

ABSTRACT
RNA methyltransferases (MTases) catalyse the transfer of a methyl group to their RNA substrates using most-often S-adenosyl-L-methionine (SAM) as cofactor. Only few RNA-bound MTases structures are currently available due to the difficulties in crystallising RNA:protein complexes. The lack of complex structures results in poorly understood RNA recognition patterns and methylation reaction mechanisms. On the contrary, many cofactor-bound MTase structures are available, resulting in well-understood protein:cofactor recognition, that can guide the design of bisubstrate analogues that mimic the state at which both the substrate and the cofactor is bound. Such bisubstrate analogues were recently synthesized for proteins monomethylating the N6-atom of adenine (m6A). These proteins include, amongst others, RlmJ in E. coli and METLL3:METT14 and METTL16 in human. As a proof-of-concept, we here test the ability of the bisubstrate analogues to mimic the substrate:cofactor bound state during catalysis by studying their binding to RlmJ using differential scanning fluorimetry, isothermal titration calorimetry and X-ray crystallography. We find that the methylated adenine base binds in the correct pocket, and thus these analogues could potentially be used broadly to study the RNA recognition and catalytic mechanism of m6A MTases. Two bisubstrate analogues bind RlmJ with micro-molar affinity, and could serve as starting scaffolds for inhibitor design against m6A RNA MTases. The same analogues cause changes in the melting temperature of the m1A RNA MTase, TrmK, indicating non-selective protein:compound complex formation. Thus, optimization of these molecular scaffolds for m6A RNA MTase inhibition should aim to increase selectivity, as well as affinity.
KEYWORDS: RNA MTases, methyltransferase, m6A, SAM analogue, inhibitor, RNA binding, TrmK, RlmJ, m1A, bisubstrate analogues
Introduction
The family of S-adenosyl methionine (SAM) dependent methyltransferases (MTases) comprises protein-, RNA- and DNA MTases. These proteins transfer methyl groups to different positions in protein side-chains, RNAs or DNAs, respectively. One subfamily of RNA MTases covers m6A RNA MTases that monomethylate the exocyclic N6-atom of adenine (m6A). This modification is found in all organisms from bacteria to human, and in both mRNA, tRNA, rRNA, small nucleolar RNA, non-coding RNA, and DNA [1,2]. m6A is involved in stabilizing RNA:RNA interactions as well as inducing RNA:protein interactions (recently reviewed in [3,4]).
A number of m6A RNA MTases have been structurally characterized as either full-length protein, e.g. Ribosomal RNA large subunit methyltransferase J (RlmJ) that methylates the 23S rRNA at position A2030 in E. coli [5,6], or as just the catalytic MTase domain as in e.g. eukaryotic methyltransferase like 16 protein (METTL16) that methylates pre-mRNAs, lncRNAs, and other ncRNAs [7–9], or METTL3 and METTL14 from the ternary complex of METTL3:METTL14 and the Wilms tumor 1 associating protein (WTAP) that introduces m6A into mRNAs [10–12]. Of the known m6A RNA MTases, only METTL16 has been crystallised in complex with substrate RNA [13]. m6A RNA MTases are functionally and structurally related to m6A DNA MTases such as T4Dam or M.TaqI, both of which are structurally characterized in complex with substrate DNA (PDB: 1YFL, 1G38) [14,15].
In the absence of a structure of E. coli RlmJ in complex with substrate RNA, biochemical studies were performed to investigate substrate binding. These studies showed that RlmJ can methylate A2030 in a hairpin fragment of 23S rRNA as short as 25 bases, with the same efficiency as for full-length substrate [6]. The m6A2030 modification was suggested to enhance long-range stacking interactions within the rRNA, to increase the overall rRNA stability [2]. RlmJ methylates deproteinized 23S rRNA, indicating that the m6A2030 modification occurs early in the ribosome biogenesis [5]. The protein is none-essential, but has been associated with the ability of bacteria to use DNA as a nutrient [16], and with repression of plasmid uptake [17].
RNA MTases, as well as protein- and DNA MTases, have been linked to various types of cancer (reviewed in [18]) or rare mitochondrial respiratory chain complex deficiencies [19,20]. Of these, only protein- and DNA MTases have so far been targets for drug development. These drugs are either non-nucleosides or explore the scaffold of the methyl-donor cofactor SAM as a starting point for inhibitor design. SAM-derivate inhibitors include e.g. azacytidine (Vidaza, Celgene), decitabine (Dacogen, SuperGen), EPZ004777 [21], and DS-437 [22], the first two of which have been approved for clinical use. Currently, no inhibitors are known for RNA MTases other than the universal nucleoside analogue sinefungin and the natural cofactor product S-adenosyl-homocysteine (SAH).
Based on extensive structural comparison with other MTase families, and emerging evidence for links to human diseases, RNA MTases are increasingly considered promising drug targets [23]. Given the similarities in SAM cofactor binding pockets across MTase families, it was suggested to design drugs that simultaneously exploit the cofactor and substrate binding pockets [23]. Recently, a series of bisubstrate analogue encompassing an analogue of the cofactor SAM covalently linked to the N6-position of an adenosine, were synthesized [24]. These molecules aim to mimic the substrate:cofactor bound state in which a methyl group is transferred from SAM to the adenosine N6-atom of substrate RNA during catalysis by m6A RNA MTases.
As a proof-of-concept, we use the m6A RNA MTase RlmJ as a model to test the binding mode of this series of bisubstrate analogues to assess their use as inhibitors against m6A RNA MTases, and their similarity in binding mode to the substrate:cofactor binding during methyl transfer. We further tested the broader capability of these bisubstrate analogues to bind to an m1A RNA MTase protein (TrmK) related to RlmJ in structure.
