

ABSTRACT
The clustered regularly interspaced short palindromic repeat (CRISPR) system is a prokaryotic adaptive defense system against foreign nucleic acids. In the methanoarchaeon Methanosarcina mazei Gö1, two types of CRISPR-Cas systems are present (type I-B and type III-C). Both loci encode a Cas6 endonuclease, Cas6b-IB and Cas6b-IIIC, typically responsible for maturation of functional short CRISPR RNAs (crRNAs). To evaluate potential cross cleavage activity, we biochemically characterized both Cas6b proteins regarding their crRNA binding behavior and their ability to process pre-crRNA from the respective CRISPR array in vivo. Maturation of crRNA was studied in the respective single deletion mutants by northern blot and RNA-Seq analysis demonstrating that in vivo primarily Cas6b-IB is responsible for crRNA processing of both CRISPR arrays. Tentative protein level evidence for the translation of both Cas6b proteins under standard growth conditions was detected, arguing for different activities or a potential non-redundant role of Cas6b-IIIC within the cell. Conservation of both Cas6 endonucleases was observed in several other M. mazei isolates, though a wide variety was displayed. In general, repeat and leader sequence conservation revealed a close correlation in the M. mazei strains. The repeat sequences from both CRISPR arrays from M. mazei Gö1 contain the same sequence motif with differences only in two nucleotides. These data stand in contrast to all other analyzed M. mazei isolates, which have at least one additional CRISPR array with repeats belonging to another sequence motif. This conforms to the finding that Cas6b-IB is the crucial and functional endonuclease in M. mazei Gö1.
Abbreviations:
sRNA: small RNA; crRNA: CRISPR RNA; pre-crRNAs: Precursor CRISPR RNA; CRISPR: clustered regularly interspaced short palindromic repeats; Cas: CRISPR associated; nt: nucleotide; RNP: ribonucleoprotein; RBS: ribosome binding site
KEYWORDS: Methanoarchaea, CRISPR-Cas system, Cas6 endonucleases, immunity of prokaryotes, regulatory RNA, Methanosarcina mazei
Introduction
All prokaryotic genomes are potential targets of invading mobile genetic elements like phages, viruses, transposable elements or plasmids. An adaptable prokaryotic defense system against those elements is the clustered regularly interspaced short palindromic repeat (CRISPR) system. Headed by a leader region, CRISPR-derived small RNAs (crRNAs) are encoded as a long precursor RNA in the CRISPR array. Within the CRISPR array, the characteristic short palindromic repeats are separated by short non-repetitive spacer sequences derived from previous offenses of foreign nucleic acids that specify the targets of CRISPR interference. The number of spacer and direct repeats in one CRISPR locus can vary considerably from one organism to another and reflects the history of the CRISPR-based immunity. Next to the CRISPR array CRISPR-associated (Cas) proteins, often organized in an operon, are located forming together the CRISPR locus. Based on the combinatory set of cas genes and protein homology, CRISPR-Cas systems are grouped into two different classes based on their RNA effector complex. These two classes harbour six main types (types I-VI), which are further divided into different subtypes [1,2]. All CRISPR types rely on similar mechanisms that are grouped into the following phases: Acquisition of new spacers into the CRISPR array (adaptation phase), generation and maturation of the small interfering RNAs (crRNAs) (transcription – and processing phase) and degradation of invading foreign nucleic acids by a crRNA effector complex (interference phase).
The endonuclease Cas6 is responsible for the crRNA biogenesis in most of type I and type III CRISPR-Cas systems. It cleaves the long precursor crRNA into the functional small crRNAs, which together with other Cas proteins form the interference complex. This ribonucleoprotein (RNP) complex is responsible for the degradation of foreign nucleic acids. Cas6-binding to the repeat RNA within the precursor crRNA often occurs at stem-loop structures formed by the repeat RNA due to its pseudo-palindromic sequence and cleavage takes place at the base of these stems. When investigating different Cas6-associated repeats, however, one surprisingly finds that the palindromic character is not mandatory and nearly half of all repeats are supposed to be unstructured. Thus, their recognition by Cas6 has to be carried out in different ways. Cleavage of Cas6 results in a crRNA containing distinct 5ʹ-handle of the repeat followed by the complete spacer sequence. The 3ʹ end of the small crRNAs varies in size among different CRISPR subtypes ranging from repeat-derived hairpin structure to a much shorter and variably trimmed 3ʹ end. In some CRISPR-Cas systems (subtype I-E and I-F), Cas6 stays bound to its cleaved crRNA and is therefore part of the interference complex, whereas in other subtypes (I-A, I-B, I-D and most subtype III) Cas6 dissociates after cleavage. Whether Cas6 stays bound to the crRNA and gets incorporated into the RNP complex can be correlated with the occurrence of a 3ʹ-hairpin structure [3].
The mesophilic methanoarchaeon Methanosarcina mazei Gö1 is able to utilize a wide spectrum of carbon sources for methanogenesis and plays a special ecological role due to its contribution to methane production [4]. It encodes two CRISPR-Cas systems which have been recognized as a subtype I-B and subtype III-C system (former subtype III-B) based on the recent nomenclature by Vestergaard et al. [5], Makarova et al. [1] and Koonin et al. [6]. Both CRISPR loci encode a Cas6 endonuclease with most structural identity to Cas6b from Methanococcus maripaludis for each protein. They were designated as Cas6b-IB (MM_0560) and Cas6b-IIIC (MM_3359, former Cas6b-IIIB) reflecting their originating CRISPR loci [7]. Both CRISPR arrays share nearly the same repeat sequence. They only differ in two nucleotides and the formation of a stable hairpin structure was identified in vitro for the repeat RNA. It was further demonstrated that each Cas6b endonuclease has the ability to specifically cleave the M. mazei repeat RNA in vitro at the same cleavage site within the repeat [7]. Here, we studied crRNA biogenesis in M. mazei by the two Cas6b endonucleases and addressed potential cross-cleavage activity using genetic and biochemical approaches.
Results
Biochemical characterization of both Cas6b endonucleases
The two Cas6b endonucleases, Cas6b-IB and Cas6b-IIIC, were heterologously expressed in Escherichia coli and fused to the maltose-binding protein (MBP) resulting in soluble proteins of total calculated masses of 67.6 kDa (MBP-Cas6b-IB) or 70 kDa (MBP-Cas6b-IIIC), respectively. By size exclusion chromatography analysis, both purified proteins were eluted with high molecular masses of over 2,000 kDa from a Superose 12 column (Figure 1). In control experiments, purified MBP migrated beside its monomeric state also up to 50% as an oligomeric protein with over 2,000 kDa (Figure S1). Thus, the oligomerization of the Cas6b fusion proteins is most likely due to their N-terminal fusion to MBP. To determine whether binding of the repeat RNA influences the oligomeric state of the Cas6b proteins, both MBP-Cas6b endonucleases were pre-incubated with a non-cleavable deoxy-repeat-IB variant prior to size exclusion chromatography. Since the repeat RNA with a dGTP at the identified cleavage site cannot be cleaved, Cas6b endonucleases should stay bound [7]. Analyzing such repeat-bound Cas6b proteins by size exclusion chromatography on a Superose 12 column revealed a second peak for each of the two proteins, eluting after 14.5 ml (MBP-Casb-IB) and 14.4 ml (MBP-Cas6b-IIIC), respectively (see Figure 1). We propose that due to binding the deoxy-repeat variant the conformation of the Cas6b proteins changes and the MBP-fusion is cleaved off. Thus, we consider the peak at 14.5 ml for Cas6b-IB as a complex of untagged monomeric Cas6b-IB protein binding the repeat-RNA and the peak at 14.4 ml (Cas6b-IIIC) as untagged monomeric Cas6b-IIIC bound to the repeat-RNA. Binding of the CRISPR repeat RNA to monomeric Cas6b-IB and Cas6b-IIIC proteins was confirmed by northern dot blot analysis after extracting potentially bound RNA from each elution fraction and probing against the repeat-IB. Repeat-IB was only detectable in protein fractions corresponding to the monomeric proteins and not in fractions belonging to the respective protein aggregations (Figure 1).
