

ABSTRACT
The purpose of this prospective cross-sectional cohort pilot study is to explore the initial microbial community of gastric aspirate fluid as collected immediately after birth and its relationships with mode of delivery and preterm birth. Twenty-nine gastric aspirate samples collected immediately after birth from infants born between 24–40 weeks gestation were analyzed for microbial composition. Total microbial content was low in many samples, with a substantial number sharing taxonomic composition with negative controls. qPCR targeting the 16S rRNA gene showed that infants delivered vaginally had a higher microbial load than infants delivered by C-section. Some pre-term samples showed high relative abundance of genus Ureaplasma, consistent with previous literature that has implicated infections with this taxon as a potential cause of pre-term birth. Vaginally born term infant samples, by contrast, had significantly higher levels of genus Lactobacillus with Lactobacillus crispatus the most dominant species. Microbial evaluation showed that vaginally born term infant gastric aspirate samples had higher levels of lactobacilli than pre-terms. Samples from many infants had low microbial load near the edge of the detection limit.
Keywords: amniotic fluid, preterm birth, prematurity, microbiome, gestational age, Lactobacillus crispatus, Lactobacillus iners
Introduction
Bacteria putatively enter the maternal fetal unit through different routes: through the maternal gastrointestinal tract via translocation and/or transport by specialized cells, through the maternal mouth and hematogenous spread, ascending from the vagina with translocation through the choriodecidual membrane, or iatrogenically (through amniocentesis).1 Microbial entry by non-commensal organisms may trigger a fetal immune response, which is associated with preterm labor.2 Early microbial exposure is important for the development of the neonatal immune system and dysbiosis has been linked to the development of multiple chronic diseases.3–6
Because traditional culture techniques frequently do not detect bacteria unless an overt infection is present, the prevailing dogma has been that a healthy fetus resides in a sterile environment. However, with the advancement of non-culture based techniques (organism specific PCR and 16s rRNA gene sequencing), efforts have been made to characterize the microbiome in all cavities of the human body. Indeed, bacterial DNA is present in the healthy human placenta, umbilical cord, and meconium obtained immediately after birth.7–10 Evolving data suggests that the placenta and fetal membranes are not sterile even in the absence of infection. These results, however, remain highly controversial. The maternal-fetal and neonatal microbiomes have low overall microbial biomass and it has been argued that inadequate attention to negative controls can explain many of the observations that oppose the “sterile womb” hypothesis.11 These issues are important to resolve, both in order to elucidate the role of infections in pre-term birth, and because the earliest exposures to the microbiome likely have a profound impact on the newly developing immune system.
In previous studies using culture based techniques, gastric fluid collected immediately after birth identified Ureaplasma species was also present in amniotic fluid, with high specificity but low sensitivity12 In addition, cytokines present in gastric fluid aspirated immediately after delivery have been associated with chorioamnionitis (indicating placental inflammation) and funisitis (indicating fetal inflammation).13 In this pilot study, we present for the first time fluid samples from gastric aspirate collected within an hour of birth from both term and pre-term infants for analysis of microbes. We utilized Illumina sequencing of the 16S rRNA gene as well as qPCR targeting the 16S rRNA gene. We find evidence of low overall microbial load, with many of our samples sharing taxonomic composition with negative controls. Furthermore, there was higher overall microbial load in vaginal than C-section births, which also was coincident in our cohort with a greater percentage of vaginal births (83%) in terms versus preterms(22%). We also find higher levels of Lactobacillus in vaginally born term births, consistent with infants acquiring Lactobacillus while traversing the birth canal.
Methods
Design
This prospective observational cross-sectional cohort pilot study was conducted between September 2014 and April 2015 at the University of Florida, UF Health Level IV NICU. The primary aim of this study was to examine the differences in the presence of microbes of very early gastric aspirate fluid using 16s rRNA gene analysis The Institutional Review Board of the University of Florida approved the human subject protocol and the study was carried out in accordance with the approved protocol (IRB #2014–00536). Written informed consent was obtained from all mothers of infants enrolled in the study.
