

ABSTRACT
Bacterial pathogens of the gastrointestinal tract alter their expression profile upon ingestion by the host and activate a variety of factors enhancing colonization and virulence. However, gene silencing during infection might be as important as gene activation to achieve full colonization fitness. Thus, we developed and successfully applied a reporter technology to identify 101 in vivo repressed (ivr) genes of the bacterial pathogen Vibrio cholerae. In depth analysis of the in vivo repressed H+/Cl− transporter ClcA revealed an inverse requirement along gastrointestinal colonization. ClcA could be linked to acid tolerance response required during stomach passage, but ClcA expression is detrimental during subsequent colonization of the lower intestinal tract as it exploits the proton-motive force in alkaline environments. The study summarized in this addendum demonstrates that constitutive expression of ivr genes can reduce intestinal colonization fitness of V. cholerae, highlighting the necessity to downregulate these genes in vivo.
KEYWORDS: Host-pathogen interactions, cholera, in vivo gene regulation, gene repression, colonization fitness, virlulence, bacterial pathogenesis, spatiotemporal gene regulation, murine model, TRIVET
Introduction
Gastrointestinal infections are among the most common diseases in primary care worldwide. The WHO estimates a global morbidity of ~1.5 million people worldwide due to diarrheal disease each year. One of the most important gastrointestinal pathogens is the Gram-negative bacterium Vibrio cholerae, the causative agent of the severe secretory diarrheal disease cholera, responsible for about 3 – 5 million cholera cases and 120 000 deaths that occur globally every year. 1–3 Thus, cholera is still a massive threat for people, especially in areas with limited fresh water supply.
As a facultative human pathogen, V. cholerae constantly transits between the intestinal tract of the human host and the aquatic environment where it persists during inter-epidemic periods. Upon oral ingestion by the human host and subsequent passage to the gastrointestinal tract, V. cholerae alters the expression profile to accommodate to the in vivo requirements and induces the virulence cascade, which is mainly mediated via activation of the ToxR regulon. 4 So far, most studies investigating gene regulation of V. cholerae during intestinal colonization focused on gene induction to identify factors contributing to virulence and in vivo survival fitness. However, two factors are already known to be downregulated in vivo. These include the outer membrane porin OmpT, which increases bile sensitivity of the bacterial cell and the mannose-sensitive hemagglutinin type IV pilus MSHA, which has adverse effects on colonization fitness of the pathogen by a non–antigen-specific binding of host immunoglobulins. 5,6 We hypothesized that OmpT and MSHA represent just the tip of the iceberg and V. cholerae needs to silence many more genes to allow proper colonization and achieve full virulence in vivo. Thus, we designed a single cell-based reporter system to identify in vivo repressed (ivr) genes of V. cholerae followed by a comprehensive characterization of the H+/Cl− transporter ClcA, which is silenced during intestinal colonization. 7 In this addendum, we provide a concise summary of the reporter system and the original results along with additional commentary, further interpretations and implications related to gene silencing as an adaptational strategy of bacterial pathogens.
How the recombination-based in vivo expression technology (RIVET) became the tetR-controlled recombination-based in vivo expression technology (TRIVET)
Single-cell based reporter technologies, like the in vivo expression technology (IVET) and RIVET have been extremely useful to identify gene induction in complex populations according to their spatial and temporal expression 8–10, but their designs limit them to identify only genes activated in a defined condition. To identify gene silencing in vivo we developed a modified version of RIVET to become TRIVET. An extensive illustration of reporter-based in vivo technologies from RIVET to TRIVET is shown in Figure 1. The original RIVET system consists of a TnpR resolvase-mediated excision of a gene reporter cassette flanked by res sequences. 11 One component of RIVET is the res cassette, which can be placed in a neutral site of the V. cholerae chromosome via homologous recombination and confers kanamycin resistance (KnR) and sucrose sensitivity (SucS). The other component of RIVET is a promoterless tnpR, encoding a site-specific DNA recombinase (resolvase), which can be randomly integrated on the chromosome to generate transcriptional fusions of V. cholerae genes to tnpR by the combinatory use of homologous recombination and the pIVET suicide plasmid-library. 12 Induction of a tnpR fusion in vivo leads to excision and irreversible loss of the res cassette that can be monitored by a phenotypic change of the resistance profile (KnS & SucR).