Figure 1.
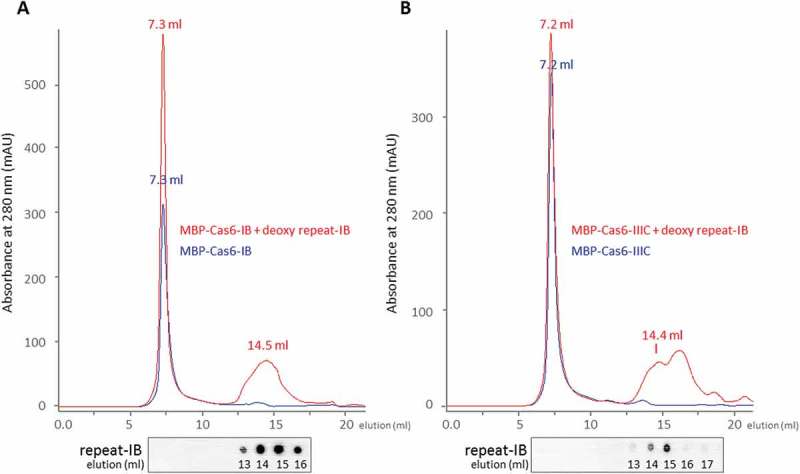
Size-exclusion chromatography of the Cas6b endonucleases of M. mazei Gö1 in the absence and presence of repeat RNA substrate.
Size-exclusion chromatography on a Superose12 column (GE Healthcare) was performed with MBP-fusion protein Cas6b-IB (a) and Cas6b-IIIC (b) from M. mazei Gö1 (17 µM). Absorbance at 280 nm is shown for the respective proteins in blue and in red for a sample containing the respective protein pre-incubated in the presence of the deoxy-repeat-IB variant (5 µM). Gel filtration was performed with a linear flow rate of 0.5 ml/min using buffer A (see Material and Methods) and elution volume (ml) is indicated. Potential co-eluting RNA was identified via northern dot blot analysis using a 5ʹ radioactive labeled repeat-IB probe. Calibration of the column was performed using the gel-filtration mass standard (Bio-Rad Laboratories) containing thyroglobulin (670 kDa), IgG (158 kDa), ovalbumin (44 kDa), myoglobulin (17 kDa) and vitamin B12 (1.35 kDa).
To confirm the results of the size exclusion chromatography, we also performed this experiment with the His-tagged version of Cas6b-IIIC. Examining purified His-tagged Cas6b-IIIC in absence of RNA, the protein elutes at 17 ml corresponding to the monomeric state of the protein (see Figure S2). In the presence of the deoxy-repeat-IB most of the Cas6b-IIIC protein elutes at 14.8 ml and only low amounts at 16.8 ml, arguing that most of the His-tagged Cas6b-IIIC forms a complex with the RNA with a higher molecular mass. Analyzing the respective individual fractions of the elution peaks confirmed the presence of the His-tagged Cas6b by SDS PAGE (silver staining) and the repeat-RNA by northern dot blot analysis (Figure S2). Taking together our findings strongly suggests that Cas6b-IB and Cas6b-IIIC apparently bind the RNA as a monomeric protein with no evidence for dimerization.
Analyzing crRNA processing in deletion mutants of both Cas6b endonucleases
Both Cas6b endonucleases in M. mazei have been demonstrated to specifically cleave the CRISPR repeat RNA in vitro at the same cleavage site [7]). Here, the specific activity of both purified his-tagged Cas6b variants was another time analyzed with in vitro cleavage assays (using subtype IB and IIIC repeat RNA) revealing that in vitro Cas6b-IIIC displays approximately five times higher specific activity (3,487 ± 1,719 U/mg) than Cas6b-IB (627+/-127 U/mg) (see Figure S3, data on IIIC repeat not shown). However, the in vivo specificity and specific activity of their cleavage activity remains unclear. To elucidate the crRNA processing of both CRISPR arrays in vivo, we generated independent single deletion mutants of both genes encoding Cas6b endonucleases by replacing the respective genes with a puromycin resistance cassette (see Materials and Methods). The respective crRNA processing was analyzed by northern blot using a probe directed against the repeat-IB.
Previous studies showed that under high salt stress (500 mM NaCl) the overall amount of crRNAs increased significantly [7], thus high salt conditions were additionally tested. For both conditions we obtained evidence showing that in the absence of cas6b-IB (∆cas6b-IB) crRNA processing did not occur, since no small crRNAs or intermediate crRNAs were detected (Figure 2). In contrast, the absence of cas6b-IIIC (∆cas6b-IIIC) had no effect on the maturation of crRNAs and reflected wild type (wt) conditions for both CRISPR arrays. The high similarity of the repeats of both CRISPR arrays does not permit distinction between the two arrays by repeat specific probes. Thus, crRNA maturation of the two different CRISPR arrays was analyzed by an RNA-Seq approach, using RNA isolated from cultures grown under high salt conditions (500 mM NaCl). Total RNA from the wt strain and the two deletion strains ∆cas6b-IB and ∆cas6b-IIIC, was enriched for transcripts that possess 5ʹ-hydroxy and 3ʹ phosphate termini by T4 polynucleotide kinase (PNK) to phosphorylate all 5ʹ ends each in two biological replicates. The crRNAs and their respective processing patterns were also analyzed for two biological replicates; one is exemplarily shown in Figure 3 for each CRISPR subtype. The abundance of individual crRNAs in the wt strain for both CRISPR arrays confirmed previous findings [7] with a higher amount of crRNAs in the front and less abundant ones in the rear part of both CRISPR arrays. In case of the ∆cas6b-IIIC strain, the same abundance pattern of the different crRNAs in the subtype I-B and the III-C CRISPR arrays occurred. Only the amount of sequencing reads for the respective small crRNAs was reduced about approximately 10% (see Figure 3). In contrast, in the ∆cas6b-IB deletion mutant only a very limited number of crRNAs (~ 20) was detectable at the very beginning of both arrays. Apart from that, no processed crRNAs were detected for both CRISPR arrays, indicating that Cas6b-IB is the main endonuclease responsible for in vivo crRNA processing of the two CRISPR-Cas systems I-B and III-C in M. mazei. Further analysis of the crRNA processing on the single nucleotide level for both deletion mutants (Figure 4), revealed that the precise processing site of the crRNAs does not differ between the wt and the ∆cas6b-IIIC mutant. This in vivo finding strongly argues for cleavage activity of Cas6b-IB for both CRISPR-Cas systems in M. mazei under standard and salt stress conditions.
Figure 2.
