

ABSTRACT
Carrier state viral infection constitutes an equilibrium state in which a limited fraction of a cellular population is infected while the remaining cells are transiently resistant to infection. This type of infection has been characterized for several bacteriophages but not, to date, for archaeal viruses. Here we demonstrate that the rudivirus SIRV3 can produce a host-dependent carrier state infection in the model crenarchaeon Sulfolobus. SIRV3 only infected a fraction of a Sulfolobus islandicus REY15A culture over several days during which host growth was unimpaired and no chromosomal DNA degradation was observed. CRISPR spacer acquisition from SIRV3 DNA was induced by coinfecting with the monocaudavirus SMV1 and it was coincident with increased transcript levels from subtype I-A adaptation and interference cas genes. However, this response did not significantly affect the carrier state infection of SIRV3 and both viruses were maintained in the culture over 12 days during which SIRV3 anti-CRISPR genes were shown to be expressed. Transcriptome and proteome analyses demonstrated that most SIRV3 genes were expressed at varying levels over time whereas SMV1 gene expression was generally low. The study yields insights into the basis for the stable infection of SIRV3 and the resistance to the different host CRISPR-Cas interference mechanisms. It also provides a rationale for the commonly observed coinfection of archaeal cells by different viruses in natural environments.
KEYWORDS: Sulfolobus, archaeal viruses, carrier state infection, CRISPR-Cas adaptation and interference, Acr proteins
1. Introduction
Viruses play important roles in nature in shaping microbial communities [1]. Many studies have been undertaken of diverse virus-prokaryote infections but they have focused mainly on those producing cell lysis or lysogens [2]. Infections producing cell lysis may eventually lead to viral loss whereas lysogenic infections impair viral multiplication by suppressing expression of most viral genes and by converting host cells to superinfection-resistant lysogens. A central question remains as to how viruses, or virus mixtures, co-exist stably within hosts in natural environments.
Carrier state infection was originally described in 1961 for bacteriophages [3] but although it has been studied periodically in bacteria almost nothing is known for archaea. Of the 100 or so archaeal viruses characterised, very few have been shown to cause cell lysis [4] while some, in particular fuselloviruses, readily integrate into host genomes [5,6]. In addition, many are released constantly into infected cultures, at a low rate, without significantly impeding host growth [7–10].
In bacteria carrier state infection has been characterised for phages that existed episomally in a small fraction of host cells and were stably maintained without causing cell lysis [11]. Recently, single cell analyses of phage P22-infected Salmonella yielded insights into the mechanism involved [12]. P22 is a temperate phage able to undergo both lytic and lysogenic cycles. Before entering the lysogenic state, Salmonella, harbouring a polarly-tethered carrier state P22, overproduced phage-encoded immunity factors to prevent superinfection. These were inherited cytoplasmically by the P22-free daughter cells and produced transient resistance against P22. On cell division, dilution of the immunity factors conferred phage susceptibility to a subpopulation. Thus, a P22-infected Salmonella culture was able to exist in multiple states, lytic, lysogenic, carrier state and P22 free [12].
Here we demonstrate a carrier state infection of S. islandicus REY15A by SIRV3, a rudivirus closely related to SIRV2 [13] that induces cell lysis in S. islandicus LAL14/1 [4]. SIRV3 only infected a subpopulation of strain REY15A cells and the fraction of actively infected cells remained relatively stable over 7 generations. In order to activate host CRISPR-Cas activity against SIRV3, and to test the effect on SIRV3 maintenance, the culture was coinfected with the bicaudavirus SMV1 [14] that stimulates CRISPR-Cas spacer acquisition against coinfecting viruses or plasmids while remaining resistant itself [14–16]. We tested the viral propagation and interplay with the three different CRISPR-Cas interference systems of strain REY15A. Both viruses were stably maintained in the culture over a 12 day period but SIRV3 was expressed more actively than SMV1. Transcript levels of the viruses and host were monitored for up to 4.5 days post-infection (dpi), and viral expression levels peaked at 2.5 – 3 dpi, coincident with activation of CRISPR-Cas adaptation when SIRV3 was the primary target for CRISPR spacer acquisition. Host genes that yielded strongly enhanced transcript levels in coinfected samples encoded, in particular, subtype I-A CRISPR-Cas proteins, diverse VapBC antitoxin-toxins and transposases. Transcriptomic and proteomic analyses were undertaken on the viruses and infected cells undergoing CRISPR-Cas adaptation.
Factors influencing the infection of strain REY15A by SIRV3 are considered including a mutated cellular receptor [17] and virus-encoded anti-CRISPR proteins (Acr) [18]. We also hypothesise on the potential benefits of viruses coinfecting host cells in natural environments.
2. Results
2.1. Genomic and proteomic features of SIRV3 and SMV1
SIRV3 was isolated from a hot spring in the Gunnuhver geothermal area of Iceland [14] and was infected in S. islandicus REY15A. dsDNA was extracted from virions and the genome was sequenced (Genbank accession number KX712143). The linear genome (Figure 1) contains 32,995 bp and carries 45 ORFs which share 80–95% amino acid sequence identity with SIRV2 homologs, except for gp06 (ORF48) and gp38 (ORF90) which show ~30% sequence identity with proteins encoded by other rudiviruses [13] and gp40 (ORF149) that yielded no significant match amongst public database sequences. The genome carries characteristic rudiviral long inverted terminal repeats [13].
Figure 1.

Genome map of SIRV3. Genes encoding virion proteins are coloured yellow and those encoding Acr proteins are green. gp02 encodes the AcrID1 that inhibits CRISPR-Cas subtype I-D activity [18]. The predicted Acr proteins gp36 and gp37 are marked.
The circular genome of SMV1 was analysed earlier (Genbank accession number HG322870.1) and it carries 48,775 bp, 51 ORFs and three transposable elements [15] but the virion proteins remain uncharacterised. The virus exhibits an exceptional life cycle that involves extracellular tail development [19].
Proteins from the purified virions of SIRV3 and SMV1 were identified, and their abundance estimated, by mass spectrometry (Table 1). SIRV3 virions carried the major rudiviral coat protein gp19 (ORF134), a minor component gp29 (ORF1070), and very minor components gp05 (ORF131) and gp17 (ORF158) (Figure 1). Homologs of gp05 and gp17 were not detected earlier as structural proteins in SIRV2 [20].
Table 1.
