

ABSTRACT
CRISPR-Cas systems provide bacteria and archaea with adaptive immunity against invading genetic elements, such as plasmids, bacteriophages and archaeal viruses. They consist of cas genes and CRISPR loci, which store genetic memories of previously encountered invaders as short sequences termed spacers. Spacers determine the specificity of CRISPR-Cas defence and immunity can be gained or updated by the addition of new spacers into CRISPR loci. There are two main routes to spacer acquisition, which are known as naïve and primed CRISPR adaptation. Naïve CRISPR adaptation involves the de novo formation of immunity, independent of pre-existing spacers. In contrast, primed CRISPR adaptation (priming) uses existing spacers to enhance the acquisition of new spacers. Priming typically results in spacer acquisition from locations near the site of target recognition by the existing (priming) spacer. Primed CRISPR adaptation has been observed in several type I CRISPR-Cas systems and it is potentially widespread. However, experimental evidence is unavailable for some subtypes, and for most systems, priming has only been shown in a small number of hosts. There is also no current evidence of priming by other CRISPR-Cas types. Here, we used a bioinformatic approach to search for evidence of priming in diverse CRISPR-Cas systems. By analysing the clustering of spacers acquired from phages, prophages and archaeal viruses, including strand and directional biases between subsequently acquired spacers, we demonstrate that two patterns of primed CRISPR adaptation dominate in type I systems. In addition, we find evidence of a priming-like pathway in type II CRISPR-Cas systems.
KEYWORDS: CRISPR-Cas, adaptation, adaptive immunity, primed, priming, spacer acquisition
Introduction
Many prokaryotes possess adaptive immune systems, termed CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR associated genes), which enable them to defend against viruses and mobile genetic elements (MGEs). CRISPR-Cas systems use genetic memories gained during previous encounters with viruses and MGE invaders to guide future immunity [1,2]. To form these memories, short sections of foreign DNA are incorporated into the host genome within CRISPR loci as spacers. There are three main stages in CRISPR-Cas immunity. During adaptation, new spacers are added to CRISPR arrays. The next stage is expression, where CRISPRs are transcribed and processed into mature CRISPR RNAs (crRNAs), which assemble with Cas protein(s) to form Cas-crRNA complexes. Finally, during interference, Cas-crRNA complexes function as sequence-specific nucleases to identify and degrade invading genetic material [1].
CRISPR-Cas systems are diverse, comprising two classes that are classified into 6 types and many subtypes [3,4]. Although the overall concept of CRISPR immunity is similar for all system types, the mechanisms involved in the adaptation, expression and interference stages differ. For example, during interference, type I, II and V systems target DNA, type VI target RNA and type III systems exhibit RNA and transcription-dependent DNA targeting. For all systems, the specificity of target (virus or MGE) recognition is primarily determined by the crRNA sequence. The target sequence of the spacer is complementary to the cognate crRNA and is termed the protospacer [5]. In addition, type I, II and V systems combine crRNA-based target recognition with protein-based sequence recognition of protospacer adjacent motifs (PAMs) [6]. Phage, archaeal virus and MGE variants with mutated protospacer or PAM sequences can escape CRISPR-Cas immunity [7], therefore, hosts must update their defences via CRISPR adaptation.
The addition of new spacers into CRISPR arrays is catalysed by Cas1 and Cas2, which are associated with most subtypes and form a Cas1-Cas2 complex [1]. New spacers are typically inserted at one end of the CRISPR array – termed the leader – which includes a promoter for CRISPR transcription and sequences that dictate the polarity of spacer addition. Cas1 is an integrase and is responsible for the insertion of the spacers, whereas Cas2 plays a structural role for DNA binding and positioning [8]. To facilitate CRISPR adaptation, prespacer substrates need to be generated for Cas1-Cas2 to integrate. The mechanisms for generation of these substrates are varied and differ between systems. In addition to Cas1 and Cas2, many systems encode accessory proteins that contribute to CRISPR adaptation. For example, in some type I and type II systems, Cas4 is involved in processing prespacer substrates [9–11] and in type II-A systems, Cas9 interacts with Cas1, Cas2 and the additional adaptation protein, Csn2 [12,13]. Spacer acquisition from RNA has also been demonstrated in a type III system with a reverse-transcriptase‒Cas1 fusion protein [14].