Results
The bisubstrate analogues increase the Tm of RlmJ
The bisubstrate analogues mimic the state at which the N6-atom of the substrate adenosine in RNA performs nucleophilic attack on the ϵ C-atom of the transferred methyl group in the natural cofactor SAM (Figure 1A). The synthesis of the bisubstrate analogues [24] entailed joining a cofactor analogue (CA) of SAM (Figure 1B) to the N6-atom of adenosine through a variety of linkers, yielding six molecules; bisubstrate analogue (BA) 1 to 6 (Figure 1C) [24]. To ensure diversity in the chemical space, the linker was synthesized as either a short chain of two C-atoms (Figure 1C, BA1 and −2), a longer chain of three C-atoms (Figure 1C, BA3 and −4), or a urea unit (Figure 1C, BA5 and −6). The bisubstrate analogues were further synthesized with (BA2, −4, −6) or without (BA1, −3, −5) the methionine group present in SAM and SAH. The binding of the CA and BA1-6, as well as the cofactor substrate SAM, cofactor product SAH, and the known inhibitor sinefungin (Figure 1D) to RlmJ, was evaluated using differential scanning fluorimetry (DSF). This technique measures the protein melting temperature (Tm), and a change in protein Tm in the presence of a bisubstrate analogue, implies formation of a protein:compound complex [25]. First, binding of the CA, SAM, SAH, and sinefungin were tested. RlmJ displayed an increased Tm-value with all four molecules, compared to apo protein (Figure 1E). The four molecules vary only by the atom or chemical group at the δ-position: NH-group in CA, S+-CH3 group in SAM, S-atom in SAH, and C-NH2 in sinefungin. The gradual increase in Tm for RlmJ with CA < SAM < SAH < sinefungin indicates that the nature of the δ-atom is important for either direct interaction with the protein or for the ligand to adapt a suitable conformation. Next, the six bisubstrate analogues were tested for binding. These experiments yielded the largest Tm shifts for RlmJ in the presence of the two compounds BA2 and BA4 (Figure 1E), indicating that these ligands form a larger number of stabilizing protein interactions, compared to BA1, −3, −5 and −6. A general Tm comparison of all compounds showed that the methionine part likely is involved in protein binding (BA1, −3, −5 compared to BA2, −4, −6) and that an aliphatic linker is preferred (BA2, −4 compared to BA6) for optimal protein interaction with RlmJ.
Figure 1.
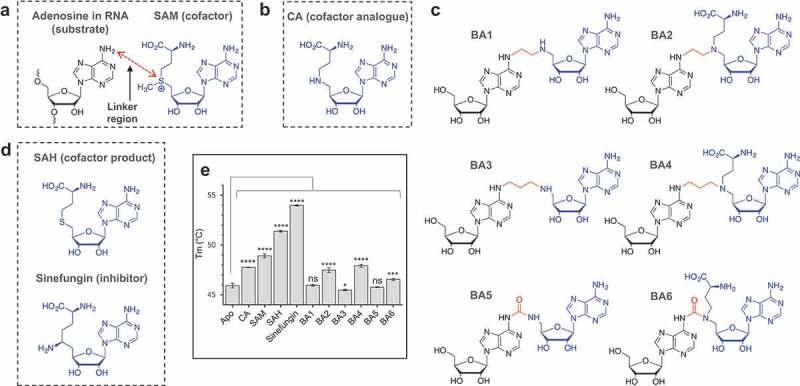
Structure of the bisubstrate analogues and binding to RlmJ. (A) The structure of the substrate adenosine in RNA (black) and natural cofactor SAM (blue). The point of linkage in the bisubstrate analogues (BA) is indicated with a double arrow (red). (B) The structure of the cofactor analogue (CA) used as building block for the BA compounds. (C) A schematic overview of the structures of the six synthesized bisubstrate analogues; BA1 to 6. (D) The structure of the natural cofactor product SAH and universal RNA MTase inhibitor sinefungin. (E) The melting temperature (Tm) for E. coli RlmJ as apo protein (no ligand) or in the presence of CA, SAM, SAH, sinefungin or one of six m6A RNA MTase bisubstrate analogues (BA1-6) from C, measured using differential scanning fluorimetry. P values are indicated as follows: ns = P > 0.05, * = P 0.05, *** = P 0.001, and **** = P 0.0001, n = 3.
BA2 and BA4 are starting scaffolds for adenine-modifying MTase inhibitors
In recent years, MTases have received considerable attention as drug targets due to their link to human disease. The bisubstrate analogues, BA2 and BA4, yielded the largest shift in Tm for RlmJ. To test the relevance of these analogues as inhibitors for RlmJ, KD-values were measured using isothermal titration calorimetry (ITC) for RlmJ in the presence of the SAM, sinefungin, BA2 or BA4. Sinefungin binds RlmJ with a KD-value of 1.7 ± 0.2 μM (Figure 2A) and SAH with a KD of 7.5 ± 0.9 μM (Figure 2B), highlighting the inhibitor potential of sinefungin. The ITC data revealed that the analogues, BA2 and BA4, bind RlmJ with low micro-molar affinity with KD-values of 25 ± 3 μM and 30 ± 4 μM, respectively (Figure 2C,D).
Figure 2.
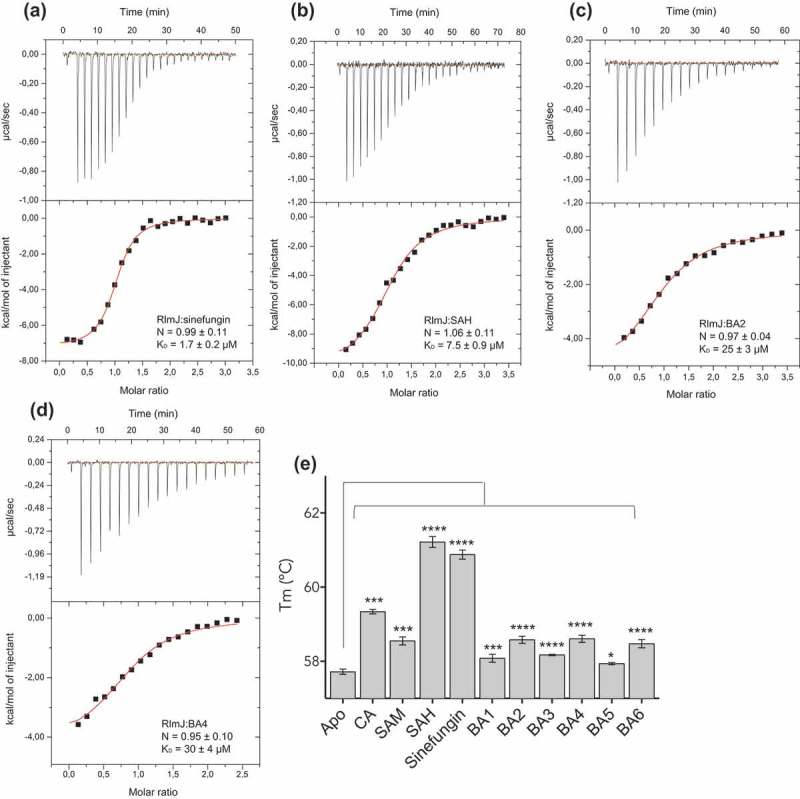
Binding of bisubstrate analogues to RlmJ and TrmK. ITC curves for RlmJ with (A) SAH, (B) sinefungin, (C) BA2, and (D) BA4. Dissociation constant (KD) -and stoichiometry (N) values are reported as mean ± standard deviation (SD). (E) The melting temperature (Tm) of TrmK as apo (no ligand) or in the presence of CA, SAM, SAH, sinefungin or one of six m6A RNA MTase bisubstrate analogues (BA1-6), measured using differential scanning fluorimetry. P values are indicated as follows: * = P 0.05, *** = P 0.001, and **** = P 0.0001, n = 3.