Mass spectroscopy analyses of the protein contents of purified virions of SIRV3 and SMV1. The proteins are ordered according to decreasing spectral count values. Intracellular spectral counts from the coinfected sample at 3.5 dpi are given for comparison. nd – not detected. SMV1 genome accession number – HG322870.1.
| gene product |
ORF size |
accession number | kDa | spectral counts | intracellular spectral counts |
homologs conserved domains |
|---|---|---|---|---|---|---|
| SIRV3 | ||||||
| gp19 | 134 | AOG61579.1 | 14 | 3160 | 100 | coat protein |
| gp29 | 1070 | AOG61587.1 | 124 | 67 | 264 | coiled-coil |
| gp17 | 158 | AOG61577.1 | 18 | 8 | 12 | dUTPase |
| gp05 | 131 | AOG61565.1 | 15 | 3 | 3 | - |
| SMV1 | ||||||
| gp09 | 2028 | CDF81336.1 | 224 | 3160 | 15 | ATV-1940 WD40 domain |
| gp05 | 752 | CDF81332.1 | 86 | 1929 | 11 | transmembrane domain |
| gp24 | 588 | CDF81351.1 | 66 | 1889 | nd | ATV-618 MoxA ATPase |
| gp11 | 153,133, 114 |
CDF81338.1 | ≤17 | 711 | 2 | ATV-131 |
| gp06 | 122 | CDF81333.1 | 14 | 625 | 2 | ATV-145 |
| gp23 | 759 | CDF81350.1 | 84 | 499 | nd | ATV-892 VWA domain |
| gp17 | 105 | CDF81344.1 | 12 | 110 | 3 | SWIM domain |
| gp16 | 156 | CDF81343.1 | 18 | 157 | nd | YidB-like domain |
| gp13 | 442 | CDF81340.1 | 45 | 62 | nd | |
| gp47 | 307 | CDF81374.1 | 36 | 46 | nd | ATV-286 |
| gp32 | 95 | CDF81359.1 | 11 | 34 | 2 | |
| gp03 | 157 | CDF81330.1 | 16 | 25 | nd | transmembrane domain |
| gp34 | 242 | CDF81361.1 | 28 | 24 | nd | ATV-241 integrase |
| gp50 | 106 | CDF81377.1 | 12 | 9 | nd | RHH domain |
| gp19 | 145 | CDF81346.1 | 16 | 4 | nd | |
| gp39 | 292 | CDF81366.1 | 34 | 0/2 | nd |
SMV1 virions were complex with 16 major, medium and minor protein components one of which gp11 (ORF114) was characterised earlier [14] (Table 1). Some of these were homologous to virion proteins of Acidianus bicaudavirus ATV [21]. They included a MoxA AAA ATPase (gp24) and a von Willebrand factor domain protein (gp23). Four SMV1 virion proteins (gp09, gp23, gp34 and gp47) were homologous to ATV-encoded proteins that were not detected in ATV virions [21].
2.2. SIRV3 undergoes carrier state infection in S. islandicus REY15A
SIRV2 induces cell lysis in S. islandicus strain LAL14/1 with virions extruding via pyramid-like structures formed in the cell membrane [4,22]. Therefore, SIRV3 was propagated in strains LAL14/1 and REY15A. Both hosts carry multiple CRISPR spacers matching SIRV3 bearing zero or one or more mismatches (Table 2). The growth curves showed that whereas SIRV3 infection at MOI 1 completely inhibited growth of strain LAL14/1, only mild growth retardation occurred for strain REY15A when infected under similar conditions (Figure 2(a)). This suggested that only cells of the former were lysed, an inference that was reinforced by flow cytometry results which demonstrated that host DNA was heavily degraded in strain LAL14/1 but not in strain REY15A (Figure S1).
Table 2.
Strain REY15A CRISPR spacers showing perfect, or near perfect, base pairing matches to the SIRV3 and SMV1 genomes. The sequence corresponding to spacer 2_115 is duplicated at the termini of the SIRV3 genome. CRISPR spacer 1_83 denotes spacer 83 of CRISPR locus 1. SIRV3-matching spacers also exist in the strain LAL14/1 genome with 0 mismatches (1 spacer), 1 mismatch (4 spacers) and two and three mismatches (2 of each).
| virus | CRISPR locus_spacer |
base pair mismatch |
protospacer location |
|---|---|---|---|
| SIRV3 | 1_83 | 0 | 15,953–15,914 |
| “ | 2_115 | 0 | 255–293/32,741–32,703 |
| “ | 2_66 | 1 | 24,473–24,513 |
| “ | 2_100 | 1 | 21,721–21,761 |
| “ | 1_84 | 2 | 8088–8050 |
| “ | 2_45 | 2 | 18,398–18,437 |
| SMV1 | 2_87 | 1 | 25,475–25,514 |
Figure 2.
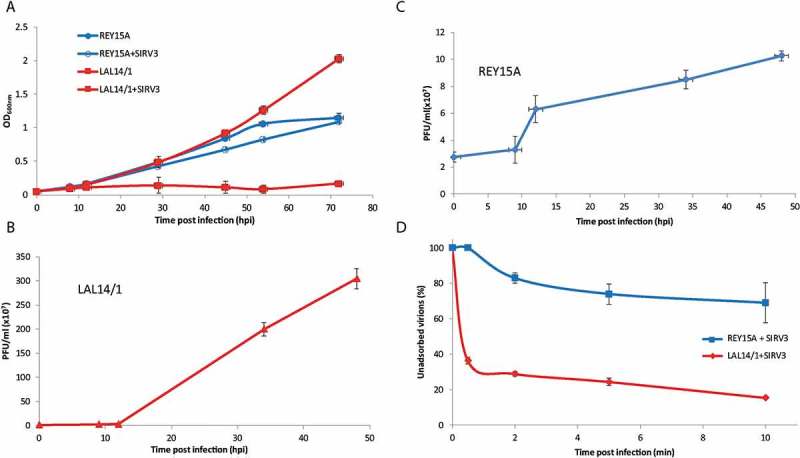
A. Growth curves of strains REY15A and LAL14/1 uninfected and infected with SIRV3. B. Virus titer in the supernatant of SIRV3-infected strain LAL14/1. C. Virus titer in the supernatant of SIRV3-infected strain REY15A. D. Unadsorbed virions of SIRV3 over time in cultures of strains REY15A and LAL14/1. Data were generated from duplicate experiments and error bars indicate standard deviations.