In the absence of existing CRISPR immunity against a phage, archaeal virus or MGE, new spacers must be acquired via naïve adaptation [15,16]. Several studies have observed ‘hotspots’ for naïve acquisition, such as double strand DNA (dsDNA) breaks that are acted upon by RecBCD, the injected ends of phage genomes, and stalled replication forks [17–20] (Figure 1(a)). If multiple spacers are acquired from the same target via hotspot-facilitated naïve adaptation, then their corresponding protospacers will appear clustered on the target genome. However, their locations will be independent of each other, because for naïve adaptation, prespacer generation is not directly influenced by existing spacers (Figure 1(b,c)). For naïve adaptation without hotspots, or for many hotspots along the same target genome, the relative distributions of protospacers will be spread out across the whole target genome.
Figure 1.
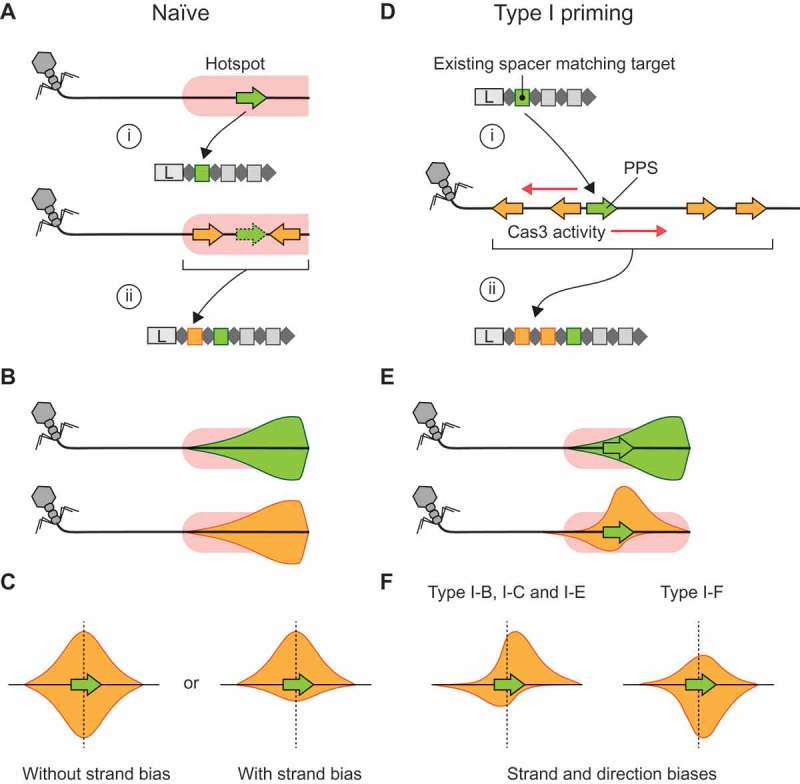
CRISPR adaptation pathways and the positional relationship between targets of multiple spacers. A) Naïve acquisition of two spacers from a ‘hotspot’ site (pink) where prespacer substrate generation frequently occurs. In this example, the hotspot represents the incoming end of a phage genome [20]. (i) The first spacer acquired (green) comes from within the hotspot. (ii) A subsequent spacer (orange) is also acquired from within the hotspot, but the location of the second spacer is not directly dependent on the location of the first spacer. B) When the first or subsequent spacers from many hotspot-facilitated naïve acquisitions are mapped to the target genome the distributions are expected to be the same (green and orange distributions). In this example, the mapping densities are skewed toward the start of the hotspot, i.e. the incoming end of the phage genome. C) If the relative positions of the first (green) and second (orange) spacer acquisitions are considered, i.e. the distance and direction between their corresponding protospacer mapping locations, the resulting relative protospacer mapping distributions will be symmetric. However, depending on the pathway for spacer substrate generation, there may be a strand bias. D) In type I primed CRISPR adaptation, an existing spacer (green) facilitates target recognition at the corresponding (priming) protospacer (PPS, green) (i) and results in Cas3 and/or Cas1-Cas2 activity initiating at this point, which generates substrates for the acquisition of additional spacers (orange) (ii). Often, multiple spacers are acquired. E) The protospacer mapping distributions for a naïve spacer (green) that subsequently triggers the primed acquisition of additional spacers (orange) differ when compared directly on the phage genome. This results in asymmetry in the relative protospacer mapping distribution (orange). F) The relative protospacer mapping distributions for type I priming are system-specific and vary in strand bias and distance from the priming protospacer. Schematic representations based on previous observations in the type I-B system of Haloarcula hispanica [26], type I-C from Legionella pneumophila [27], type I-E system from Escherichia coli [40] and the type I-F system in Pectobacterium atrosepticum [19] are shown.