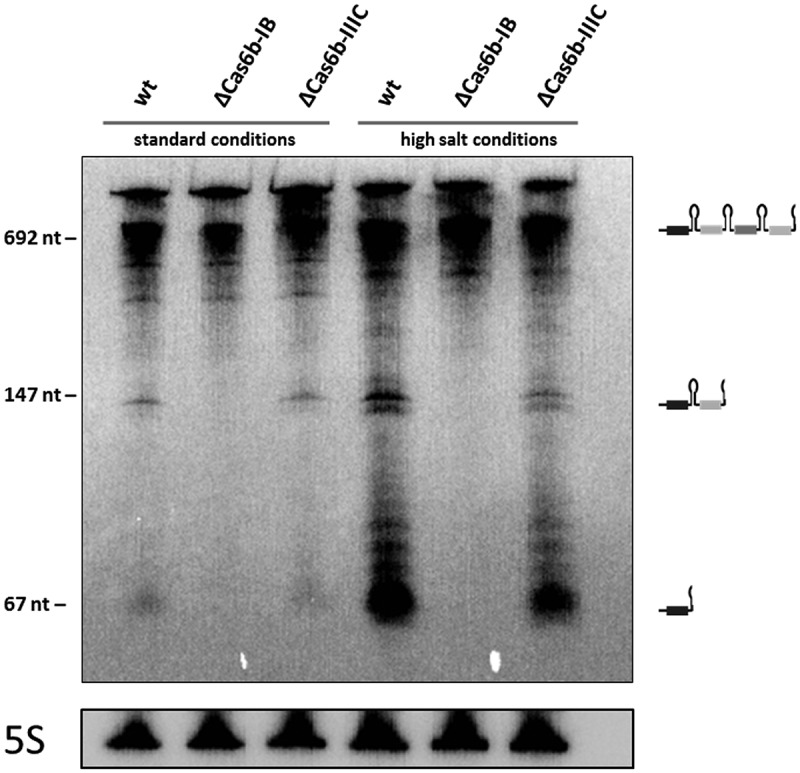
crRNA abundance in M. mazei cas6b deletion strains.
Total RNA was isolated from the M. mazei wild type strain and the deletion strains ∆cas6b-IB and ∆cas6b-IIIC growing under standard and high salt conditions (500 mM NaCl) as described in the Materials and Methods. Subsequent northern blot analysis was performed, using a 5ʹ radioactively labeled subtype IB-repeat probe. In addition, the expression of 5S rRNA of the respective RNA preparation is shown. The pUC mix8 standard marker (Thermo Scientific) was used (fragment sizes are indicated at the left side). On the right side, observed crRNA processing is depicted schematically.
Figure 3.
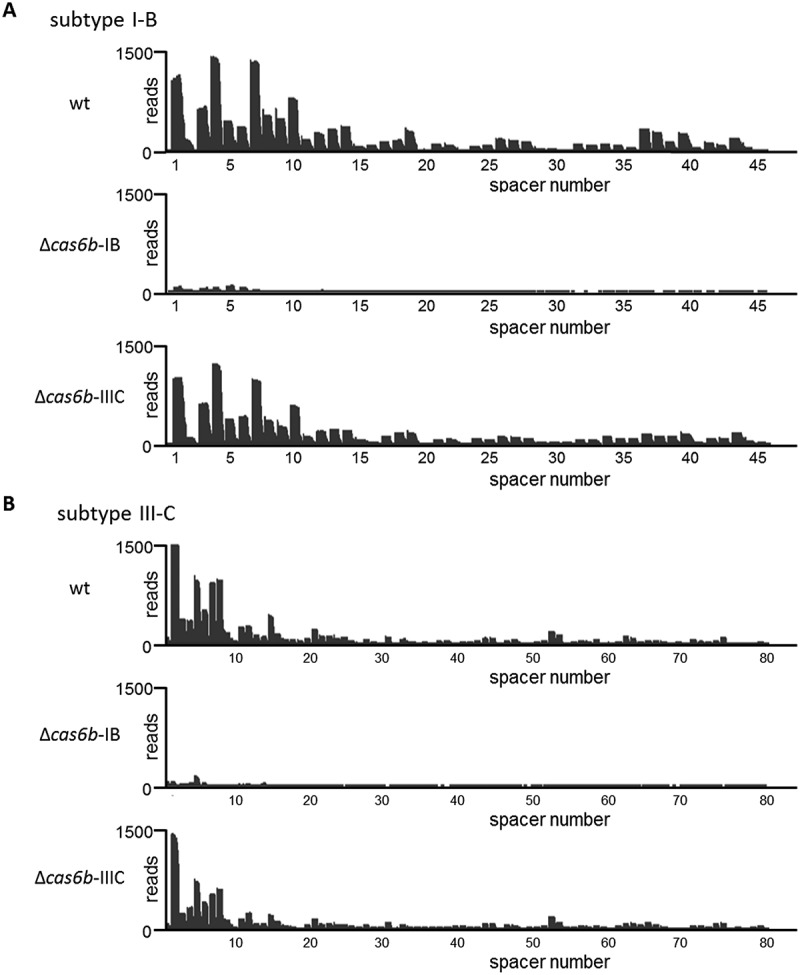
crRNA processing in M. mazei wt and cas6b deletion strains for CRISPR-Cas subtypes I-B and III-C analyzed by an RNA-Seq approach.
Normalized sequencing reads (HiSeq) for the CRISPR array subtype I-B (a) and subtype III-C (b) regions are mapped to the M. mazei genome. Comparison of analyzed RNA-Seq reads derived from exponentially growing cultures from wt and respective M. mazei deletion strains ∆cas6b-IB and ∆cas6b-IIIC. Cells were grown under high salt conditions (500 mM NaCl) and total RNA was treated with T4 polynucleotide kinase (PNK) as described in Materials and Methods.
Figure 4.

crRNA 5ʹ and 3ʹ end processing on single nucleotide level in the M. mazei cas6b deletion strains.
Normalized RNA-Seq reads for the CRISPR array subtype I-B and subtype III-C are mapped to the M. mazei genome. Comparison of analyzed RNA-Seq reads derived from the wt and from the M. mazei deletion strains ∆cas6b-IB and ∆cas6b-IIIC. Cells were grown under high salt conditions (500 mM NaCl) as described in Materials and Methods. Depicted is the crRNA containing spacer 4 from the subtype-IB array and the crRNA for the spacer 8 belonging to the subtype-IIIC array. The crRNA processing is shown on single nucleotide level, with one bar representing a single nucleotide. The position of spacer (grey box) and repeat (dark grey line) sequences are indicated according to the RNA-Seq reads.
Expression patterns
To clarify whether different expression levels of both endonucleases were responsible for the highly reduced activity of Cas6b-IIIC in pre-crRNA cleavage, the absolute transcript numbers of both cas6b endonucleases in the wt were determined by quantitative (q) RT-PCR (Figure S4). Although the absolute transcript numbers varied between the three biological replicates due to slight differences in time points of cell harvest or cell fitness, there were no significant differences between the two genes encoding endonucleases cas6b-IB and cas6b-IIIC. Furthermore, the deletion of one of the two cas6b genes did not affect the transcription of the particular other nor the crRNA of subtype I-B or III-C under either standard or high salt conditions (Figures S5 and S6). In general, no significant changes in the transcript levels were observed under high salt stress (Figure S5), excluding that the experimental setup has an impact on the results obtained. In addition, assessment of the different Cas proteins present in M. mazei wt cells was determined by re-analyzing the proteomic data sets from Cassidy et al. [8]. In this study, Tholey and colleagues combined Bottom-up (2D-LC-MS) and Semi-top-down (GelFree–LC-MS) methodologies to provide a comprehensive qualitative assessment of the detectable proteins in M. mazei Gö1. For a protein to be clearly identified it requires the presence of at least two peptides of high confidence (< 1% FDR), of which at least one has to be unique. Under these strict criteria, Cas8 (MM_0564), Cas7 (MM_0563) and Cas5 (MM_0562) were identified for the subtype I-B CRISPR locus. In addition, Cas10 (MM_3356), Cmr4 (MM_3355), SS (MM_3354) and Cas5 (MM_3353) were identified for the subtype III-C CRISPR locus. Although tentative (medium confidence), evidence for all other proteins for the type I-B and III-C system were found with the exception of the adaption protein Cas2 (MM_0558) from the type I-B CRISPR locus for which no evidence was identified at all (Table 1).
Table 1.