Patient population
Inborn neonates admitted to the NICU immediately after birth and who had a nasogastric (NG) or orogastric (OG) tube placed were included. Exclusion criteria included: (1) a known gastrointestinal malformation, (2) maternal age less than 18, and (3) a non-English speaking mother. Several infants were not eligible for the study because they required orogastric or nasopharyngeal suctioning during resuscitation. Routine deep suctioning of all neonates during resuscitation is no longer frequently used at the time of delivery. Frequent reasons for placing an OG or NG tube upon admission to the NICU include, respiratory distress caused by respiratory distress syndrome or transient tachypnea of the newborn, perinatal resuscitation, and congenital heart disease.
Sample collection
Gastric aspirate samples were collected within 1°hour after birth during routine placement of a nasogastric or orogastric tube upon admission to the NICU. All tubes were placed and advanced to the appropriate depth based on current nursing literature and when fluid was aspirated to confirm gastric placement, at least 0.5 mL of gastric fluid was obtained, placed into sterile collection tubes and stored in a −80°Celsius freezer.14
DNA isolation and microbial DNA quantification
After the gastric aspirate samples thawed, an equal amount of sterile phosphate buffered solution was added to the sample tube to facilitate aspiration due to the viscous nature of the samples. Microbial DNA was isolated using the PowerLyzer Power Soil DNA Isolation Kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions. The presence of microbial DNA was detected and quantified by real-time PCR using microbial universal primers in triplicates with SYBR Green Dye assay on CFY96 Real time system (Bio-Rad Laboratories, Inc, Hercules, CA, USA).
Microbial 16S rRNA gene sequencing
For microbiome analyses, gastric aspirate DNA was amplified by Illumina MiSeq compatible bar-coded primers 27F/534R, targeting the 16s rRNA gene V1-V3 region. PCR reactions were performed with Taq DNA Polymerase (Invitrogen Corp., Carlsbad, CA, USA) under the following condition: initial melting step at 94°C for 2 minutes, followed by 30 cycles of 94°C for 20°seconds, 56°C for 30 seconds, and 72°C for 45°seconds. Amplicons were individually identified by 2% agarose gel and cleaned by PureLink Quick PCR Purification kit (Invitrogen Corp., Carlsbad, CA, USA). Cleaned amplicons were quantified by qPCR using KAPA Library Quantification Kit Illumana Platforms (Kapa Biosystems, Woburn, MA, USA). Equal amount of amplicons were pooled and then purified by Agencourt Ampure bead (Beckman Coulter, Indianapolis IN, USA). Pooled and purified amplicons were mixed with 10% of Phix. Miseq v3 reagent kit (Illumina, Inc., San Diego, CA, USA) was used to run the pooled samples on the Illumina Miseq machine. Negative controls included a mock sample (water) run through the full isolation process (DNA isolation-PCR-purification/quantification-multiplex (hereafter referred to as the “water” control) and water added to the PCR step control (hereafter referred to as the “neg” control). Both negative controls were PCR grade sterilized-purified water. One “neg” control and one “water” control were tested in the study. Triplicates were used in real-time PCR for all samples and controls.
DNA quantification using universal primers
DNA amplification was carried out in triplicates with SYBR Green Dye assay on CFX384 Real time system (Bio-Rad Laboratories, Inc, Hercules, CA, USA). 16S rRNA universal primers were used to quantify microbial DNA: forward AGAGTTTGATCCTGGCTCAG, reverse ACTGCTGCCTCCCGTAGGAG. Primer concentration was 0.3 µM for all forward and reverse primers. Reactions were performed in 12 µl final volume with 2 µl of DNA (2 to 20°ng) at an initial denaturation at 95°C, followed by temperature cycling of 95°C for 10 s, 55°C for 10 s, 72°C for 30°s with total 40 cycles.
Sequencing and data processing
Taxonomic ranks were assigned for the forward reads using the RDP (ribosomal database project) classifier version 2.2 with confidence set to 50%. Reads were grouped by genera and the counts were normalized as simple proportions and, in some analyses log10 transformed using the following formula:
where RC is the read count for a particular OTU in a particular sample, n is the total number of reads in that sample, the sum of x is the total number of reads in all samples and N is the total number of samples.15 This normalization attempts to standardize the effect of the pseudo-count on samples of different sequencing depth. Multi-Dimensional Scaling (MDS) was performed with the Bray-Curtis distance of the normalized and log10 transformed counts using the capscale function in the vegan R package.
