

ABSTRACT
The large genome of the migratory locust (Locusta migratoria) genome accumulates massive amount of accumulated transposable elements (TEs), which show intrinsic transcriptional activities. Hampering the ability to precisely determine full-length RNA transcript sequences are exonized TEs, which produce numerous highly similar fragments that are difficult to resolve using short-read sequencing technology. Here, we applied a 5ʹ-Cap capturing method using Nanopore long-read direct RNA sequencing to characterize full-length transcripts in their native RNA form and to analyze the TE exonization pattern in the locust transcriptome. Our results revealed the widespread establishment of TE exonization and a substantial contribution of TEs to RNA splicing in the locust transcriptome. The results of the transcriptomic spectrum influenced by Piwi expression indicated that TE-derived sequences were the main targets of Piwi-mediated repression. Furthermore, our study showed that Piwi expression regulates the length of RNA transcripts containing TE-derived sequences, creating an alternative UTR usage. Overall, our results reveal the transcriptomic characteristics of TE exonization in the species characterized by large and repetitive genomes.
KEYWORDS: Nanopore, direct RNA sequencing, transposable element, piwi, insects
Introduction
Transposable elements (TEs), which persist through the independent accumulation and movement of their sequences, are ubiquitous in eukaryotic genomes [1]. TEs show widespread transcriptional activities in metazoan species, and their expressions are restricted by Piwi protein through two major silencing modes, namely transcriptional silencing and posttranscriptional silencing [2,3]. A large portion of TE copies are remnants of relatively ancient insertions and are deprived of the capability for transposition. The process by which TEs are inserted within introns is recognized by splicing machinery and recruited into RNA transcripts as exon is termed exonization [1,4]. The exonized TEs contain numerous splice donor and acceptor sites and contribute to the formation of alternative RNA transcripts [5]. TE exonization inserted within host genes that generates alternative splicing of the new exons may be co-opted for novel gene functions [6–8]. However, few studies have investigated the transcriptional landscape of TEs at the genome-wide scale, mainly due to the technical difficulties in accurately assembling and mapping of repetitive sequences using short-read sequencing technology [9].
Accurate determination of full-length RNA transcript sequences is a foundation for transcriptome studies. However, currently transcriptomic approaches based on complementary DNA (cDNA) synthesis are plagued by the under-representation of 5ʹ ends of the gene structure due to degraded transcripts and natural drop-off of reverse transcriptase during first-strand synthesis [10]. High-throughput RNA-seq sequencing using short-read data has been the most popular strategy for exploring eukaryotic transcriptomes, providing an important tool to identify the gene structure, novel isoforms, alternative splicing and alternative polyadenylation [11]. The technological shortcoming limits the application of short-read RNA-seq data to complex transcriptomic events, such as isoform reconstruction of transcripts containing TE sequences. RNA-seq sequencing is based on either polydeoxythymidine priming or random hexamer priming, followed by cDNA synthesis. Due to the involvement of reverse transcription and PCR amplification of cDNAs, the inherent biases of RNA-seq sequencing include reduced complexity of cDNA library, inadequate representation of RNA species and distortion of relative expression quantification [12,13]. Furthermore, PCR amplification of repetitive sequences generate PCR-mediated artifacts such as recombination or chimera formation [14]. The more recently developed approach for full-length cDNA sequencing using PacBio long-read sequencing technology is based on a template-switching reaction that relies on adding a non-templated poly-cytosine tail when the reverse transcriptase reaches the 5ʹ end of the RNAs [15]. The imperfection of this approach includes nonspecific priming of template switching oligonucleotides, producing artificial chimeric cDNAs and converting degraded RNAs containing a poly-A tail into cDNAs [16,17]. Additionally, reverse transcription, which is included in PacBio transcriptome sequencing, produces artifacts of splicing sites, due to template switching between repetitive sequences of TEs [18]. Therefore, the transcriptomic approaches based on cDNA synthesis have intrinsic limits to determine the RNA transcripts containing TE sequences.
The migratory locust, Locusta migratoria, possesses a huge genome (6.5Gb, ~35-fold larger than the Drosophila melanogaster genome), which has experienced massive accumulation of repetitive elements [19]. The polyphenism of the locusts is resulted from epigenetic differences during the transition between solitary and gregarious phases [20–22]. TE sequences accounts for more than 65% of the locust genome, and a large number show transcriptional activation [19,23,24]. In our previous study, we identified 105 TEs in the locust transcriptome using short-read RNA-seq data and found only seven of them are in their full-length forms [23]. The transcriptional abundance of TEs across a wide range of divergence rates suggests diverse and abundant TE copies are expressed in the locust transcriptome. Thus, the migratory locust is an idea model for study on TE structure and function. Nanopore direct RNA sequencing uses a voltage-based nanoscale pore in a membrane to capture the very long nucleotide sequences from RNA molecules [25]. Nanopore direct RNA sequencing sequences a linear, single-stranded RNA molecule by detecting ionic current changes when nucleic acid threads pass through the nanopore. The rapid development of Nanopore long-read RNA sequencing technology extends the opportunity to gain an accurate representation of RNA transcript structure because each sequencing read encompasses a full RNA transcript. This knowledge is particularly critical for species with a large genome size because a considerable fraction of RNA transcripts is derived from repetitive sequences of TEs.
In this study, we characterized the locust transcriptome in its native RNA form using Nanopore direct RNA sequencing, a sequencing technology that does not require reverse transcription during library preparation. We applied a 5ʹ-Cap capturing method using Nanopore direct RNA sequencing to determine the full-length sequences of RNA transcripts in the locusts. We analyzed the TE exonization pattern in the locust transcriptome and determined the influences of Piwi expression on the spectrum of RNA transcripts containing TE-derived sequences. Taken together, our results demonstrate the usefulness of direct RNA sequencing by 5ʹ-Cap capturing in characterizing full-length RNA transcripts containing repetitive sequences, and reveal the transcriptomic characteristics of TE exonization in a species with large and highly repetitive genomes.