Consistent with these results, the plaque assay revealed that SIRV3 propagated at a high level in strain LAL14/1 (Figure 2(b)). In contrast, the virus titre in the supernatant of strain REY15A had only doubled 12 hours post infection (hpi) and required a further 36 hours to redouble (Figure 2(c)). Thus, SIRV3 propagated inefficiently in strain REY15A.
Next, we estimated the adsorption rates of SIRV3 to both strains. Whereas about 70% of SIRV3 virions bound to cells of strain LAL14/1 within 30 seconds, the adsorption rate for strain REY15A was significantly lower with only about 25% of virions adsorbed after 10 hrs incubation (Figure 2(d)). The results are consistent with SIRV3 undergoing a lytic infection in strain LAL14/1, similar to SIRV2 [4], whereas infection of strain REY15A was inefficient.
Then we estimated the cell fraction of strain REY15A carrying active SIRV3 over 84 hpi by expressing the core virus-encoded single-strand DNA binding protein gp12 using fluorescence microscopy [23,24]. Samples were taken at regular time intervals after infecting with SIRV3 at MOI 1. Cells were then fixed and subjected to immunohybridization with an antibody raised against SIRV2 gp17, a close homolog of SIRV3 gp12, as illustrated in Figure S2.
The proportion of SIRV3-infected cells in rich medium over 84 hpi fluctuated between 2% and 17% (Figure 3(a)). Importantly, there was no increase in the infection level with time and, given that the lytic life cycle of rudiviruses is about 12 hours [4,24,25], the results indicate that active SIRV3 was stably maintained. Consistent with this inference, the growth curves and cellular DNA contents of the SIRV3-infected cultures, showed no significant differences from those of the uninfected control culture (Figure 3(b,c)). In summary, stable maintenance of SIRV3, and the unimpeded growth of host cells over 4 days, supported the presence of an active viral infection in a small fraction of the culture.
Figure 3.
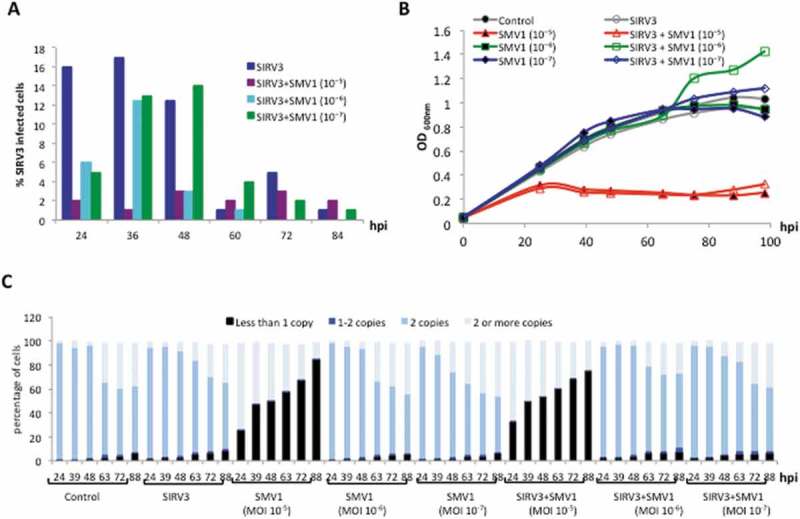
Characterization of strain REY15A cells infected by active SIRV3 (MOI 1) and SMV1 (MOIs in brackets), separately and together. A. Percentage of cells infected by SIRV3 alone, or coinfected at increasing levels of SMV1. B. Growth curves of strain REY15A cells infected with SIRV3 or coinfected. C. DNA content of strain REY15A cells infected by the single viruses or the mixture and analysed by flow cytometry. hpi – hrs post infection. A duplicated dataset is presented in Figure S3.
2.3. Effect of SMV1 coinfection on the level of SIRV3 infection
SMV1 can activate CRISPR spacer acquisition against a coinfecting virus or conjugative plasmid but not against itself [14–16]. Therefore, since no spacer acquisition activity was detected in SIRV3-infected cells (see below), we examined the effect of SMV1 coinfection first on the level of SIRV3 in infected cells.
SMV1 infection of strain REY15A was tested at MOIs in the range 10−8 – 1. At MOIs 10−8, 10−7 and 10−6 no growth retardation was observed over 100 hpi. Growth inhibition was first observed at MOI 10−5 (Figure 3(b)) and it was coincident with strong degradation of cellular DNA in about 80% of infected cells at 88 hpi, determined by flow cytometry (Figure 3(c)). Furthermore, very low amounts of extracellular virions (< 104 PFU/ml) were detected at 16 hpi by plaque assays indicating that most of the cell culture had died without releasing virions.
Next, cultures were coinfected with SIRV3 (MOI 1) and SMV1 (MOI 10−5, 10−6 and 10−7), adding first SIRV3 and then SMV1, and the proportion of cells infected with active SIRV3 was monitored over 84 hours. At SMV1 MOIs 10−6 and 10−7, the infection level of SIRV3 fluctuated between 0 and 14%, similar to the range of 2 – 17% observed for the culture infected with SIRV3 alone (Figure 3(a)). Notably, the active SIRV3 infection level always remained low (< 3%) in the culture coinfected with SMV1 (MOI 10−5) possibly due to cell death caused by the relatively high SMV1 MOI (Figure 3(a)). Repeated experiments under the same conditions (SIRV3 alone and coinfected at three different SMV1 MOIs) produced minor fluctuations at the same time points but infection levels were always less than 20% (Figure S3). It was concluded that whereas coinfection with SMV1 at low MOI did not significantly reduce the SIRV3 infection level, at higher MOIs some cell death occurred that limited SIRV2 infection.
Therefore, we infected SIRV3 at about MOI 1 and then SMV1 at MOI below 10−5, and the densities of uninfected and coinfected cells were measured at A600 with dilutions of fresh medium every 3–4 days over 12 days. Growth retardation occurred at 2–3 dpi and it increased over 3.5–7.5 dpi. Subsequent growth recovery at 8–9 dpi preceded strong retardation at 9.5–11 dpi (Figure S4A). Viral concentrations were determined by diluting samples to the same DNA concentration and then viral genes (gp40 of SIRV3 and gp11 of SMV1) were PCR amplified and analysed by visually estimating intensities of the PCR bands (Figure S4B). The results demonstrated that SIRV3 DNA levels remained fairly constant until 9 dpi and then increased while SMV1 DNA levels were fairly constant over 12 days (Table 3).