In addition to genus level classifications with the RDP classifier, we used the DADA 2 algorithm to perform de-novo clustering.16 The DADA 2 tutorial (https://benjjneb.github.io/dada2/tutorial.html) was followed to generate clusters. Sequences associated with each cluster (supplementary File 4) were classified with the RDP pipeline (shown in supplementary File. 3) and by using NCBI BLAST 2.6.0+ to search the Silva database (SILVA_132_SSURef_Nr99_tax_silva.fasta; supplementary File 5). Only hits across the entire read (> 90 basepairs) with no more than one mismatch were kept in the comparison to the Silva database.
Significant genera were detected using R to perform a two-way ANOVA of linear models comparing the normalized relative abundance of each taxa to birth group (a numeric variable from 1–5) and delivery mode.17 The P-values were then adjusted for multiple hypothesis testing using the method of Benjamini & Hochberg for all taxa plus diversity.18 Birth group was defined as a scalar variable with the following: Birth group 1: gestational age of 23–26 weeks; Birth group 2: 27–30 weeks; Birth group 3: 31–34 weeks, Birth group 4: 35–37 weeks, Birth group 5 (term): ≥ 38 weeks. Unlike our RDP pipeline, there were no significant taxa associated with birth group or delivery mode in our DADA 2 pipeline, presumably because our sample size was not large enough to overcome the splitting of genera into the sparse pattern of OTUs observed within each genra (for example, Figure 3 for Lactobacillus).
Figure 3.
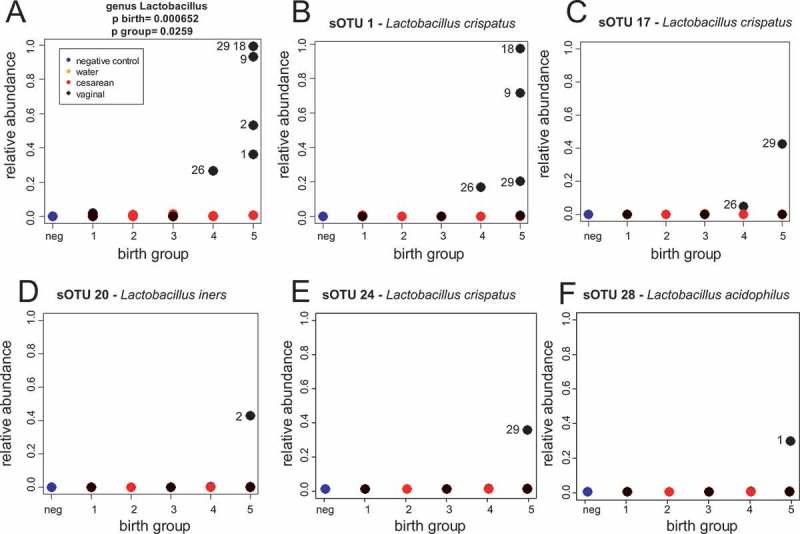
Lactobacillus crispatus is the dominant species in our cohort. (A) Relative abundance of genus Lactobacillus as determined by our RDP pipeline. (B-F) Relative abundance of clusters determined from DADA 2 that matched Lactobacillus in the Silva database. Numbers in all panels next to symbols are subject IDs.
PCR quantification for lactobacillus crispatus (L. crispatus)
To quantitate the abundance of L. crispatus, qPCR was performed on the DNA extracted from gastric aspirate samples as described above. DNA amplification was carried out in triplicates with SYBR Green Dye assay on CFX384 Real time system (Bio-Rad Laboratories, Inc, Hercules, CA, USA). The primers 46F/190R for L.crispatus (Gen Bank: MH327495) were forward 5´-AACTAACAGATTTACTTCGGTAATGA-3´ and reverse 5´-AGCTGATCATGCGATCTGC-3´. The primer set was highly specific for L.crispatus by checking with PRIMER BLAST (http://www. ncbi.nlm.nih.gov/tools/primer-blast). The product length was 145°bp. Primer concentration was 0.3 µM for all forward and reverse primers. Reactions were performed in 12 µl final volume with 2 µl of DNA (2-20°ng) at an initial denaturation at 95°C, followed by temperature cycling of 95°C for 15 s, 55°C for 1 min with total 40 cycles.