Results
Enrichment of full-length RNA transcripts by 5ʹ-Cap capturing
Because Nanopore direct RNA sequencing is a newly developed sequencing technology which only has been validated in a few species, we first measured the quality metrics including transcript length, sequence identity, isoform coverage and detected gene number to assess its performance for transcriptome sequencing in locusts. These data suggested that Nanopore direct RNA sequencing demonstrates an inherent capacity to accurately sequence RNA transcripts for the locust transcriptome (Supplementary Texts). All full-length RNA transcripts transcribed by RNA Polymerase II (Pol II) possess a characteristic Cap structure, which consists of an inverted guanosine at their 5ʹ-end [26]. A modified oligo-capping method (Figure 1(a)), which depends on enzymatic replacement of the Cap structure with a biotinylated RNA adaptor, was used to enrich RNA transcripts that contain the 5ʹ-Cap [27]. The 5ʹ-biotinylated RNA transcripts were captured using Streptavidin-coupled Dynabeads. This procedure allows the preferential enrichment for full-length RNA transcripts. The synthetic RNA adaptor in the 5ʹ-end of RNA transcripts served as a sequence tag to assess the completeness of the RNA transcripts. The full-length RNA transcripts were specifically enriched using both a short RNA adaptor (16 nt in length, short-adaptor) and a long RNA adaptor (50 nt in length, long-adaptor). We generated 457,470 and 269,712 high-quality reads in the short-adaptor library and long-adaptor library, respectively. The adaptors in the 5ʹ-end of RNA transcripts were detected using the Smith-Waterman algorithm, as implemented in the SSEARCH program [28]. To validate the 5ʹ-Cap capturing method, we examined the normalized coverage of the adaptor position from the 5ʹ-end to 3ʹ-end. As expected, in the long-adaptor library, most of the identified adaptors were located at the most 5ʹ-end of the adaptor-containing RNA transcripts (Figure 1(b)). However, no adaptor enrichment in the 5ʹ-end was observed in the short-adaptor library. A truncated adaptor approach based on global to local alignment searches was used to determine the sequence identity distribution along the long adaptor. Using the full-length long adaptor as the global query, the adaptor sequences could only be detected in the 17.8% of the adaptor-containing transcripts. Using the different adaptors truncated toward the 3ʹend, both the sequence identity and transcript percentage showed a gradual increase (Figure 1(c)). The most dramatic increases in the sequence identity and transcript percentage were observed between the alignment searches using the full-length long adaptor and those using the truncated adaptor corresponding to the 11th to 50th nt (R40). The transcript percentage even decreases to 4.3% using the truncated adaptor corresponding to the 1st to 30th nt (L30) as a global query. These results indicate that direct RNA sequencing shows poor performance of sequencing accuracy at the extreme 5ʹ-end sequencing of RNA transcripts. Therefore, the low sequencing accuracy at the extreme 5ʹ-end results in the failure of sequence alignment in adaptor detection for the short-adaptor library.
Figure 1.
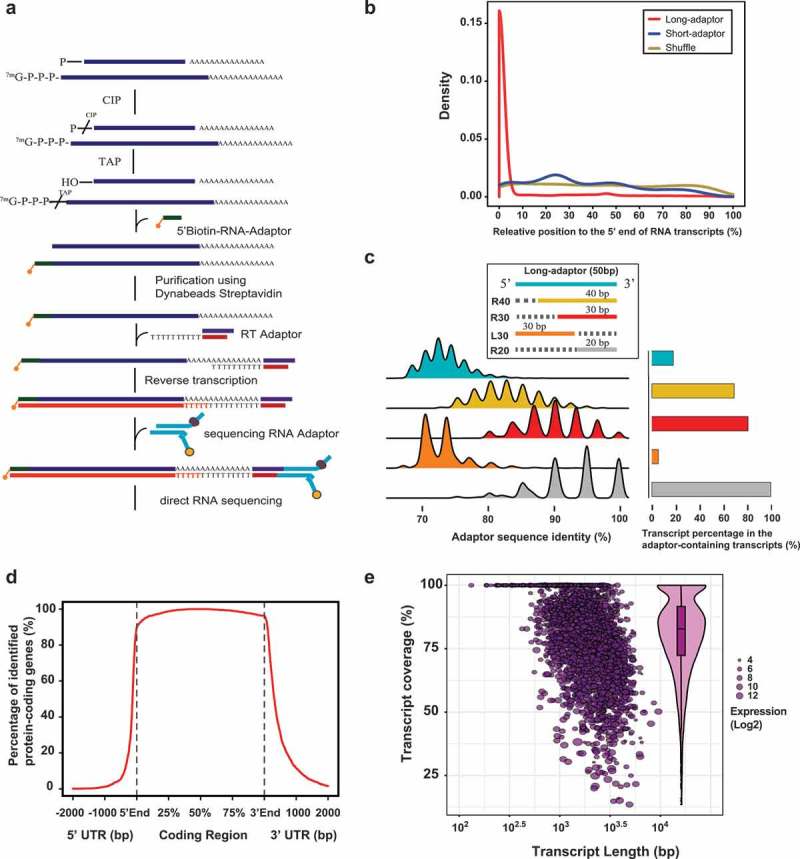
Enrichment of full-length RNA transcripts by 5ʹ-Cap capturing. (a) Flowcharts illustrating the protocols to enrich full-length RNA transcripts by 5ʹ-Cap capturing. (b) Enrichment of the long-adaptor sequences in the 5ʹend of RNA transcripts. The tool uShuffle shuffles RNA transcript sequences while preserving the k-let counts. (c) Distribution of the adaptor sequence identity and percentage of adaptors in the adaptor-containing transcripts. The adaptor sequences were identified by the GLSEARCH program using a global-local alignment algorithm. L30, the truncated adaptor sequences ranging from 1 to 30 in the 5ʹ end of the long adaptor; R40, the truncated adaptor sequences ranging from −40 to −1 in the 3ʹ end of the long adaptor. Using R20 results in a maximum number of adaptor-containing transcripts and thus are considered 100%. (d) Gene coverage evaluation by aligning RNA transcripts to the protein-coding genes. The relative coverage was summed and plotted along each 1/100 portion for each protein-coding transcript containing the long-adaptor sequences. (E) Transcript integrity assessment of individual RNA transcripts. For each protein-coding gene, the transcript coverage was calculated as the average coverage of each individual RNA transcript against its corresponding CAP transcript.