Figure 1.
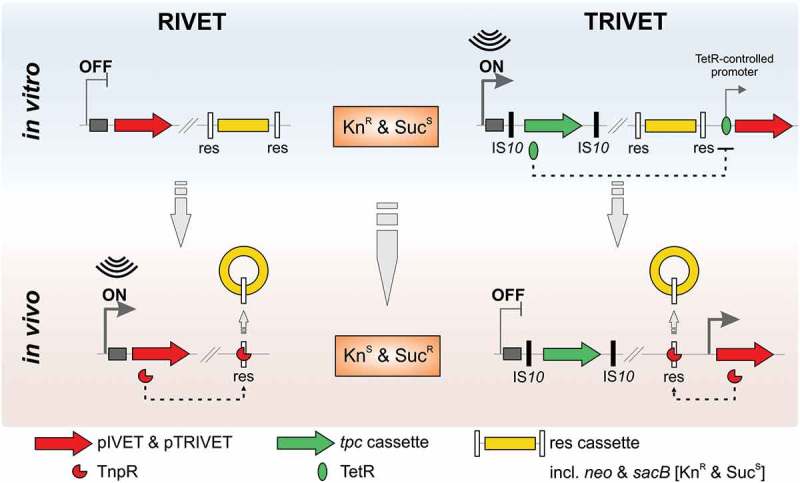
Comparative illustration of RIVET (left) and TRIVET (right) fused to a hypothetical geneX (dark grey) in the ON/OFF scenario of its promotor. Additional chromosomal sequences are highlighted in light gray, the res cassette parts in yellow, the integrated suicide vectors (pIVET and pTRIVET) harbouring tnpR in red, and tetR-phoA-cat (tpc) cassette in green flanked by IS10 sites (black). In case of RIVET, the pIVET suicide vector is integrated into V. cholerae hypothetical geneX via homologous recombination resulting in a merodiploid in which geneX and tnpR (resolvase) are transcriptionally fused and controlled by the chromosomal promotor of geneX. TnpR expression via activation of the geneX promotor results in an irreversible excision of the res cassette marked by a change in the resistance profile of the cell [kanamycin resistant (KnR) and sucrose sensitive (SucS) to kanamycin sensitive (KnS) and sucrose resistant (SucR)]. Thus, resolved strains (loss of the res cassette) can be selected by their ability to grow in sucrose.
The principle of an irreversible excision of the res cassette upon alteration in gene expression is conserved in TRIVET. However, the promotorless tnpR was replaced by a tnpR allele controlled by a TetR-controlled promoter, which originates from the tetracycline-resistance gene tetA. Thus, tnpR, which is integrated next to the res cassette in the V. cholerae genome via the suicide plasmid pTRIVET is now tightly controlled by the repressor TetR. The tetR-phoA-cat (tpc) cassette is the new element of TRIVET harbouring a promotorless tetR and phoA, The tpc cassette can by randomly integrated into the chromosome to generate transcriptional fusions of V. cholerae genes to tetR and phoA via the transposable element Tn10. TRIVET strains with sufficient tetR expression via the tpc cassette, repression of tnpR will keep the res cassette stably integrated and sustain the respective resistance profile (KnR & SucS). Upon silencing of a tetR fusion in vivo, insufficient expression of tetR leads to induction of tnpR resulting in excision and irreversible loss of the res cassette (KnS & SucR) allowing the identification of in vivo repressed genes.
Reduced colonization fitness of strain overexpressing ivr genes highlights the impact of gene silencing
Using a TRIVET-library comprising ~ 10,000 independent tpc-fusions in combination with the infant mouse model of cholera we identified 101 ivr genes, which can be allocated into diverse functional groups (Figure 2). Gene ontology analyses (http://pantherdb.org) which supports enrichment analysis using pathway classifications from the reactome resource 13 did not reveal any category to be significantly overrepresented in the identified ivr genes with regard to their abundance in the V. cholerae genome. This could indicate that gene silencing during infection involves global changes of several pathways and networks instead of only a few specific candidates within a defined functional group.