Detection of Cas proteins by LC-MS/MS analysis. Protein extracts (standard growth conditions) were analyzed by 2DLC-MS/MS and GelfreeLC-MS/MS analysis by Cassidy et al. [8]. Depicted are the merged identification results for Cas proteins of the I-B and III-C CRISPR loci. For a protein to be classified as identified, it required the presence of at least two peptides of high confidence (< 1% false discovery rate) of which at least one was unique. Proteins that met the criteria are shaded in grey. Medium confidence (< 5% false discovery rate) search settings were utilized to enable a more complete picture of the possible proteins present within the samples. Cas6b proteins are highlighted in bold.
| Protein Name | ORF Number | Σ Coverage | Σ# Peptides (Unique) | High (medium) Confidence |
|---|---|---|---|---|
| Cas4 | MM_0557 | 22.11 | 3 (2) | 0 (3) |
| Cas2 | MM_0558 | - | - | - |
| Cas1 | MM_0559 | 7.74 | 3 (3) | 1 (2) |
| Cas6bIB | MM_0560 | 2.78 | 1 (1) | 0 (1) |
| Cas3 | MM_0561 | 6.91 | 4 (4) | 1 (3) |
| Cas5 | MM_0562 | 27.59 | 4 (4) | 2 (2) |
| Cas7 | MM_0563 | 44.67 | 12 (12) | 8 (4) |
| Cas8 | MM_0564 | 15.82 | 9 (9) | 5 (4) |
| Cas5 | MM_3353 | 51.28 | 13 (12) | 8 (5) |
| SS | MM_3354 | 67.42 | 5 (5) | 3 (2) |
| Cmr4 | MM_3355 | 46.35 | 11 (11) | 8 (3) |
| Cas10 | MM_3356 | 26.85 | 22 (22) | 12 (10) |
| Cmr6 | MM_3357 | 32.43 | 5 (4) | 1 (4) |
| Cas7 | MM_3358 | 39.07 | 10 (9) | 1 (9) |
| Cas6bIIIC | MM_3359 | 28.27 | 5 (5) | 0 (5) |
Despite not meeting the strict criteria both Cas6b-IB and Cas6b-IIIC were detected in the proteomic data set. To evaluate potential differences in translation ribosome binding sites (RBS) were analyzed, demonstrating the presence of a strong RBS (TGGAGA) in the 5ʹ upstream region of cas6b-IB, whereas only a week RBS was detectable for cas6b-IIIC (Figure S7). This might argue for a potentially reduced level of Cas6b-IIIC in the cell.
Conservation of Cas6 proteins, repeat and leader sequences in different M. mazei strains
The repeat sequences from different free accessible genomes of M. mazei strains (Gö1, SarPi, WWM610, LYC, C16, Tuc01 and S-6) were extracted and a phylogenetic tree was created based on sequence conservation (Figure 5(a)). Overall, two different repeat sequence motifs were identified for all repeats from the CRISPR arrays of the analyzed M. mazei strains. Both repeats from the two CRISPR arrays from M. mazei Gö1 (subtype I-B and III-C) cluster, within the same repeat motif, whereas all other M. mazei strains contain CRISPR arrays with repeat sequences belonging to different sequence motifs. Further phylogenetic analysis of the leader sequences of the respective M. mazei strains (Figure 5(b)) showed a strong correlation between repeat and leader sequences.
Figure 5.
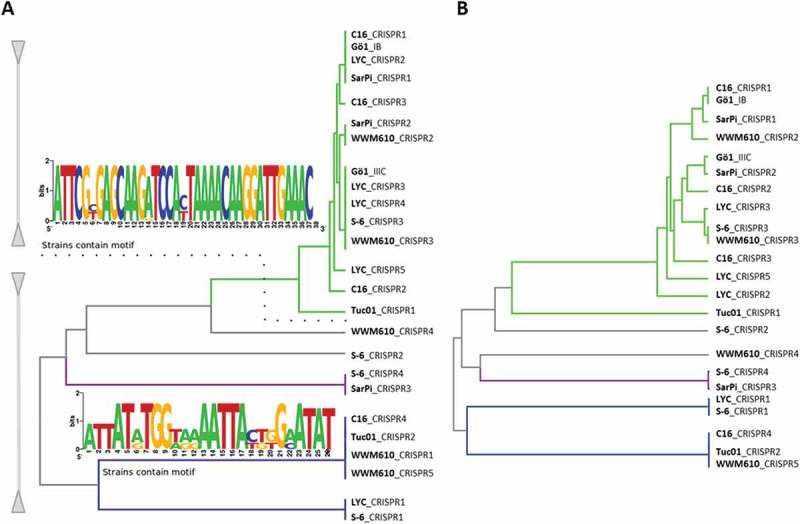
Repeat and leader conservation among different M. mazei strains.
The repeat (a) and leader (b) sequences from different free accessible genomes of M. mazei strains (Gö1, SarPi, WWM610, LYC, C16, Tuc01 and S-6) were extracted and a phylogenetic tree was generated based on sequence conservation. Conserved repeat motifs (Weblogo [49],) of each cluster are shown.
The Cas6 protein sequences from different M. mazei strains were extracted and conservation, based on amino acid level, was analyzed (Figure 6(a)). Cas6b-IB from M. mazei Gö1 showed the highest conservation to the Cas6 endonucleases from M. mazei SarPi (MSMAP_713) and C16 (MSMAC_738) with amino acid similarities of 99.54% each. Cas6b-IIIC on the other hand shared 100% similarity with a Cas6 protein (MSMAS_1326) from M. mazei S-6, or shared 91.98% similarity with the endonuclease from M. mazei WWM610 (MSMAW_1296). Overall, all analyzed Cas6 proteins showed an enormous variety on amino acid level, also emphasized in a hierarchical cluster tree (Figure 6(b)).
Figure 6.
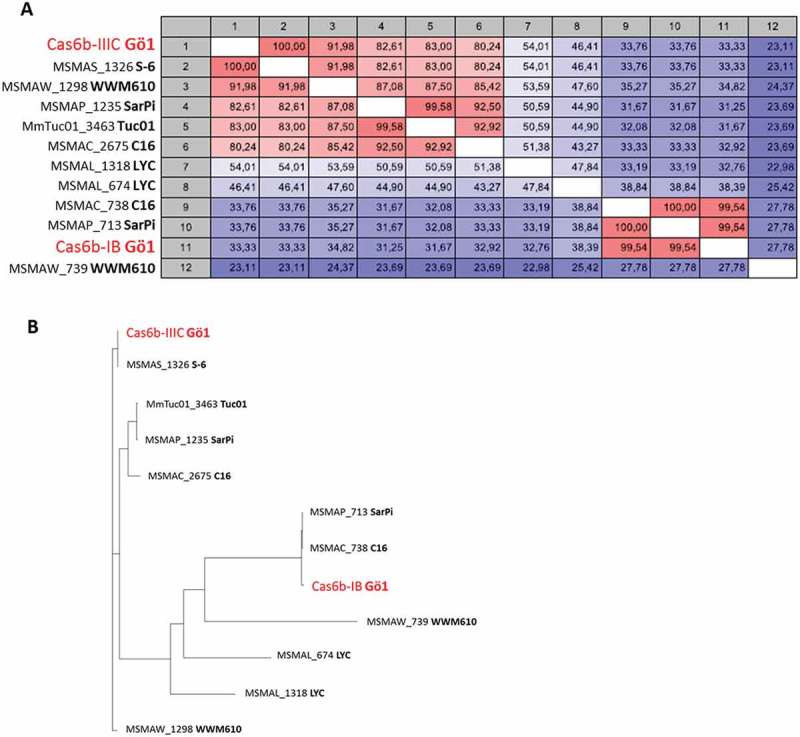
Cas6 conservation among different M. mazei strains.