The gene coverage is an important performance benchmark for transcriptome sequencing. Four additional 5ʹ-Cap-enriched libraries using the long-adaptor were further generated to assess the gene coverage of RNA transcripts. For each protein-coding gene, the RNA transcript that contains the long-adaptor sequence in its 5ʹ-end (CAP transcript) was considered as a full-length RNA transcript. The gene coverage was evaluated by aligning the CAP transcript to the protein-coding genes and examining the coverage percentage of coding sequences (CDS). All the CAP transcripts were divided into 100 portions to display the general gene coverage of the RNA transcripts. The good overall coverage of the coding region and substantial inclusion of the untranslated region indicate that the direct RNA sequencing by 5ʹ-Cap capturing can represent the entire length of full-length CDS (Figure 1(d)).
Because transcript fragmentation is not required in direct RNA sequencing, the adaptor sequences can serve as a tag for full-length transcript reference to assess the transcript integrity. We compared the transcript integrity of RNA transcripts in the standard libraries (combining polyAControl-1, polyAControl-2 and polyAControl-3 together) with that of RNA transcripts in the 5ʹ-Cap-enriched libraries. To assess transcript integrity, the average full-length transcript coverage for each gene was calculated by aligning each of the RNA scripts in the standard libraries to its corresponding CAP transcript in the 5ʹ-Cap-enriched libraries. To further assess the effects of RNA expression on transcript integrity, the CAP transcripts were divided into five expression categories. The bubble distribution in Figure 1(e) suggests that the average transcript coverage is largely independent of the expression level and transcript length. As shown in the violin-boxplots in Figure 1(e), the average full-length transcript coverage ranges from 13.55% to 100% with an average of 80.64%. Thus, direct RNA sequencing by 5ʹ-Cap capturing achieves a better full-length transcript coverage than direct RNA sequencing, although the latter one can sequence a large portion of the full-length transcript.
A substantial fraction of RNA transcript contains exonized transposable element sequences
The redundant RNA transcripts were collapsed based on their mapping positions, and the longest transcript for each transcription unit was selected as a representative RNA transcript. The transposable elements (TEs) in the representative RNA transcripts were inferred by calculating the fraction of RNA transcripts for which exons were overlapped with TE deposited in Repbase update 23.03. Among the 60,908 representative RNA transcripts, 51.88% (31,599) of them contained at least one TE-derived sequences. A total of 19,950,829 bp in the representative RNA transcripts was covered by TEs, comprising 19.94% (19,950,829 bp in 100,066,907 bp) of the locust transcriptome. Comparing genome-wide TE coverage (more than 65%) and transcriptomic TE coverage, the RNA transcripts were depleted for TE contribution [19]. This may reflect pervasive selection against TE invasion into the transcriptional region. TEs can be divided into three major categories: DNA transposons, non-long terminal repeat (non-LTR) retrotransposons and LTR retrotransposons, and each of these categories comprises multiple families. Roughly, DNA transposons (37.45%), non-LTR retrotransposons (32.93%) and LTR retrotransposons (29.63%) each occupied one-third of the transcript sequence coverage (Figure 2(a)). Mariner/Tc1family, Penelope family and Gypsy family represented the largest fraction in the DNA transposons, non-LTR retrotransposons and LTR retrotransposons, respectively. At least two different TE families in the same transcript (TE co-occurrence) could be detected in 56.16% (16,482 in 31,599) of the representative RNA transcripts that contain TE-derived sequences. The percentage of the TE family involved in TE co-occurrence is generally correlated with the transcript sequence coverage. The frequency analysis between TE families showed that there was no specific TE family mainly facilitating the TE co-occurrence of other TE family members (Figure 2(b)). Taken together, these results show that the exonization of TE-derived sequences was observed in a substantial fraction of RNA transcripts and that different TE families have varied contributions to RNA transcripts in locusts.
Figure 2.
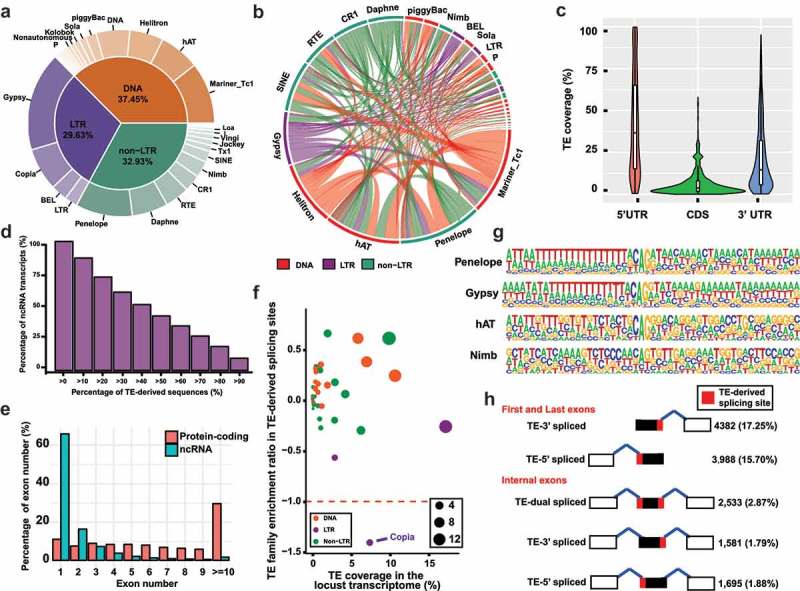
TE occurrence in the locust transcriptome. (a) Pie charts summarizing the annotation of the TE family in the locust transcriptome. (b) Co-occurrence frequency of TE families in the locust transcriptome. The length in the outer circle indicates the occurrence frequency for each TE family. The line width between two TE families is proportional to the co-occurrence frequency of two TE families. (c) TE coverage in the UTR and CDS regions. The longest transcript in each transcription unit is selected for coverage calculation. (d) Distribution of the percentage of TE-derived sequences in ncRNA transcripts. (e) Number of exons of ncRNA transcripts and protein-coding genes. (f) TE family enrichment ratio in TE-derived splicing sites. The TE family enrichment ratio is calculated as log2-scale (PS + 1/PT + 1). PS = Percentage of TE family in the TE-derived splicing site, and PT = Coverage percentage of TE family in the locust transcriptome. (g) Examples of enriched motifs in the TE-derived splicing acceptor sites in different TE families. (h) Summary of TE-derived splicing sites in the first/last exons and in internal exons.