Figure 2.
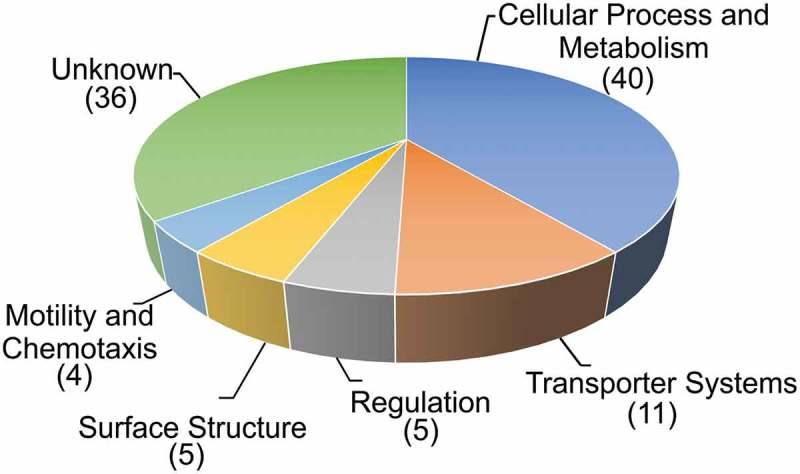
Functional distribution of in vivo repressed (ivr) genes of V. cholerae. Shown are ivr genes identified with the TetR-controlled recombination-based screening technology, allocated in functional groups by their proposed function according to KEGG (http://www.genome.jp/kegg/). The number of ivr genes in the respective group is indicated in parenthesis. In case of TRIVET, the pTRIVET suicide vector harbouring a TetR-controlled tnpR allele is integrated next to the res cassette. The tpc cassette is mobilized into the chromosome via Tn10 mutagenesis resulting in transcriptional fusion and control by the chromosomal promotor of geneX. Loss of TetR expression via repression of the geneX promotor results in de-repression of tnpR and irreversible excision of the res cassette causing the same resistance profile as described above (KnR & SucS) to (KnS & SucR).
Based on our hypothesis microbial pathogens like V. cholerae silence genes during in vivo passage to achieve full colonization fitness. To challenge this idea, we tested the impact of gene repression during murine colonization by competing V. cholerae strains constitutively expressing ivr genes against the isogenic wildtype in the infant mouse model of cholera. In six out of nine cases, constitutive expression of an ivr gene resulted in significant attenuation in vivo compared to the wildtype. Thus, silencing of ivr genes seems to be crucial to achieve full colonization fitness.
For most ivr genes, ongoing and future studies aim to elucidate the exact mechanism why silencing of these ivr genes is required for proper colonization. However, some ivr candidates already connect in vivo gene silencing to other important networks and pathways. To briefly touch two examples, we could show that overexpression of the VC0704–2 operon causes a 10-fold attenuation during intestinal colonization. Notably, the operon has been previously implicated to modulate biofilm formation of V. cholerae. 14,15 VC0704 and VC0703, also known as nspS and mbaA, have been recently reported to encode a polyamine signaling system affecting biofilm formation via modulation of c-di-GMP levels. 16,17 The current model suggests that NspS binds norspermidine and spermidine causing distinct conformational changes, which in turn affects the interaction of MbaA enhancing or decreasing its c-di-GMP phosphodiesterase activity resulting in high or low exopolysaccharide production and consequently increased or decreased biofilm formation. In V. cholerae levels of the second messenger need to be tightly controlled and vary along the life cycle as c-di-GMP positively regulates biofilm formation through induction of the vps genes 18,19, but represses motility and virulence. 20–22 Although the exact molecular mechanism causing attenuation of strain overexpressing the operon needs to be elucidated one could easily imagine that interference in the c-di-GMP signaling might disturb coordinated virulence gene expression.