The Cas6 protein sequences from different free accessible genomes of M. mazei strains (Gö1, SarPi, WWM610, LYC, C16, Tuc01 and S-6) were extracted and (A) a heatmap and (B) a hierarchical cluster tree based on amino acid sequence conservation was generated. The both Cas6 proteins from M. mazei Gö1 are highlighted in red.
Conservation analysis based on the repeat sequence and the Cas6 endonucleases showed that all studied M. mazei strains harbor at least two different CRISPR arrays with repeat sequences clustering into two separate sequence motifs and at least two distinct Cas6 proteins, showing conservation with one of the two endonucleases in M. mazei Gö1. In this study, M. mazei Gö1 is the only strain which harbors CRISPR-Cas systems with repeat sequences belonging solely to one sequence motif, supporting the finding that in M. mazei Gö1 only one Cas6 protein is required for the crRNA precursor cleavage of both CRISPR-Cas systems.
Discussion
Small crRNAs are incorporated into large protein complexes where they are part of the interference complex. Mature crRNAs are generated from a long precursor crRNA by a variety of different molecular mechanisms depending on their respective CRISPR-Cas subtype. The fundamental role of crRNAs during the CRISPR interference emphasizes the importance of crRNA maturation and their loading into the ribonucleoprotein (RNP) complex. In most type I and type III CRISPR-Cas systems, the endonuclease Cas6 is responsible for crRNA cleavage. However, it is poorly understood how different Cas6 endonucleases in organisms with multiple CRISPR-Cas systems distinguish between their target RNAs and insert them into the respective interference complex [9].
Repeat binding behavior of Cas6b endonucleases in M. mazei
Cas6 endonucleases bind pre-crRNA with high affinity and specificity. While the majority of Cas6 proteins bind their RNA substrate as a monomeric protein, dimerization especially for Cas6 proteins from thermophilic organisms (reviewed in [9]), have been reported as well. For example, Cas6a from Pyrococcus horikoshii binds single stranded RNA and dimerizes in a RNA sequence specific manner [10]. Cas6b from Methanococcus maripaludis, which has been shown to be the nearest structural homolog of Cas6b-IB and Cas6b-IIIC of M. mazei Gö1 [7], binds non-hydrolysable repeat RNA in a dimeric conformation and mature crRNA as a monomeric protein [11] strongly indicating monomerization upon cleavage. In the current study, we showed that the non-cleavable deoxy-repeat variant binds and stays bound to monomeric M. mazei Cas6b proteins (Figures 1 and S2). Since the repeats from M. mazei and M. maripaludis have considerable differences regarding their sequence and structure and are thus not grouped into the same repeat class defined by the CRISPRmap tool [12,13], it is most likely that their respective RNA binding and cleavage behaviors vary between each other.
Promiscuous crRNA processing in different M. mazei strains
Conservation and phylogenetic analysis of Cas6 proteins, repeat and leader sequences of several M. mazei strains revealed an enormous variety regarding the Cas6 endonucleases (Figure 6), although the repeats can be grouped in only two main sequence motifs, which nicely correlate with respective leader sequences (Figure 5). These findings strongly argue for a more promiscuous manner of crRNA processing in these organisms other than the tight coevolution between Cas6 proteins and their cognate repeat RNAs as it was suggested for Synechocystes sp. PCC6803 and Streptococcus thermophilus [14,15]. This is in line with our results where both endonucleases, Cas6b-IB and Cas6b-IIIC, from M. mazei Gö1 cleave the repeat RNA from each CRISPR array in vitro [7].
Cas6b-IB is the crucial endonuclease for crRNA processing of both CRISPR loci
Despite the hypothesis of a more promiscuous manner of crRNA processing, we obtained clear evidence by a genetic approach that in vivo Cas6b-IB is mainly responsible for crRNA cleavage of both arrays in M. mazei. The obtained high specific activity of Cas6b-IIIC in vitro might be due to the in vitro assay using the repeat without flanking spacers, whereas in vivo flanking spacers most likely influence the repeat structure and thus Cas6b activities. Such a contrary finding of in vitro and in vivo cleavage activity was also shown for Cas6 proteins in the cyanobacteria Synechocystis sp. PCC 6803. Here, an in vitro promiscuity of the two studied Cas6 endonucleases to process the crRNA, not only from their own but also from the respective other CRISPR array, was detected [16]. These observation also stay in contrast to the in vivo specificity of these proteins, determined by deletion mutants for Synechocystis sp. PCC 6803 [14].
In M. mazei Gö1, both proteins, Cas6b-IB and Cas6b-IIIC, were detected, albeit under medium stringency, during standard growth conditions in the wildtype proteome (Table 1). Quantitative determination of Cas6b proteins was not possible due to a generally low amount of Cas proteins in M. mazei Gö1. However, analyzing the RBS of the two cas6b genes regarding their similarity with the consensus sequence for the M. mazei strain Gö1 RBS (AGGAGG [17,18]), we identified a strong RBS for cas6b-IB but only a weak RBS for cas6b-IIIC. Most likely this leads to lower protein level of Cas6b-IIIC [8]. From the genetic approach we know that Cas6b-IB is the main active endonuclease for processing of crRNA of both CRISPR loci in vivo. Considering that on the other hand Cas6b-IIIC showed a higher specific cleavage activity in vitro (Figure S3), this finding might argue for an additional potential regulation of Cas6-IIIC in vivo. Possible mechanisms are small RNAs or proteins that influence the translation or the activity of Cas6b-IIIC in vivo.
A tight co-evolution was suggested for Cas6 proteins and their corresponding repeat substrates from early on. This is nicely reflected by the comparative analysis for the different M. mazei strains in this study. However, it can only be speculated which system has been acquired more recently. Type III CRISPR-Cas systems are known to exclusively co-exist together with another CRISPR-Cas system, in which they often share the adaptation module and one Cas6 endonuclease, which are then both encoded by the respective other CRISPR-Cas system [19,20]. For type III CRISPR-Cas systems, different arrangements of cas genes have been reported, which might be due to frequent horizontal gene transfer and recombination between different CRISPR loci. For example, the type III-D system from Synechocystis sp. PCC 6803 was shown to be a combination of a type I and a type III CRISPR-Cas system [21]. Thus, type III CRISPR-Cas systems are thought to have evolved later than type I systems, which explains why they are often found to rely upon additional systems. Moreover, M. mazei shows the same dependence regarding the adaptation module, which is part of the I-B operon. However, as demonstrated in this study, Cas2, one of the two main adaptation proteins (Cas1 and Cas2), was not detected in the proteomic data of the wt strain even with our medium stringency search settings (Table 1) [8], arguing for an inactive adaptation module. Such an inactive adaptation module was also observed in Sulfolobus islandicus (reviewed in [22]). Here, the adaptation genes are activated by a CRSPR associated transcriptional regulator Csa3 [23]. Thus, we propose a tightly regulated adaption module for the CRISPR-Cas system in M. mazei as well. In terms of a missing adaptation module and the fact that Cas6b-IB processes the type III crRNAs more effective it is tempting to speculate that the type III system in M. mazei Gö1 is evolutionary younger. Nevertheless, evolutionary analysis regarding CRISPR-Cas systems by Mohanraju et al. (2016) [3] identified type III CRISPR systems as a very ancestral one.
Specification of crRNAs after Cas6b-IB cleavage
With repeats from two different CRISPR arrays (I-B and III-C) sharing a very similar sequence and being cleaved via the same Cas6 endonuclease, it remains unclear how the subsequent assembly of the two different RNPs and their complementary crRNAs occurs. Furthermore, it is unknown if the two interference complexes share the crRNAs from both CRISPR arrays.