Table 3.
Relative viral DNA levels of coinfected strain REY15A, estimated qualitatively over 1 – 12 dpi by PCR band intensities. The relative levels are only valid for the comparison between different time points for the same virus, not for the comparison between SIRV3 and SMV1 DNA. The band intensities were estimated visually as indicated in Figure S3B.
| dpi | 1 | 2 | 2.5 | 3 | 3.5 | 4 | 5 | 5.5 | 6 | 7 | 7.5 | 8 | 8.5 | 9 | 9.5 | 10 | 10.5 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIRV3 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ++ | ++ | ++ | ++ | ++ |
| SMV1 | - | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | + | ++ | ++ | ++ | ++ |
2.4. CRISPR spacer acquisition from SIRV3 induced by SMV1
Strain REY15A carries three different CRISPR-Cas interference systems, subtype I-A, subtype III-Bα and subtype III-Bβ and a single CRISPR-Cas adaptation module [26]. Therefore next we tested whether SMV1 coinfection activated CRISPR-Cas adaptation from SIRV3. The leader-end regions of CRISPR loci 1 and 2 from SIRV3-infected and the coinfected samples were PCR amplified. The appearance of new larger PCR products in agarose gels occurred only from the coinfected sample indicating the onset of spacer acquisition in the CRISPR loci at 3–3.5 dpi (Figure S4C) and this effect coincided with growth retardation of the coinfected sample at 3–3.5 dpi (Figure S4A).
Next, we performed a transcriptome analysis of the uninfected and infected samples with the aim to determine: (a) identities of new CRISPR spacers acquired from viral DNA, and (b) transcript levels of the CRISPR-Cas machinery. Total RNA was extracted from samples taken from the same culture at time points 2, 2.5, 3, 3.5 and 4.5 dpi and from an uninfected and SIRV3-infected culture at 4 dpi carrying a very low level of virus (Table 4). Libraries were prepared and sequenced yielding in total about 123 × 106 paired-end 51 bp reads. Transcript sequences from each sample were aligned with the viral and host genomes (Table 4).
Table 4.
Total host and viral transcript yields. Total numbers of transcript reads after subtracting rRNA reads are given with the percentage of reads matching host and viral genomes.
| sample | dpi | total reads | SiRe (%) | SIRV3 (%) | SMV1 (%) |
|---|---|---|---|---|---|
| uninfected | 5 | 10,520,901 | 100 | 0 | 0 |
| SIRV3 | 4 | 12,241,903 | 99.90 | 0.10 | 0 |
| SIRV3 + SMV1 |
2 | 14,132,316 | 99.74 | 0.22 | 0.04 |
| 2.5 | 12,883,680 | 99.55 | 0.35 | 0.10 | |
| 3 | 53,000,092 | 84.38 | 14.85 | 0.77 | |
| 3.5 | 17,546,875 | 92.01 | 7.10 | 0.89 | |
| 4.5 | 14,433,285 | 89.63 | 4.75 | 5.62 |
Transcript abundance levels of both viruses in the coinfected samples rose sharply after 2.5 dpi, albeit at about a 20-fold higher level for SIRV3 (Table 4). However, whereas the SIRV3 transcript levels peaked at 3 dpi, they remained fairly constant for SMV1 with an increase at 4.5 dpi (Table 4). SIRV3 transcript levels were very low in the SIRV3-infected sample (4 dpi) (Table 4) while the host transcript profile was closely similar to that of the uninfected sample (Table 4).
First, we examined the CRISPR leader-end transcripts for newly acquired spacers in each coinfected sample. An increasing fraction of transcripts (0.7% to 2.2%) carried newly acquired spacers from SIRV3, in both CRISPR loci, throughout the late infection period (Table 5).
Table 5.
Total numbers of newly acquired SIRV3 CRISPR spacers detected amongst total leader-end transcript reads for all samples. The percentage of CRISPR transcript reads carrying new spacer sequences is given in parentheses.
| virus | dpi | CRISPR leader-end reads | new SIRV3 spacers |
|---|---|---|---|
| none | 5 | 133 | 0 |
| SIRV3 | 4 | 128 | 0 |
| SIRV3 + SMV1 |
2 | 984 | 2 (0.2%) |
| 2.5 | 592 | 0 | |
| 3 | 13,224 | 89 (0.7%) | |
| 3.5 | 766 | 9 (1.2%) | |
| 4.5 | 820 | 18 (2.2%) |
The seven samples were then divided into three groups and a differential transcription analysis was performed. The groups reflect the observed phases of infection development: (a) an uninfected and minimally SIRV3-infected samples as control group; (b) 2 and 2.5 dpi samples represent the ‘early’ infection phase; and (c) 3, 3.5 and 4.5 dpi samples where viral transcript levels increased and CRISPR-Cas adaptation was active constitute the ‘late’ infection stage. The global transcriptional profile comparison shows a clear segregation between the three groups, strongest between the ‘control’ and coinfected groups and with a smaller time-course difference underpinning the division between the early and late infected groups (Figure S5A).
Transcripts were detected from the CRISPR-Cas adaptation cassette and the three interference cassettes (subtypes I-A, III-Bα and III-Bß) in all samples (Table 6). However, coinfection yielded strongly increased transcript abundance in the late infection phase from the CRISPR loci, cas1, essential for CRISPR adaptation, and from the subtype I-A interference gene cassette (Table 6). In contrast, transcript levels from some type III-Bβ interference genes were strongly reduced (Table 6).
Table 6.
