fetal bovine serum (FBS; Life Technologies, Grand Island, NY) about 8–12 hours before the experiment. No institutional approval was required for this study. The study was not pre-registered.
2.3. Assay of trans-endothelial electrical resistance (TEER)
For evaluation of the endothelial barrier function, hBMVECs were plated on fibronectin-coated polycarbonate filters (Transwell system, Corning, Midland, NC) containing Endothelial Growth Medium (Angio-Proteomie Cat#: cAP-02). The medium was renewed every other day. Five daysm after seeding, the medium was replaced with Endothelial Basal Medium (Angio-Proteomie Cat#: cAP-03) containing 0.5% FBS and incubated for 2 days. Following drug treatments, transendothelial electrical resistance (TEER) was measured by EVOM2 (Word Precision Instruments) as described previously [35].
2.4. RhoA activity assay
RhoA activity in hBMVECs was analyzed by RhoA Activation Assay Kit (Abcam Cat#: ab211164, Cambridge, MA). Briefly, following drug treatments, the cells were lysed with 1XAssay buffer provided in the kit. Lysates were centrifuged (14,000 x g for 10 sec), and supernatants were incubated with agarose beads coupled to GST-Rhotekin-Rho binding domain (RBD) for 2 h at 4 °C. Beads were then washed with 1XAssay buffer and GTP-bound RhoA was eluted with 2X SDS-PAGE sample buffer. Amounts of active (GTP-bound) RhoA were determined by Western blot analysis using antibody specific to RhoA (Abcam).
2.5. Assay for F-actin stress fiber development and endothelial cell contraction
hBMVECs were cultured on fibronectin-coated chamber slides (BD Bioscience). Following drug treatments, the cells were fixed with 4% (wt/vol) paraformaldehyde, permeabilized by the addition of 0.25% Triton X-100, and blocked by 2% bovine serum albumin (BSA) in phosphate buffered saline (PBS). The slides were immunostained for phospho-MLC (Ser19) as well as stained with Phalloidin for F-actin (F-actin Visualization Biochem kit, Cytoskeleton, Inc, Cat#: BK005, Denver, CO) and DAPI for nucleus (4′,6-diamidino-2-phenylindole; ThermoFisher Scientific, Houston, TX). The cells were imaged by BX60 Olympus fluorescent/light microscope equipped with DP-70 digital camera (Olympus, Tokyo, Japan). The density of fluorescence was analyzed by ImageJ (NIH, Bethesda, MD).
2.6. Assay for intracellular Ca2+ influx
Intracellular Ca2+ concentration ([Ca2+]i) was measured with Fluo-4 Direct Calcium Assay Kit (Thermo Fisher Scientific, Cat#: F10471, Grand Island, NY). Briefly, culture medium in the 96-well plate was replaced with a Ca2+ sensitive dye Fluo-4 in an endothelial basal medium. After 30 min incubation, the dye was removed and cells were incubated with the original medium with or without the drugs at 37°C for 15 min. Following thrombin treatment, time course changes of fluorescent intensity were quantified using a CLARIOstar multi-well fluorometer (BMG Labtech, Cary, NC).
2.7. Assay of intracellular NO production
The intracellular NO production was analyzed by DAF-FM fluorescence intensity. hBMVECs were cultured in 96-well plates and treated with 10µM DAF-FM diacetate for 60min. Following the washing of the cells with fresh medium, the cells were incubated with thrombin (0.1 unit/ml) for 20min. The intracellular NO production was quantified using a CLARIOstar multi-well fluorometer (495nm for excitation and 515nm for emission).
2.8. Cell viability assay
Cell viability was determined by mitochondrial conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan as described previously [36]. hBMVECs were cultured in 96-well plates and treated with 1/20 media volume of MTT reagent (5 mg/ml; Sigma-Aldrich). Following incubation for 2 hours, the cells were washed with PBS and dissolved in isopropanol, including 0.1N HCl. The index of cell viability was measured at an optical density of 570 nm by SpectraMax 190 (Molecular Devices, Sunnyvale, CA).
2.9. Western blot analysis
Western immunoblot analysis was performed by standard method using 50µg of cell lysates. Following the SDS-PAGE electrophoresis, proteins were transferred from the gel onto the Polyvinylidene fluoride membrane (GE Healthcare Life Sciences, Marlborough, MA). Membranes were blocked with non-fat dry milk (Santa Cruz Biotechnology) or I-Block™ (ThermoFisher Scientific, Waltham, MA) for detection of phospho-proteins and incubated with primary antibodies, such as MBP (Santa Cruz Biotech Cat#: sc13914; RRID: AB_648798), phospho-(Ser19) MLC (Abcam, Cat#: ab2480; RRID: AB_303094), MLC (Abcam, Cat#: ab79935; RRID:AB_1952220), β-actin (Santa Cruz Biotechnology, Cat#: sc-47778; RRID:AB_2714189), phospho-eNOS (Ser1177) (Cell Signaling, Cat#: 9571; RRID: AB_329837, Danvers, MA), eNOS (Cell Signaling, Cat#: 32027), RhoA (Santa Cruz Biotechnology; Cat#: sc418; RRID: AB_628218) or 3-nitrotyrosine (Abcam, Cat#: ab61392; RRID: AB_942087). Following washing, the membranes were incubated with horseradish peroxidase conjugated secondary antibody (Jackson Immunoresearch Lab, West Grove, PA), washed and then incubated with ECL reagent (Amersham Life Science, Pittsbrugh, PA), and exposed to Amersham Hyperfilm ECL film.
2.10. Assay for protein-associated nitrotyrosine
Cellular levels of protein-associated 3-nitrotyrosine were analyzed by ELISA Kit (Abcam, Cat#: ab116691) and Western blot analysis using antibody specific to 3-nitrotyrosine (Abcam, Cat#: ab61392). For the ELISA, hBMVECs were lysed in extraction buffer provided with the kit followed by centrifugation at 16,000 x g 4°C. The cell lysate supernatants were subjected to protein quantification with Bio-Rad DC protein assay kit (Bio-Rad, Hercules, CA) and the equal amounts of proteins (500 µg) were loaded onto 96 well microplate coated with 3-nitrotyrosine capture antibody and followed by incubation with biotin-conjugated 3-nitrotyrosine detector antibody. Following washing, the plates were incubated with HRP-conjugated streptoavidin and the levels of 3-nitrotyrosine were measured by incubation with 3,3’,5,5’-tetramethylbenzidine solution and colorimetric analysis at 600 nm using SpectraMax 190 Microplate Reader (Molecular Devices, Sunnyvale, CA). For analysis of degree of RhoA tyrosine nitration, the cell lysates were immunoprecipiated with antibody specific to 3-nitrotyrosine (Abcam) and the levels of tyrosine nitrated RhoA were analyzed by Western analysis for RhoA.
2.11. Assay for protein-associated S-nitrosylation
Protein S-Nitrosylation was analyzed by using biotin-switch method as described in our previous reports [16; 36]. hBMVECs were lysed in 250 mM HEPES, pH 7.7, 1 mM EDTA, 0.1 mM neocuproine, 1% Nonidet P-40, 150 mM NaCl, 1 mM phenylmethanesulfonylfluoride, 20mM methyl methanethiosulfonate (MMTS), 80 µM carmustine, protease inhibitor mixture (Sigma-Aldrich), and mixed with an equal volume of 25 mM HEPES, pH 7.7, 0.1 mM EDTA, 10 µM neocuproine, 5% SDS, 20 mM MMTS and incubated at 50°C for 20 min. Following acetone precipitation, the precipitates were resuspended in 25 mM HEPES, pH 7.7, 0.1 mM EDTA, 10 µM neocuproine, 1% SDS and mixed with two volumes of 20 mM HEPES, pH 7.7, 1 mM EDTA, 100 mM NaCl, 0.5% Triton X-100. The S-nitrosylated proteins were then modified with biotin in 25 mM HEPES, pH 7.7, 0.1 mM EDTA, 1% SDS, 10 µM neocuproine, 10 mM ascorbate sodium salt, and 0.2 mM N-[6-(biotinamido)hexyl]-30-(20-pyridyldithio) propionamide (biotin-HPDP, Pierce). Following acetone precipitation, biotinylated (S-nitrosylated) proteins were analyzed by Western analysis. For detection of S-nitrosylated RhoA, the biotinylated proteins were pull down with neutravidin-agarose and followed by Western analysis for RhoA.
2.12. Controlled cortical impact (CCI) rat model of focal TBI
All animals used in this study received humane care in compliance with the Medical University of South Carolina’s (MUSC) guidance and the National Research Council’s criteria for humane care. Animal procedures were approved by the institutional animal care and use committee of MUSC (AR# 2703). For generation of CCI model of TBI, young adult male (~3–4 months old) Sprague Dawley rats weighing between 260–300 g were randomly divided into four groups: 1) TBI animals treated with vehicle (TBI; n=13), 2) TBI with GSNO (0.05 mg/kg body weight/i.v.) treatment (TBI+GSNO; n=13), 3) TBI with FeTPPS (3 mg/kg body weight, i.v.) treatment (TBI+FeTPPS; n=13), 4) sham-operated treated with vehicle (Sham; n=13). The group size was determined by power analysis based on our previous data [37; 38]. Ketamine (90 mg/kg body weight) and xylazine (10 mg/kg body weight) as surgical anesthesia were administered intraperitoneally. Analgesic buprenorphine was administered pre-emptively to alleviate pain following surgery. Utilizing aseptic techniques, CCI injury was produced as previously described from our laboratory [37; 38] and others [39; 40]. A cortical contusion was produced on the exposed cortex using a controlled impactor device as described in our previous TBI studies [37; 38]. Immediately after injury, the skin incision was closed with nylon sutures. Lidocaine jelly (2%) was applied to the lesion site to minimize any possible infection/discomfort. Sham animals had no cortical impact but underwent the same procedure otherwise.