In M. mazei, the CRISPR repeats from both CRISPR arrays are able to form a distinct hairpin structure, which can also be form in the respective spacer context. The Cas6 recognition and binding to the stem loop structure was discussed previously in detail [7]. Comparison of all repeats from both arrays revealed that the stem regions are highly conserved in all repeats and with the exception of a handful of point mutations through the sequences, the 5ʹ handles are also the same for all CRISPR repeats in M. mazei [7]. The hairpin structure was shown to be important for crRNA recognition by Cas6 in Sulfolobus solfataricus [24] and other bacterial systems [20,25]. In S. solfataricus, two CRISPR repeat families were described, which are variable in their stem loop but highly conserved in their 8 nt 5ʹ handle [24,26]. Here, Cas6 specificity and interference complex assembly was discussed to result from different hairpin-binding of the particular Cas6 proteins and a subsequent handover between Cas6 and the interference complex. This hypothesis was supported by the finding of Cas6 as an integral subunit of the interference complex in type I-E and I-F systems and also in the archaeal type I-B system [27–30]. In contrast, Cas6 is not part of any type III interference complex [31,32]. Crystal structures of two Cas6 proteins from Thermus thermophilus together with their RNA substrates revealed two binding sites within the repeat RNA, one at the hairpin stem and one at the unstructured region upstream of the stem-loop structure, being a hint for an evolutionary relationship between Cas6 endonucleases with different RNA recognition modes [33].
Since both mature crRNAs are cleaved via a single Cas6b protein in M. mazei resulting in the specific 5ʹ handle, and given that the sequences from both repeats are identical regarding this crRNA 5ʹ handle, a differentiation between the two CRISPR-Cas systems based on its sequence information is unlikely. The two most apparent differences in the repeat sequences are two exchanged nucleotides, of which one is found in the 5ʹ region of the repeat RNA becoming the 3ʹ end of the crRNA and one in the loop region of the hairpin structure. We propose that based on these differences, the Cas6b-IB protein stays bound to the cleaved type I-B related crRNAs, guiding them to the I-B interference complex and is possibly part of this complex itself. For type III-C related crRNAs, we hypothesize that Cas6b-IB dissociates from the crRNA after cleavage, which might be due to an opening of the hairpin structure of this particular repeat RNAs or a different mode of binding due to the two different nucleotides. Binding of Cas6b-IB to only one part of the type III-C repeat RNA is possible, where it might be able to bind to an additional region of the type I-B RNA, for example to the unstructured region upstream of the stem loop where the two repeat variants differ in their conserved sequence. Consistent with such a scenario is the obtained difference in binding affinity (Figure 1). We cannot completely rule out the possibility of shared crRNAs among both subtypes. However, the identified 3ʹ trimming of crRNAs in the type III CRISPR arrays [7,32,34,35] strongly argues for a crRNA specificity of the interference complexes in M. mazei.
In conclusion, this study demonstrated that although the genetic information of two Cas6 endonucleases is present in M. mazei, the type I-B derived protein is solely responsible for the crRNA maturation of both CRISPR subtypes under standard and high salt conditions. Moreover, this is the first report of a Cas6 endonuclease not responsible for the in vivo processing of its corresponding CRISPR array, although its respective crRNA is actively transcribed. Conservation and phylogenetic analysis of Cas6 proteins together with the repeat and leader sequences from other M. mazei strains argue for a more promiscuous manner of crRNA processing in these organisms, supporting the hypothesis of post-translational regulation or a function beyond the adaptive defense system of cas6b-IIIC in M. mazei Gö1. Differentiation and distribution of crRNAs between the two interference complexes are still not clear. In M. mazei, precise determination of the binding affinity of the two Cas6b endonucleases to the different crRNAs and the isolation of crRNAs from the respective interference complexes followed by sequencing of the respective cDNAs might help to clarify those questions. Furthermore, the regulation of important CRISPR-Cas compounds, such as the adaptation module, needs to be extensively studied in the future.
Materials and methods
Strains and plasmids
Strains and plasmids, which were used in this study, are listed in Supplementary Table S1. Plasmid DNA was transformed into M. mazei as described by Ehlers et al. [36]. Plasmid DNA was in general transformed into E. coli DH5α [37] or JM109 λpir [38] according to the method of Inoue et al. [39].
Generation of plasmids and construction of M. mazei mutant strains
Chromosomal deletion mutants were constructed by the replacement of the respective gene with the puromycin resistance (pac) cassette based on homologous recombination (Gebhardt, 1990). The up- and downstream fragments (~ 1,000 nt) for the construction of the deletion mutants ∆cas6b-IB (MM560) and ∆cas6b-IIIC (MM3359) were constructed by polymerase chain reaction (PCR) amplification of the respective fragments from genomic M. mazei DNA using primers ∆MM560_1_for and ∆MM560_1_rev as well as ∆MM3359_1_for and ∆MM3359_1_rev for the upstream fragments and ∆MM560_2_for and ∆MM560_2_rev as well as ∆MM3359_2_for and ∆MM3359_2_rev for the respective downstream fragments (Table S2). All four fragments were TA cloned into the pCR4-TOPO (Invitrogen Karlsruhe, Germany). The upstream fragments were then cloned from the respective pCR4-TOPO vectors into the multiple cloning site of the pBlueskript SK+ vector (Stratagene) after restriction with ApaI and SacI. The downstream fragments were cloned using BamHI and SacI from the pCR4-TOPO vector (Invitrogen) into the pBlueskript SK+ vectors (Stratagene, La Jolla, US) containing the upstream fragments resulting in pRS1040 for ∆cas6b-IB and pRS1042 for ∆cas6b-IIIC. The pac cassette from Streptomyces alboniger [40] was restricted with EcoRI from pRS207 and blunt ends were created by mung bean nuclease. pRS1040 and pRS1042 were opened with BamHI followed by a mung bean nuclease treatment and were then ligated with the pac-cassette resulting in pRS1041 (∆cas6b-IB) and pRS1043 (∆cas6b-IIIC). These plasmids were linearized with AhdI and transformed into M. mazei* by liposome mediated transformation as previously described [36]. Puromycin-resistant transformants were selected as colonies that grew on minimal medium plates with trimethylamine as the carbon and energy source plus 5 µg puromycin/ml during incubation. Pac-cassette insertion was verified via southern blot analyzes of genomic DNA from puromycin-resistant transformants as described by Ehlers et al. [36] and the deletion of the respective gene was tested by PCR amplification of cas6b-IB using the primers MM560_for and MM560_rev and cas6b-IIIC with the primers MM3359_for and MM3359_rev (Table S2) with genomic DNA of the respective deletion strain and with wild type DNA as a positive control.
Growth of M. mazei
M. mazei strain Gö 1 was grown under anaerobic conditions at 37°C under a gas atmosphere of 80% N2 plus 20% CO2 in 70 ml closed bottles in minimal medium that contained 150 mM methanol and 40 mM acetate as sole energy and C-source and with 10 mM ammonium chloride for cultivation under nitrogen sufficiency according to Ehlers et al. [36]. The medium was supplemented with 100 μg/mL ampicillin to prevent bacterial contamination. For high salt stress additional 500 mM NaCl was added to the minimal medium right after inoculation. Cultures were grown until cells reached a turbidity at 600 nm of 0.3 (high salt) or 0.5 and were harvested by 4°C via centrifugation.