Results
Gastric fluid samples were obtained from 29 neonates (Table 1). Ordination of the results of 16S rRNA sequences classified to the genus level revealed a substantial overlap with a sequenced negative control (water added to the PCR sequencing step) and water control (water run through the full DNA isolation-sequencing process; Figure 1A). In our pilot dataset, 5 of the 6 term (group 5) samples were vaginally delivered, and vaginally delivered samples clustered distinctly in a PCoA visualization when compared to C-section samples (Figure 1). To more directly explore the impact of microbial load in our samples, we performed qPCR targeting the 16S rRNA gene. Consistent with our sequencing results, microbial load was significantly higher for the infants delivered by vaginal birth (Figure 2A-B). Microbial abundance as measured by qPCR targeting the 16S rRNA gene was well correlated with distance of each sample from the average position of the negative and water controls along the first MDS axis (Figure 2C). Taken together, these data suggest that there was a minimum threshold of microbial abundance, associated with a PCR cycle number of < 25, that was required before sequencing results were substantially different from background and that many, but not all, of our samples were indistinguishably close to this background.
Table 1.
Sample demographics.
| Group 1 (23–26 6/7 weeks) | Group 2 (27–30 6/7 weeks) | Group 3 (31–34 6/7 weeks) | Group 4 (35–37 6/7 weeks) | Group 5 (38+ weeks) | |
|---|---|---|---|---|---|
| Number of Samples | 5 | 5 | 7 | 6 | 6 |
| Average Gestational age (days) | 184 | 209 | 230 | 251 | 274 |
| Male sex | 3 (60%) | 2 (40%) | 4 (57%) | 1 (17%) | 6 (100%) |
| African American | 2/4* | 3 (60%) | 3 (42%) | 0 | 3 (50%) |
| Caucasian | 2/4* | 2 (40%) | 3 (42%) | 4 (80%) | 3 (50%) |
| Asian | 0/4* | 0 | 1 (14%) | 1 (20%) | 0 |
| Spontaneous Preterm birth | 3(60%) | 2 (40%) | 4 (57%) | 3 (50%) | 0 |
| Antenatal antibiotics | 4 (80%) | 3 (60%) | 3(42%) | 3(50%) | 2(33%) |
| Antenatal corticosteroids | 5(100%) | 5(100%) | 5(71%) | 1 (17%) | 0 |
| Chorioamnionitis | 1(20%) | 1(20%) | 0 | 1(17%) | 0 |
| Rupture of membranes > 18 hours | 3(16%) | 2 (40%) | 0 | 1(17%) | 0 |
| Average length of ROM (hours) | 228 | 234 | 1 | 9 | 6 |
| Vaginal Birth | 2(40%) | 1 (20%) | 1 (14%) | 1 (17%) | 5 (83%) |
| Respiratory Distress Syndrome | 5 (100%) | 5(100%) | 6 (86%) | 5(83%) | 6 (100%) |
| Intracranial hemorrhage | 0 | 0 | 0 | 0 | 0 |
| Necrotizing Enterocolitis (St. II or greater) | 2(40%) | 0 | 0 | 0 | 0 |
| *one person declined to associate with a race |
Figure 1.