Furthermore, strains constitutively expressing VC2137, also known as flrA, exhibit a 7-fold attenuation during intestinal colonization. The transcriptional activator FlrA is the key class-I regulator for flagella gene expression in V. cholerae. 23 Importantly, ON/OFF-control of the polar flagellum of V. cholerae and virulence regulation are connected during intestinal colonization in a spatio-temporal manner. In the initial phase of infection, the flagellar motility is required to attach to and penetrate through the intestinal mucosal layer. 24,25 During this mucosal penetration the flagella breaks, which results in secretion of the anti-sigma factor FlgM and consequently releases the alternative sigma factor FliA, belonging to class II flagella genes and essential to activate flagella class IV genes for assembling a functional flagellum. 26,27 Once V. cholerae has digged into the mucus, flagellar motility becomes dispensable and flagellar gene expression is tuned down by a Lon-dependent proteolysis of FliA to allow full virulence gene expression. 28–30 At the late stage of infection V. cholerae activates a RpoS-dependent mucosal escape response to again induce flagella and chemotaxis genes, which facilitates detachment from the epithelium and transition into the aquatic environment. 8,31 Our data suggests that constitutive expression of FlrA can be as detrimental for colonization fitness as presence of FliA, which is a potent inhibitor for virulence gene expression. 28 Thus, tight spatio-temporal control of the flagellar gene cascade seems to be crucial for proper colonization fitness.
Overexpression of VCA0526 (ClcA, H+/Cl− transporter, CLC family) also resulted in a pronounced 50-fold colonization defect in vivo. In general, the CLC family comprises several integral membrane proteins involved in the translocation of chloride ions through biological membranes. Moreover, these chloride channel proteins were previously connected to the acid tolerance response (ATR) against hydrochloric acid in bacteria. 32 Interestingly, the ATR was already identified to be induced during infection and crucial for survival in acidic conditions, which are encountered during passage of the stomach. 33,34 Intrigued by these facts, ClcA was prioritized for a more detailed analysis to reveal molecular explanation for required downregulation during intestinal colonization.
H+/Cl− transporter of V. cholerae requires different regulation and temporal expression through gastrointestinal tract
Gastrointestinal pathogens, such as V. cholerae, have to pass the acidic stomach barrier to reach their primary colonization sites in the intestine. Thus, bacteria have evolved protective mechanisms known as ATR to enhance survival under low pH conditions.35,36 The most common ATR pathway of bacteria is the amino acid-dependent decarboxylation system, found in several gastrointestinal pathogens, including Escherichia coli, Shigella flexneri, Salmonella typhimurium, Listeria monocytogenes and V. cholerae. 37 Excessive protons are detoxified by cytoplasmic amino acid-decarboxylases converting glutamate, arginine or lysine to CO2 and γ-amino butyric acid, agmatine or cadaverine, respectively (Figure 3). This is coupled with an antiporter importing the amino acid in exchange to the amine. Thus, bacteria can stabilize the intercellular pH for survival conditions from life-threatening acidic environment. Two open questions arise: First, the antiporter process is electrogenic and effectively moves a positive charge outward (e.g.: glutamate− exchange for GABA°, arginine1+ exchange for agmatine2+, or lysine1+ exchange for cadaverine2+). Hence, an electrical shunt is required to prevent the excessive inner-membrane hyperpolarization. Second, the counterion of the acid, e.g. chloride in case of hydrochloric acid, accumulates and needs to be removed. H+/Cl− transporters of the CLC family were implicated to fulfil these roles, especially in the context of the hydrochloric acid-rich stomach, by export of two chloride ions for one proton. 32
Figure 3.