A second pathway to spacer acquisition is observed in type I systems and is termed primed CRISPR adaptation (priming) [1]. Priming is a positive feedback loop where target recognition, facilitated by existing spacers, enhances the acquisition of new spacers, thereby reinforcing immunity (Figure 1(d)) [21,22]. This can occur even when there are mismatches between the existing spacer and the invader genome, allowing hosts to update their immunity to counter escape mutants or heavily divergent threats [21–23]. During interference in type I systems, the helicase-nuclease Cas3 generates prespacer substrates that can be integrated by Cas1-Cas2 [19,24,25]. As such, new spacers are typically obtained close to the original target site, i.e. centred at the priming protospacer (PPS) (Figure 1(e)). Owing to the 3ʹ to 5ʹ helicase activity of Cas3, most spacers are acquired asymmetrically on each given strand relative to the PPS. Experimental studies of priming in type I systems (subtypes B, C, E and F) have shown that primed adaptation can occur at high frequencies and that the distribution of the positions of the protospacers differs between subtypes [19,22,26–28] (Figure 1(f)). These differences are thought to result from variations of how substrates for Cas1-Cas2 are generated between systems, such as the strand loading and processivity of Cas3 [1].
Primed CRISPR adaptation has not yet been observed in class 2 systems. However, we predict two potential pathways that would constitute priming in the DNA-targeting class 2 systems, types II and V. Firstly, dsDNA breaks caused by CRISPR-Cas interference might act as hotspots for host-specific mechanisms that produce prespacer substrates, such as DNA repair processes [1,18]. Secondly, the accessory proteins in some class 2 systems might function directly in CRISPR-Cas‒specific prespacer generation mechanisms, such as for Cas3-facilitated priming in type I systems. In both cases, target recognition by an existing spacer might initiate a pathway that ultimately leads to the acquisition of additional immunity, thereby constituting primed CRISPR adaptation. The relative protospacer distributions for predicted type II/V priming will depend on the prespacer generation pathways, potentially resembling either the symmetric hotspot-facilitated naïve or the asymmetric type I primed distributions (Figure 1(c,f)).
So far, priming has been demonstrated for four different type I subtypes in laboratory experiments. However, it is not known if primed CRISPR adaptation occurs in other types and subtypes in nature. Moreover, most work on type I priming has focused on specific model organisms and it is unclear if priming occurs similarly in different hosts harbouring the same CRISPR-Cas subtype. To address these gaps in knowledge, we have analysed publicly-available host, phage, archaeal virus and prophage genomes, along with a viral metagenome dataset, to understand the distributions of spacer acquisition events. We find that priming by type I systems occurs in diverse hosts and that the currently proposed mechanisms of type I primed adaptation are likely conserved between systems. We also uncover evidence of strand and directional biases for spacer acquisition in type II systems that are indicative of priming-like behaviour, suggesting that priming might also occur in class 2 CRISPR-Cas systems.