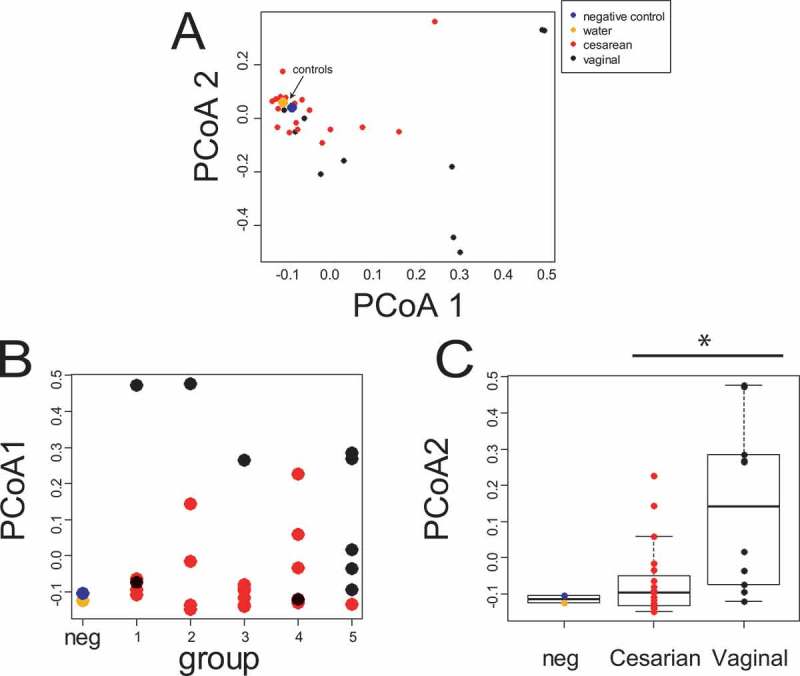
Sample derived from C-section share considerable microbial content with our negative control. (A) MDS ordination based on logged RDP calls to the genus level. The arrow indicates the position of the water and negative controls. (B) MDS 1 broken down by birth group. (C) The same data as (B) broken down by birth mode. A two-way ANOVA for MDS1 reports significant difference for birth mode (p = 0.002) but not birth group (p = .31). Birth group 1: 23–26 weeks; Birth group 2: 27–30 weeks; Birth group 3: 31–34 weeks, Birth group 4: 35–37 weeks, Birth group 5 (term): ≥ 38 weeks.
Figure 2.
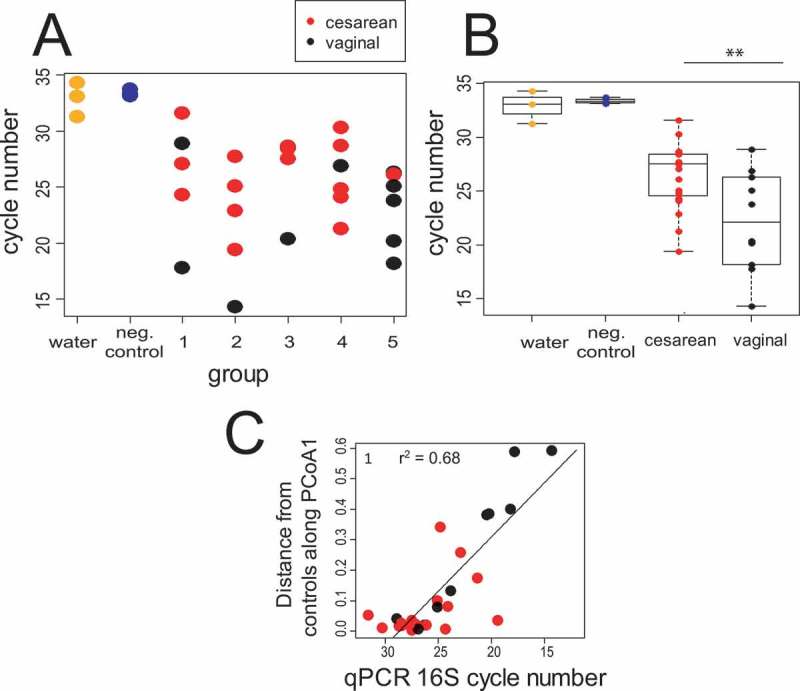
qPCR targeting the 16S rRNA gene shows a higher microbial load in vaginal vs. C-section births. (A: qPCR data broken down by group; (B) the same data collapsed by birth mode. Birth groups are defined as in Figure1. A two-way ANOVA for reports significant difference for birth mode (p = 7.248e-07) but not birth group (p = 0.6378). (C) qPCR data compared to the distance of each sample from the average of the water and negative control along MDS axis from the MDS ordination shown in Figure 1. The line is from a linear regression fit (p = 3.32e-08; r2 = 0.68). For all panels, qPCR data is an average of three technical replicates. In all but 4 of the samples, the SD was less than 1 cycle number (supplementary File 6).