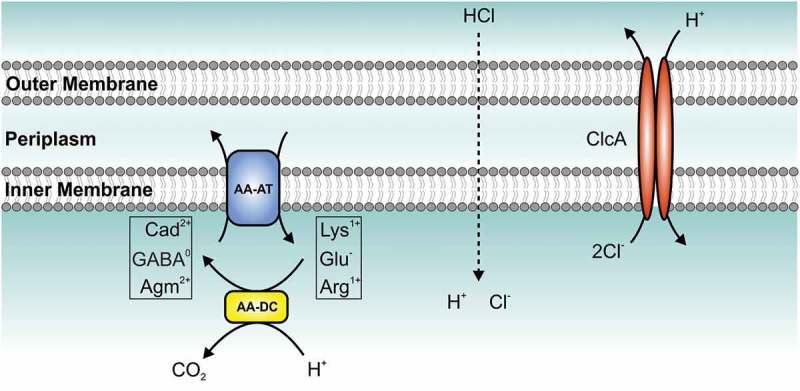
Model of the amino acid-dependent decarboxylation system and ClcA (H+/Cl− transporter) within the bacterial acid tolerance response. Upon exposure to hydrochloric acid (HCl), bacteria activate the amino acid-dependent decarboxylation system: amino acid decarboxylase (AA-DC, yellow) convert amino acids [lysine (Lys1+), glutamate (Glu−) or arginine (Arg1+)] to their decarboxylated versions [cadaverine (Cad2+), γ-aminobutyric acid (GABA°), or agmatine (Agm2+)] thereby consuming a proton (H+) and producing carbon dioxide (CO2). A coupled, electrogenic amino acid-amine antiporter (AA-AT, blue) located in the membrane removes the product and provides new substrate via exchange of Cad2+ and Lys1+, GABA° and Glu− or Agm2+ and Arg1+, respectively. The H+/Cl− transporter (ClcA, red) exports two chloride-ions (Cl−) in exchange of one proton (H+) to detoxify the chloride and acts as electrical shunt to prevent excessive inner-membrane hyperpolarization, which would paralyze the system. However, under alkaline conditions an active H+/Cl− transporter would exploit the proton motive force thereby depleting the energy of the bacterial cell. Thus, expression of clcA is high in the stomach, but V. cholerae represses clcA during colonization of the intestine representing an alkaline environment.
In concordance to this proposed mechanism, we can show that a loss-of-function mutant of the H+/Cl− transporter (ΔclcA) is attenuated during passage of the acidic stomach as well as in vitro conditions with low pH due to high levels of hydrochloric acid. Moreover, our study reveals a tight spatial regulation of the ClcA-system in vivo. V. cholerae silences clcA in alkaline environments, such as the intestinal tract representing the primary site of colonization for V. cholerae, allowing its identification as ivr gene along the study. In such alkaline environments an active H+/Cl− transporter exploits the proton-motive force of the bacterium resulting in severe energy depletion. Concordantly, our data indicate that constitutive expression of clcA becomes detrimental under alkaline conditions, e.g. during colonization of the lower gastrointestinal tract (Figure 4).
Figure 4.
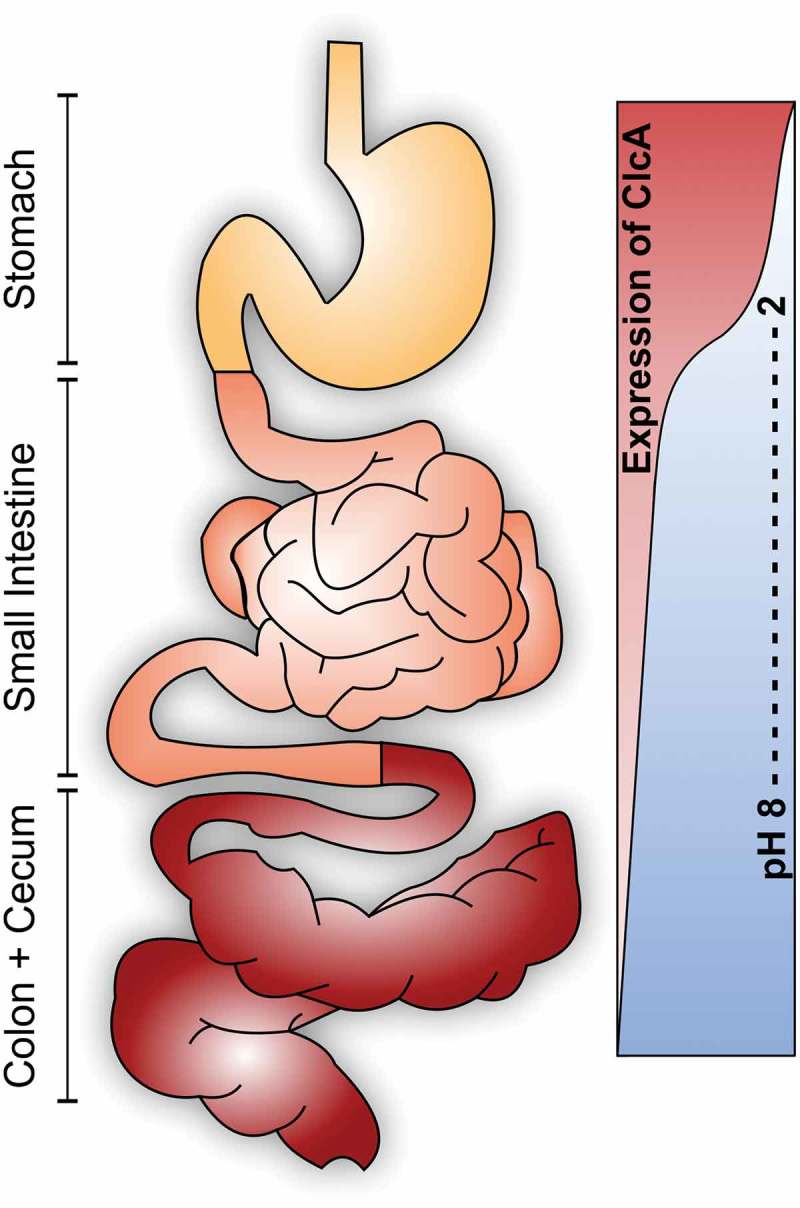