For the 3,524 protein-coding genes whose transcripts harbor TE-derived sequences, the average coverage values of the TE-derived sequences in the 5ʹ UTR, coding region and 3ʹ UTR were 42.01%, 6.48% and 21.86%, respectively. The TE coverage in the coding region showed less variance than that in the 5ʹ UTR and 3ʹ UTR (Mann–Whitney U test, Ps < 0.05), showing a stronger selection against TE exonization in the coding region than those in the 5ʹ UTR and 3ʹ UTR regions (Figure 2(c)). The 5ʹ UTR showed more tolerance to TE exonization than the 3ʹUTR (Mann–Whitney U test, P < 0.05). Among the 50,729 representative ncRNA transcripts, RepeatMasker analysis showed that 55.34% (28,075 in 50,729) of them contained at least one TE-derived sequence. The median length of TE-derived sequences was 290 bp, and the maximum length was 6,197 bp. We found that 2,534 (9.02%) of 28,075 ncRNA transcripts comprised more than 90% of TE-derived sequences, indicating that only a minor portion of ncRNA transcripts containing TE-derived sequences comprised a major percentage of TE-derived sequences (Figure 2(d)).
The numbers of representative RNA transcripts harboring only one single exon, two exons and multiple exons were 35,499, 9,044 and 16,365, respectively. The protein-coding genes were greater in exon number than the ncRNA transcripts, and the dominant portion of the ncRNA transcripts was single exons (Figure 2(e)). Among the 25,409 representative RNA transcripts with splicing sites, 27.21% (6,913 in 25,409) of them contained at least one splicing site overlapped with TE-derived sequences. To determine whether any specific TE family contributes to generating splicing sites, the TE family enrichment ratios in TE-derived splicing sites were calculated as log2-scale (PS + 1/PT + 1) values, where PS = Percentage of TE family in the TE-derived splicing site, and PT = Coverage percentage of TE family in the locust transcriptome. A greater than two-fold change was not observed in almost all the TE families, except for the LTR/Copia family (Figure 2(f)). The TE family percentage in the TE-derived splicing site was significantly and positively associated with the coverage TE percentage in the locust transcriptome (Pearson product-moment correlation test, P = 2.2E-12, R = 0.92). The motif enrichment analysis indicated that the polypyrimidine tracts to be required for splicing are presented in the TE-derived splicing site in most of the locust TE families, except for a few TE families, including hAT and Nimb (Figure 2(g) and Supplementary Figure 3). BLAST searches showed that the polypyrimidine tracts were not present in the consensus sequences of the TE families (data not shown), suggesting that no locust TE is similar to the Alu element to provide polypyrimidine tracts that can be recognized by spliceosome. Thus, no dominant TE family contributed substantially to splicing in the locust genome.
To examine the TE-spliced exon pattern, the exons in the transcripts of protein-coding genes and ncRNAs were classified into the first/last exons and internal exons. The representative RNA transcripts harboring only one single exon (35,499) were removed, and the representative RNA transcripts harboring two (9,044) and more (16,365) exons were kept in this analysis. In the first/last exons, the splicing site in 17.25% (4,382 in 25,408) of TE-3ʹ spliced exons and 15.71% (3,988 in 25,408) of TE-5ʹ spliced exons were overlapped with TE-derived sequences. The internal exons, which were defined as an exon flanked by two introns, were identified as those following the previous studies [29,30]. The total number of internal exons is 89,649 with an average size is 216 bp in length. According to the splicing position of TEs within internal exons, the internal exons were classified into five types: TE-dual spliced exons (regardless of the presence or absence of TEs in the middle region of internal exons), TE-5ʹ spliced exons (donor splicing site), TE-3ʹ spliced exons (acceptor splicing site), TE-spliced-free exons and non-TE exons (Figure 2(h)). The numbers of internal exons absent and present in the TE-derived sequences were 80,891 (91.61%) and 7,408 (8.39%), respectively. In 2.87% (2,533 in 88,299) of the TE-dual spliced exons, 1.88% (1,695 in 88,299) of the TE-5ʹ spliced exons and 1.79% (1,581 in 88,299) of the TE-3ʹ spliced exons, the exonized TE-derived sequences provided acceptor splicing site and/or donor splicing site sequences. Comparing the first/last exons (32,94%, 8,370 in 25,408) with the internal exons (6.70%, 5,909 in 88,299), the TE-derived sequences contributed more splicing sites in the first/last exons than in the internal exons (Chi-Square test, P < 0.01). The splicing sites in 27.21% (6,913 in 25,408) of the representative RNA transcripts harboring multiple exons (two exons or more) were provided from TE-derived sequences.
Piwi expression influences the length of RNA transcripts containing TE-derived sequences
Because Piwi is a gene essential for the defense system against the expression of transposable elements, we investigate the Piwi role in TE exonization [2]. Double-stranded RNA (dsRNA) interference was used to de-repress TE activity by knocking down Piwi expression. To alleviate the influence of individual genomic variation, we isolated total RNAs from dsPiwi and dsGFP offspring that are derived from the same parents. The qPCR results showed that Piwi expression levels were significantly depleted by double-stranded RNA injection (Supplementary Figure 4). The expression levels were determined by counting the number of mapped reads on each protein-coding gene. Principal component analysis showed a clear separation (PC1 explains 61% of the variance) between the dsPiwi and dsGFP samples (Figure 3(a)). The RNA transcripts with more than 80% of TE-derived sequences were considered as locust transposons. A substantial number of transposons showed elevated expression in the dsPiwi samples (Figure 3(b)), suggesting that the transcriptional silencing of transposons was de-repressed by the RNA interference of Piwi expression. Following a previous study [31], the DESeq2 method was applied to estimate the statistic of protein-coding gene expression change, which is summarized in the MA plot in Supplementary Figure 5. Gene ontology (GO) enrichment analysis showed that the down-regulated genes were enriched for GO terms, such as ribosome biogenesis, RNA splicing, positive regulation of translation, base excision repair and fatty acid beta oxidation (Supplementary Figure 6). In the up-regulated genes, the enriched GO terms included oxidation reduction, amino acid biosynthetic process, glucose catabolic process and insulin-like growth factor binding. So, the RNA interference of Piwi expression has a substantial impact on the expression of protein-coding genes in locust transcriptome.