Heterologous expression of proteins in E. coli and purification
For the heterologous expression of the two Cas6b endonucleases in E. coli both genes cas6b-IB and cas6b-IIIC were PCR amplified out of chromosomal M. mazei DNA. The primer sets MM560_for and MM5601_rev as well as MM3359_for and MM3359_rev were used (Table S2). Both fragments were TA-cloned into pCR4-TOPO (Invitrogen) resulting in pRS1078 (cas6b-IB) and pRS1080 (cas6b-IIIC) followed by subsequent cloning via XmnI and HindIII from these plasmids to pMAL-cs (New England BioLabs). The received plasmids were designated pRS1079 (cas6b-IB) and pRS1081 (cas6b-IIIC) and were each transformed into E. coli BL21-CodonPlus®-RIL (Stratagene) for the heterologous protein expression as MBP (maltose-binding protein) fusion proteins.
The two E. coli BL21-CodonPlus®-RIL strains (Stratagene) containing pRS1079 and pRS1081 were grown under standard growth conditions in LB medium. Expression of MBP-Cas6b-IB and MBP-Cas6b-IIIC was induced at a turbidity at 600 nm of 0.6 with 50 µM Isopropyl β-D-thiogalactoside for 3 h. Cells were harvested and disrupted in MBP-buffer (20 mM Tris/HCl pH 7.4, 200 mM NaCl, 1 mM EDTA) with a French pressure cell followed by centrifugation. MBP-fusion proteins were purified from the supernatants by affinity chromatography with amylose resin according to the manufacturer’s instructions (New England BioLabs). The purified proteins were dialyzed against 10 mM Tris/HCL pH 7.6, followed by subsequent purification by anion-exchange chromatography using a Q-Seharose FF (XK26, Pharmacia Biotech) as described before [41]. The purified proteins were again dialyzed against 10 mM Tris/HCL pH 7.6.
The his-tagged version of Cas6b-IIIC (pRS833 in E. coli pRIL) and Cas6b-IB (pRS714 in E. coli Rosetta) was purified as described by Nickel et al. 2013 [7] with the exception of addition of 3% glucose to the LB media and that expression of proteins was induced after addition of 10 µM Isopropyl β-D-thiogalactoside for 16 h at 18°C.
Size-exclusion chromatography
Oligomeric states of MBP-Cas6b-IB and MBP-Cas6b-IIIC were determined by size-exclusion chromatography. Purified proteins (17 µM) in buffer A (10 mM Tris/HCl pH 7.6, 150 mM NaCl) separately or after 30 min incubation at 37°C in the presence of 15 µl of 100 µM type I-B deoxy-repeat RNA (5′-AUUCGCGAGCAAGAUCCAUUAAAACAA(dG)GAUUGAAC-3ʹ) were applied to a Superose12 column (10/30) (GE Healthcare) and fractionated using an Äkta-Purification system (GE Healthcare). Proteins were detected by monitoring the absorbance at 280 nm and were eluted from the column in 1 ml fractions, using a flow rate of 0.5 ml· min−1. Calibration of the column was performed using the gel-filtration mass standard (Bio-Rad Laboratories) containing thyroglobulin (670 kDa), IgG (158 kDa), ovalbumin (44 kDa), myoglobulin (17 kDa) and vitamin B12 (1.35 kDa).
In vitro endonuclease activity assay
Endonuclease activity assay was conducted on 5′-end-labeled CRISPR repeat RNA (type I-B repeat 5´-AUUCGCGAGCAAGAUCCAUUAAAACAAGGAUUGAAC-3´, type III-C 5ʹ-AUUCGUGAGCAAGAUCCACUAAAACAAGGAUUGAAAC-3ʹ) like described before [7]. Initially, RNA was denatured 1 min at 95°C, chilled on ice for 5 min and incubated with different indicated concentrations of heterologously expressed and purified his-tagged Cas6b-IB and Cas6b-IIIB protein in reaction puffer (20 mM Hepes pH8, 1 mM DTT, 250 mM KCl, 1.875 mM MgCl2,) for 5 min at 37°C. All reactions were stopped and precipitated with 70% ethanol. Samples were denatured for 5 min at 95°C prior to separation on 8% polyacrylamide/7 M urea sequencing gels in TBE buffer. Gels were analyzed using a PhosphorImager (FLA-3000 Series, Fuji) and AIDA software (Raytest).
RNA extraction
Total RNA was isolated by phenol extraction as described before [42] but using the Isol-RNA Lysis Reagent (5ʹPRIME GmbH, Cat. No. 2302700) and was followed by DNase I treatment as described in Nickel et al. [7].
For the identification of protein-bound RNA from size-exclusion chromatography, potential bound RNA was isolated from all fractions by phenol chloroform precipitation [17] and was subsequently used for dot blot analysis (see below).
Northern blot and dot blot analyzes
Northern blot analyzes of total RNA with [γ-32P]-ATP end-labeled oligodeoxynucleotide crRepeat-IB (Table S2) directed against the IB-repeat were performed as described before [17]. Isolated RNA from size-exclusion fractions was identified via dot blot analysis, by dropping 2 µl of isolated RNA onto a HybondXL membrane (GE Healthcare). After UV-crosslinking hybridization of oligo probes was performed as described before [17].
Quantitative (q) RT-PCR
Transcript levels of genes in different strains or during different growth conditions were determined by qRT-PCR as described before [42,43]. The assays were performed using QuantiTect Probe RT-PCR Kit (Qiagen) and the ViiA™ 7 Real-Time PCR System (Applied Biosystems). All primers used in this study are listed in Table S2. The fold change in transcript abundance for each gene of interest was determined by comparison with the Cycle threshold (Ct) of three control genes (MM1215, MM1621 and MM2181) which were known to remain constant during different growth phases and growth conditions [42].
Absolut transcript numbers were calculated by absolute qRT-PCR with the QuantiTect Probe RT-PCR Kit (Qiagen) with the primers qRT_MM560_for and qRT_560_rev for cas6b-IB and qRT_MM3359_for and qRT_MM3359_rev for cas6b-IIIC. Standard curves were generated with serial 10-fold dilutions (107 copies to 10 copies) of one DNA template strand from the plasmid pRS1078 for cas6b-IB and pRS1080 for cas6b-IIIC each in three technical replicates. Absolute transcript numbers in the M. mazei wild type strain grown under normal growth conditions were determined for 50 ng of total RNA each for three biological replicates and in technical replicates.
Construction of cDNA libraries for dRNA-Seq and Illumina sequencing
Libraries for Illumina sequencing of cDNA were constructed by vertis Biotechnology AG, Germany (http://www.vertis-biotech.com/) from PNK treated RNA using a HiSeq 2000 machine (Illumina) as described before [7].
The Illumina reads in FASTQ format were trimmed with a cut-off phred score of 20 by the program fastq_quality_trimmer from FASTX toolkit version 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/). The following steps were performed using the subcommand ‘create’, ‘align’ and ‘coverage’ of the tool READemption [44] version 0.3.5. The poly(A)-tail sequences (introduced by the library construction protocol) were removed and a size filtering step was applied in which sequences shorter than 12 nt were eliminated. The collections of remaining reads were mapped to the reference genome sequences (NC_003901.1) using segemehl version 0.2.0 [45]. Coverage plots in wiggle format representing the number of aligned reads per nucleotide were generated based on the aligned reads and visualized in the Integrated Genome Browser [46].
Conservation and phylogenetic analysis of M. mazei Cas6 endonucleases, repeat and leader sequences
All current M. mazei genomes analyzed in this study were downloaded from NCBI FTP site (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/) in January 2017. CRISPR arrays were predicted using CRISPER-identifier tool (in preparation for publication) and using default parameter values (at least two repeats within a CRISPR array, repeat and spacer lengths were set to a range between 18 to 52 bp). We then generated a consensus repeat for each CRISPR array based on multiple alignments of all repeats using MAFFT program [47]. In this way, we obtained a CRISPR repeat dataset with 25 consensus repeats. CRISPR arrays orientations were predicted using CRISPRstrand [13]. Consensus repeats were clustered into related families using the CRISPRmap method [12] and a hierarchical cluster tree was generated by UPGMA [48] based on distance matrix from the alignment. The multiple sequence alignment was converted into a logo by WebLogo version 3 [49]. Finally, 23 leader sequences were identified and analyzed in all M. mazei genomes using CRISPRleader [50].