2.13. Evaluation of BBB disruption by Evans blue (EB) extravasation
BBB leakage was assessed as previously described from our laboratory [37; 38]. The rats received 100 μl of a 5% solution of EB in saline administered intravenously 4 hours following CCI. At 24 hours, cardiac perfusion was performed under deep anesthesia with 200 ml of saline to clear the cerebral circulation of EB. The brain was removed, photographed, and sliced. The brain tissues were homogenized in 750 μl of N, N-dimethylformamide (DMF) and centrifuged at 10,000 x g for 25 minutes, and EB content in supernatant was fluorimetrically analyzed (λex 620 nm, λem 680 nm).
2.14. Measurement of edema (brain water content)
At 24 h following CCI, animals were euthanized to determine brain water content (edema) as described earlier [37; 41]. The cortices, excluding the cerebellum, were quickly removed, and the contralateral and ipsilateral hemispheres separately weighed. Each hemisphere was dried at 60°C for 72 hours, and the dry weight was determined. Water content was calculated in ipsilateral hemisphere as: water content (%) = (wet weight – dry weight)/wet weight x 100.
2.15. EAE induction
EAE was induced as described previously [42]. Animal procedures were approved by the institutional animal care and use committee of MUSC (AR# 1644). Briefly, female C57BL/6J mice of 8–12 weeks of age weighing 18–22g (The Jackson Laboratory, Bar Harbor, ME, USA) were randomly divided into four groups: 1) EAE animals treated with vehicle (EAE; n=8), 2) EAE with GSNO (1 mg/kg body weight per day; i.p.) treatment (EAE+GSNO; n=12), 3) EAE with FeTPPS (30 mg/kg body weight per day; i.p.) treatment (EAE+FeTPPS; n=8), 4) control with vehicle (Ctrl; n=8). The group size was determined by power analysis based on our previous data [42].Then, the mice were immunized subcutaneously in the flank regions with MOG35–55 peptide (MOG; 200ug; Peptide International) emulsified (1:1) in 100ul complete Freund’s adjuvant (CFA) on day 0 and day 7. Additionally, 200 ng of Pertussis toxin (PTX; Sigma-Aldrich, St Louis, MO) was given on day 0 and day 2 by i.p. injection. PTX used as per the standardized protocol reported by us and other investigators for the induction of EAE [42]. Similarly, control group received subcutaneous injection of CFA emulsion and PTX. Clinical signs of EAE were scored in animal facility in a blinded fashion to experimenter between 2 and 4 pm daily by examiners blinded to experimental treatments using the following scale: 0 = no clinical signs of disease; 1 = limp tail or waddling gait with tail tonicity; 2 = waddling gait with limp tail (ataxia); 2.5 = ataxia with partial limb paralysis; 3 = full paralysis of one limb; 3.5 = full paralysis of one limb with partial paralysis of second limb; 4 = full paralysis of two limbs; 4.5 = moribund stage; 5 = death. Starting the day of disease onset (with clinical score between 1 and 2), the animals were given daily treatment with drugs and vehicle (phosphate buffered saline).
2.16. Histological and immuno-histological analysis
Animals were anesthetized and fixed with cardiac perfusion of 4% paraformaldehyde [43]. Tissue samples (lumbar spinal cords) were paraffin-embedded and sectioned transversely (4-µm-thick). Haemotoxylin and Eosin (H&E) staining was performed to assess infiltration of mononuclear cells. To assess the status of myelin, the sections were stained with antibody specific to MBP and detected with secondary antibody conjugated with immunofluorescent analysis. DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) was used for staining of nuclei. All digital images were taken using BX-60 microscope equipped with DP70 camera unit (Olympus, Tokyo, Japan).
2.17. Statistical analysis
Statistical analysis was performed with Graphpad Prism5. Values are expressed as mean ± standard deviation (SD). Comparisons among means of groups were made with a two-tailed Student’s t-test for unpaired variables. Multiple comparisons were performed using one-way ANOVA followed by Bonferroni test. A value of p < 0.05 was considered statistically significant.
3. Results
3.1. Thrombin induced cell signaling for endothelial F-actin stress fiber formation and barrier disruption in cultured hBMVECs
RhoA/ROCK activation and [Ca2+]i influx leading to MLC phosphorylation is a critical event in thrombin-induced F-actin stress fiber formation and actomyosin contraction in endothelial cells [29]. Fig. 1A shows time- and concentration-dependent activation of RhoA by thrombin treatment in hBMVECs where 0.1 unit of thrombin increased maximum activity of RhoA at 5 min after treatment. Fig. 1B shows time lapse (i) and cumulative value (ii) of [Ca2+]i influx where thrombin increased [Ca2+]i influx in a concentration dependent manner in hBMVECs. Along with the inductions of [Ca2+]i influx and RhoA activation, thrombin also induced cellular levels of phospho-MLC (Ser19) in time- and concentration-dependent manners (Fig. 1C). Accordingly, thrombin treatment induced the formation of robust long F-actin filaments (Phalloidin staining), which contained higher amount of phospho-MLC (Fig. 1D-i), so called stress fibers. Thrombin treatment also decreased trans-endothelial electrical resistance (TEER), thus indicating endothelial barrier disruption (Fig. 1D-ii). Inhibition of thrombin-induced MLC phosphorylation by pretreatment with RhoA inhibitor I (C3 transferase/C3-Tr) or [Ca2+]I chelator (BAPTA) (Fig. 1E) indicates a causal relationship between thrombin-induced [Ca2+]i influx or RhoA activation and MLC phosphorylation in hBMVECs.
Figure 1. Thrombin induces cell signaling for endothelial barrier disruption in cultured hBMVECs.
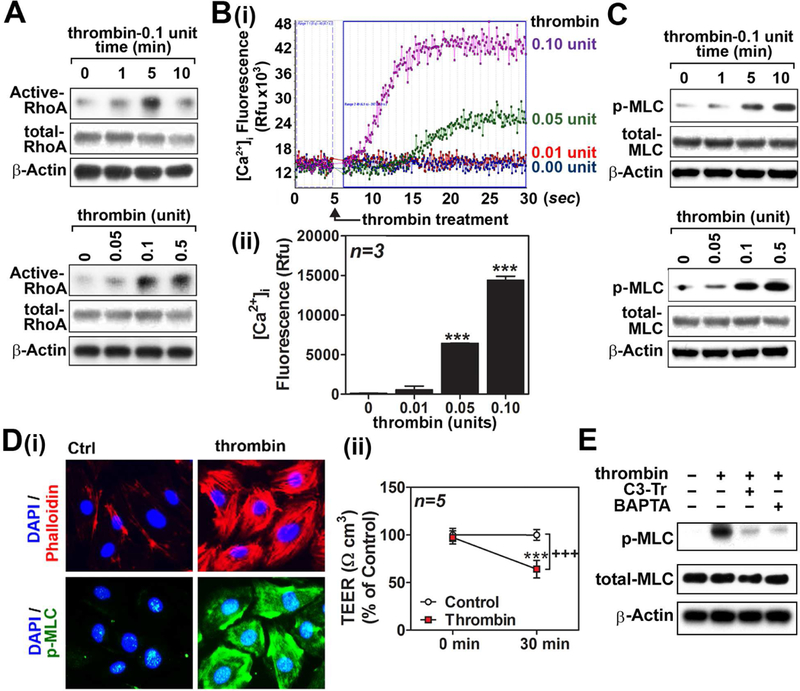
Human brain microvessel endothelial cells (hBMVECs) were treated with thrombin (0.1unit/ml) and time dependent activation of RhoA activity was analyzed (left panel). The cells were also treated with various concentrations of thrombin and a dose dependent activation of RhoA activation was analyzed at 5 min following the treatment as described in method section (A). hBMVECs were treated with various concentrations of thrombin and intracellular Ca2+ ([Ca2+]i) influx was analyzed by fluorometric assay as described in method section (B-i). Twenty five seconds following thrombin treatment, the increased [Ca2+]i influxes were represented by bar graph (B-ii). In another set of experiment, thrombin-induced time- and concentration-dependent phosphorylation of myosin light chain (Ser19) was analyzed in hBMVECs by Western analysis. β-actin was used for internal loading control for Western analysis (C). hBMVECs were treated with thrombin (0.1unit/ml for 30 min) and development of F-actin stress fiber was analyzed by immunofluorescent staining of F-actin bundles by Phalloidin (red) and phosphorylated MLC (p-MLC; green). Nuclei were stained by DAPI (blue) (D-i). For endothelial barrier study, hBMVECs cultured on transwell plates were analyzed for transendothelial electric resistance (TEER) in the absence or presence of thrombin (0.1unit/ml for 30 min) treatment (D-ii). To investigate causal relationships between RhoA activation or [Ca2+]i influx and MLC phosphorylation, hBMVECs were pretreated with RhoA inhibitor I (C3 transferase/C3-Tr; 1µg/ml) or [Ca2+]i chelator BAPTA (100µM) for 30min, followed by thrombin treatment (0.1unit/ml) for 10 min, and then cellular levels of phospho- and total-MLC levels were analyzed by Western analysis (E). The vertical bars (B-ii) and dots (D-ii) are means of individual data set (n=3) and T-bars are standard deviation. *** p ≤ 0.001 as compared to control group. All experiments were repeated at least three times and representative data are shown.