Figure 3.
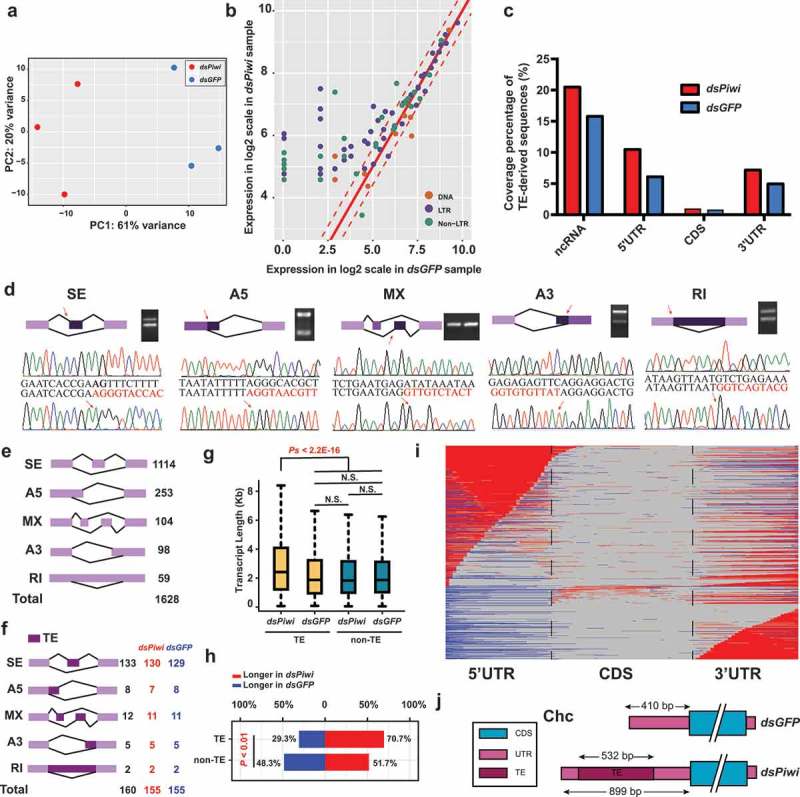
Burst of TE expression activity upon RNAi silencing of Piwi. (a) Principal component analysis plot of individual libraries of dsPiwi and dsGFP samples. (b) Scatterplot comparing TE expression (log2-scale TPM + 1) between dsPiwi and dsGFP samples. RNA transcripts with more than 80% TE-derived sequences are shown. The dashed lines in red indicate a 1.5-fold change in TE expression. To improve visualization clarity, the RNA transcripts with TPM > 20 are shown. (c) Coverage percentage of TE-derived sequences in the four categories, including ncRNA, 5ʹUTR, CDS and 3ʹUTR. The coverage percentage was calculated as the ratio of the total length of TE-derived sequences in each category and total length of its corresponding category. (d) Random validation of alternative splicing events using PCR reactions and Sanger sequencing of PCR products purified from agarose gels. (e) Summary of alternative splicing events. Skipped exon, SE; Mutually exclusive exons, MX; Alternative 5′ or 3′ splice site, A5 or A3; Retained intron, RI. (f) Summary of alternative splicing events that are overlapped with TE exonization. (g) Length distribution of the representative RNA transcripts in dsPiwi and dsGFP samples. The transcriptional units whose representative RNA transcripts could be detected in both the dsPiwi and dsGFP samples were used for comparison. N.S., not significant. (h) Comparison of the transcript length ratios between the dsPiwi and dsGFP samples. The portion in red represents the percentage of the genes whose transcript length in the dsPiwi samples is longer than that in the dsGFP samples. (i) Distribution of sequence variants along the protein-coding genes that are longer in the dsPiwi samples than in the dsGFP samples. For each representative RNA transcript, all the exons in the dsPiwi and dsGFP samples were merged into a concatenated set and were normalized into 100 bins. Red and blue indicate the bins are only covered by the dsPiwi samples and dsGFP samples, respectively. Light grey indicates the bins are covered by both the dsPiwi and the dsGFP samples. (j) An example diagram shows the length difference of protein-coding transcripts (Clathrin heavy chain gene, homologous to CG9012 in Drosophila melanogaster) in the dsPiwi and dsGFP samples.
The coverage percentage of TE-derived sequences was determined to compare the transcriptomic composition of TE-derived sequences between the dsPiwi and dsGFP samples. The representative RNA transcripts in each transcriptional unit in the dsPiwi and dsGFP samples were divided into four categories (ncRNA, 5ʹUTR, CDS and 3ʹUTR). The transcriptional units, among which representative RNA transcripts could be detected in both the dsPiwi and dsGFP samples (dual-expressed representative RNA transcripts), were used in the coverage percentage comparison. The coverage percentage of TE-derived sequences was calculated as the ratio of the total length of TE-derived sequences in each category and total length of its corresponding category. In all four categories, the coverage percentage of TE-derived sequences in the dsPiwi samples was greater than that in the dsGFP samples (Figure 3(c)), showing a higher inclusion level of TE-derived sequences in the dsPiwi sample.
The five main modes of alternative splicing were quantified to ascertain the relative contribution of alternative splicing to the inclusion of TE-derived sequences. The quantification of intron retention (RI), exon skipping (SE), alternative 3ʹ-acceptor (A3), alternative 5ʹ-donor (A5) and mutually exclusive exon (MX) was performed by using the SUPPA2 program. Random PCR validation for each alternative splicing mode was performed to exclude the possibility that these alternative splicing events were artifacts (Figure 3(d)). In total, 1,628 alternative splicing events were identified (Figure 3(e)), and most of these events were SE events (68.43%, 1,114 in 1,628). The A5 event (15.54%, 253 in 1,628) was the second most prevalent mode, whereas the RI event (5.05%, 59 in 1,628) was the least frequent. To determine whether alternative splicing events created by TE-derived sequences contributed to the higher inclusion of TE-derived sequences in the dsPiwi samples, the TE-derived sequences in the alternative exons were identified. Only a minor portion (9.82%, 160 in 133) of alternative splicing events was associated with TE-derived sequences. Most (96.88%, 155 in 160) of the TE-derived exons could be detected in both the dsPiwi and dsGFP samples (Figure 3(f)), so that alternative splicing is not the major contributor for the high inclusion of TE-derived sequences in the dsPiwi samples.