Lactobacillus characterizes the dominant microbial community of vaginally born term infants
The low microbial load present in many of our pre-term samples lead to substantial challenges in data analysis. In our negative control sample, we noticed that two most abundant genera, Escherichia/Shigella and Clostridium sensu stricto, made up over 51% of all the reads in our negative control. These taxa were abundant in many of our samples as well (supplementary File 1), which we took as further evidence of a low microbial biomass causing detection of contamination. When analyzed with a two-way ANOVA at a significance threshold of FDR adjusted p < .05 with fixed terms of group (1–5) and birth mode (C-section or vaginal), 5 genera (Lactobacillus, Clostridium XI, Clostridium sensu stricto, Sarcina, and Enterococcus) as well as Shannon diversity were significantly different between C-section and vaginal delivery (supplementary Table 1; supplementary File 1). However, Shannon diversity and all of these taxa (except for Lactobacillus) were also more abundant in our negative control than in our C-section samples, suggesting that all of these taxa except Lactobacillus likely reflect contaminants19 that more substantially affect our low microbial biomass C-section than our vaginal samples. By contrast, Lactobacillus is significantly associated with both vaginal delivery and birth group but is largely absent from our negative controls (Figure 3A). Indeed, Lactobacillus was the only taxa significantly different by birth group number (FDR adjusted p < .05) in our dataset (supplementary Table 1). Lactobacillus is therefore associated with full-term, vaginal delivery, and this association is unlikely to be due to background contamination.
To further confirm associations with Lactobacillus, and allow a finer level of taxonomic resolution, we analyzed the 16S rRNA dataset with DADA2,16 a pipeline designed to infer exact nucleotide sequences via a clustering procedure that explicitly models variance in sequence in relationship to quality score distributions (supplementary files 3–4). Comparison of reads assigned to genus Lactobacillus by our RDP pipeline (Figure 3A) to DADA2 clusters with matches to Lactobacillus species in the Silva database (Figure 3B-3F) reveals that L. crispatus makes up the dominant species of the Lactobacillus that we observed. Two subjects (subjects 18 and 9) had a single DADA 2 cluster assigned to L. crispatus (Figure 3B) while other subjects (26 and 29) had multiple DADA 2 L. crispatus clusters (Figure 3B, 3C and 3E). The presence of L. crispatus in subjects 9, 18, 26 and 29 above a background observed in other subjects was confirmed with qPCR (supplementary Figure 1). In addition to L. crispatus, one subject (subject 2) had a cluster with a match to L. iners in the Silva database (Figure 3D) and another had (subject 1) a cluster with a match to L. acidophilus. Taken together, these data demonstrate that in vaginally delivered subjects, L. crispatus is a common dominant Lactobacillus species.
Ureaplasma is present in a subset of our pre-term cohort
Ureaplasma species have been previously associated with pre-term birth.12 A cluster with an exact match to Ureaplasma was the 2nd most abundant DADA2 cluster in our pipeline (Supplementary File. 3, p. 2). All of our Ureaplasma clusters from our DADA2 pipeline had exact matches to more than one species in the Silva database (supplementary table 5) suggesting a longer sequence read would be required to achieve species resolution within the Ureaplasma genus. At the genus level, Ureaplasma was elevated in some of our pre-term samples and not in negative controls (Figure 4). It therefore seems likely that Ureaplasma may have been present in the gastric aspirate fluid of some but not all pre-term infants. While this study was not powered to detect differences between the relative abundance of Ureaplasma in term vs. pre-term gastric aspirate samples, our observations are consistent with previous literature in suggesting that some, but not all, pre-term births are linked to Ureaplasma infection.
Figure 4.