Results
A comprehensive non-redundant dataset of host-target spacer matches
To investigate CRISPR adaptation patterns in diverse systems, we first identified all CRISPR-Cas systems in sequenced host genomes. For each of the 35,240 unique archaeal and bacterial strains in RefSeq83, we searched the most complete genome assembly of each strain for CRISPR-Cas systems (Figure 2(a)). We identified CRISPR arrays in 71.9% of archaea and 52.2% of bacteria, and cas genes in 75.4% of archaea and 63.4% of bacteria. These occurrences are consistent with previous reports [29,30]. Most hosts possess single subtypes, but type III systems frequently co-reside with type I systems (Figure 2(b)) [31]. Co-residing CRISPR-Cas systems can share communal CRISPR arrays with similar repeats [32–34] or can engage in cross-talk with CRISPR arrays belonging to other systems [35,36]. Therefore, to allow an accurate assessment of CRISPR adaptation by specific subtypes, we focused on hosts possessing single CRISPR-Cas systems. We identified putative protospacer matches (hits) for many of these hosts’ spacers in target genomes (consisting of phage, prophage and archaeal viruses), then filtered the matches to reduce false positive hits and reduce redundancy caused by similar host or target genome sequences (Figure 2(a)) (Methods). This resulted in 1,448 unique host-target pairs with 3,228 spacer/protospacer hits. The CRISPR-Cas subtypes I-B, I-C, I-E, I-F, II-A and II-C were all represented by at least 50 non-redundant host-target pairs, which was our cut-off for further analyses (Figure 2(c)). Overall, this non-redundant dataset of host-target spacer matches represented a diverse range of hosts (Figures 2(c) & S1).
Figure 2.
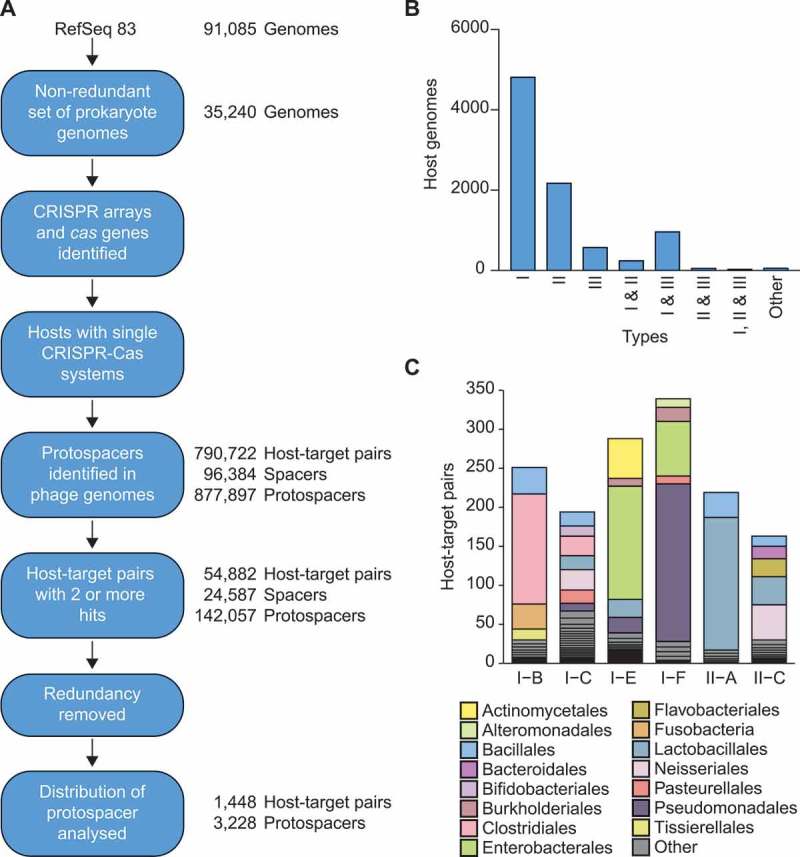
Identification of archaeal virus, phage and prophage protospacers. A) An overview of the steps used to generate the non-redundant host-target dataset of spacer-protospacer matches. For details see Methods. B) The number of genomes possessing each designated CRISPR-Cas type or multiple co-occurring types, based on the cas genes identified. Due to their low occurrences, all other types and combinations are included in ‘other’ (i.e. types IV, V and VI along with any other subtype co-occurrences). C) Taxonomic overview of the final dataset. Species-level diversity is shown in Fig. S1.