To test the specificity of the bisubstrate analogues against m6A RNA MTases, their binding to another adenine-modifying RNA MTase family was examined. The N1-monomethylation of adenine (m1A) has been extensively studied (reviewed in [26]), and thus we chose the m1A RNA MTase, TrmK, from Mycoplasma capricolum, that catalyzes formation of m1A in position 22 in tRNAs [27]. TrmK is essential for many pathogenic bacteria [28,29], and therefore serves as an appropriate target for inhibitor development. All six bisubstrate analogues, as well as CA, SAM, SAH and sinefungin were tested against TrmK using DSF as described. Tm-values were increased in the presence of the CA, SAM, SAH, and sinefungin (Figure 2E). In contrast to the result for RlmJ, the CA here resulted in a higher ΔTm than SAM, suggesting that the N-atom in this molecule could be directly bound to residues from TrmK but not from RlmJ. Alternatively, binding of the CA could induce stabilizing conformational changes in TrmK, not occurring in RlmJ. For the bisubstrate analogues, BA2, BA4 and BA6, resulted in the largest Tm increase for TrmK (Figure 2E). The binding of BA2 and BA4 increased the Tm for both RlmJ and TrmK, suggesting that these analogues would not be inhibitory specific for m6A RNA MTases, but could instead be used as starting scaffolds for synthesis for both m1A and m6A RNA MTase inhibitors.
Structure determination of RlmJ and Trmk bound to SAH, BA2, or BA4
To understand the Tm and KD differences between bisubstrate analogues, the molecular binding mode of BA2 -or BA4 to protein was investigated. Crystallisation assays were performed with RlmJ or TrmK and each of these analogues. Experiments were also performed for each protein with SAH for later direct comparison with the cofactor part of BA2 and BA4. Crystals were obtained for RlmJ bound to SAH, BA2, or BA4. TrmK crystallised only in the presence of SAH. Structures were solved for RlmJ:SAH to 1.61 Å, RlmJ:BA2 to 1.39 Å, RlmJ:BA4 to 2.10 Å, and TrmK:SAH to 2.36 Å (Table 1), each with one ligand bound per active site. In RlmJ:BA4, the ligand adopted two conformations, where one occupied chain A and C, and one occupied chain B. The ligand in Chain D presented poor electron density and could only be partly fitted. The multiple binding modes for BA4 compared to the single mode for BA2 could explain the lower KD-value determined for BA4, compared to BA2. Interaction surface analysis [30] of the structures suggested, that both RlmJ and TrmK were monomers in solution, confirmed by size exclusion chromatography yielding a monomeric molecular weight (MW) for both proteins (Supplementary Figure 1A). The crystallised RlmJ structures display a class I MTase Rossmann-fold and a helical subdomain inserted at residues 47–98 (Figure 3A), similar to what was previously described [6]. The structure of TrmK:SAH displayed a class-I MTase Rossmann-fold core with an additional C-terminal extension (residues 179–245) (Figure 3B) comparable to TrmK from Bacillus subtilis (PDB: 6Q56). The core class-I Rossmann-fold of TrmK and RlmJ align with an RMSD of 2.9 Å2 over 160 Cα. Such a similarity agrees with the DSF data suggesting that BA2 and BA4 could bind both proteins, and consistently, an alignment of TrmK:SAH with both RlmJ:BA2 and -BA4 shows that both ligands can be spatially accommodated in the active site of TrmK (Supplementary Figure S1B).
Table 1.
Crystallographic data. Processing and refinement statistics for the crystallised RlmJ protein bound to SAH, BA2, or BA4, and TrmK bound to SAH.
| RlmJ:SAH | RlmJ:BA2 | RlmJ:BA4 | TrmK:SAH | |
|---|---|---|---|---|
| Data collection | ||||
| Beamline | ESRF ID23-2 | Soleil PX2 | Soleil PX2 | ESRF ID23-2 |
| Space group | P1 | P 1 21 1 | P 1 21 1 1 |
P 1 21 1 |
| a, b, c | 46.218, 46.224, 71.361 | 47.82, 58.40, 49.23 | 66.42, 170.73, 66.59 | 40.76, 131.09, 58.47 |
| α, β, γ | 79.583, 77.595, 70.349 | 90, 97.32, 90 | 90, 118.32, 90 | 90, 103.88, 90 |
| Molecule/ASU | 2 | 1 | 4 | 2 |
| Wavelength (Å) | 0.87313 | 0.98010 | 0.98007 | 0.87313 |
| Resolution range (Å) | 43.23–1.61 (1.67–1.61) | 37.46–1.39 (1.44–1.39) | 48.24–2.10 (2.18–2.10) | 42.91–2.36 (2.44–2.36) |
| Total reflections | 236,903 (23,207) | 340,809 (32,392) | 428,800 (39,500) | 167,982 (17,199) |
| Unique reflections | 67,854 (6668) | 53,277 (5084) | 75,173 (7404) | 24,468 (2448) |
| Multiplicity | 3.5 (3.5) | 6.4 (6.4) | 5.7 (5.3) | 6.9 (7.0) |
| Completeness (%) | 97.11 (95.82) | 98.68 (92.40) | 99.00 (97.48) | 99.75 (99.43) |
| I/σ(I) | 9.89 (2.26) | 12.71 (0.52) | 5.63 (1.40) | 5.47 (1.09) |
| CC1/2 | 0.997 (0.808) | 0.999 (0.43) | 0.98 (0.675) | 0.996 (0.823) |
| Rmerge(%) | 0.07581 (0.4406) | 0.0759 (2.033) | 0.2072 (0.7769) | 0.2719 (1.5) |
| Structure refinement | ||||
| Rwork (%) | 0.1888 (0.2571) | 0.2244 (0.4486) | 0.2094 (0.3084) | 0.1847 (0.2645) |
| Rfree (%) | 0.2217 (0.3153) | 0.2657 (0.4859) | 0.2512 (0.3084) | 0.2514 (0.3555) |
| RMSDbond (Å) | 0.006 | 0.006 | 0.008 | 0.008 |
| RMSDangle (ᵒ) | 0.94 | 0.91 | 1.06 | 0.91 |
| No. of non-hydrogen atoms | 4735 | 2552 | 9598 | 3964 |
| Macromolecules | 4450 | 2237 | 8699 | 3776 |
| Solvent | 285 | 268 | 724 | 188 |
| Ramachandran favored | 96.81 | 97.45 | 96.84 | 98.21 |
| Ramachandran outliers | 0.00 | 0.00 | 0.00 | 0.00 |
| Rotamer outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 |
| Average B-factor (Å2) of | 17.11 | 30.19 | 29.36 | 41.06 |
| Macromolecules | 16.81 | 29.31 | 28.69 | 41.04 |
| Solvent | 21.88 | 37.66 | 35.14 | 41.46 |
| PDB entry ID | 6QE5 | 6QE0 | 6QDX | 6QE6 |
Figure 3.