3.2. Thrombin activated eNOS causes increased protein nitration (3-nitrotyrosine) but not protein-associated S-nitrosothiols in hBMVECs
Endothelial cells predominantly express eNOS but they are also known to express nNOS under certain conditions [44]. In this study, we confirmed that cultured hBMVECs predominantly express eNOS and do not express any other NOS isoforms, such as nNOS or iNOS (Fig. 2A-). Stimulation of hBMVECs with thrombin (0.1 unit/ml) induced intracellular NO production as detected by increased DAF-FM fluorescence, a fluorescent dye for imaging nitric oxide (Fig. 2A-ii). Accordingly, thrombin induced eNOS activation by phosphorylation at Ser1177 (Fig. 2A-iii).
Figure 2. Effect of thrombin on endothelial eNOS activity and NO metabolism in hBMVECs.
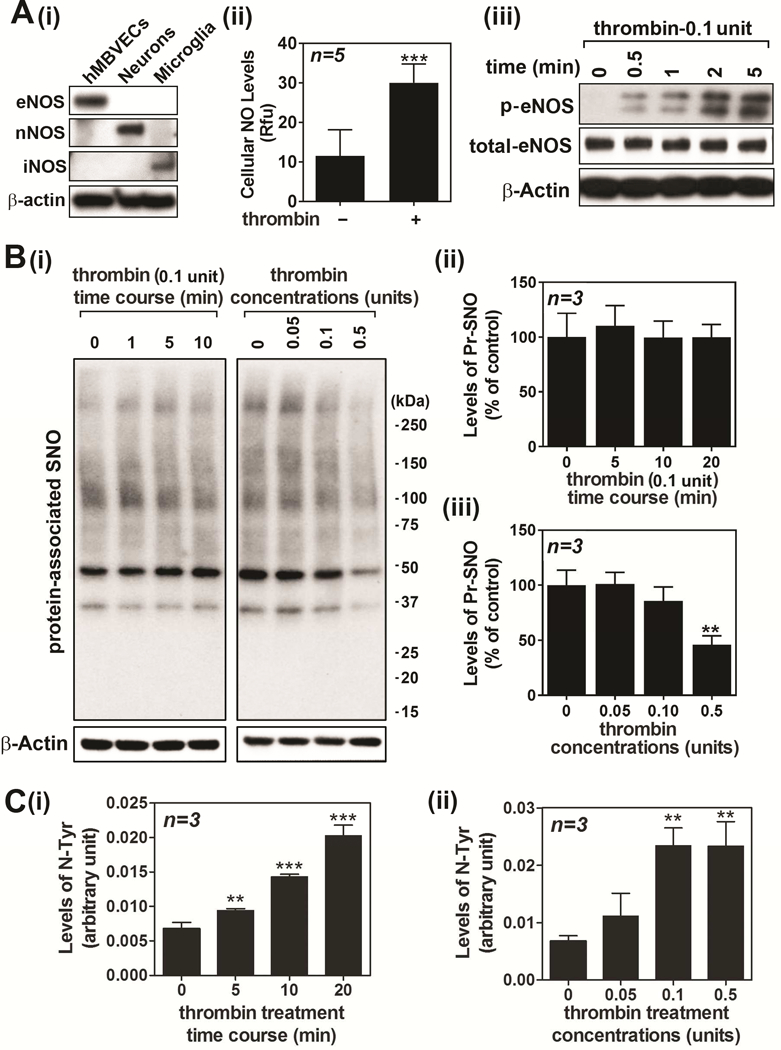
Cell lysates from cultured human brain microvessel endothelial cells (hBMVECs), neurons, and activated microglia were analyzed for expression levels of eNOS, nNOS, and iNOS (A-i). hBMVECs were treated with thrombin (0.1 unit/ml) and the cellular levels of NO was analyzed by fluorometric analysis using dye DAF-FM (A-ii). hBMVECs were treated with thrombin (0.1 unit/ml) and time course activation of eNOS was analyzed by Western analysis using antibody specific to phospho (Ser1177) eNOS (A-iii). β-actin was used for internal loading control and lysate extracted from glutamate treated cultured neurons was used for positive control for nNOS activation. hBMVEC were treated with thrombin and time and concentration dependent accumulation of protein-associated S-nitrosothiols (Pr-SNO) (B) or protein-associated 3-nitrotyrosine (N-Tyr) (C) or were analyzed by biotin switch assay or ELISA, respectively. The vertical columns represent means of individual data set and T-bars are standard deviation. ** p ≤ 0.01 and *** p ≤ 0.001 as compared to the control group. All experiments were repeated at least three times and representative data are shown.
NO is a short-lived molecule and its longer effect can be achieved by formation of secondary redox metabolites, such as GSNO and ONOO−, and subsequent modifications of protein thiols (S-nitrosylation) or tyrosines (tyrosine nitration) [10; 12]. Fig. 1 shows that 0.1 unit of thrombin is effective for activation of cell signaling for endothelial barrier disruption. However, the same concentration of thrombin treatment had no effect on the cellular levels of protein-associated S-nitrosothiol (Pr-SNO) (Figs 2B-i and ii), which is in dynamic equilibrium with cellular levels of GSNO [45]. However, we also observed that higher concentration of thrombin (0.5 unit) significantly reduced cellular levels of Pr-SNO (Figs. 2B-i and iii). On the other hand, thrombin treatment resulted in increased cellular levels of protein-associated 3-nitrotyrosine (Fig. 2C), which is formed by nitration of protein tyrosine residues by ONOO−. Therefore, these data indicate that thrombin induces eNOS activation for de novo synthesis of ONOO− instead of GSNO.
3.3. Thrombin-induced eNOS activation for ONOO− production is involved in endothelial barrier disruption in hBMVECs
Next, we investigated the role of thrombin-induced eNOS activation and ONOO− production in cell signaling pathways for MLC phosphorylation. Fig. 3A shows that inhibition of thrombin-induced eNOS activation by NOS inhibitor L-NIO (10 µM) inhibited thrombin-induced induction of MLC phosphorylation. In addition, L-NIO treatment also attenuated thrombin-induced production of 3-nitrotyrosine (Fig. 3B). Next, we assessed the role of ONOO− in thrombin-induced phosphorylation of MLC by treatment of the cells with ONOO− scavenger FeTPPS (10 µM). As shown in Figs. 3B and C, FeTTPS treatment inhibited thrombin-induced increases in 3-nitrotyrosine levels (ONOO−) and MLC phosphorylation, indicating the role of eNOS-mediated ONOO− production in thrombin-induced MLC phosphorylation. Next, we examined the effects of L-NIO and FeTPPS on thrombin-induced RhoA activation and [Ca2+]i. influx. Figs. 3D shows that treatment of hBMVECs with either L-NIO or FeTPPS decreased thrombin-induced RhoA activation. However, L-NIO and FeTTPs treatment had no effect on thrombin-induced [Ca2+]i influx (Fig. 3E). As [Ca2+]I influx is a critical step for activation of eNOS [46], [Ca2+]I chelator BAPTA, but not RhoA inhibitor I (C3-transferase), inhibited thrombin-induced eNOS activation (Fig. 3F). These data indicate that thrombin-induced [Ca2+]I influx is an early and upstream event to eNOS activation and ONOO− synthesis as well as RhoA activation and MLC phosphorylation. Figure 3G shows that DETA-NO (free NO donor) treatment reversed the L-NIO-mediated inhibition of thrombin-induced MLC phosphorylation, thus indicating a role for eNOS produced NO in regulation of MLC phosphorylation.
Figure 3. Effects of eNOS inhibitor and peroxynitrite scavenger on thrombin-induced cell signaling for endothelial barrier disruption in hBMVECs.