Many CRISPR-Cas subtypes acquire multiple spacers from non-random locations
Both hotspot-facilitated naïve and primed CRISPR adaptation are expected to result in distinct spacer mapping (protospacer) distributions, exhibiting strand and/or positional biases (Figure 1). To visualise these distributions for our host-target dataset, the protospacers corresponding to the oldest spacers matching each target, i.e. furthest from the CRISPR leader, were assigned as the PPS(Figure 3(a)). Each PPS was set at the middle of the distribution plot on the upper (target) strand and the protospacers corresponding to more recently acquired spacers (designated PS+1, PS+2 etc.) were then mapped relative to this point (Figure 3(b)).
Figure 3.
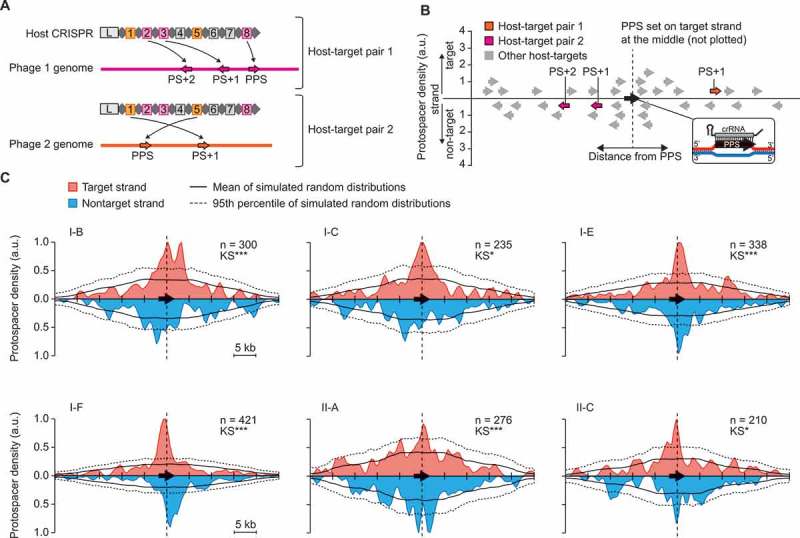
Type I and II CRISPR-Cas subtypes display preferential acquisition of spacers from non-random locations. A) Assignment of the presumed priming (oldest) protospacer (PPS) specific to each host-target pair by taking the protospacer corresponding to the spacer furthest from the CRISPR leader (L). A single host with spacers matching two phage genomes is assigned as two independent host-target pairs. For example, spacer 8 in the host CRISPR is the oldest spacer targeting the magenta phage, so the corresponding protospacer on the phage genome is assigned as the PPS. The target of spacer 3 is assigned as PS+1 and spacer 2 as PS+2. For spacers matching the orange phage, the target of spacer 5 is assigned as the PPS and spacer 1 as PS+1. Grey spacers do not match either phage. B) For each host-target pair the DNA strand with the PPS was designated as the target strand, and the PPS position was set at the middle of the plot. Protospacers on the target or non-target strands were plotted above or below the x-axes, respectively. A smoothing window (width 500 bp) was then applied to the data. C) The relative protospacer mapping distributions for each CRISPR-Cas subtype in our dataset. The mean mapping density for 1000 simulated random distributions are shown as a black line and the 95th percentile is indicated by a dotted black line – each is based on the underlying data represented for the specific subtype. The significance of the difference between the observed data and simulated data were determined using a Kolmogorov–Smirnov (KS) test; p < 0.05 *, <0.01 **, <0.001 ***. (n) indicates the number of unique protospacers mapped (excluding the PPS for each host-target pair).