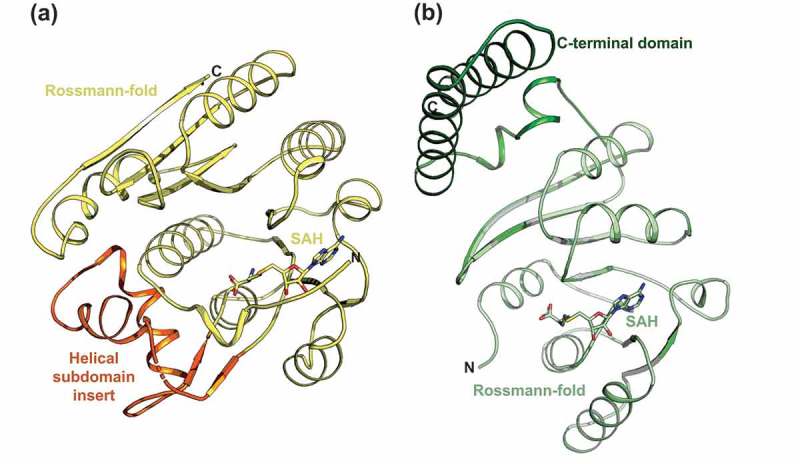
Crystal structures of RlmJ and TrmK. (A) RlmJ bound to SAH. The core Rossman-fold is shown in yellow. The helical subdomain insert is shown in orange. SAH is shown in stick representation in yellow. (B) The crystal structure of TrmK bound to SAH. The core Rossman-fold is shown in light green. The C-terminal domain is shown in dark green. The SAH ligand is shown in stick representation.
The bisubstrate analogues bind in two conformations and near catalytic residues
In the RlmJ:BA4 structure, two distinctively different molecular conformations are observed for the BA4 molecule: a folded conformation, hereon named BA4(a) (Figure 4A,C), and an extended conformation, hereon named BA4(b) (Figure 4B,C). Each conformation was compared to the binding mode of SAH in RlmJ:SAH to analyze their applicability to mimic the substrate:cofactor binding state during methyl transfer. In RlmJ:SAH, the ligand binds in a pocket lined with hydrophobic residues and forms hydrogen bonds to residues H19, H42 and S100 via its methionine part, and E118, D143 and G144 via its adenosine part (Supplementary Figure S2A). Compared to SAH, most parts of BA4(a) binds out of the usual cofactor binding pocket (Figure 4A). π-π stacking of the two adenine moieties induces a folded conformation of the compound that displaces the central δ N-atom 1.7 Å from the expected position. The substrate part and the cofactor adenine of BA4(a) binds in between the cofactor and the expected substrate binding pocket, and as a result, BA4(a) shares only its interaction between the methionine part and residues H19 and H42 with SAH (Supplementary Figure S2C). A similar folded conformation was also assumed by BA2 in RlmJ:BA2 that, however, additionally shares the interaction with S100 with SAH (Supplementary Figure S2B,S3A,B). Contrary to BA4(a), BA4(b) occupies both the presumed substrate binding pockets and parts of the cofactor binding pocket. In fact, relative to a possible real substrate:cofactor binding mode with RNA and SAM, BA4(b) deviates only by the orientation of the adenosine in the cofactor part, which is rotated 120° out of the canonical binding pocket for SAH due to a shift in R/S-configuration around the central δ-atom (Figure 4B). The substrate part of BA4(b) binds in the presumed substrate binding pocket, lined with hydrophobic and aromatic residues, and forms hydrogen bonds with residues N12, K18, and D164 (Supplementary Figure S2D). Two of these residues were shown to be catalytically important: the strictly conserved K18 residue, and the D164 residue from a conserved D/N-P-P-Y/F/W motif [6,15,31]. D164 forms a hydrogen bond of 3.0 Å to the N6-atom of the substrate base, and of 2.8 Å to the cofactor part of BA4(b) (Figure 4D). It is stabilized by a hydrogen bond to K18 (2.7 Å), that further binds to the N1-atom (3.2 Å) of the substrate base (Figure 4D). In this position, D164 could act as the suggested proton acceptor during catalysis by increasing the electron density of the N6-atom [6,15], and K18 is positioned such that it fixates D164 for catalysis. The N6-atom is positioned 2.5 Å away from the ϵ C-atom (Figure 4B). Such a distance, in two non-linked moieties, would allow for an SN2 methyl transfer to the N6-atom after its deprotonation [31].
Figure 4.