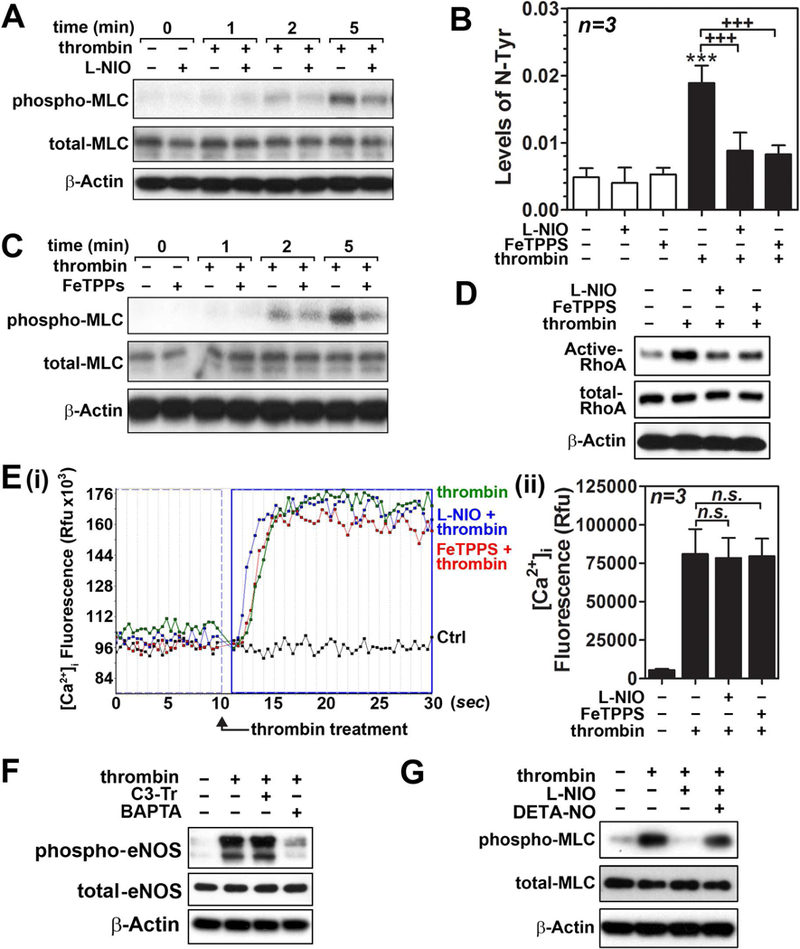
Human brain microvessel endothelial cells (hBMVECs) in the presence or absence of NOS inhibitor L-NIO (10µM; pretreated for 30min) were treated with thrombin (0.1 unit/ml for 5min) and MLC phosphorylation (Ser19) was analyzed by Western analysis with β-actin as internal loading control (A). hBMVECs were treated with thrombin (0.1 unit/ml for 20min) in the presence or absence of L-NIO (10µM; pretreated for 30min) or ONOO− scavenger FeTTPS (10µM; pretreated for 30min) and cellular levels of protein-associated 3-nitrotyrosine (a protein adduct formed by ONOO−) was analyzed by ELISA (B). hBMVECs were treated with thrombin (0.1 unit/ml for 5min) in the presence or absence of FeTPPS or L-NIO and MLC phosphorylation (C), RhoA activity (D), and intracellular Ca2+ ([Ca2+]i) influx (E) were analyzed. To investigate causal relationship between RhoA activation or [Ca2+]i influx and eNOS phosphorylation (Ser1177), hBMVECs were pretreated with RhoA inhibitor I (C3 transferase/C3-Tr; 1µg/ml) or [Ca2+]i chelator BAPTA [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; 100µM] for 30min, followed by thrombin treatment (0.1unit/ml) for 10 min, then cellular levels of phospho and total eNOS levels were analyzed by Western analysis (F). To confirm the role of eNOS in regulation of MLC phosphorylation, hBMVECs were treated with NOS inhibitor L-NIO (10µM), in the presence or absence of DETA-NO (free NO donor; 1mM), for 30min and effect of thrombin (0.1 unit/ml for 5min) on MLC phosphorylation was analyzed by Western analysis. The vertical bars are means of individual data and T-bars are standard deviation. *** p ≤ 0.001 as compared to the control group. +++ p ≤ 0.001 as compared to thrombin treated group. All experiments were repeated at least three times and representative data are shown.
3.4. Opposing roles of GSNO vs. ONOO− in thrombin-induced cell signaling for endothelial barrier disruption in hBMVECs
Next, we assessed the role of GSNO vs. ONOO− treatments on thrombin-induced cell signaling for endothelial barrier disruption. Figure 4A show that GSNO treatment of hBMVECs increased the cellular levels of protein-associated S-nitrosothiols, while SIN-1 (a donor of ONOO−) treatment increased the cellular levels of protein-associated 3-nitrotyrosine. In addition, GSNO also increased RhoA S-nitrosylation while SIN-1 increased RhoA tyrosine nitration. RhoA activity is reported to be down-regulated by S-nitrosylation and up-regulated by 3-nitrotyrosinylation [17; 18; 19]. Accordingly, we observed that GSNO treatment inhibited the thrombin-induced RhoA activation while SIN-1 treatment enhanced the thrombin-induced RhoA activation (Fig. 4B). Interestingly, thrombin-induced [Ca2+]i influx in hBMVECs was also inhibited by GSNO treatment, while enhanced by SIN-1 treatment (Figs. 4C-i and ii). Under these experimental conditions, neither GSNO nor SIN-1 induced any obvious cell death observed by MTT assay (Fig. 4C-iii). Thrombin-induced MLC phosphorylation was also attenuated by GSNO treatment but enhanced by SIN-1 treatment (Fig. 4D). On the other hand, decomposed GSNO and SIN-1 in culture media under ambient light and temperature for 48hr had no effect on thrombin-induced MLC phosphorylation, indicating roles for S-nitrosylation and tyrosine-nitration mediated mechanisms in cell signaling for MLC phosphorylation.
Figure 4. Opposing roles of GSNO vs. ONOO− in thrombin-induced cell signaling for endothelial barrier disruption in hBMVECs.
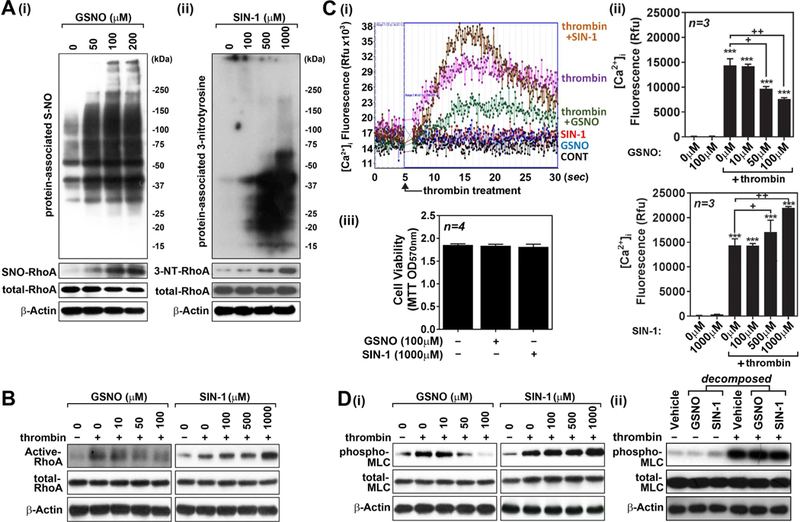
Human brain microvessel endothelial cells (hBMVECs) were treated with various concentrations of GSNO or SIN-1 (ONOO− donor), incubated for 2hr, and cellular levels of S-nitrosylated proteins and RhoA (A-i) and tyrosine-nitrated proteins and RhoA (A-ii) were analyzed as described in method section. hBMVECs were treated with thrombin (0.1 unit/ml for 5min), in the presence or absence of various concentrations GSNO or SIN-1 (pretreated for 2hr), and RhoA activity was analyzed as described in method section (B). hBMVECs were treated with thrombin (0.1 unit/ml) in the presence or absence of various concentrations GSNO or SIN-1 and intracellular Ca2+ ([Ca2+]i) influx (C-i and ii) and cell viability (MTT assay) (C-iii) were analyzed. hBMVECs were treated with thrombin (0.1 unit/ml for 5min), in the presence or absence of various concentrations GSNO or SIN-1 (D-i) or decomposed GSNO (100µM) or SIN-1 (1000µM) (D-ii), and MLC phosphorylation was analyzed by Western analysis. β-actin was used for internal loading control for Western analysis. The vertical bars are means of individual data and T-bars are standard deviation. *** p ≤ 0.001 as compared to the control group. + p ≤ 0.05 and ++ p ≤ 0.01 as compared to thrombin treated group. All experiments were repeated at least three times.
Next, we investigated the effect of GSNO vs. SIN-1 (ONOO−) on thrombin-induced F-actin stress fiber formation and endothelial barrier disruption. Figures 5A and B describe that GSNO treatment inhibited thrombin-induced development of F-actin stress fiber formation (phalloidin staining and MLC-phosphorylation) as well as thrombin-induced loss of TEER (Fig. 5C). On the other hand, SIN-1 (ONOO−) treatment enhanced the thrombin-induced development of F-actin stress fiber formation and loss of TEER. Taken together, these data document regulation of endothelial barrier by different redox-dependent NO metabolites (GSNO vs. ONOO−) in opposing signaling mechanisms.
Figure 5. Opposing roles of GSNO vs. ONOO− in thrombin-induced cell signaling for endothelial barrier disruption in hBMVECs.
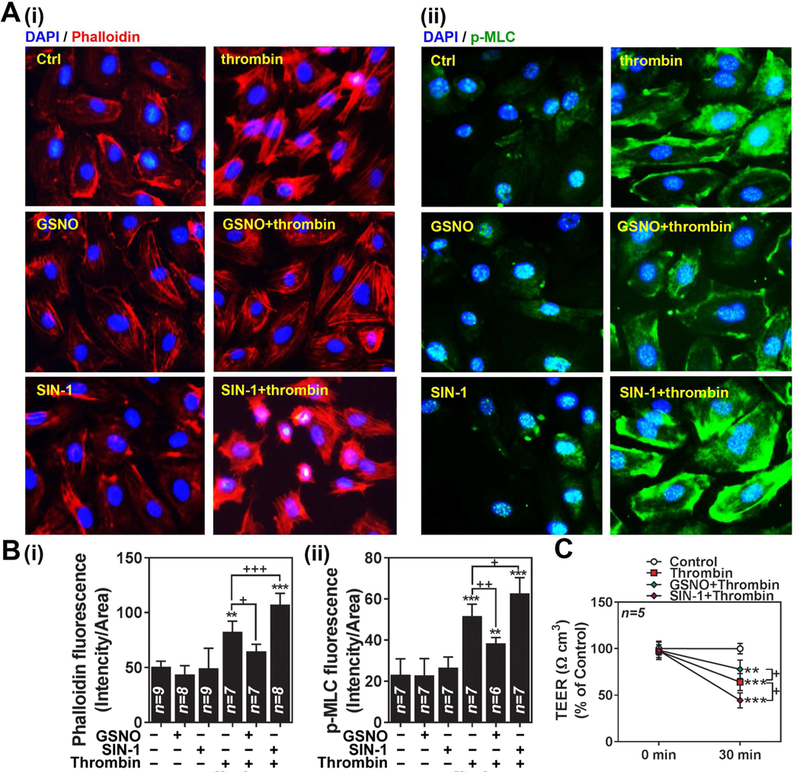
A. Human brain microvessel endothelial cells (hBMVECs) were treated with thrombin (0.1 unit/ml for 30min) in the presence or absence of GSNO (100µM; pretreated for 2hr) or SIN-1 (100µM; pretreated for 2hr) and development of F-actin stress fiber was analyzed by immunofluorescent staining of F-actin bundles by Phalloidin (red-i) and phosphorylated MLC (p-MLC; green-ii). Nuclei were stained by DAPI (blue). B. The resulting digital images were used for quantification of fluorescence and the data is represented by RFU (relative fluorescence unit). C. hBMVECs were cultured on transwell plates and transendothelial electric resistance (TEER) was analyzed. The cells were treated with thrombin (0.1 unit/ml for 5min) in the absence or presence of GSNO (100µM; pretreated for 2hr) or SIN-1 (500µM; pretreated for 2hr). The vertical bars and dotted lines are means of individual data and T-bars are standard deviation. ** p ≤ 0.01 and *** p ≤ 0.001 as compared to the control group. + p ≤ 0.05, ++ p ≤ 0.01, and +++ p ≤ 0.001 as compared to thrombin treated group. All experiments were repeated at least three times.