Differential requirement of ClcA (H+/Cl−-transporter) along the human gastrointestinal tract. Upon oral ingestion, V. cholerae has to pass the acidic stomach barrier. ClcA facilitates survival in presence of hydrochloric acid via detoxification of chloride and acts as an electrical shunt to prevent membrane hyperpolarization. Once V. cholerae reaches the alkaline environment of the intestine an active ClcA would exploit the proton motive force thereby disrupting the energy metabolism. Concordantly, clcA is induced during the stomach passage, but silenced during colonization of the intestine.
Concluding remarks
Reporter-based systems, such as IVET and RIVET, delivered valuable insights how bacterial pathogens adapt to in vivo conditions during host colonization. In our recent study, we extended the application of these single-cell expression technologies to identify gene silencing during in vivo colonization of V. cholerae. The necessity to silence these genes in vivo became evident by subsequent analyses, which revealed that constitutive expression of ivr genes can be detrimental for the colonization fitness of V. cholerae. Our results highlight the potential of studying gene silencing by bacterial pathogens during infection, which has been so far fairly neglected in the field. It is quite likely that gene silencing has a similar impact for other bacterial pathogens. For example, repression of flagellins at later stages of intestinal colonization seems to be a common theme for several gastrointestinal pathogens, although flagellar motility is generally important at early stages to initiate infections. In addition to V. cholerae, L. monocytogenes and Salmonella enterica serovar Typhimurium downregulate their flagellar motility to avoid host detection, as their flagellins are potent stimulators of the human immune system. 38,39 Moreover, it might be worthwhile to study gene silencing during other stages of the pathogen’s lifecycle. For example, we recently adapted RIVET to identify genes induced during biofilm formation of V. cholerae, representing its aquatic lifestyle. 40 Amongst other interesting candidates, extracellular nucleases were found to be induced during biofilm formation, which resulted in the characterization of extracellular DNA as a novel matrix component of V. cholerae biofilms. 41 Similarly, identification of genes silenced during biofilm formation could provide new physiological insights in this persistence mode. The design of the reporter-technology TRIVET offers the possibility for such investigations in the future, as it could be adapted to different conditions and other genetically engineerable bacterial pathogens.