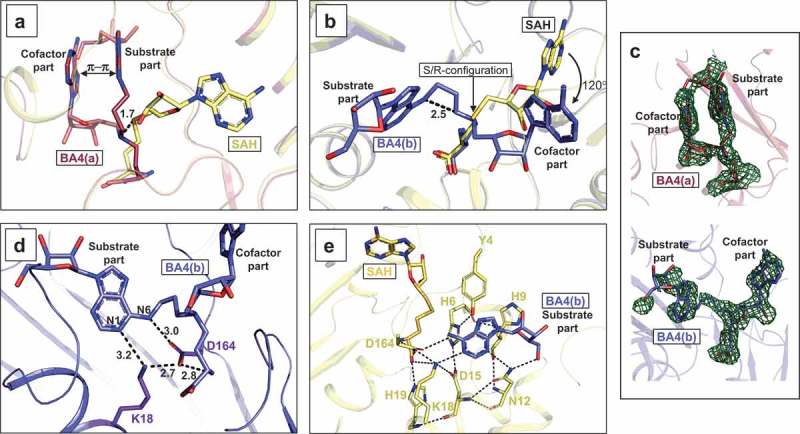
Binding conformations adopted by BA4. (A) RlmJ protein bound to the folded conformation of BA4 (BA4(a)) (red). RlmJ:SAH is aligned with RlmJ:BA4 and shown in yellow. The parts of BA4(b) involved in π-stacking are indicated, as well as the parts of the compound corresponding to the natural substrate -and cofactor. The 1.7 Å displacement of the central δ N-atom from the expected position is indicated in as a dashed black line. (B) As for A, but presenting the extended conformation of BA4 (BA4(b)) (blue). The point of rearrangement from S-configuration in SAH to R-configuration in BA4(b) is indicated, along with the resulting degree of rotation (120°) for the cofactor part of BA4(b). The parts of the compound corresponding to the natural substrate -and cofactor are indicated. The distance between the N6-atom of adenine and the ϵ C-atom from the cofactor part (dashed black line) is given in Å. (C) Fo-Fc of BA4(a) (top, red) and BA4(b) (bottom, blue) contoured at 2.0 σ. The substrate – and cofactor parts are indicated for clarity. (D) RlmJ bound to BA4(b) (blue). Hydrogen bonds are indicated as dashed lines and distances are shown in Å between both the N1-/N6-atoms of the BA4(b) substrate base, the cofactor part of BA4(b) that is bound similarly to SAH, and the catalytically important residues; K18 and D164 (stick representation, purple). BA4(b) is shown in blue stick representation. (E) Superposition of RlmJ:BA4(b) (blue) with RlmJ:SAH (yellow). The substrate base from BA4(b) is fitted into the closed conformation of RlmJ, which is adopted when the protein binds to SAH. Dashed lines indicate the hydrogen bonds formed between different RlmJ residues (stick representation, yellow) in the closed conformation around the substrate base from BA4(b). Distances are shown in Å.
Y4 and H6 from the N-terminal region of RlmJ were also shown to be important for activity, and suggested to contribute to substrate binding [6]. The first 19 residues at the N-terminal are highly flexible and rotate up to 88º between the apo open conformation and the SAH-bound closed conformation [6]. A closed conformation of the N-terminal would sterically clash with parts of the BA molecules, and thus these ligand-bound structures assume the open conformation. For BA4(b), these clashes are solely with the rotated adenosine from the cofactor part, and thus the substrate base alone can be fitted nicely into the closed conformation of RlmJ:SAH (Figure 4E). In this fit, the two residues Y4 and H6 point towards the substrate base. The side-chain O-atom of Y4 was shown to be more important for activity than the aromatic ring [6]. In agreement with this, no π-π stacking occurs between Y4 and the substrate base. Instead, this O-atom is part of an intricate network of hydrogen bonds that ultimately binds to the base via the catalytically important K18 (Y4-H6-D15-L18-base) (Figure 4E). The close proximity of the substrate base from BA4(b) to the catalytically important residues (D164 and K18) of RlmJ, implies, that the extended conformation of BA4(b) resembles the real m6A RNA MTase substrate:cofactor bound state.
Ba4(b) is applicable for studying RNA recognition in all m6a RNA and DNA MTases
Structural similarities of the active site across m6A RNA MTase suggest that the substrate analogues, tested here for RlmJ, could also be used to investigate RNA recognition of other m6A RNA MTases. The crystal structure of the m6A RNA MTase METTL16 has been solved in complex with substrate RNA [13] without the cofactor, and the Rossmann-fold of RlmJ aligns with this protein (PDB: 6DU4) with an RMSD of 2.9 Å2 over 287 Cα[32]. In the METTL16:RNA structure, the substrate adenine binds in a similar plane and position as the substrate adenine in BA4(b) (Figure 5A). In METTL16, the adenine is swapped 180° relative to the BA4(b) base, but the N6-atom to be methylated are located only 1.5 Å apart. Compared to the 4.0 Å distance between the N6-atom and δ N-atom in BA4(b), the N6-atom in METTL16 is at a distance of 4.6 Å to the corresponding S-atom of a modelled SAH molecule (Figure 5A). Apart from similarities at the level of the substrate base, the RNA from METTL16:RNA does not model well onto RlmJ with multiple clashes observed with the core Rossmann-fold. Taken together, a comparison of BA4(b) with RNA-bound METTL16 indicates, that the substrate part of BA4(b) could bind METTL16 in the position of the RNA substrate, and might thus be used broadly to study RNA binding in m6A MTases.
Figure 5.
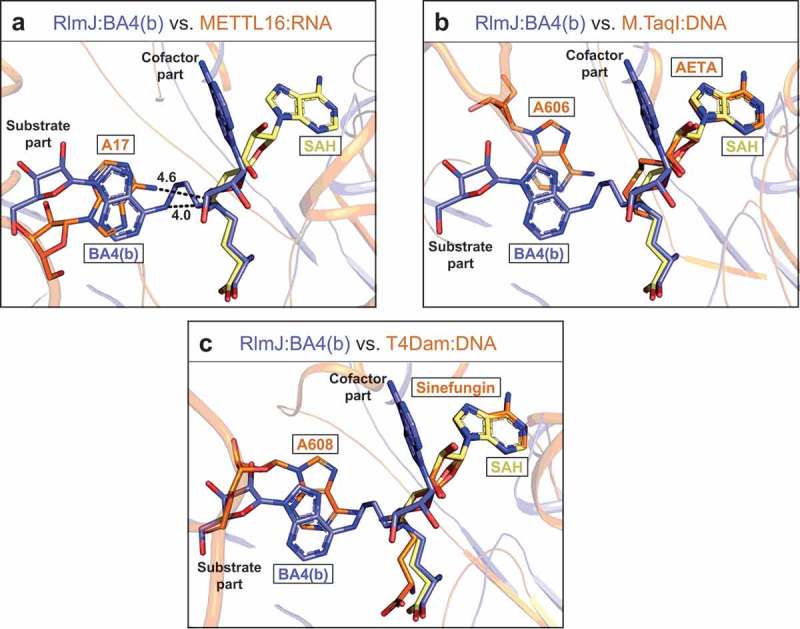
Comparison of RlmJ:BA4(b) with RNA-bound m6A RNA and DNA MTases. (A) Comparison of the position of the substrate base of BA4(b) (blue) from RlmJ:BA4 and the corresponding base (A17) in the human homologue METTL16 co-crystallised with an RNA substrate (PDB: 6DU4) (orange). SAH (yellow) from RlmJ:SAH, aligned with RlmJ:BA4, is shown for comparison. The methylated bases and all cofactors are shown in stick representation. (B) As for A, but compared to the corresponding base (A606) from the DNA co-crystallised with m6A DNA MTase M.TaqI (PDB: 1G38) bound to the cofactor analogue 5′-[2-(amino)ethylthio]-5′-deoxyadenosine (AETA) (orange). (C) As for A, but compared to the corresponding base (A608) from the DNA co-crystallised with m6A DNA MTase T4Dam (PDB: 1YFL) bound to the cofactor analogue sinefungin (orange).