3.5. Roles of GSNO vs. ONOO− in regulation of endothelial barrier function in TBI model
Based on the observed opposing effects of GSNO vs. ONOO− in endothelial barrier disruption, we next investigated the roles of GSNO vs. ONOO− in regulation of vascular pathology leading to edema in rat model of TBI. TBI was induced by controlled cortical impact in adult male rats. GSNO (0.05 mg/kg/i.v./day) or FeTPPS (ONOO− scavenger; 3 mg/kg/i.v./day) was administered at right after the impact. Next day, BBB leakage and degree of edema were assessed by Evan’s blue extravasation and brain water content. Figs. 6A and B show that TBI-induced increases in Evan’s blue extravasation and degree of brain water content were reduced with GSNO as well as FeTTPS treatment, indicating the opposing roles of different redox dependent NO metabolites (GSNO vs. ONOO−) in post-traumatic BBB leakage and edema formation. It is of interest to note that GSNO treatment, in addition to FeTTPs treatment, reduced the brain levels of 3-nitrotyrosine in rat brains with TBI (Fig. 6C), indicating that GSNO-mediated mechanisms also protect cerebrovascular nitrosative stress under TBI conditions.
Figure 6. Roles of GSNO and FeTPPS on BBB leakage, edema and the expression of 3-NT in TBI rat model.
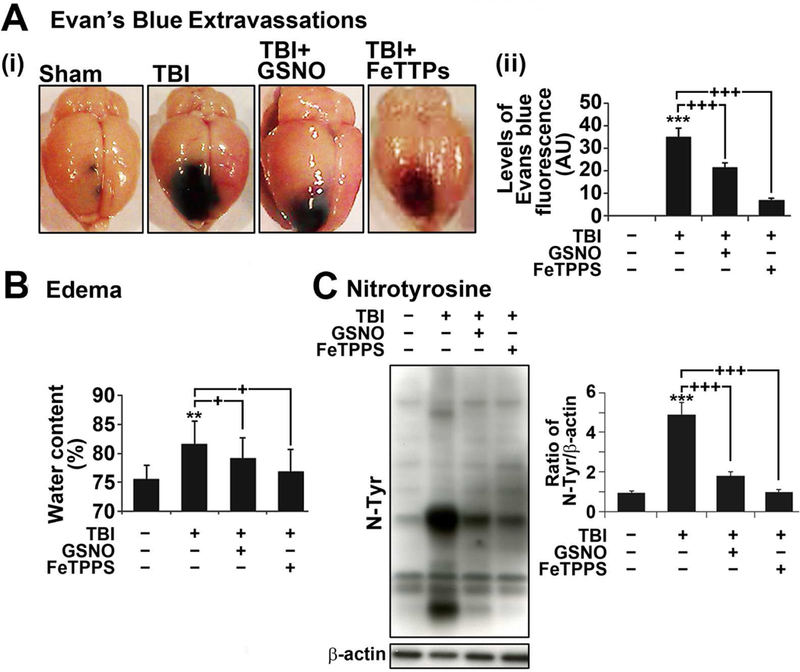
A. Photographs showing Evan’s blue (EB) extravasations in brain starting at 4 hr after TBI. Animals were sacrificed at 24 hr, the brain was photographed (i) and the intensity of EB (ii) was determined by spectrofluorometric estimation. EB extravasations were not observed in sham brain. B. Edema (tissue water content) was measured at 24 hr after TBI. C. The levels of nitrotyrosine (N-Tyr) as an index of ONOO− were also measured at 24 hr in the traumatic penumbra region using Western and its quantitation by densitometry. Data are expressed as mean ± standard deviation from five different experiments for Evan’s blue and edema each and three different experiments for western blot. * p ≤ 0.05, *** p ≤ 0.001 vs. Sham and + p ≤ 0.05, ++ p ≤ 0.01, and +++ p ≤ 0.001 vs. TBI.
3.6. Roles of GSNO vs. ONOO− in regulation of endothelial barrier function in EAE model:
MS is induced by peripheral activation of myelin specific autoreactive lymphocytes and their CNS infiltration across the leaky BBB leading to encephalitogenic inflammatory disease [31]. To investigate the role of GSNO vs ONOO− in endothelial barrier disruption, EAE mice were treated with daily dose of GSNO (1 mg/kg/i.p./day) or FeTPPS (30 mg/kg/i.p./day) at the onset of disease with clinical score between 1 and 2 (day 13 post immunization) (Fig. 7A). Similar to our previous study [47], GSNO treatment provided great efficacy against clinical disease of EAE (Figs. 7A-i and -ii). FeTTPS treatment also provided significant efficacy but to a lower degree than GSNO treatment (Figs. 7A-i and -ii).
Figure 7. Roles of GSNO and FeTPPS on clinical disease, expression of 3-nitrotyrosine, BBB leakage, and spinal cord demyelination in mouse EAE model.
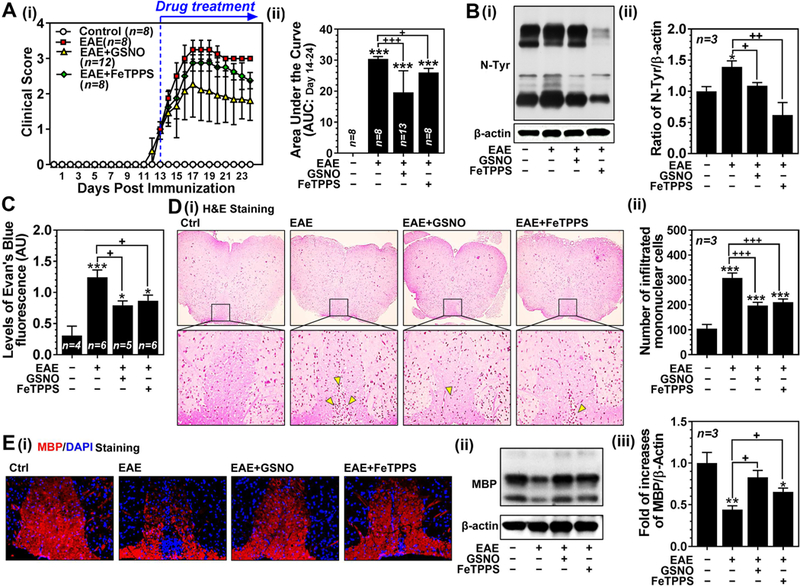
A. Clinical score of control C57BL/6 mice (Ctrl: n=8), C57BL/6 mice immunized with MOG35–55 peptide (EAE: n=8), EAE mice treated with 1mg/kg/day of GSNO (EAE+GSNO: n=12) or 30 mg/kg/day of FeTPPS (EAE+FeTPPS: n=8) was determined daily as described in Materials and Methods (A-i). All drugs were administered starting at the day of disease onset (day 13 post-immunization) via intraperitoneal routes. The area under the curve (AUC) between post immunization day 14 and 24 of the overall disease severity was calculated and represented as bar graph (A-ii). B. At 24 day post-immunization, the mice (n=3) were sacrificed and the levels of 3-nitrotyrosine (N-Tyr), as an index of ONOO−, were measured by Western (B-i) and densitometry analysis (B-ii). C. In addition, another set of mice were injected with Evans blue for analysis of BBB leakage. D. Spinal cord infiltration of mononuclear cells was analyzed by H&E staining of paraffin-embedded spinal cord section (D-i). The number of mononuclear cells (dark-brown nuclei aggregates indicated by yellow triangles) was counted manually and represented by bar graph (D-ii). E. The spinal cord sections and tissue lysates were also subjected to immunofluorescent staining (E-i) and Western analysis for MBP (E-ii and -iii) for degree of demyelination. Data are expressed as mean ± standard deviation. *p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 vs. control and + p ≤ 0.05, ++ p ≤ 0.01, and +++ p ≤ 0.001 vs. EAE.
Next, degree of tissue levels of ONOO− (protein nitrotyrosine levels in Fig. 7B), BBB leakage (Evan’s blue extravasation assay in Fig. 7C), peripheral mononulcear cell infiltration (H&E staining in Figs. 7D-i and ii), and spinal cord demyelination (myelin basic protein/MBP staining in Fig. 7E-i and Western analysis in Figs. 7E-ii and iii) were analyzed. Consistent with effects on clinical disease, GSNO and FeTPPS treatments also significantly decreased the EAE-induced nitrotyrosine levels in spinal cords as well as extravasation of Evan’s blue dye and peripheral mononuclear cells into the CNS. Accordingly, both treatments also protected myelin in the spinal cord from EAE disease.