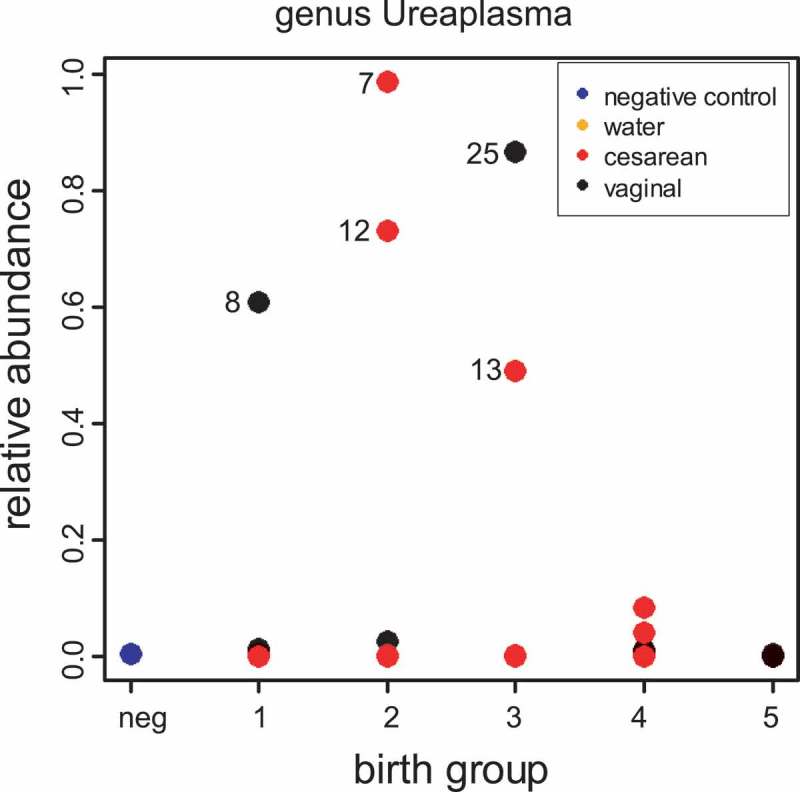
Ureaplasma is higher in some pre-term infants. Shown is relative abundance of Ureaplasma as determined by our RDP pipeline. Numbers in all panels next to symbols are subject IDs.
Discussion
An accurate description of the first microbes to which the infant gut is exposed is crucial to understanding the earliest events in the development of the microbiome, which in turn heavily influences the development of the immune system. The relationships between preterm birth, mode of delivery, and the neonatal microbiome are just starting to be understood. Our study collecting gastric aspirate within an hour of birth from term and pre-term infants provides a unique view of the initial newborn stomach microbiota.
As we demonstrate in this study, and has been previously reviewed the study of the newborn microbiota is highly challenging because low biomass samples are prone to background contamination, which can be naively misinterpreted as belonging to the infant microbiome.11 Placental samples have shown to have low biomass and in several studies placental samples were similar to contamination controls.20,21 A strength of our study is the careful measurement of negative controls and our interpretation of our results with respect to these negative controls. With these precautions taken, our results clearly indicate that, when compared to pre-term infants, our term cohort – primarily vaginally delivered – displayed a microbial signal strongly dominated by genus Lactobacillus. Our study supports the hypothesis that in term vaginally delivered babies, Lactobacillus is an initial inhabitant of the infant stomach, although future work will be required to determine if these initial Lactobacillus are in fact viable and therefore can act as colonizers.
A limitation of our study is that 83% of our term (group 5) cohort were delivered vaginally, in contrast to our pre-term cohort, in which only 22% were delivered vaginally, although our statistical modeling did find Lactobacillus significant for both birth group and mode of delivery, suggesting that not all of our observations of the differences between pre-term vs. term status for Lactobacillus can be explained by delivery mode. In addition to Lactobacillus, we found in some preterm infants an increased relative abundance of Ureaplasma, which has been associated with premature birth, although the sample size in our study is too small to allow for rigorous assessment of this correlation.
Whether Ureaplasma or Lactobacillus is prenatally acquired is not clear. It is certainly plausible that the exposure of the infant through the birth canal at delivery would colonize the newborn’s gastric fluids. While the clustering of many of our caesarian deliveries with our negative controls lends support to the hypothesis that there is little to no colonization prior to birth, larger cohorts with additional term vaginal and C-section delivered samples and paired maternal sampling will be required to discriminate the source of the gastric aspirate microbes we detected in our study.
Conclusions
In conclusion, the microbial community composition of preterm infant gastric fluid immediately after birth is significantly different in microbial composition from vaginally born term samples. Future research should include a larger sample size, including infants who were not admitted to an NICU. Analysis of vaginal and intestinal samples from the mother obtained throughout pregnancy and at the time of delivery would aid in identification of the source of bacterial invasion. Analysis of the first neonatal stool would be important to elucidate the relationship between amniotic fluid, gastric fluid and meconium.
Funding Statement
National Institutes of Health grant NRO14019 supported this work.