In addition to resembling each other, the active site of m6A RNA MTases also align well with active site components of m6A DNA MTases. Thus, the bisubstrate analogues might also be used to study substrate binding in this subfamily of proteins. A DALI search comparing RlmJ pairwise with the m6A DNA MTases T4Dam (PDB: 1YFL) or M.TaqI (PDB: 1G38) yielded RMSD-values of 3.2 Å2 and 2.8 Å2 over 281 Cα, respectively [32]. T4Dam and M.TaqI were previously crystallised in complex with substrate DNA, and one of the cofactor analogues; sinefungin or 5′-[2-(amino)ethylthio]-5′-deoxyadenosine (AETA), respectively. A superposition of their cofactor analogues (sinefungin, AETA) with SAH and BA4(b) in RlmJ reveals that the substrate adenine of BA4(b) locates in the same area as the substrate base in M.TaqI (Figure 5B) and T4Dam (Figure 5C), resembling most that of T4Dam. Interestingly, in these proteins the adenine base is bound in the same plane as in BA4(b), emphasizing the potential of BA4 to also mimic the substrate:cofactor bound state in DNA MTases, further broadening the applicability of this molecule in DNA binding studies.
Discussion
In this study, we successfully identify two bisubstrate analogues that bind to the active site of RlmJ: BA2 or BA4. ITC data for RlmJ with these compounds showed KD-values in the low micromolar range. The values for BA2 and BA4 were about 15-fold higher than that of sinefungin, but comparable to KD-values found for the dual inhibitor DS-437 of 25 and 30 μM against the human protein MTases hPRMT5:hMEP50 and hPRMT7, respectively [22]. Based on their corresponding IC50-values of 5.9 ± 1.4 μM and 6.0 ± 0.5 μM, and the inactivity of DS-437 towards 29 other human protein-, DNA- and RNA MTases, this probe was deemed a promising template for the development of potent and selective inhibitors for hPRMT5 and hPRMT7. We thus hypothesize that the BA2 and BA4 compounds could similarly serve as template scaffolds for inhibitor design against m6A RNA MTases.
Binding investigation with BA2, BA4 and the m1A RNA MTase protein TrmK revealed a similar pattern for this protein, compared to RlmJ. Thus, inhibitory optimization of these compounds for m6A RNA MTases should aim to increase not just affinity, but also specificity.
For optimization of affinity, some hints were provided from the Tm of RlmJ in the presence of each substrate analogue. Analogues which included the methionine part of the cofactor displayed a larger increase in protein thermostability, compared to analogues lacking this cofactor part. This indicates that the methionine part of the analogues interacts directly with the protein, as confirmed by our RlmJ:BA crystals structures. A similar larger increase was also consistently observed for the longer linker of three C-atoms (BA3,-4) compared to the linker of two C-atoms (BA1,-2), or a urea unit (BA5,-6). Comparison of the structure of BA2 containing a two C-atom linker (folded conformation) with those of BA4 containing a three C-atom linker (folded and extended conformation), indicates that a longer linker is required to allow the molecule to adopt the extended form. We thus suggest that future designs should include the SAM methionine group and a linker of at least three C-atoms length. A successfully optimized compound would ultimately be used either as a small-molecule drug, or as a cell-active chemical compound, to investigate the therapeutic potential of targeting N6-monomethylation pathways. Such a compound becomes even more interesting with the discovery of the human m6A RNA MTase METTL16 and METTL3:METTL14 complex, all related to RlmJ in structure, as these targets have been associated with a number of cancers [18].
Structures of proteins in complex with RNA and cofactors are difficult to obtain for a number of reasons: (1) the often-low protein affinity for the modified base itself excludes the use of just single nucleotides such as AMP or GMP for structural studies, (2) RNA-binding often involves concomitant binding of cofactors, complicating complex reconstitution, (3) the RNA has to be correctly folded and homogeneous for facile crystallisation, and (4) incubation in crystallisation trials requires completely RNase free conditions. SAM-dependent MTases often co-crystallise easily with the cofactor. Therefore, directly linking the substrate base to this moiety at the point of nucleophilic attack to mimic the substrate:cofactor bound state, facilitates structural studies by increasing RNA substrate base affinity, reducing flexibility, and by-passing RNase contamination issues. Using such linked molecules, we solved the co-crystal structure of RlmJ with either of two compounds: BA2 or BA4. Structure analysis revealed that the adenine base in an extended conformation of BA4 likely binds in the real substrate binding pocket for substrate RNA. Thus, this molecule might be used more generally as a tool to determine the substrate binding site in other m6A RNA MTases. Structural similarities between m6A RNA and DNA MTases suggests that the here tested bisubstrate analogues could also be used for m6A DNA MTases, further broadening the applicability of these compounds.
The newly synthesized bisubstrate analogues [24] appear with both the R- and S-configuration with respect to the δ N-atom. The cofactor adenosine of the extended form of BA4 binds outside of the natural cofactor binding pocket due to an R/S-configurational rearrangement. Using a BA containing a carbon instead of the δ N-atom would allow to synthesize BA with S-configuration and should favor the good positioning of the cofactor moiety into the catalytic site of the methyltransferase. A C-atom is already present in the only known RNA MTase inhibitor; sinefungin, which displayed the highest Tm for RlmJ, of all tested compounds. The characteristics of the atom at this position (the δ-atom) was shown throughout our experiments to be generally important for protein thermostability, and thus likely also for the KD.
Overall, we present here the first utilization of bisubstrate analogues to study substrate recognition of m6A MTases. The substrate parts of these molecules occupy the substrate binding pockets and might be used to study substrate binding and mechanisms in protein throughout the m6A RNA and DNA MTase families. Chemistry that allows coupling of the cofactor to different positions in the RNA base or sugar, would facilitate the production of a full co-crystallisation library of MTase bisubstrate analogues, matching each family of proteins catalyzing post-transcriptional modifications. Such a library would strongly forward the fields understanding of substrate recognition and enzymatic mechanisms of these protein families.