Taken together, in vitro cell culture studies and in vivo studies with animal models of TBI and EAE document that redox-dependent metabolites of eNOS produced NO (GSNO vs. ONOO−) play critical roles in cell signaling pathways for endothelial barrier integrity (e.g. RhoA/ROCK, [Ca2+]i influx, and MLC phosphorylation) and thus BBB disruption under traumatic and inflammatory neurological disease conditions.
4. Discussion
BBB disruption, a characteristic feature of numerous neurological disease conditions [1], causes brain edema as well as greater influx of blood-borne cells and substances into brain parenchyma, thus exacerbating neuroinflammation and brain injuries [48]. Recent studies report that early events of BBB permeability (e.g. cell signaling pathways for F-actin stress fiber formation and junctional protein redistribution) may be partially reversible [3; 4; 5]. Therefore, early events of BBB permeability have been the potential targets for therapeutic interventions of various neurological diseases [5]. Here, we report that early events of BBB permeability, especially RhoA mediated MLC phosphorylation and endothelial F-actin stress fiber formation, is regulated by eNOS-derived NO metabolites (ONOO− and GSNO) in opposing manners, thus highlighting the potential therapeutic importance of redox dependent NO metabolites in BBB protection. These conclusions are supported by in vitro mechanistic studies using thrombin-induced endothelial hyper-permeability model and studies using animal models of traumatic and inflammatory brain injuries (TBI and MS).
Thrombin-induced endothelial hyper-permeability has been a useful model to investigate cellular mechanisms for MLC phosphorylation mediated barrier dysfunction induced by RhoA activation and [Ca2+]i influx [49; 50]. Thrombin induces endothelial cell signaling pathways for hyper-permeability via PAR1 activation and subsequent induction of [Ca2+]i influx and activation of RhoA/ROCK [29]. As a result, the activated MLC kinase and inactivated MLC phosphatase increase phosphorylation of MLC and induce actomyosin stress fiber formation [29] and thus alteration in endothelial cell shape, adhesion, and intercellular permeability [51]. As expected, thrombin induced MLC phosphorylation-mediated endothelial F-actin stress fiber formation and endothelial barrier disruption via inducing RhoA activity and [Ca2+]i influx in in vitro hBMVEC culture model (Fig. 1). Thrombin induced eNOS activation resulted in synthesis of NO as well as ONOO− (protein-associated 3-nitrotyrosine) (Figs. 2A and C), but not GSNO (Fig. 2B). Conversion of NO to ONOO− or GSNO requires O2¯ or GSH, respectively, and therefore, cellular redox potential is critical for metabolic fate of NO. Thrombin was reported to induce cellular oxidative stress by activating NADPH oxidase [52]. Therefore, thrombin may induce eNOS activation and shift eNOS-produced NO metabolism toward ONOO− synthesis via inducing oxidative stress, while limiting GSNO de novo synthesis.
In hBMVECs, thrombin induced eNOS activation and ONOO− synthesis correlated with the increased RhoA activation (Figs. 1 and 2) whereas pretreatment of hBMVECs with eNOS inhibitor or ONOO− scavenger inhibited the thrombin-induced RhoA activation (Fig. 3), indicating a role for eNOS-mediated ONOO− synthesis in thrombin-induced RhoA activation. Previous studies reported that tyrosine nitration of RhoA at Tyr34 enhances RhoA activity and accelerates endothelial barrier disruption [18]. On the other hand, S-nitrosylation of RhoA at Cys16, Cys20, and Cys159 is reported to inhibit its activity in endothelial cells [19]. Accordingly, we observed that exogenous GSNO treatment increased the S-nitrosylation of RhoA and inhibited its activation by thrombin (Figs. 4A and B). On the other hand, treatment of hBMVECs with SIN-1 (ONOO− donor) increased tyrosine nitration of RhoA and enhanced its activation by thrombin (Figs. 4A and B). These data document opposing roles of ONOO− (enhancement) vs. GSNO (inhibition) in thrombin-induced RhoA activation, thus highlighting the importance of endothelial redox-dependent NO metabolism in RhoA and MLC phosphorylation-dependent early event of endothelial barrier disruption.
ONOO− and GSNO also regulated thrombin-induced [Ca2+]i influx but in opposing manners. In hBMVECs, thrombin-induced [Ca2+]i influx was enhanced by SIN-1 (ONOO− donor) pretreatment while inhibited by GSNO pretreatment (Fig. 4C). Though these data suggest possible roles of NO metabolites (ONOO− and GSNO) as feedback regulators of [Ca2+]i influx for eNOS activation, we observed that thrombin-induced [Ca2+]i influx was not affected by eNOS inhibition by L-NIO as well as ONOO− scavenge by FeTTPS (Fig. 3E). These data indicate that NO and ONOO− produced by eNOS may not serve as feedback regulators for thrombin-induced [Ca2+]I influx. The timing of [Ca2+]I influx (earlier event) and eNOS activation (later event) following the thrombin treatment may account for these differences [53]. In addition, we observed that thrombin-induced NO synthesis did not increase GSNO de novo synthesis (Fig. 2B), thus indicating lack of GSNO mediated feedback regulation of [Ca2+]I influx. At present, ir is not fully understood how ONOO− and GSNO regulate thrombin-induced [Ca2+]i. However, the previous studies with smooth muscle cells suggested possible roles of L-type voltage-gated Ca2+ channels in ONOO− induced [Ca2+]i influx [54] and inositol-1,4,5-trisphosphate (IP3) in GSNO induced inhibition of [Ca2+]i influx [55]. These data suggest potential role of endothelial preconditioning by GSNO or ONOO− in thrombin-induced [Ca2+]i influx and early events of BBB disruption. [Ca2+]I influx in endothelial cells is known to activate eNOS as well as other cell signaling including Ca2+/calmodulin dependent protein kinases, protein kinase C, and NADPH oxidase [56]. Therefore, participation of other cellular mechanisms in endothelial pathobiology cannot be excluded.
Brain edema, especially vasogenic edema caused by BBB disruption, is a significant challenge in clinical management of TBI during the acute period of diseases. If edema reaches a critical point, it leads to severe morbidity or death if left untreated. Current therapies for management of post-traumatic edema include osmotherapy, diuretics, corticosteroids, barbiturates, propofol, and/or hyperventilation. However, endothelial mechanism underlying the vasogenic brain edema is still elusive and thus no specific mechanism-based-therapy is currently available. Our laboratory has studied the efficacy of GSNO treatment during the acute disease of stroke and TBI to attenuate brain endothelial barrier disruption, abnormal BBB permeability, edema formation, and vascular inflammation in rat models [37; 57]. Later on, we also reported that GSNO treatment attenuates neurodegeneration and accelerates neovascularization and neuro-repair and thus improved functional outcome in TBI animals [38; 58; 59]. In the present study, we have described the opposing roles of redox-dependent NO metabolites (ONOO− vs. GSNO) in regulation of RhoA activation and [Ca2+]i influx and thus MLC phosphorylation leading to endothelial stress fiber formation and barrier disruption in hBMVECs. We previously reported increased oxidative stress in the brains of TBI animal models, observed as decreased ratio of GSH/GSSG and increased levels of 4-hydroxynonenal [59]. Accordingly, we also observed increased levels of ONOO− in the brains of TBI animals (Fig. 6C). Although we have not measured GSNO levels in the TBI brain tissue, we expect decreased GSNO synthesis due to increased ONOO− synthesis as a result of oxidative stress. Consistent with in vitro studies, we observed in animal model of TBI that treatment with GSNO or ONOO− scavenger (FeTPPS) shifts balance of NO metabolites (GSNO vs. ONOO−) toward GSNO, thus ameliorated TBI-induced BBB leakage and edema formation (Fig. 6).
In MS, CNS infiltration of myelin specific autoreactive lymphocytes across the disrupted BBB is one of the critical pathological events leading to inflammatory demyelination [31]. Brain imaging studies have shown that patients with relapsing-remitting MS (RRMS), the most common type of MS (> 80 %), have generally increase in BBB permeability [60; 61]. Accordingly, EAE mice had increased nitrosative stress and BBB permeability in the spinal cords (Figs. 7B and C), and treatment of these mice with GSNO or ONOO− scavenger (FeTPPS) attenuated BBB leakage as well as CNS infiltration of mononuclear cells (Figs. 7C and D). Consequently, GSNO or FeTPPS treatment ameliorated inflammatory demyelination as well as clinical EAE disease (Figs. 7A AND E). These data document the roles of GSNO vs. ONOO− mediated mechanisms for maintenance of BBB integrity during the course of traumatic and inflammatory neurological diseases.
Recent studies underscore the pathological role of thrombin in BBB disruption and CNS inflammation [62; 63; 64; 65; 66]. In this study, we observed that GSNO and FeTPPS treatments inhibited endothelial cell signaling for early event of BBB disruption (MLC phosphorylation) as well as BBB disruption under conditions of TBI and EAE diseases. Pathoanatomical consequences of contusion TBI include mechanical stress associated blood vessel injuries as well as hemorrhages [62]. Following vascular damage and hemorrhage, thrombin controls blood loss [67], but its excessive production is also known to cause BBB disruption leading to edema formation [21]. In MS, proteomic analysis of chronic active lesions identified several dysregulated coagulation factors, highlighting a potential link between the coagulation cascade and MS pathology [68]. Moreover, observations of high levels of activated platelets, which are activated by thrombin, in MS and EAE lesions/bloods and amelioration of EAE disease by platelet depletion [69; 70; 71], and spatial correlation of increased thrombin activity to local BBB disruption, CNS inflammation, and neurodegeneration [63; 72] indicate potential role of thrombin in BBB disruption and subsequent CNS disease of MS (EAE). Overall, these studies document pathological role of thrombin-induced BBB disruption in CNS disease of TBI and MS, thus suggesting the potential efficacies of GSNO and FeTTPS in thrombin associated pathologies under these disease conditions.