Global length distribution analysis showed that the lengths of the dual-expressed representative RNA transcripts containing TE-derived sequences in the dsPiwi samples are significantly longer than those in the dsGFP samples (Mann–Whitney U test; P < 2.2E-16; Figure 3(g)). However, there were no significant length differences between the dual-expressed representative RNA transcripts containing TE-derived sequences in the dsGFP sample and the dual-expressed representative RNA transcripts without TE-derived sequences in the dsPiwi and dsGFP samples (Mann–Whitney U tests; P = 0.456, P = 0.911 and P = 0.446). Pairwise length comparison showed that, for the dual-expressed representative RNA transcripts containing TE-derived sequences, the dsPiwi samples had longer transcripts than the dsGFP samples (Chi-Square test; P < 0.01; Figure 3(h)). To ascertain the location of the sequence variation along the protein-coding transcripts, relative coverage analysis was performed for the dual-expressed representative RNA transcripts that were longer in the dsPiwi sample than in the dsGFP sample. The length differences in the protein-coding transcripts were attributable to the sequence variations in the 5ʹUTR and 3ʹUTR regions but not in the CDS region. The lengthening at the 5ʹ head and 3ʹ tail of RNA transcripts could largely explain the observed length differences of the protein-coding transcripts between the dsPiwi and dsGFP samples (Figure 3(i)), implying alternative UTR usage regulation mediated by Piwi expression. An example illustration for Clathrin heavy chain gene is shown in Figure 3(j).
Discussion
In this study, we applied the 5ʹ-Cap capturing method using Nanopore direct RNA sequencing to characterize the full-length sequences of RNA transcripts in the locusts. Unlike to Nanopore direct RNA sequencing with its inability to determine the 5ʹ end of RNA transcripts, full-length sequencing of RNA transcripts could be achieved by the 5ʹ-Cap capturing method using Nanopore direct RNA sequencing. We found that widespread TE exonization in the locust transcriptome and exonized TEs are prone to inserting into the ncRNAs and untranslated region of protein-coding genes. We determined the spectrum of RNA transcripts that are influenced by Piwi expression. Consistent with previous studies examining Piwi transcriptional silencing, our results confirmed that TE transcripts are the main targets of Piwi-mediated repression [32]. Our study distinctly indicated that Piwi expression regulates the length of RNA transcripts containing TE-derived sequences, creating alternative UTR usage.
Regarding the huge locust genome, because TEs produce numerous highly similar fragments that are difficult to be resolved using short-read sequencing data, the read length limitation further hamper the ability to precisely determine the full-length transcripts [33]. The high level of sequence similarity in genome-wide spread TE-derived sequences results in the generation of identical short reads that are expressed from more than one genomic location [34]. The repetitive nature of TEs makes TE-derived short-reads challenging to map onto a reference assembly in a precise manner. Therefore, sequence reconstruction of RNA transcripts containing TE-derived sequences in the locusts is a computational challenge using short-read sequencing technology. In this study, we demonstrated that the 5ʹ-Cap capturing method using Nanopore direct RNA sequencing can determine the full-length sequences of RNA transcripts containing TE-derived sequences. Furthermore, we used the representative RNA transcripts instead of all full-length RNA transcripts in the length comparison between the dsPiwi and dsGFP samples. This is because Nanopore RNA sequencing produces truncated reads that are caused by electronic signal noise associated with enzyme motor stalls during strand translocation or by stray current spikes of unknown origin [35]. However, there were no significant length differences between the representative RNA transcripts containing TE-derived sequences in the dsGFP sample and the representative RNA transcripts without TE-derived sequences in the dsPiwi and dsGFP samples, indicating that the length differences were not caused by read length fluctuation or read truncation of Nanopore RNA sequencing.
Because TEs, particularly L1 and Alu element, contain good substrate sequences for the TE exonization process, transposable elements were considered as major contributors to recruit TE into exons in vertebrate genomes [4,30]. Alu elements inserted into a gene can provide both splicing acceptor and donor sites, creating new alternative splicing exons [36,37]. Our results in locusts indicate that considering neither TE-derived exons nor TE-derived splicing sites, no TE exonization preference exists for a specific element. Motif enrichment of the splicing sites showed that the presence of polypyrimidine tracts upstream of 3ʹ splicing sites of introns was observed in the TE-derived exons. However, the polypyrimidine tracts were provided from the locust TE families. These results showed that each TE element has equal potential to be recognized as exons with assistance from additional flanking signals of polypyrimidine tracts. However, the exonization level of primate-specific Alu (SINE) elements is approximately three times that of the LINE, MIR and CR1 elements in the human genome [38]. Similar to that in humans, 84.4% of SINE-containing exons in the zebrafish genome are derived from HE1 elements, which comprise almost 10% of the zebrafish genome [4]. The overrepresentation of these specific TE elements in TE exonization is mainly due to its special structure within these TE elements. Because of the existence of polypyrimidine tracts in these TE elements, an enrichment of these TE elements within intron sequences produces a greater level of TE exonization. These results suggest that, although both the locust and vertebrate genomes contain a large portion of TEs, the formation of TE-derived exons in locusts depends on a mechanism that is distinct from that in vertebrates.
In addition to the transcriptional de-repression of the locust transposons that are not expressed in the dsGFP samples, we observed an over-representation of transcriptomic composition of TE-derived sequences in the dsPiwi samples. Transcriptional de-repression of transposons and TE-derived sequences by knockdown of Piwi expression have been observed in previous studies using short-read sequencing technology [2,32,39]. Recovering full-length transcript sequences using short-read sequencing data is a challenge due to computational complexity and experimental drawbacks. This fragmentary transcript assembly problem is prominent in TE-harboring transcripts due to the inherent assembly difficulty of diverse TE copies [33]. Because the long-read sequencing showing the ability to sequence the full-length transcripts provides an advantage over assembly-based sequencing using short-read data, the development of Nanopore long-read RNA sequencing provides an opportunity to compare the transcript length without the assembly of fragments to resolve full-length transcripts. We found that the RNA transcripts containing TE-derived sequences in the dsPiwi samples are significantly longer than those in the dsGFP samples. Regarding the protein-coding transcripts, the sequence variation in the UTR region, but not in the CDS region, is largely responsible for the transcript lengthening in the dsPiwi samples. This observation is consistent with our results that, compared with the UTR region, the CDS region contains only a minor portion of TE-derived sequences. TE exonization in the CDS region might disrupt open reading frames and tends to be disadvantageous [36]. The increased portion of TE-derived sequences in ncRNAs and UTRs suggests that ncRNAs and UTRs are under a lower selection constraint of TE accumulation than CDSs. A mechanistic explanation for these findings may be that Piwi-interacting RNAs guide Piwi complexes to destroy TE-containing transcripts by endonucleolytic cleavage [2]. The knockdown of Piwi expression shows a specific depletion in the biogenesis of Piwi-interacting RNAs [40]. Because UTRs contain a substantial portion of TE-derived sequences, the increased UTR length results in an increased proneness to include TE-derived sequences. The RNA transcripts containing TE-derived sequences in longer UTRs may escape from the endonucleolytic cleavage of Piwi-interacting RNA upon knockdown of Piwi expression. This issue should be further addressed by more sophisticated experiments.