Methods/materials
Cloning, expression and purification of RlmJ and TrmK
DNA fragments encoding the entire proteins of RlmJ (residues 1–280) or TrmK (residues 1–245) were subcloned into the pET15b vector bearing an N-terminal His6-tag followed by a thrombin protease cleavage site. Plasmids were transformed into E. coli BL21(DE3) competent cells, cultured in Lysogeny Broth at 37°C, and protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside for 3 h growth at 37°C. Cell pellets from 1–2 L culture were lysed by sonication, clarified by centrifugation, and the clarified lysate was applied to immobilized metal affinity column (IMAC) for purification. After IMAC, TrmK purity necessary for crystallisation assays was reached (Supplementary Figure S4) and TrmK was concentrated before storage at −80°C in a buffer of 50 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, 1 mM dithiothreitol and 1 mM ethylenediaminetetraacetic acid. Further purification of RlmJ was performed by size exclusion chromatography (SEC) on Superdex S75 PG (GE Healthcare). Parts of the expressed protein was further treated with Thrombin protease for 4 h at 22°C to remove the His6-tag and purified by SEC on Superdex S75 PG (GE Healthcare). After these steps, RlmJ purity necessary for crystallisation assays was reached (Supplementary Figure S4). RlmJ was concentrated and stored at −80°C in a buffer of 20 mM Tris pH 7.0, 150 mM NaCl, 5% glycerol and 5 mM β-mercaptoethanol.
Analytical size exclusion chromatography (analytical SEC)
100 μL protein sample of 90 μM for RlmJ and 30 μM for TrmK was injected onto a 24 mL Superdex Increase S75 10/300 GL column (GE Healthcare). MW standards from Bio-Rad containing thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (13.5 kDa) were used for calibration. MWs for RlmJ and TrmK were determined from a linear plot of LOG [10](MW) with the MW of each Bio-Rad standard proteins against their elution volume.
Differential scanning fluorimetry (DSF)
DSF was performed in a 96-well plate using a CFX96Touch real-time PCR detection system (Bio-Rad) with excitation and emission filters of 450–490 and 515–530 nm, respectively. Each well consisted of 2 µL protein in a buffer of 20 mM HEPES pH 7.5, 150 mM NaCl to a final concentration of 5 µM, 2 µL of SYPRO ORANGE diluted 5000-fold in buffer from the manufacturer’s stock (Invitrogen), and (if applicable) 2 µL ligand to 1 mM. Fluorescence intensities were measured from 25 to 85°C with a ramp rate of 1°C/min. Tm was determined by curve-fitting using GraphPad Prism v.5.01 software [25]. Data were evaluated using a standard P-test according to standard procedure performed using the GraphPad Prism v.5.01 software.
Isothermal titration calorimetry (ITC)
ITC measurements for the binding affinities between RlmJ and SAH, sinefungin, BA2 or BA4 were performed at 25°C using an ITC200 titration calorimeter (MicroCal/Malvern Panalytical). The molecules were dissolved in protein buffer of 20 mM Tris pH 7.0, 150 mM NaCl, 5% glycerol and 40 μL of this was placed in a syringe. The molecules were titrated into 200 μL of the protein in the cell. The first injection (0.2 μL) was followed by 23 injections for SAH and sinefungin and 19 injections for BA2 and BA4, each of 1.6 µL or 2 µL, respectively. The heat of dilution for the molecules was measured for background subtraction. The titration curves were analyzed using the Origin software (MicroCal) with a single binding-site model, after exclusion of the first injection point. Standard errors were estimated from the data spread and from the uncertainty of the titrant concentration determination as previously described [33]. Dissociation constant (KD) -and stoichiometry (N) values are reported as mean ± standard deviation (SD).
Crystallisation, data collection, and structure determination
Crystals of RlmJ (with BA2, BA4) or His6-RlmJ (with SAH) were grown by vapor diffusion at 20°C, from sitting drops composed of 200 nL of protein and 40 μL of reservoir solution. For His6-RlmJ bound to SAH, the protein concentration was 13 mg/ml; pre-incubated with 760 µM SAH and crystals grew with a reservoir solution containing 0.1M KSCN, 30% (w/v) polyethylene glycol methyl ether (PEG MME) 2000. RlmJ bound to BA2 crystallised with a protein concentration of 9 mg/ml, pre-incubated with 2.2 mM BA2 in a reservoir solution containing 0.1M BIS-Tris pH 6.5, 25% (w/v) PEG3350. RlmJ bound to BA4 crystallised with a protein concentration of 9 mg/ml, pre-incubated with 2.7 mM BA4 in a reservoir solution of 0.1 M MMT pH 6.0, 25% (w/v) PEG1500. For TrmK bound to SAH, crystals were grown by vapor diffusion at 20°C, from sitting drops composed of 150 nL of protein and 40 μL of reservoir solution, with a protein concentration of 1.1 mg/ml, pre-incubated with 2 mM SAH in a reservoir solution of 0.2 M Li2SO4, 0.1 M HEPES pH 7.5, 25% (w/v) PEG3350. Crystals were cryoprotected with reservoir solution supplemented with 15% (v/v) glycol, and flash-cooled in liquid nitrogen. Diffraction data were collected on the Synchrotron Soleil beamline PX2 to 1.39 Å for RlmJ:BA2 and 2.10 Å for RlmJ:BA4, or at the European beamline ID23-2 to 1.61 Å for RlmJ:SAH and 2.36 Å for TrmK:SAH. Phases were determined by molecular replacement in PHASER [34] in the CCP4 Suite of programs [35], using an existing RlmJ structure as search model (PDB: 4BLV) for RlmJ complexes, and TrmK from Bacillus subtilis (PDB: 6Q56) for TrmK. Modelling and refinement were carried out using Refmac [36], COOT [37] and Phenix [38]. Density was visible for all residues aside from the tag and loopy parts covering residues 52–58 and 231–233 in chain A of RlmJ:SAH, residues 52–58 in chain B of RlmJ:SAH, residues 52–56 in RlmJ:BA2, and residues 1–2 and 53–57 in all chains of RlmJ:BA4.
Funding Statement
This work was supported by the Centre National de la Recherche Scientifique [UMR8261]; Labex Dynamo [UMR8261].