Transcript level changes of host CRISPR loci and cas genes of the subtype I-A CRISPR-Cas system of strain REY15A during coinfection. log2 fold-changes are shown in the early and late infection stages relative to the control samples, and to one another. Standard Sulfolobus cas gene annotations are employed [26].
| host gene ID | cas/cmr genes | amino acids | log2 fold changes | ||
|---|---|---|---|---|---|
| spacer acquisition | early | late | late/early | ||
| SiRe_0761 | cas1 | 290 | + 3.4 | ||
| I-A interference | |||||
| SiRe_0769 | cas3’ | 501 | + 2.7 | + 1.9 | |
| SiRe_0770 | cas3ʹ’ | 236 | + 2.5 | + 2.1 | |
| SiRe_0768 | cas5 | 240 | + 3.1 | + 2.0 | |
| SiRe_0772 | cas6 | 298 | + 2.7 | + 2.1 | |
| SiRe_0767 | cas7 | 321 | + 2.4 | ||
| SiRe_0771 | cas8’ | 345 | + 2.8 | + 2.0 | |
| SiRe_0766 | cas8ʹ’ | 143 | + 1.6 | + 2.9 | |
| III-B Cmr-β interference | |||||
| SiRe_0602 | cmr4 | 286 | −1.2 | ||
| SiRe_0601 | cmr5 | 155 | −1.7 | −1.2 | |
| SiRe_0599 | cmr6 | 283 | −1.4 | −1.1 | |
| CRISPR-115 locus 1 | + 3.2 | ||||
| CRISPR-93 locus 2 | + 2.3 | ||||
2.5. Cellular transcript abundance changes in coinfected samples
Significant fold changes in transcript abundance were observed for many host genes, especially in the late infection phase. In particular, transcripts from five of the 21 vapBC toxin-antitoxin gene pairs were significantly (p < 0.05) enhanced (Table S2). The strong activation closely paralleled earlier observations on virus-infected S. islandicus strains undergoing CRISPR-Cas activity [27,28] suggesting that it is a general virus infection response in Sulfolobus.
Large changes in transcript abundance, mainly increases, were also observed from IS element-encoded genes, five of which corresponded to IS605-orfB genes, or fragments thereof, including two genes (SiRe0752 and SiRe0773) neighbouring CRISPR loci (Table S2). In contrast to the toxin-antitoxin results many of the latter changes were detected at the early infection stage (Table S2).
Transcript levels of the cell division cdv operon (SiRe1173-SiRe1175), and most DNA replication genes, were not altered significantly, consistent with normal host growth. In contrast, transcript abundance levels from several transcriptional regulator genes were significantly up- or down-regulated (data not shown).
2.6. Viral proteome analysis of coinfected samples
Proteome analyses were performed on the uninfected sample and the coinfected sample undergoing CRISPR-Cas adaptation at 3.5 dpi, from the same culture as used in the transcriptome study. Approximately 70% of the total host-encoded proteins were detected (data not shown). Most SIRV3-encoded proteins yielded positive spectral counts (Table S1) and the major virion proteins gp19 and gp29 were especially abundant (Table 1). In general, protein levels for SMV1 were low, consistent with the transcription data (Table 4), and only five SMV1 proteins were detected three of which were virion proteins (Table S1).
3. Discussion
Archaea host a wide variety of viruses exhibiting diverse morphotypes and genometypes and many of those characterised are stably maintained intracellularly [7–10]. However, little is currently known about the factors influencing this stability. Here we studied two different viruses. The rudivirus SIRV3 contains characteristic linear dsDNA genome with inverted terminal repeats and covalently linked ends [13,29] and the virions exhibit a major coat protein gp19/ORF134 [13] and three minor components (Table 1); some differences were observed in the minor protein components from those described earlier for SIRV2 [20]. SMV1 carries a circular dsDNA genome [15] and 16 virion proteins (Table 1) and is related to the bicaudavirus ATV [21]. Both viruses undergo exceptional extracellular virion tail development [14,19,21] inferred to be induced by a co-chaperone complex of a MoxA AAA-ATPase and a von Willebrand domain protein [30].
On infecting strain REY15A with SIRV3 alone, virions were adsorbed weakly to host cells relative to strain LAL14/1 and minimal cell lysis was detected. The weak adsorption may result from the occurrence of truncations in host genes encoding protein components of one putative rudiviral receptor [2,17]. SIRV3 also propagated stably in a small fraction of the cellular population and did not induce a detectable CRISPR-Cas adaptation response.
In order to test the influence of CRISPR-Cas activity on the level of SIRV3 infection, it was activated on coinfecting with SMV1 that induces spacer acquisition against coinfecting genetic elements [14,15] possibly as a result of increased cellular stress generated by two genetic elements. The specific targeting of SIRV3 may partly reflect that spacer acquisition is dependent on active viral replication [14,31] and rudiviruses produce exceptionally complex replication intermediates [23].
Spacer acquisition from SIRV3 occurred in the late infection phase of the coinfected samples coincident with enhanced transcript levels from the spacer acquisition gene cas1 and subtype I-A interference genes (Table 6). This activity coincided with a partial decrease in the total level of SIRV3 transcripts (Table 4) but both viruses remained in the culture throughout the 12 day period (Table 3) although their transcriptional activities peaked at different times (Table 4). The transcript levels of SMV1 were lower than those of SIRV3 overall but also showed a marked increase at 3 dpi, albeit 19-fold lower than SIRV3, with a second stronger increase at 4.5 dpi (Table 4). These results are consistent with the proteome data obtained at 3.5 dpi when most SIRV3-encoded proteins were detectable in vivo whereas only a few SMV1 proteins including three of the 16 virion proteins were present in low yields (Table S1).
The presence of multiple SIRV3-matching spacers in CRISPR loci of strain REY15A (Table 2), together with the new spacers added in the coinfected cultures after 3 dpi (Table 5), were expected to provide a robust viral defence [32,33]. A likely explanation for the survival of the active viruses is that CRISPR-Cas interference was inhibited by Acr proteins encoded by SIRV3. One Acr protein (gp02) was recently shown to inhibit subtype I-D CRISPR-Cas interference of strain LAL14/1, and two further SIRV3-encoded homologs (gp36 and gp37) were predicted to perform anti-CRISPR functions [18]; moreover, all three proteins were expressed in the coinfected cells (Table S1).
The latter hypothesis is strengthened by a recent study of strain REY15A infection with SMV1 alone [34]. Guo et al. demonstrated that SMV1 was susceptible to attack from subtype III-B but not the subtype I-A interference complex, where the former degrades RNA and DNA and the latter dsDNA [34]. They inferred that the virus encoded an Acr protein specific for the latter subtype I-A complex. Potentially, that, together with SIRV3-encoded Acrs, could, have protected DNA of both SMV1 and SIRV3 from degradation in our coinfected samples, despite the strongly increased transcript levels observed for the subtype I-A interference proteins (Table 6). Furthermore, the observation that transcript yields from the subtype III-B interference genes were strongly reduced in the coinfected samples (Table 6) suggests that a second mechanism was activated to repress the latter immune system. The susceptibility of SMV1 to subtype III-B interference also provides an explanation for the low levels of SMV1 transcripts observed in our coinfected cultures because they would be a target for subtype III-B interference.