Materials and methods
Insect rearing
Locusts (the migratory locust, Locusta migratoria) were reared in large, well-ventilated cages (40 cm × 40 cm × 40 cm) at a density of 500–1000 insects per container. These locust colonies were reared under a 14:10 light/dark photo regimen at 30°C and were fed fresh wheat seedlings and bran.
Enrichment of full-length RNA transcripts by 5ʹ-Cap capturing
The locust testes were dissected from the adults at 7 days after eclosion. Total RNAs were isolated using TRIzol regent (Invitrogen) according to the manufacturer’s instructions. Contaminant DNAs were removed by DNase I (Invitrogen) treatment. Contaminant DNAs were removed by incubation of the RNA with DNase I (Thermo Fisher Scientific). The DNA-free total RNAs were poly-A enriched using Oligo (dT)25 Dynabeads (Thermo Fisher Scientific) twice. The poly-A RNAs were treated with calf intestinal alkaline phosphatase (CIP) at 37°C for 1 hour. The reaction mixture was purified using 2.2 × Agencourt RNAClean XP beads (Beckman Coulter). Next, CIP-treated RNAs were treated with Tobacco Acid Pyrophosphatase (TAP) at 37°C for 1 hour and purified with 2.2 × RNAclean XP beads. Finally, the biotin-labeled RNA adaptors were ligated to the RNAs using T4 RNA ligase at 25°C for 4 hours, and the free biotin-labeled RNA adaptors were removed with 1.8× RNAclean XP beads (Beckman Coulter). The RNAs containing biotin-labeled RNA adaptors in their 5ʹ-end were captured with Streptavidin C1 beads (Thermo Fisher Scientific) and then were eluted in 10 mM EDTA (pH8.2) with 95% formamide at 65°C for 5 minutes. The elution was purified using 2.2 × RNAClean XP beads.
RNA isolation, library preparation and sequencing
In RNA inference experiments, double-stranded RNAs of Piwi and green fluorescent protein (GFP) were prepared using the T7 RiboMAX Express RNAi system (Promega) according to the manufacturer’s protocols. To knock down Piwi expression, 15 μg of double-stranded RNAs was injected at the dorsal site near the locust testis in the adults at 3 and 5 day after eclosion by using a nanoliter injector 2000 (World Precision Instruments). The abdomens of the adults at 7 day after eclosion were vertically opened, and the testes were separated from other tissues for total RNA isolation. Total RNAs were isolated using TRIzol reagent (Thermo Fisher Scientific). Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega) were used to prepare the Oligo (dT)-primed cDNAs. The mRNAs were subjected to qPCR using the SYBR Green gene expression assays on a LightCycler® 480 instrument (Roche). The direct RNA libraries were prepared using the Direct RNA Sequencing Kit SQK-RNA001 (Oxford Nanopore Technologies) according to the manufacturer’s protocol. Briefly, the poly-A RNAs were enriched using the Dynabeads mRNA Purification Kit (Thermo Fisher Scientific) with Dynabeads Oligo (dT)25. Next, 100–500 ng of the poly-A enriched RNAs were ligated to the reverse transcriptase adaptor using T4 DNA ligase, followed by reverse transcription. The reverse-transcribed RNAs were ligated to the sequencing adaptor and were purified using Agencourt RNAClean XP beads (Beckman Coulter). Finally, 200 ng of the RNA libraries were loaded on FLO-MIN106 (R9.4) flowcells and were run on a MinION or a GridION X5 (Oxford Nanopore Technologies) over 48 hours at Nextomics Biosciences (Wuhan, China). A total of 12 libraries, including five standard libraries and seven capturing libraries, were generated. Data were archived at NCBI Sequence Read Archive under accession PRJNA517220.
Data analysis
FAST5 files were base called using Albacore 2.2.7 (Oxford Nanopore Technologies) by executing the read_fast5_basecaller.py script. The adaptors in the 5ʹ-end of RNA transcripts were detected using the Smith-Waterman algorithm, as implemented in the SSEARCH program [28]. The sequencing errors in direct RNA reads were corrected by Illumina reads using LoRDEC version 0.8 [41], and the error-corrected reads were aligned to the locust genome using GMAP version 2018-03-25 [42] and the cross-species parameter as described in a previous study [13]. The RepeatMasker-open-4-0-7_2 program was applied to screen RNA transcript sequences and locust genome sequence for interspersed repeats and transposable elements using the locust repetitive elements deposited in Repbase update 23.03 [43]. To avoid bias introduced by the sequencing data amount, an equal amount of Nanopore RNA sequencing data in both the dsPiwi and dsGFP samples was used in the difference analysis upon knockdown of Piwi expression. The overlaps between TEs and RNA transcripts were identified using BEDTools version 2.25 package [44]. For alternative splicing analysis, the raw direct RNA sequencing reads were polished and collapsed using Racon version 1.3.1 and the pinfish (github.com/nanoporetech/pinfish) pipeline with minimap2 version 2.12 [45,46]. The alternative splicing events were identified using SUPPA2 version 2.3 [47]. All the statistics were implemented in R (www.r-project.org).
Funding Statement
This work was supported by the National Natural Science Foundation of China [31672353]; the open project of The State Key Laboratory of Integrated Management of Pest Insects and Rodents of China [ChineseIPM1708]; National Natural Science Foundation of China [31702060].