All type I systems analysed exhibited relative protospacer mapping distributions that were significantly different from simulated random distributions (Figure 3(c)). Each observed type I distribution displayed a higher density of protospacers close to the PPS than seen in >95% of the simulated random distributions. Moreover, this clustering was typically asymmetric and system-specific, indicative of primed CRISPR adaptation. In general, our results are consistent with experimental data. For example, the type I-F distribution shows asymmetric clustering around the PPS that begins to tail off after ~ 3 kb, as observed experimentally in P. atrosepticum, P. aeruginosa and E. coli (Figure 3(c)) [19,37–39]. For the E. coli type I-E system, which has previously been shown to acquire spacers with a strand bias and/or also clustering nearby the priming protospacer [21,22,40], our data is largely in agreement (Figure 3(c)). Our results also corroborate experimental data of protospacer distributions for the type I-B and I-C systems in Haloarcula hispanica [26] and Legionella pneumophila [27], respectively.
For class 2 CRISPR-Cas systems, we had sufficient data to analyse type II-A and II-C systems. The observed protospacer mapping distributions for these systems were both significantly different from the simulated random sampling along target genomes (Figure 3(c)). Furthermore, clustering of protospacers mapping near the PPS was apparent, although less pronounced than for type I systems (Figure 3(c)). The type II-A distribution appeared relatively symmetric, whereas the type II-C distribution appeared asymmetric, indicative of a priming-like mechanism. Overall, these protospacer mapping distributions indicate that for members of both class 1 and 2 CRISPR-Cas systems (types I and II), spacers are typically not obtained randomly along target genomes and that hotspot-facilitated naïve and/or primed CRISPR adaptation contribute significantly to the acquisition of multiple spacers.
Strand and directional protospacer biases indicate priming in type I and II systems
Having established that all six subtypes with sufficient data to analyse showed significant non-random acquisition of multiple spacers from targets, we asked whether the observed clustering of protospacers close to the PPS resulted from either hotspot-facilitated naïve or primed CRISPR adaptation. To do this, we focused on spacers acquired within 5 kb either side of the PPS and examined the strand and directional biases for PS+1, PS+2 etc. relative to the PPS (Figure4). For the type I systems, I-B, I-C and I-E exhibited biases toward the acquisition of new spacers that target the same strand as the priming protospacer (Figure 4(a)). This is consistent with experimental data of hosts possessing these systems (Fig. S2) [26–28]. We also uncovered a directional bias in the 3ʹ direction (for protospacers) for both I-B and I-C systems (Figure 4(b)), supporting previous experimental data (Fig. S2) [26,27]. There was also a significant direction bias for type I-E systems, albeit less pronounced, mostly in the 3ʹ direction on the target strand. A different acquisition pattern was observed in the type I-F systems and included no overall strand bias (Figure 4(a)), but a significant bias in the 5ʹ direction on each strand away from the PPS (Figure 4(b)). These distributions closely resemble the acquisition measured experimentally during type I-F priming (Fig. S2) [19,39].
Figure 4.