Abbreviations
- L. crispatus
Lactobacillus crispatus
- L. iners
Lactobacillus iners
- NEC
necrotizing enterocolitis
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Ethics approval and consent to participate
The Institutional Review Board of the University of Florida approved the human subject protocol and the study was carried out in accordance with the approved protocol (IRB #2014-00536). Written informed consent was obtained from all mothers of infants enrolled in the study.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Funkhouser LJ, Bordenstein SR.. Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 2013;11:e1001631. doi: 10.1371/journal.pbio.1001631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM.. The fetal inflammatory response syndrome. Am J Obstet Gynecol. 1998;179:194–202. [DOI] [PubMed] [Google Scholar]
- 3.Houghteling P, Walker W. Why is initial bacterial colonization of the intestine important to infantsʼ and childrenʼs health? J Pediatr Gastroenterol Nutr. 2015;60(3):294–307. doi: 10.1097/MPG.0000000000000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neu J. Developmental aspects of maternal-fetal, and infant gut microbiota and implications for long-term health. Matern Health Neonatol Perinatol. 2015;1:6. doi: 10.1186/s40748-015-0007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neu J. The microbiome during pregnancy and early postnatal life. Semin Fetal Neonatal Med. 2016;21:373–379. doi: 10.1016/j.siny.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Neu J, Lorca G, Kingma SD, Triplett EW. The intestinal microbiome: relationship to type 1 diabetes. Endocrinol Metab Clin North Am. 2010;39:563–571. doi: 10.1016/j.ecl.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J.. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ardissone AN, de la Cruz DM, Davis-Richardson AG, Rechcigl KT, Li N, Drew JC, Murgas-Torrazza R, Sharma R, Hudak ML, Triplett EW , et al. Meconium microbiome analysis identifies bacteria correlated with premature birth. PLoS One. 2014;9. doi: 10.1371/journal.pone.0090784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiménez E, Fernández L, Marín ML, Martín R, Odriozola JM, Nueno-Palop C, Narbad A, Olivares M, Xaus J, Rodríguez JM. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section Curr Microbiol. 2005;51:270. doi: 10.1007/s00284-005-0020-3. [DOI] [PubMed] [Google Scholar]
- 10.Stout MJ, Conlon B, Landeau M, Lee I, Bower C, Zhao Q, Roehl KA, Nelson DM, Macones GA, Mysorekar IU. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol. 2013;208:226.e1-7. doi: 10.1016/j.ajog.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez-Muñoz ME, Arrieta M-C, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome. 2017;5:48. doi: 10.1186/s40168-017-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SM, Romero R, Lee J, Chaemsaithong P, Docheva N, Yoon BH Gastric fluid versus amniotic fluid analysis for the identification of intra-amniotic infection due to Ureaplasma species. J Maternal-Fetal & Neonatal Med. 2016;29:2579–2587. doi: 10.3109/14767058.2015.1098614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bry KJ, Jacobsson B, Nilsson S, Bry K. Gastric fluid cytokines are associated with chorioamnionitis and white blood cell counts in preterm infants. Acta Paediatrica (Oslo, Norway: 1992) 2015;104:575–580. doi: 10.1111/apa.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan XC, Huttenhower C. Chapter 12: human microbiome analysis. PLoS Comput Biol. 2012;8:e1002808. doi: 10.1371/journal.pcbi.1002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCafferty J Mühlbauer M, Gharaibeh RZ, Arthur JC, Perez-Chanona E, Sha W, Jobin C, Fodor AA. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. Isme j. 2013;7:2116–2125. doi: 10.1038/ismej.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7). doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kusters JG, Reuland EA, Bouter S, Koenig P, Dorigo-Zetsma JW. A multiplex real-time PCR assay for routine diagnosis of bacterial vaginosis. Eur J Clin Microbiol Infect Dis. 2015;34:1779–1785. doi: 10.1007/s10096-015-2412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. [DOI] [PubMed] [Google Scholar]
- 19.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lauder AP, Roche AM, Sherrill-Mix S, Bailey A, Laughlin AL, Bittinger K, Leite R, Elovitz MA, Parry S, Bushman FD. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. 2016;4:29. doi: 10.1186/s40168-016-0172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leon LJ, Doyle R, Diez-Benavente E, Clark TG, Klein N, Stanier P, Moore GE. Enrichment of clinically relevant organisms in spontaneous preterm-delivered placentas and reagent contamination across all clinical groups in a large pregnancy cohort in the United Kingdom. Appl Environ Microbio. 2018. July 15;84(14). doi: 10.1128/AEM.00483-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.