Acknowledgments
The computational resources were provided by the Research Network of Computational Biology and the Supercomputing Centre at Beijing Institutes of Life Science, Chinese Academy of Sciences.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kapusta A, Kronenberg Z, Lynch VJ, et al. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013;9:e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Le Thomas A, Rogers AK, Webster A, et al. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013;27:390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sienski G, Donertas D, Brennecke J.. Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell. 2012;151:964–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sela N, Kim E, Ast G. The role of transposable elements in the evolution of non-mammalian vertebrates and invertebrates. Genome Biol. 2010;11:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Belancio VP, Hedges DJ, Deininger P. LINE-1 RNA splicing and influences on mammalian gene expression. Nucleic Acids Res. 2006;34:1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Abascal F, Tress ML, Valencia A. Alternative splicing and co-option of transposable elements: the case of TMPO/LAP2alpha and ZNF451 in mammals. Bioinformatics. 2015;31:2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351:aac7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lavi E, Carmel L. Alu exaptation enriches the human transcriptome by introducing new gene ends. RNA Biol. 2018;15:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Criscione SW, Zhang Y, Thompson W, et al. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15:583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cartolano M, Huettel B, Hartwig B, et al. cDNA library enrichment of full length transcripts for SMRT long read sequencing. PLoS One. 2016;11:e0157779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kozarewa I, Ning Z, Quail MA, et al. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods. 2009;6:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Garalde DR, Snell EA, Jachimowicz D, et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods. 2018;15:201–206. [DOI] [PubMed] [Google Scholar]
- [14].Hommelsheim CM, Frantzeskakis L, Huang M, et al. PCR amplification of repetitive DNA: a limitation to genome editing technologies and many other applications. Sci Rep. 2014;4:5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Picelli S, Faridani OR, Bjorklund AK, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. [DOI] [PubMed] [Google Scholar]
- [16].de Klerk E, Den Dunnen JT, t Hoen PA. RNA sequencing: from tag-based profiling to resolving complete transcript structure. Cell Mol Life Sci. 2014;71:3537–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Matz M, Shagin D, Bogdanova E, et al. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999;27:1558–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cocquet J, Chong A, Zhang G, et al. Reverse transcriptase template switching and false alternative transcripts. Genomics. 2006;88:127–131. [DOI] [PubMed] [Google Scholar]
- [19].Wang X, Fang X, Yang P, et al. The locust genome provides insight into swarm formation and long-distance flight. Nat Commun. 2014;5:2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang X, Kang L. Molecular mechanisms of phase change in locusts. Annu Rev Entomol. 2014;59:225–244. [DOI] [PubMed] [Google Scholar]
- [21].Jiang F, Liu Q, Liu X, et al. Genomic data reveal high conservation but divergent evolutionary pattern of Polycomb/Trithorax group genes in arthropods. Insect Sci. 2019;26:20–34. [DOI] [PubMed] [Google Scholar]
- [22].Jiang F, Liu Q, Wang Y, et al. Comparative genomic analysis of SET domain family reveals the origin, expansion, and putative function of the arthropod-specific SmydA genes as histone modifiers in insects. GigaScience. 2017;6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jiang F, Yang M, Guo W, et al. Large-scale transcriptome analysis of retroelements in the migratory locust, Locusta migratoria. PLoS One. 2012;7:e40532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guo W, Wang XH, Zhao DJ, et al. Molecular cloning and temporal-spatial expression of I element in gregarious and solitary locusts. J Insect Physiol. 2010;56:943–948. [DOI] [PubMed] [Google Scholar]
- [25].Deamer D, Akeson M, Branton D. Three decades of nanopore sequencing. Nat Biotechnol. 2016;34:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pelechano V, Wei W, Jakob P, et al. Genome-wide identification of transcript start and end sites by transcript isoform sequencing. Nat Protoc. 2014;9:1740–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Maruyama K, Sugano S. Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene. 1994;138:171–174. [DOI] [PubMed] [Google Scholar]
- [28].Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith-Waterman and FASTA algorithms. Genomics. 1991;11:635–650. [DOI] [PubMed] [Google Scholar]
- [29].Kim D-S, Huh J-W, Kim Y-H, et al. Bioinformatic analysis of TE-spliced new exons within human, mouse and zebrafish genomes. Genomics. 2010;96:266–271. [DOI] [PubMed] [Google Scholar]
- [30].Sela N, Mersch B, Gal-Mark N, et al. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu‘s unique role in shaping the human transcriptome. Genome Biol. 2007;8:R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jenjaroenpun P, Wongsurawat T, Pereira R, et al. Complete genomic and transcriptional landscape analysis using third-generation sequencing: a case study of Saccharomyces cerevisiae CEN.PK113-7D. Nucleic Acids Res. 2018;46:e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sytnikova YA, Rahman R, Chirn GW, et al. Transposable element dynamics and PIWI regulation impacts lncRNA and gene expression diversity in Drosophila ovarian cell cultures. Genome Res. 2014;24:1977–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Loomis EW, Eid JS, Peluso P, et al. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013;23:121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Workman RE, Tang A, Tang PS, et al. Nanopore native RNA sequencing of a human poly (A) transcriptome. bioRxiv. 2018;459529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cowley M, Oakey RJ. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet. 2013;9:e1003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schwartz S, Gal-Mark N, Kfir N, et al. Alu exonization events reveal features required for precise recognition of exons by the splicing machinery. PLoS Comput Biol. 2009;5:e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gal-Mark N, Schwartz S, Ast G. Alternative splicing of Alu exons–two arms are better than one. Nucleic Acids Res. 2008;36:2012–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rozhkov NV, Hammell M, Hannon GJ. Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev. 2013;27:400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rajasethupathy P, Antonov I, Sheridan R, et al. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell. 2012;149:693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Salmela L, Rivals E. LoRDEC: accurate and efficient long read error correction. Bioinformatics. 2014;30:3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. [DOI] [PubMed] [Google Scholar]
- [43].Tarailo‐Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;25(1):4.10. 1–4.10.14. [DOI] [PubMed] [Google Scholar]
- [44].Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vaser R, Sovic I, Nagarajan N, et al. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Trincado JL, Entizne JC, Hysenaj G, et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018;19:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.