In conclusion, we show that SIRV3 undergoes a carrier state infection in strain REY15A whereby active virus is maintained in a low fraction of cells over longer periods. The presence of the second co-infecting virus SMV1 appeared to facilitate the stability of this state, despite the activation of the CRISPR-Cas immune response. This suggests that there is an ongoing, and unresolved, conflict between virus and host that is affected by inefficient cell entry of SIRV3 and specific blockage of the CRISPR-Cas interference complexes by Acr proteins with different specificities, potentially from both viruses. Thus, a likely explanation for the stable coinfection observed here, and for the similar coinfections frequently observed in natural thermophilic environments, is that the coinfecting viruses complement one another by providing Acr proteins which target the different types of CRISPR-Cas interference encoded by the host.
4. Materials and methods
4.1. Sulfolobus cultures and nucleic acid preparation
S. islandicus REY15A was cultured in Sulfolobus medium supplemented with 0.2% trypton, 0.1% yeast extract and 0.2% sucrose (TYS medium) at 78ºC. The rudivirus SIRV3 and monocaudavirus SMV1 were propagated and isolated as described [14,35]. The viruses were coinfected into Sulfolobus cultures and cell densities were monitored at A600 in 50 mL batches and diluted to A600 = 0.05 with fresh medium in every 3–4 days when uninfected control cultures exceeded A600 = 1 in order to prevent cell death from nutrient deficiency. Samples were taken every 24 hr and cells were harvested by centrifugation (6000 g, 10 min). SIRV3 infection was detected by PCR amplifying gp40 (ORF149) using primers forward 5ʹ-CTAATAAGACAAGAACATCAG-3ʹ and reverse 5ʹ-CATATATAGTATTGGTGAAAAG-3ʹ; SMV1 infection was detected by PCR amplifying gp11 (ORF153) using primers forward 5ʹ-GCGGATTCGCCCCTGCAGGTACG-3ʹ and reverse 5ʹ-GTCTCTCTCATATTTGCAATC-3ʹ
DNA was isolated using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and sequencing of the SIRV3 DNA was performed by GATC Biotech (Konstanz, Germany) and the genome was assembled using the CLC genomics workbench (Qiagen). Total RNA was extracted using the TRIzol reagent (Sigma Aldrich, Copenhagen, Denmark) following the manufacturer’s protocol.
4.2. Plaque assay
Approximate virus titres (PFU/ml) were determined by plaque assays after purification, infection and coinfection. Serial dilutions (10.0 μL) of viral preparations were mixed with 2.0 mL of exponentially growing host culture (2 x 108 cells); S. islandicus ΔCRISPR_1 LAL14/1 for SIRV3 infection [35] and S. islandicus ΔC1C2 for SMV1 infection [36]. The mixture was incubated for 30 min at 78°C to allow for adsorption of viruses onto host cells. Immediately after adding 1 mL of 0.4% (w/v) Gelrite (78%) or 2 mL of 0.4% (w/v) Gelrite (78%) for SIRV3 and SMV1 mixtures, respectively, the sample was layered on a premade 0.7% (w/v) Gelrite plate (78°C). Plates were incubated for 2–3 days at 78°C. SIRV3 plaques were small and turbid whereas SMV1 plaques appeared as small clear halozones. Single plaques were counted and PFU values were determined.
4.3. Determination of SIRV3 burst size and adsorption rate
The SIRV3 burst size in strain REY15A was measured according to Bize et al. [4]. Briefly, REY15A cells were infected with SIRV3 at MOI 0.1 and the virus titer (PFU/ml) in the supernatant at 12 hpi was measured by the plaque assay. The cell density was estimated as 108 cells/ml at A600 0.15. The burst size was estimated by dividing the virus titer at 12 hpi with the cell density. SIRV3 adsorption rates in strains REY15A and LAL14/1 were determined as described by Uldahl et al. [19]. The cells were infected with the virus at MOI 1 and the virus titer (PFU/ml) in the supernatant, i.e. the virus remaining unadsorbed at 30 seconds post-infection, was determined by the plaque assay. The level of viruses adsorbed to cells was estimated relative to the amount initially added.
4.4. Flow cytometry analysis
Strain REY15A cells were infected with SIRV3 (MOI ≈ 1) and SMV1 (MOI variable) with samples taken at regular intervals from uninfected, single infected and coinfected cultures. 300 μl culture was mixed with 700 μl of 100% ethanol and stored at 4°C. After all samples were collected, the fixed cells were centrifuged at 8000 rpm for 4 min and resuspended in 1 ml of 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2. Samples were recentrifuged under the same conditions and pellets were resuspended in 150 μl fresh staining solution containing ethidium bromide (20 μg/ml) and mithramycin (100 μg/ml) for 1 hr. Samples were kept cold at all steps and analyzed in an ApogeeFlow A-40 flow cytometer illuminating with a 405 nm laser (Apogee Flow Systems, Hemel Hempstead, UK). Statistical analyses of the qualitative flow cytometry results were performed by using Flowing Software 2 [http://flowingsoftware.btk.fi].
4.5. Immunofluorescence staining
Cells recovered by centrifugation at 8000 rpm for 3 min. were fixed by suspending in 4% paraformaldehyde for 10 min at room temperature. They were washed three times with PBS buffer (10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4, 137 mM NaCl, 2.7 mM KCl) and were pelleted at 8000 rpm for 3 min and resuspended in PBS. Samples were permeabilized with PBS + 0.1% Tween®20 (PBST) for 15 min at room temperature and washed three times with PBS. 10 μl cell-suspension was spotted onto poly-L-lysine-coated slides (Sigma-Aldrich) and air-dried. Slides were rinsed with PBS and incubated with PBST + 1% bovine serum albumin (BSA) for 15 min at room temperature. Thereafter, slides were incubated with the corresponding primary antibody diluted in PBST + 1% BSA for 1 hr at room temperature. Guinea rabbit anti-gp17 polyclonal antibody was used at a dilution of 1:5000. After three washes of 2 min each with PBS, the slides were incubated with the corresponding secondary antibody diluted in PBST + 1% BSA for 1 hr at room temperature. The commercial secondary antibody Alexa Fluor® 488 goat anti-guinea rabbit was used at a dilution of 1:1000. Slides were washed three times with PBS and incubated with 10 μM 4ʹ, 6-diamino-2-phenylindole (DAPI) (Sigma-Aldrich) for 5 min at room temperature and then washed a further three times with PBS and mounted with PVP mounting medium (1x PBS, 78% glycerol and 0.2% polyvinyl pyrrolidone). Finally, slides were analyzed using a Nikon Eclipse Ti-E inverted microscope coupled to an Andor Zyla 5.5 sCMOS camera. Images were processed using Adobe Photoshop CS6.