Cas6 endonucleases were identified using the MegaBLAST search tool [51]. All extracted sequences were analyzed and a heat map based on amino acid similarity was created using CLC Genomics Workbench 9 (Qiagen).
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [BA-2186/5-2 and Schm1052/12-2].
Acknowledgments
We thank members of the Schmitz group for useful discussions, as well as Claudia Kießling for technical assistance. This work was supported by the German Research Council (DFG) ‘Forschergruppe FOR1680ʹ Unravelling the prokaryotic immune system’ [Schm1052/12-2 to R.S. and BA2168/5-2 to R.B.].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13(11):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60(3):385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mohanraju P, Makarova KS, Zetsche B, et al. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353(6299):aad5147. [DOI] [PubMed] [Google Scholar]
- [4].Rogers JE, Whitman WB, editors. Microbial production and consumption of greenhouse gases: methane, nitrogen oxides and halomethanes. Washington (DC): ASM Press; 1991. [Google Scholar]
- [5].Vestergaard G, Garrett RA, Shah SA.. CRISPR adaptive immune systems of Archaea. RNA Biol. 2014;11(2):156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nickel L, Weidenbach K, Jäger D, et al. Two CRISPR-Cas systems in Methanosarcina mazei strain Gö1 display common processing features despite belonging to different types I and III. RNA Biol. 2013;10(5):779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cassidy L, Prasse D, Linke D, et al. Combination of bottom-up 2D-LC-MS and semi-top-down GelFree-LC-MS enhances coverage of proteome and low molecular weight short open reading frame encoded peptides of the archaeon methanosarcina mazei. J Proteome Res. 2016;15(10):3773–3783. [DOI] [PubMed] [Google Scholar]
- [9].Hochstrasser ML, Doudna JA. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem Sci. 2015;40(1):58–66. [DOI] [PubMed] [Google Scholar]
- [10].Wang R, Zheng H, Preamplume G, et al. The impact of CRISPR repeat sequence on structures of a Cas6 protein-RNA complex. Protein Sci. 2012;21(3):405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Richter H, Lange SJ, Backofen R, et al. Comparative analysis of Cas6b processing and CRISPR RNA stability. RNA Biol. 2013;10(5):700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lange SJ, Alkhnbashi OS, Rose D, et al. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 2013;41(17):8034–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alkhnbashi OS, Costa F, Shah SA, et al. CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformatics. 2014;30(17):i489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Scholz I, Lange SJ, Hein S, et al. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One. 2013;8(2):e56470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Carte J, Christopher RT, Smith JT, et al. The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in streptococcus thermophilus. Mol Microbiol. 2014;93(1):98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Reimann V, Alkhnbashi OS, Saunders SJ, et al. Structural constraints and enzymatic promiscuity in the Cas6-dependent generation of crRNAs. Nucleic Acids Res. 2017;45(2):915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jäger D, Sharma CM, Thomsen J, et al. Deep sequencing analysis of the Methanosarcina mazei Gö1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci USA. 2009;106(51):21878–21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Karlin S, Mrázek J, Ma J, et al. Predicted highly expressed genes in archaeal genomes. Proc Natl Acad Sci USA. 2005;102(20):7303–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Haurwitz RE, Jinek M, Wiedenheft B, et al. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329(5997):1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Garrett RA, Shah SA, Erdmann S, et al. CRISPR-Cas adaptive immune systems of the sulfolobales: unravelling their complexity and diversity. Life (Basel). 2015;5(1):783–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu T, Li Y, Wang X, et al. Transcriptional regulator-mediated activation of adaptation genes triggers CRISPR de novo spacer acquisition. Nucleic Acids Res. 2015;43(2):1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shao Y, Li H. Recognition and cleavage of a nonstructured CRISPR RNA by its processing endoribonuclease Cas6. Structure. 2013;21(3):385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gesner EM, Schellenberg MJ, Garside EL, et al. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat Struct Mol Biol. 2011;18(6):688–692. [DOI] [PubMed] [Google Scholar]
- [26].Sokolowski RD, Graham S, White MF. Cas6 specificity and CRISPR RNA loading in a complex CRISPR-Cas system. Nucleic Acids Res. 2014;42(10):6532–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wiedenheft B, Van Duijn E, Bultema JB, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci USA. 2011;108(25):10092–10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wiedenheft B, Lander GC, Zhou K, et al. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature. 2011;477(7365):486–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sashital DG, Jinek M, Doudna JA. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol. 2011;18(6):680–687. [DOI] [PubMed] [Google Scholar]
- [30].Sternberg SH, Haurwitz RE, Doudna JA. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. Rna. 2012;18(4):661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hale CR, Zhao P, Olson S, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139(5):945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang J, Rouillon C, Kerou M, et al. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol Cell. 2012;45(3):303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Niewoehner O, Jinek M, Doudna JA. Evolution of CRISPR RNA recognition and processing by Cas6 endonucleases. Nucleic Acids Res. 2014;42(2):1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hale C, Kleppe K, Terns RM, et al. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. Rna. 2008;14(12):2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11(3):181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ehlers C, Weidenbach K, Veit K, et al. Development of genetic methods and construction of a chromosomal glnK(1) mutant in Methanosarcina mazei strain Gö1. Mol Genet Genomics. 2005;273:290–298. [DOI] [PubMed] [Google Scholar]
- [37].Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557–580. [DOI] [PubMed] [Google Scholar]
- [38].Miller VL, Mekalanos JJ. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol. 1988;170(6):2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96(1):23–28. [DOI] [PubMed] [Google Scholar]
- [40].Metcalf WW, Zhang JK, Apolinario E, et al. A genetic system for Archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc Natl Acad Sci USA. 1997;94(6):2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ehlers C, Veit K, Gottschalk G, et al. Functional organization of a single nif cluster in the mesophilic archaeon Methanosarcina mazei strain Gö1. Archaea. 2002;1:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Veit K, Ehlers C, Schmitz RA. Effects of nitrogen and carbon sources on transcription of soluble methyltransferases in Methanosarcina mazei strain Gö1. J Bacteriol. 2005;187(17):6147–6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Weidenbach K, Glöer J, Ehlers C, et al. Deletion of the archaeal histone in Methanosarcina mazei Gö1 results in reduced growth and genomic transcription. Mol Microbiol. 2008;67(3):662–671. [DOI] [PubMed] [Google Scholar]
- [44].Förstner KU, Vogel J, Sharma CM. READemption-a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics. 2014;30(23):3421–3423. [DOI] [PubMed] [Google Scholar]
- [45].Hoffmann S, Otto C, Kurtz S, et al. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol. 2009;5(9):e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Freese NH, Norris DC, Loraine AE. Integrated genome browser: visual analytics platform for genomics. Bioinformatics. 2016;32(14):2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Katoh K, Misawa K, Kuma K-I, et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gronau I, Moran S. Neighbor joining algorithms for inferring phylogenies via LCA distances. J Comput Biol. 2007;14(1):1–15. [DOI] [PubMed] [Google Scholar]
- [49].Crooks GE, Hon G, Chandonia J-M, et al. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Alkhnbashi OS, Shah SA, Garrett RA, et al. Characterizing leader sequences of CRISPR loci. Bioinformatics. 2016;32(17):i576–i585. [DOI] [PubMed] [Google Scholar]
- [51].Morgulis A, Coulouris G, Raytselis Y, et al. Database indexing for production MegaBLAST searches. Bioinformatics. 2008;24(16):1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.