Acknowledgments
We thank the staff at the European Synchrotron Radiation Facility (ESRF) beamline ID23-2 and the Synchrotron Soleil beamline Proxima-2 (PX2) for support during data collection. We further thank the Centre national de la recherche scientifique (CNRS) and LabEx DYNAMO for project funding.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data Availability
Atomic coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank (PDB) under accession number 6QE5, 6QE0, 6QDX, 6QE6.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Boccaletto P, MacHnicka MA, Purta E, et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46:D303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cantara WA, Crain PF, Rozenski J, et al. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sergiev PV, Golovina AY, Osterman IA, et al. N6-methylated adenosine in RNA: from bacteria to humans. J Mol Biol. 2016;428:2134–2145. Internet. [DOI] [PubMed] [Google Scholar]
- [4].Huang J, Yin P.. Structural insights into N6-methyladenosine (m6A) modification in the Transcriptome. Genomics Proteomics Bioinf [Internet] 2018; 16:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Golovina AY, Dzama MM, Osterman IA, et al. The last rRNA methyltransferase of E. coli revealed: the yhiR gene encodes adenine-N6 methyltransferase specific for modification of A2030 of 23S ribosomal RNA. RNA. 2012;18:1725–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Punekar AS, Liljeruhm J, Shepherd TR, et al. Structural and functional insights into the molecular mechanism of rRNA m6A methyltransferase RlmJ. Nucleic Acids Res. 2013;41:9537–9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Warda AS, Kretschmer J, Hackert P, et al. Human METTL16 is a N 6 -methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. [Internet] 2017; 18:2004–2014. Available from: http://embor.embopress.org/lookup/doi/10.15252/embr.201744940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pendleton KE, Chen B, Liu K, et al. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169:824–835.e14. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ruszkowska A, Ruszkowski M, Dauter Z, et al. Structural insights into the RNA methyltransferase domain of METTL16. Sci Rep. 2018;8:1–13. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell [Internet] 2016; 63:306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Śledź P, Jinek M. Structural insights into the molecular mechanism of the m6A writer complex. Elife. 2016;5:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang X, Feng J, Xue Y, et al. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–578. [DOI] [PubMed] [Google Scholar]
- [13].Doxtader KA, Wang P, Scarborough AM, et al. Structural basis for regulation of METTL16, an S-adenosylmethionine homeostasis factor. Mol Cell. [Internet] 2018; 71:1001–1011.e4. Available from: https://linkinghub.elsevier.com/retrieve/pii/S109727651830594X [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Horton JR, Liebert K, Hattman S, et al. Transition from nonspecific to specific DNA interactions along the substrate-recognition pathway of Dam methyltransferase. Cell. 2005;121:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goedecke K, Pignot M, Goody RS, et al. Structure of the N6-adenine DNA methyltransferase M center dot Taql in complex with DNA and a cofactor analog. Nat Struct Biol. 2001;8:121–125. [DOI] [PubMed] [Google Scholar]
- [16].Palchevskiy V, Finkel SE. Escherichia coli competence gene homologs are essential for competitive fitness and the use of DNA as a nutrient. J Bacteriol. 2006;188:3902–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Matsuda A, Kurono N, Kawano C, et al. Genome-wide screen for Escherichia coli genes involved in repressing cell-to-cell transfer of non-conjugative plasmids. Biochem Biophys Res Commun. 2012;428:445–450. [Internet]. [DOI] [PubMed] [Google Scholar]
- [18].Boriack-Sjodin PA, Ribich S, Copeland RA. RNA-modifying proteins as anticancer drug targets. Nat Rev Drug Discov. 2018;17:435–453. Internet. [DOI] [PubMed] [Google Scholar]
- [19].Metodiev MD, Thompson K, Alston CL, et al. Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am J Hum Genet. 2016;98:993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Van Haute L, Dietmann S, Kremer L, et al. Deficient methylation and formylation of mt-tRNAMet wobble cytosine in a patient carrying mutations in NSUN3. Nat Commun. 2016;7:1–10. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smil D, Eram MS, Li F, et al. Discovery of a dual PRMT5-PRMT7 inhibitor. ACS Med Chem Lett. 2015;6:408–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schapira M. Structural chemistry of human RNA methyltransferases. ACS Chem Biol. 2016;11:575–582. [DOI] [PubMed] [Google Scholar]
- [24].Atdjian C, Iannazzo L, Braud E, et al. Synthesis of SAM-adenosine conjugates for the study of m6A-RNA methyltransferases. European J Org Chem. 2018:4411–4425 Internet. doi: 10.1002/ejoc.201800798 [DOI] [Google Scholar]
- [25].Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. [Internet] 2007; 2:2212–2221. Available from. http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=17853878&retmode=ref&cmd=prlinks [DOI] [PubMed] [Google Scholar]
- [26].Oerum S, Dégut C, Barraud P, et al. m1A post-transcriptional modification in tRNAs. Biomolecules. [Internet] 2017; 7:E20 Available from. http://www.mdpi.com/2218-273X/7/1/2028230814 [Google Scholar]
- [27].Roovers M, Kaminska KH, Tkaczuk KL, et al. The YqfN protein of Bacillus subtilis is the tRNA: m1A22 methyltransferase (TrmK). Nucleic Acids Res. 2008;36:3252–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thanassi JA, Hartman-Neumann SL, Dougherty TJ, et al. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 2002;30:3152–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hutchison III CA, Chuang R, Noskov VN, et al. Design and synthesis ofa minimal bacterial genome. Synth Biol. 2016;351:1–12. [DOI] [PubMed] [Google Scholar]
- [30].Krissinel E, Henrick K. Interference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. [DOI] [PubMed] [Google Scholar]
- [31].Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol. 1995;253:618–632. [DOI] [PubMed] [Google Scholar]
- [32].Holm L, Laakso LM. Dali server update. Nucleic Acids Res. 2016;44:W351–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tellinghuisen J, Chodera JD. Systematic errors in isothermal titration calorimetry: concentrations and baselines. Anal Biochem [Internet] 2011; 414:297–299. [DOI] [PubMed] [Google Scholar]
- [34].McCoy AJ, Grosse-Kunstleve RW, Adams PD, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D Biol Crystallogr. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Vagin AA, Steiner RA, Lebedev AA, et al. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2184–2195. [DOI] [PubMed] [Google Scholar]
- [37].Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- [38].Afonine PV, Grosse-Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr Sect D Biol Crystallogr. 2012;68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank (PDB) under accession number 6QE5, 6QE0, 6QDX, 6QE6.