4.6. Transcriptome analysis
Paired-end HiSeq reads were analysed for 7 libraries including one uninfected control culture, another infected with a low level of SIRV3 and five samples coinfected with SIRV3 and SMV1 taken at times 2, 2.5, 3, 3.5 and 4.5 dpi. Total RNA libraries were constructed from each sample and 51 bp from each end was sequenced in an Illumina HiSeq 2500 at GATC Biotech AG (Konstanz, Germany). All reads were mapped independently on the viral and host genomes strain REY15A (CP002425), SIRV3 (KX712143) and SMV1 (NC_023585) using ERANGE within the CLC Genomics Workbench (CLC Bio, version 7.0, Qiagen) [37]. Default settings for the algorithm for sequence matching were: mismatch cost 2, gap opening and extension costs 3, minimal alignment/similarity region 80%. Total numbers of transcripts were used for differential host transcription analysis using EdgeR version 3.16.5 [38]. General transcriptional profiles were compared before the differential analyses to verify that sample groups were clustering together and thus coherent (Figure S5). Three groups were compared pairwise: a control group including uninfected or minimally SIRV3 infected libraries (n = 2), an early coinfection group including 2 and 2.5 dpi samples (n = 2), and a late infection group comprising the 3, 3.5 and 4.5 dpi samples (n = 3). Genes were considered differentially transcribed below a false discovery rate (FDR)-corrected p-value of 0.05 [39]. For the analysis the total transcript counts for each gene were employed after excluding rRNA and ncRNAs reads (counts per million). In order to remove noise from low-transcription genes, only those with > 1 count per million in 5 or more libraries (from a total of 7) were retained for the analysis.
4.7. Quantifying new spacer acquisition
CRISPR spacer acquisition was monitored over an 8 day period by PCR amplifying leader-proximal regions of both CRISPR loci using 5ʹ-GTCCATAGGAG GACCAGCTTTC-3ʹ and 5ʹ-CCAACCCCTTAGTTCCTCCTCTATAG-3ʹ for locus 1, and 5ʹ- GTTCCTTCCACTATGGGACTAGGAAC-3ʹ and 5ʹ-CGTCACTGACACCATATTTATAC-3ʹ for locus 2. PCR products were resolved in 1% agarose gels and detected by ethidium bromide staining under UV light (Doc-ItLS; UVP, Cambridge, UK).
CRISPR RNA reads were collected by selecting records containing a 36 nt sequence corresponding to the full repeat sequence CTTTCAATTCTATAGTAGATTAGC, using the Biopieces bioinformatics toolset (www.biopieces.org). New CRISPR spacers and their protospacer origins were identified by BLAST alignments to a local database of the host and viral genomes using word size 28 nt for spacer sequences, mismatch cost 3 and gap cost 2. The overall level of CRISPR adaptation activity was estimated from the proportion of transcripts carrying de novo virus-matching spacers relative to the number of wild-type leader-end transcripts in each sample. The protospacer origin of the de novo CRISPR spacers was analyzed using BLAST. Only forward reads of each library insert were used for quantification.
4.8. Proteome analysis
Virus particles were purified by CsCl density gradient as described earlier [40]. For mass spectrometric analysis, proteins were in-gel-digested with trypsin as described previously [41]. Extracted peptides were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) on an Orbitrap XL instrument (Thermo Fisher Scientific) under standard conditions. Peptide fragment spectra were searched against open reading frames of SIRV3 and SMV1. The proteome data were analysed with the MaxQuant package which yielded peptide counts, sequence coverage, total spectral counts, intensity and PEP score values [42]. The data obtained for proteins isolated from SIRV3 and SMV1 were analysed against a database comprising only virus proteins, whereas results obtained for the host, and infected host, proteins were analysed against a database for all host and viral proteins.
Funding Statement
The work was supported by Danish Council for Independent Research/Technology and Production (grant number DFF–7017-00060) to XP and by grants from Copenhagen University to SE, CL-S and RAG. The proteomics analysis was supported by the Deutsche Forschungsgemeinschaft (DFG - FOR 1680).