Funding Statement
This work was supported by the Austrian Science Fund (FWF) grants: W901 (DK Molecular Enzymology) to F.C., F.G.Z. and S. S., as well as P27654 and P25691 to S. S
References
- 1.Clemens JD, Nair GB, Ahmed T, Qadri F, Holmgren J.. Cholera Lancet. 2017;390:1539–1549. doi: 10.1016/S0140-6736(17)30559-7. [DOI] [PubMed] [Google Scholar]
- 2.Christian KA, Iuliano AD, Uyeki TM, Mintz ED, Nichol ST, Rollin P, Staples JE, Arthur RR. What we are watching-top global infectious disease threats, 2013–2016: an update from CDC’s global disease detection operations center. Health Secur. 2017;15:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization Diarrhoeal disease. 2018. [accessed 2018 April30]. http://www.who.int/mediacentre/factsheets/fs330/en/
- 4.Reidl J, Klose KE. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol Rev. 2002;26:125–139. [DOI] [PubMed] [Google Scholar]
- 5.Wibbenmeyer JA, Provenzano D, Landry CF, Klose KE, Delcour AH. Vibrio cholerae OmpU and OmpT porins are differentially affected by bile. Infect Immun. 2002;70:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsiao A, Liu Z, Joelsson A, Zhu J. Vibrio cholerae virulence regulator-coordinated evasion of host immunity. Proc Natl Acad Sci U S A . 2006;103:14542–14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cakar F, Zingl FG, Moisi M, Reidl J, Schild S. In vivo repressed genes of vibrio cholerae reveal inverse requirements of an H(+)/Cl(-) transporter along the gastrointestinal passage. Proc Natl Acad Sci U S A. 2018;115:E2376–E85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, Camilli A. Genes induced late in infection increase fitness of vibrio cholerae after release into the environment. Cell Host Microbe. 2007;2:264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angelichio MJ, Merrell DS, Camilli A. Spatiotemporal analysis of acid adaptation-mediated vibrio cholerae hyperinfectivity. Infect Immun. 2004;72:2405–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Angelichio MJ, Spector J, Waldor MK, Camilli A. Vibrio cholerae intestinal population dynamics in the suckling mouse model of infection. Infect Immun. 1999;67:3733–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camilli A, Beattie DT, Mekalanos JJ. Use of genetic recombination as a reporter of gene expression. Proc Natl Acad Sci U S A. 1994;91:2634–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osorio CG, Crawford JA, Michalski J, Martinez-Wilson H, Kaper JB, Camilli A. Second-generation recombination-based in vivo expression technology for large-scale screening for Vibrio cholerae genes induced during infection of the mouse small intestine. Infect Immun. 2005;73:972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karatan E, Duncan TR, Watnick PI. NspS, a predicted polyamine sensor, mediates activation of vibrio cholerae biofilm formation by norspermidine. J Bacteriol. 2005;187:7434–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bomchil N, Watnick P, Kolter R. Identification and characterization of a vibrio cholerae gene, mbaA, involved in maintenance of biofilm architecture. J Bacteriol. 2003;185:1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobe RC, Bond WG, Wotanis CK, Zayner JP, Burriss MA, Fernandez N, Bruger EL, Waters CM, Neufeld HS, Karatan E. Spermine inhibits vibrio cholerae biofilm formation through the NspS-MbaA polyamine signaling system. J Biol Chem. 2017;292:17025–17036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cockerell SR, Rutkovsky AC, Zayner JP, Cooper RE, Porter LR, Pendergraft SS, Parker ZM, McGinnis MW, Karatan E. Vibrio cholerae NspS, a homologue of ABC-type periplasmic solute binding proteins, facilitates transduction of polyamine signals independent of their transport. Microbiology. 2014;160:832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beyhan S, Tischler AD, Camilli A, Yildiz FH. Differences in gene expression between the classical and El Tor biotypes of vibrio cholerae O1. Infect Immun. 2006;74:3633–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tischler AD, Camilli A. Cyclic diguanylate (c-di-GMP) regulates vibrio cholerae biofilm formation. Mol Microbiol. 2004;53:857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krasteva PV, Fong JC, Shikuma NJ, Beyhan S, Navarro MV, Yildiz FH, Sondermann H. Vibrio cholerae VpsT regulates matrix production and motility by directly sensing cyclic di-GMP. Science. 2010;327:866–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamayo R, Pratt JT, Camilli A. Roles of cyclic diguanylate in the regulation of bacterial pathogenesis. Annu Rev Microbiol. 2007;61:131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tischler AD, Camilli A. Cyclic diguanylate regulates vibrio cholerae virulence gene expression. Infect Immun. 2005;73:5873–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prouty MG, Correa NE, Klose KE. The novel sigma54- and sigma28-dependent flagellar gene transcription hierarchy of vibrio cholerae. Mol Microbiol. 2001;39:1595–1609. [DOI] [PubMed] [Google Scholar]