In summary, the present study demonstrates the role of redox-based NO metabolites (ONOO– vs. GSNO) in endothelial barrier disruption leading to vasogenic edema formation and peripheral immune cell infiltration under traumatic and inflammatory neurological disease conditions. ONOO− accelerates endothelial barrier disruption via enhancing cell signaling for MLC phosphorylation (e.g. RhoA activation and [Ca2+]i influx) and endothelial stress fiber formation whereas GSNO inhibits endothelial barrier disruption via inhibiting these cell-signaling mechanisms, thus documenting that ONOO− and GSNO levels mechanistically antagonize each other in endothelial barrier disruption. This study also shows that BBB disruption in animal model of TBI and EAE are inhibited by ONOO− scavenger (FeTPPS) or GSNO treatment and thus identifying redox-dependent NO metabolites (ONOO− vs. GSNO) as a potential therapeutic targets for neurovascular integrity in neurological disorders.
Highlights.
eNOS/NO controls brain endothelial actin structure and thus barrier integrity.
Different redox metabolites of NO differently regulate actin stress fiber formation.
ONOO− increases RhoA and Ca2+ dependent actin stress fiber formation.
On contrary, GSNO inhibits RhoA and Ca2+ dependent actin stress fiber formation.
These NO metabolites are potential targets for vascular diseases of CNS.
Acknowledgements
This work was supported in part by U.S. Department of Veterans Affairs (BX002829 and BX003401) and National Institutes of Health (NS037766).
List of Abbreviations:
- [Ca2+]i
intracellular calcium ion
- BBB
blood–brain barrier
- CCI
Controlled cortical impact
- CFA
complete Freund’s adjuvant
- DAPI
4’,6-Diamidino-2-Phenylindole, Dihydrochloride
- EAE
experimental autoimmune encephalomyelitis
- EB
Evans blue
- eNOS
endothelial nitric oxide synthase
- FeTTPS
5,10,15,20-Tetrakis(4-sulfonatophenyl)porphyrinato Iron (III)
- Cl
Fig.,figure
- GSH
glutathione
- GSNO
S-nitrosoglutathione
- H&E
hematoxylin and eosin
- hBMVEC
human brain microvessel endothelial cell
- IP3
inositol-1,4,5-trisphosphate
- L-NIO
N5-(1-Iminoethyl)-L-ornithine dihydrochloride
- MBP
myelin basic protein
- MLC
myosin lingh chain
- MLCK
myosin lingh chain kinase
- MLCP
myosin lingh chain phosphatase
- MS
multiple sclerosis
- NO
nitric oxide
- ONOO−
peroxynitrite
- PAR
protease-activated receptor
- Pr-SNO
protein-associated S-nitrosothiol
- PTX
pertussis toxin
- SEM
standard error mean
- SIN-1
3-morpholinosydnonimine chloride
- TBI
traumatic brain injury
- TEER
trans-endothelial electrical resistance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- [1].Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR, Engaging neuroscience to advance translational research in brain barrier biology, Nat Rev Neurosci 12 (2011) 169–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Obermeier B, Daneman R, Ransohoff RM, Development, maintenance and disruption of the blood-brain barrier, Nat Med 19 (2013) 1584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Neumann-Haefelin T, Kastrup A, de Crespigny A, Yenari MA, Ringer T, Sun GH, Moseley ME, Serial MRI after transient focal cerebral ischemia in rats: dynamics of tissue injury, blood-brain barrier damage, and edema formation, Stroke 31 (2000) 1965–72; discussion 1972–3. [DOI] [PubMed] [Google Scholar]
- [4].Olah L, Wecker S, Hoehn M, Secondary deterioration of apparent diffusion coefficient after 1-hour transient focal cerebral ischemia in rats, J Cereb Blood Flow Metab 20 (2000) 1474–82. [DOI] [PubMed] [Google Scholar]
- [5].Kaur J, Tuor UI, Zhao Z, Petersen J, Jin AY, Barber PA, Quantified T1 as an adjunct to apparent diffusion coefficient for early infarct detection: a high-field magnetic resonance study in a rat stroke model, Int J Stroke 4 (2009) 159–68. [DOI] [PubMed] [Google Scholar]
- [6].Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, Xu N, Stetler RA, Zhang F, Liu X, Leak RK, Keep RF, Ji X, Chen J, Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury, Nat Commun 7 (2016) 10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lahteenvuo J, Rosenzweig A, Effects of aging on angiogenesis, Circ Res 110 (2012) 1252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kraehling JR, Sessa WC, Contemporary Approaches to Modulating the Nitric Oxide-cGMP Pathway in Cardiovascular Disease, Circ Res 120 (2017) 1174–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Arora D, Jain P, Singh N, Kaur H, Bhatla SC, Mechanisms of nitric oxide crosstalk with reactive oxygen species scavenging enzymes during abiotic stress tolerance in plants, Free Radic Res 50 (2016) 291–303. [DOI] [PubMed] [Google Scholar]
- [10].Pacher P, Beckman JS, Liaudet L, Nitric oxide and peroxynitrite in health and disease, Physiol Rev 87 (2007) 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].He W, Frost MC, Direct measurement of actual levels of nitric oxide (NO) in cell culture conditions using soluble NO donors, Redox Biol 9 (2016) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gaston BM, Carver J, Doctor A, Palmer LA, S-nitrosylation signaling in cell biology, Mol Interv 3 (2003) 253–63. [DOI] [PubMed] [Google Scholar]
- [13].Haldar SM, Stamler JS, S-nitrosylation: integrator of cardiovascular performance and oxygen delivery, J Clin Invest 123 (2013) 101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Radomski MW, Rees DD, Dutra A, Moncada S, S-nitroso-glutathione inhibits platelet activation in vitro and in vivo, Br J Pharmacol 107 (1992) 745–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Won JS, Kim J, Annamalai B, Shunmugavel A, Singh I, Singh AK, Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion, J Alzheimers Dis 34 (2013) 621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Prasad R, Giri S, Nath N, Singh I, Singh AK, GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions, Glia 55 (2007) 65–77. [DOI] [PubMed] [Google Scholar]
- [17].Di Lorenzo A, Lin MI, Murata T, Landskroner-Eiger S, Schleicher M, Kothiya M, Iwakiri Y, Yu J, Huang PL, Sessa WC, eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases, J Cell Sci 126 (2013) 5541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rafikov R, Dimitropoulou C, Aggarwal S, Kangath A, Gross C, Pardo D, Sharma S, Jezierska-Drutel A, Patel V, Snead C, Lucas R, Verin A, Fulton D, Catravas JD, Black SM, Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA, J Biol Chem 289 (2014) 4710–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen F, Wang Y, Rafikov R, Haigh S, Zhi WB, Kumar S, Doulias PT, Rafikova O, Pillich H, Chakraborty T, Lucas R, Verin AD, Catravas JD, She JX, Black SM, Fulton DJ, RhoA S-nitrosylation as a regulatory mechanism influencing endothelial barrier function in response to G+-bacterial toxins, Biochem Pharmacol 127 (2017) 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Crawley JT, Zanardelli S, Chion CK, Lane DA, The central role of thrombin in hemostasis, J Thromb Haemost 5 Suppl 1 (2007) 95–101. [DOI] [PubMed] [Google Scholar]
- [21].Popovic M, Smiljanic K, Dobutovic B, Syrovets T, Simmet T, Isenovic ER, Thrombin and vascular inflammation, Mol Cell Biochem 359 (2012) 301–13. [DOI] [PubMed] [Google Scholar]
- [22].Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton JW 2nd, Malik AB, Thrombin-induced increase in albumin permeability across the endothelium, J Cell Physiol 128 (1986) 96–104. [DOI] [PubMed] [Google Scholar]
- [23].Nishino A, Suzuki M, Ohtani H, Motohashi O, Umezawa K, Nagura H, Yoshimoto T, Thrombin may contribute to the pathophysiology of central nervous system injury, J Neurotrauma 10 (1993) 167–79. [DOI] [PubMed] [Google Scholar]
- [24].O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF, Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1, J Biol Chem 275 (2000) 13502–9. [DOI] [PubMed] [Google Scholar]
- [25].Gerszten RE, Chen J, Ishii M, Ishii K, Wang L, Nanevicz T, Turck CW, Vu TK, Coughlin SR, Specificity of the thrombin receptor for agonist peptide is defined by its extracellular surface, Nature 368 (1994) 648–51. [DOI] [PubMed] [Google Scholar]
- [26].Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R, Proteinase-activated receptors, Pharmacol Rev 53 (2001) 245–82. [PubMed] [Google Scholar]
- [27].Buhl AM, Johnson NL, Dhanasekaran N, Johnson GL, G alpha 12 and G alpha 13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly, J Biol Chem 270 (1995) 24631–4. [DOI] [PubMed] [Google Scholar]
- [28].Majumdar M, Seasholtz TM, Buckmaster C, Toksoz D, Brown JH, A rho exchange factor mediates thrombin and Galpha(12)-induced cytoskeletal responses, J Biol Chem 274 (1999) 26815–21. [DOI] [PubMed] [Google Scholar]
- [29].van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW, Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases, Circ Res 87 (2000) 335–40. [DOI] [PubMed] [Google Scholar]
- [30].Price L, Wilson C, Grant G, Blood-Brain Barrier Pathophysiology following Traumatic Brain Injury. in: Laskowitz D, Grant G, (Eds.), Translational Research in Traumatic Brain Injury, Boca Raton (FL), 2016. [PubMed] [Google Scholar]