Acknowledgments
M. Raabe and U. Pleßmann are thanked for technical assistance with the proteomics analysis
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Suttle A. Viruses - major players in the global ecosystem. Nat Rev Microbiol. 2007;5(10):801–812. [DOI] [PubMed] [Google Scholar]
- [2].Hobbs Z, Abedon ST. Diversity of phage infection types and associated terminology with “Lytic or lysogenic” FEMS. Microbiol Lett. 2016;363(7):fnw047. [DOI] [PubMed] [Google Scholar]
- [3].Li K, Barksdale L, Garmise LJ. Phenotypic alterations associated within the bacteriophage carrier state of Shigella dysenteriae. Gen Microbiol. 1961;24:355–367. [DOI] [PubMed] [Google Scholar]
- [4].Bize A, Karlsson EA, Ekefjärd K, et al. A unique virus release mechanism in the Archaea. Proc Natl Acad Sci USA. 2009;106:11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Reiter WD, Palm P, Transfer YS. RNA genes frequently serve as integration sites for prokaryotic genetic elements. Nucleic Acids Res. 1989;17(5):1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wiedenheft B, Stedman K, Roberto F, et al. Comparative genomic analysis of hyperthermophilic archaeal fuselloviridae viruses. J Virol. 2004;78:1954–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zillig W, Arnold HP, Holz I, et al. Genetic elements in the extremely thermophilic archaeon Sulfolobus. Extremophiles. 1998;2:131–140. [DOI] [PubMed] [Google Scholar]
- [8].Prangishvili D, Forterre P, Garrett RA. Viruses of the Archaea: a unifying view. Nat Rev Microbiol. 2006;4:837–848. [DOI] [PubMed] [Google Scholar]
- [9].Pina M, Bize A, Forterre P, et al. The archeoviruses. FEMS Microbiol Rev. 2011;35:1035–1054. [DOI] [PubMed] [Google Scholar]
- [10].Wang H, Pen N, Shah SA, et al. Archaeal chromosomal elements. Microbiol Mol Biol Rev. 2015;79(1):117–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Siringan P, Connerton PL, Cummings NJ, et al. Alternative bacteriophage life cycles: the carrier state of Campylobacter jejuni. Open Biol. 2014;4:130200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cenens W, Makumi A, Govers SK, et al. Viral transmission dynamics at single-cell resolution reveal transiently immune subpopulations caused by a carrier state association. PLoS Genet. 2015;11(12):e1005770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peng X, Blum H, She Q, et al. Sequences and replication of the archaeal rudiviruses SIRV1 and SIRV2: relationships to the archaeal lipothrixvirus SIFV and some eukaryal viruses. Virology. 2001;291:226–234. [DOI] [PubMed] [Google Scholar]
- [14].Erdmann S, Le Moine Bauer S, Garrett RA. Inter-viral conflicts that exploit host CRISPR immune systems of Sulfolobus. Mol Microbiol. 2014;91:900–917. [DOI] [PubMed] [Google Scholar]
- [15].Erdmann S, Shah SA, Garrett RA. SMV1 virus-induced CRISPR spacer acquisition from the conjugative plasmid pMGB1 in Sulfolobus solfataricus P2. Biochem Soc Trans. 2013;41:1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Erdmann S, Garrett RA. Selective and hyperactive uptake of foreign DNA by adaptive immune systems of an archaeon via two distinct mechanisms. Mol Microbiol. 2012;85: 1044–1056. Corrigendum Mol Microbiol 2012;86:757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Deng L, He F, Bhoobalan_Chitty Y, et al. Unveiling cell surface and type IV secretion proteins responsible for archaeal rudivirus entry. J Virol. 2014;88(17):10264–10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].He F, Bhoobalan-Chitty Y, Van LB, et al. Anti-CRISPR proteins against a type I-D CRISPR-Cas immunity encoded by archaeal lytic viruses. Nat Microbiol. 2018;3(4):461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Uldahl KB, Jensen SB, Bhoobalan-Chitty Y, et al. Life cycle characterization of Sulfolobus monocaudavirus 1, an extremophilic spindle-shaped virus with extracellular tail development. J Virol. 2016;90:5693–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vestergaard G, Shah SA, Bize A, et al. Stygiolobus rod-shaped virus and the interplay of crenarchaeal rudiviruses with the CRISPR antiviral system. J Bacteriol. 2008;190(20):6837–6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prangishvili D, Vestergaard G, Häring M, et al. Structural and genomic properties of the hyperthermophilic archaeal virus ATV with an extracellular stage of the reproductive cycle. J Mol Biol. 2006;359:1203–1216. [DOI] [PubMed] [Google Scholar]
- [22].Brumfield SK, Ortmann AC, Ruigrok V, et al. Particle assembly and ultrastructural features associated with replication of the lytic archaeal virus Sulfolobus turreted icosahedral virus. J Virol. 2009;8:5964–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Martinez-Alvarez L, Bell SD, Peng X. Multiple consecutive initiation of replication producing novel brush-like intermediates at the termini of linear viral dsDNA genomes with hairpin ends. Nucleic Acids Res. 2016;44(18):8799–8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Martinez-Alvarez L, Deng L, Peng X. Formation of a viral replication focus in Sulfolobus cells infected by the rudivirus Sulfolobus islandicus rod-shaped virus 2. J Virol. 2017;91(13):e00486–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Okutan E, Deng L, Mirlashari S, et al. Novel insights into gene regulation of the rudivirus SIRV2 infecting Sulfolobus cells. RNA Biol. 2013;10(5):875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vestergaard G, Garrett RA, Shah SA. CRISPR adaptive immune systems of Archaea. RNA Biol. 2014;11:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].León-Sobrino C, Kot WP, Garrett RA. Transcriptome changes in STSV2-infected Sulfolobus islandicus REY15A undergoing continuous CRISPR spacer acquisition. Mol Microbiol. 2016;99:719–728. [DOI] [PubMed] [Google Scholar]
- [28].Quax TEF, Voet M, Sismeiro O, et al. Massive activation of archaeal defense genes during viral infection. J Virol. 2013;87:8419–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bautista MA, Black JA, Youngblut ND, et al. Differentiation and structure in Sulfolobus islandicus rod-shaped virus populations. Viruses. 2017;9(5):E120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Scheele U, Erdmann E, Ungewickell EJ, et al. Chaperone role for proteins p618 and p892 in the extracellular tail development of Acidianus two-tailed virus. J Virol. 2011;85:4812–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Levy A, Goren MG, Yosef I, et al. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Manica A, Zebec Z, Steinkellner J, et al. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2013;41:10509–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mousaei M, Deng L, She Q, et al. Major and minor cRNA annealing sites facilitate low stringency DNA protospacer binding prior to Type I-A CRISPR-Cas interference in Sulfolobus. RNA Biol. 2016;13(11):1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Guo T, Han W, She Q. Tolerance of Sulfolobus SMV1 virus to the immunity of I-A and III-B CRISPR-Cas systems in Sulfolobus islandicus. RNA Biol. 2018. DOI: 10.1080/15476286.2018.1460993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].He F, Vestergaard G, Peng W, et al. CRISPR-Cas type I-A Cascade complex couples viral infection surveillance to host transcriptional regulation. Nucleic Acids Res. 2017;45:1902–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Erdmann S, Garrett RA. Archaeal viruses of the Sulfolobales: isolation, infection and CRISPR spacer acquisition. In CRISPR: methods and Protocols (Lundgren M. et al. eds.). Methods Mol Biol. 2015;1311:223–232. [DOI] [PubMed] [Google Scholar]
- [37].Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. [DOI] [PubMed] [Google Scholar]
- [38].Robinson MD, McCarthy DJ, Smyth GK. EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- [40].Erdmann S, Chen B, Huang X, et al. A novel single-tailed fusiform Sulfolobus virus STSV2 infecting model Sulfolobus species. Extremophiles. 2014;18:51–60. [DOI] [PubMed] [Google Scholar]
- [41].Shevchenko A, Wilm M, Vorm O, et al. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. [DOI] [PubMed] [Google Scholar]
- [42].Schaab C, Geiger T, Stoehr G, et al. Analysis of high accuracy quantitative proteomics data in the MaxQB database. Mol Cell Proteom. 2012;11:M111.014068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.