- 24.Freter R, Jones GW. Adhesive properties of vibrio cholerae: nature of the interaction with intact mucosal surfaces. Infect Immun. 1976;14:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitner DR, Feichter S, Schild-Prufert K, Rechberger GN, Reidl J, Schild S. Lipopolysaccharide modifications of a cholera vaccine candidate based on outer membrane vesicles reduce endotoxicity and reveal the major protective antigen. Infect Immun. 2013;81:2379–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Correa NE, Barker JR, Klose KE. The Vibrio cholerae FlgM homologue is an anti-sigma28 factor that is secreted through the sheathed polar flagellum. J Bacteriol. 2004;186:4613–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Miyashiro T, Tsou A, Hsiao A, Goulian M, Zhu J. Mucosal penetration primes vibrio cholerae for host colonization by repressing quorum sensing. Proc Natl Acad Sci U S A. 2008;105:9769–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syed KA, Beyhan S, Correa N, Queen J, Liu J, Peng F, Satchell KJ, Yildiz F, Klose KE. The vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J Bacteriol. 2009;191:6555–6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pressler K, Vorkapic D, Lichtenegger S, Malli G, Barilich BP, Cakar F, Zingl FG, Reidl J, Schild S. AAA+ proteases and their role in distinct stages along the vibrio cholerae lifecycle. Int J Med Microbiol. 2016;306:452–462. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen AT, Dolganov NA, Rasmussen T, Otto G, Miller MC, Felt SA, Torreilles S, Schoolnik GK. A bistable switch and anatomical site control vibrio cholerae virulence gene expression in the intestine. PLoS Pathog. 2010;6:e1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen AT, Dolganov NA, Otto G, Miller MC, Wu CY, Schoolnik GK. RpoS controls the vibrio cholerae mucosal escape response. PLoS Pathog. 2006;2:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer R, Iverson TM, Accardi A, Miller C. A biological role for prokaryotic ClC chloride channels. Nature. 2002;419:715–718. [DOI] [PubMed] [Google Scholar]
- 33.Merrell DS, Camilli A. The cadA gene of vibrio cholerae is induced during infection and plays a role in acid tolerance. Mol Microbiol. 1999;34:836–849. [DOI] [PubMed] [Google Scholar]
- 34.Merrell DS, Camilli A. Regulation of vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcriptional regulators. J Bacteriol. 2000;182:5342–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2:898–907. [DOI] [PubMed] [Google Scholar]
- 36.Merrell DS, Camilli A. Acid tolerance of gastrointestinal pathogens. Curr Opin Microbiol. 2002;5:51–55. [DOI] [PubMed] [Google Scholar]
- 37.Lund P, Tramonti A, De Biase D. Coping with low pH: molecular strategies in neutralophilic bacteria. FEMS Microbiol Rev. 2014;38:1091–1125. [DOI] [PubMed] [Google Scholar]
- 38.Shen A, Higgins DE. The MogR transcriptional repressor regulates nonhierarchal expression of flagellar motility genes and virulence in listeria monocytogenes. PLoS Pathog. 2006;2:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. In vivo, fliC expression by salmonella enterica serovar typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol Microbiol. 2006;61:795–809. [DOI] [PubMed] [Google Scholar]
- 40.Seper A, Pressler K, Kariisa A, Haid AG, Roier S, Leitner DR, Reidl J, Tamayo R, Schild S. Identification of genes induced in vibrio cholerae in a dynamic biofilm system. Int J Med Microbiol. 2014;304:749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seper A, Fengler VH, Roier S, Wolinski H, Kohlwein SD, Bishop AL, Camilli A, Reidl J, Schild S. Extracellular nucleases and extracellular DNA play important roles in vibrio cholerae biofilm formation. Mol Microbiol. 2011;82:1015–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]