- [31].Compston A, Coles A, Multiple sclerosis, Lancet 359 (2002) 1221–31. [DOI] [PubMed] [Google Scholar]
- [32].Ortiz GG, Pacheco-Moises FP, Macias-Islas MA, Flores-Alvarado LJ, Mireles-Ramirez MA, Gonzalez-Renovato ED, Hernandez-Navarro VE, Sanchez-Lopez AL, Alatorre-Jimenez MA, Role of the blood-brain barrier in multiple sclerosis, Arch Med Res 45 (2014) 687–97. [DOI] [PubMed] [Google Scholar]
- [33].Kinoshita K, Traumatic brain injury: pathophysiology for neurocritical care, J Intensive Care 4 (2016) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gordge MP, Hothersall JS, Noronha-Dutra AA, Evidence for a cyclic GMP-independent mechanism in the anti-platelet action of S-nitrosoglutathione, Br J Pharmacol 124 (1998) 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li B, Zhao WD, Tan ZM, Fang WG, Zhu L, Chen YH, Involvement of Rho/ROCK signalling in small cell lung cancer migration through human brain microvascular endothelial cells, FEBS Lett 580 (2006) 4252–60. [DOI] [PubMed] [Google Scholar]
- [36].Kim J, Won JS, Singh AK, Sharma AK, Singh I, STAT3 regulation by S-nitrosylation: implication for inflammatory disease, Antioxid Redox Signal 20 (2014) 2514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Khan M, Im YB, Shunmugavel A, Gilg AG, Dhindsa RK, Singh AK, Singh I, Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact, J Neuroinflammation 6 (2009) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Khan M, Dhammu TS, Matsuda F, Annamalai B, Dhindsa TS, Singh I, Singh AK, Targeting the nNOS/peroxynitrite/calpain system to confer neuroprotection and aid functional recovery in a mouse model of TBI, Brain Res 1630 (2016) 159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kline AE, Hoffman AN, Cheng JP, Zafonte RD, Massucci JL, Chronic administration of antipsychotics impede behavioral recovery after experimental traumatic brain injury, Neurosci Lett 448 (2008) 263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kline AE, Wagner AK, Westergom BP, Malena RR, Zafonte RD, Olsen AS, Sozda CN, Luthra P, Panda M, Cheng JP, Aslam HA, Acute treatment with the 5-HT(1A) receptor agonist 8-OH-DPAT and chronic environmental enrichment confer neurobehavioral benefit after experimental brain trauma, Behav Brain Res 177 (2007) 186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hoda N, Singh I, Singh AK, Khan M, Reduction of lipoxidative load by secretory phospholipase A2 inhibition protects against neurovascular injury following experimental stroke in rat, J Neuroinflammation 6 (2009) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S, Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis, J Immunol 182 (2009) 8005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nath N, Giri S, Prasad R, Singh AK, Singh I, Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy, J Immunol 172 (2004) 1273–86. [DOI] [PubMed] [Google Scholar]
- [44].Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, Marsden PA, Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels, Arterioscler Thromb Vasc Biol 17 (1997) 2479–88. [DOI] [PubMed] [Google Scholar]
- [45].Broniowska KA, Diers AR, Hogg N, S-nitrosoglutathione, Biochim Biophys Acta 1830 (2013) 3173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fleming I, Busse R, Signal transduction of eNOS activation, Cardiovascular Research 43 (1999) 532–541. [DOI] [PubMed] [Google Scholar]
- [47].Nath N, Morinaga O, Singh I, S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis, J Neuroimmune Pharmacol 5 (2010) 240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nishikawa H, Suzuki H, Implications of periostin in the development of subarachnoid hemorrhage-induced brain injuries, Neural Regen Res 12 (2017) 1982–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Satpathy M, Gallagher P, Lizotte-Waniewski M, Srinivas SP, Thrombin-induced phosphorylation of the regulatory light chain of myosin II in cultured bovine corneal endothelial cells, Exp Eye Res 79 (2004) 477–86. [DOI] [PubMed] [Google Scholar]
- [50].van Hinsbergh VW, van Nieuw Amerongen GP, Intracellular signalling involved in modulating human endothelial barrier function, J Anat 200 (2002) 549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bogatcheva NV, Garcia JG, Verin AD, Molecular mechanisms of thrombin-induced endothelial cell permeability, Biochemistry (Mosc) 67 (2002) 75–84. [DOI] [PubMed] [Google Scholar]
- [52].Holland JA, Meyer JW, Chang MM, O’Donnell RW, Johnson DK, Ziegler LM, Thrombin stimulated reactive oxygen species production in cultured human endothelial cells, Endothelium 6 (1998) 113–21. [DOI] [PubMed] [Google Scholar]
- [53].Touyz RM, Regulation of endothelial nitric oxide synthase by thrombin, Hypertension 49 (2007) 429–31. [DOI] [PubMed] [Google Scholar]
- [54].Pan BX, Zhao GL, Huang XL, Zhao KS, Mobilization of intracellular calcium by peroxynitrite in arteriolar smooth muscle cells from rats, Redox Rep 9 (2004) 49–55. [DOI] [PubMed] [Google Scholar]
- [55].Nalli AD, Kumar DP, Al-Shboul O, Mahavadi S, Kuemmerle JF, Grider JR, Murthy KS, Regulation of Gbetagammai-dependent PLC-beta3 activity in smooth muscle: inhibitory phosphorylation of PLC-beta3 by PKA and PKG and stimulatory phosphorylation of Galphai-GTPase-activating protein RGS2 by PKG, Cell Biochem Biophys 70 (2014) 867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].De Bock M, Wang N, Decrock E, Bol M, Gadicherla AK, Culot M, Cecchelli R, Bultynck G, Leybaert L, Endothelial calcium dynamics, connexin channels and blood-brain barrier function, Prog Neurobiol 108 (2013) 1–20. [DOI] [PubMed] [Google Scholar]
- [57].Khan M, Dhammu TS, Sakakima H, Shunmugavel A, Gilg AG, Singh AK, Singh I, The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke, J Neurochem 123 Suppl 2 (2012) 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Khan M, Dhammu TS, Baarine M, Kim J, Paintlia MK, Singh I, Singh AK, GSNO promotes functional recovery in experimental TBI by stabilizing HIF-1alpha, Behav Brain Res (2016). [DOI] [PMC free article] [PubMed]
- [59].Khan M, Sakakima H, Dhammu TS, Shunmugavel A, Im YB, Gilg AG, Singh AK, Singh I, S-nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats, J Neuroinflammation 8 (2011) 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Stone LA, Smith ME, Albert PS, Bash CN, Maloni H, Frank JA, McFarland HF, Blood-brain barrier disruption on contrast-enhanced MRI in patients with mild relapsing-remitting multiple sclerosis: relationship to course, gender, and age, Neurology 45 (1995) 1122–6. [DOI] [PubMed] [Google Scholar]
- [61].Cramer SP, Simonsen H, Frederiksen JL, Rostrup E, Larsson HB, Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI, Neuroimage Clin 4 (2014) 182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chodobski A, Zink BJ, Szmydynger-Chodobska J, Blood-brain barrier pathophysiology in traumatic brain injury, Transl Stroke Res 2 (2011) 492–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Davalos D, Baeten KM, Whitney MA, Mullins ES, Friedman B, Olson ES, Ryu JK, Smirnoff DS, Petersen MA, Bedard C, Degen JL, Tsien RY, Akassoglou K, Early detection of thrombin activity in neuroinflammatory disease, Ann Neurol 75 (2014) 303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Grammas P, Martinez JM, Targeting thrombin: an inflammatory neurotoxin in Alzheimer’s disease, J Alzheimers Dis 42 Suppl 4 (2014) S537–44. [DOI] [PubMed] [Google Scholar]
- [65].Cannon JR, Hua Y, Richardson RJ, Xi G, Keep RF, Schallert T, The effect of thrombin on a 6-hydroxydopamine model of Parkinson’s disease depends on timing, Behav Brain Res 183 (2007) 161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Chen B, Cheng Q, Yang K, Lyden PD, Thrombin mediates severe neurovascular injury during ischemia, Stroke 41 (2010) 2348–52. [DOI] [PubMed] [Google Scholar]
- [67].Xi G, Reiser G, Keep RF, The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective?, J Neurochem 84 (2003) 3–9. [DOI] [PubMed] [Google Scholar]
- [68].Shapiro PE, “Cello scrotum” questioned, J Am Acad Dermatol 24 (1991) 665. [DOI] [PubMed] [Google Scholar]
- [69].Brass LF, Thrombin and platelet activation, Chest 124 (2003) 18S–25S. [DOI] [PubMed] [Google Scholar]
- [70].Langer HF, Choi EY, Zhou H, Schleicher R, Chung KJ, Tang Z, Gobel K, Bdeir K, Chatzigeorgiou A, Wong C, Bhatia S, Kruhlak MJ, Rose JW, Burns JB, Hill KE, Qu H, Zhang Y, Lehrmann E, Becker KG, Wang Y, Simon DI, Nieswandt B, Lambris JD, Li X, Meuth SG, Kubes P, Chavakis T, Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis, Circ Res 110 (2012) 1202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sheremata WA, Jy W, Horstman LL, Ahn YS, Alexander JS, Minagar A, Evidence of platelet activation in multiple sclerosis, J Neuroinflammation 5 (2008) 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Stolz L, Derouiche A, Devraj K, Weber F, Brunkhorst R, Foerch C, Anticoagulation with warfarin and rivaroxaban ameliorates experimental autoimmune encephalomyelitis, J Neuroinflammation 14 (2017) 152. [DOI] [PMC free article] [PubMed] [Google Scholar]