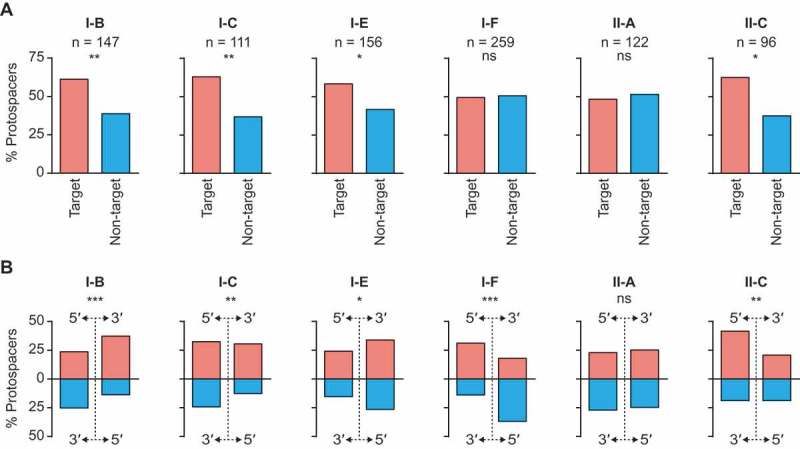
Type I and II systems display spacer acquisition biases consistent with priming. A) The proportion of protospacers present on each strand for hits within 5 kb either side of their respective priming protospacers. The significance of the differences between strands were determined using a binomial test; p > 0.05 non-significant (ns), p < 0.05 *, <0.01 **, <0.001 ***. B) Directional bias for hits within 5 kb either side of the priming protospacers, summarised by plotting the proportion of protospacers found in each quadrant (strand and direction). The significance of the distributions were determined using a multinomial test; p > 0.05 non-significant (ns), p < 0.05 *, <0.01 **, <0.001 ***. The same analyses were also performed on all data, regardless of distance from the PPS (Fig. S3).
Although clustered (Figure 3(c)), the data for type II-A systems did not exhibit significant strand or direction biases (Figure 4). Therefore, we cannot rule out that the protospacer clustering resulted from hotspot-facilitated naïve adaptation for type II-A (Figure 1(c)). However, type II-C systems exhibited a bias toward the target strand and in the 5ʹ direction along this strand relative to the PPS (Figure 4). This significant asymmetry provides support to reject the null hypothesis that hotspot-facilitated naïve adaptation is responsible for the clustering of acquired spacers in type II-C systems. Instead, we propose that a priming-like pathway can occur in type II-C systems.
Overall, the type I systems exhibited directional and/or strand biases that support the current models for primed adaptation, previously derived from a limited number of model systems. Since our data is derived from a large number of phage/archaeal virus-prokaryote pairs (Figure 2(c), S1), our findings show that these biases may be generalised to many prokaryotic hosts. In addition, type II-C systems exhibited both strand bias and direction biases that could be the result of a priming-like CRISPR adaptation pathway.
Examples of priming in diverse CRISPR-Cas systems
Within the dataset of host-target pairs we identified many examples of overlapping host-target pairs that further support the presence of widespread priming in type I systems and priming-like spacer acquisition patterns in type II hosts. For instance, we observed closely-related host species possessing different spacers targeting the same phage (Figure 5). In many of these cases we observed clustering of spacers/protospacers within, but not between, prokaryotes that match the same target genome. For example, the protospacers corresponding to the Corynebacterium type I-E strains form clusters at different locations along the Corynebacterium jeikeium prophage (Figure 5). These patterns are indicative of primed CRISPR adaptation. In other cases, we observed clustering between related hosts in the same region of the target genome, e.g. the Acinetobacter type I-F hosts targeting the Acinetobacter calcoaceticus prophage (Figure 5). This might be an example of a naïve hotspot, which is followed by primed acquisition of subsequent spacers. There were also examples of related hosts that possess different types of CRISPR-Cas system targeting the same phage, e.g. the Bifidobacterium hosts with type I-C, I-E or II-A systems (Figure 5), which shows that diverse CRISPR-Cas systems can be effective in defence against the same targets.
Figure 5.
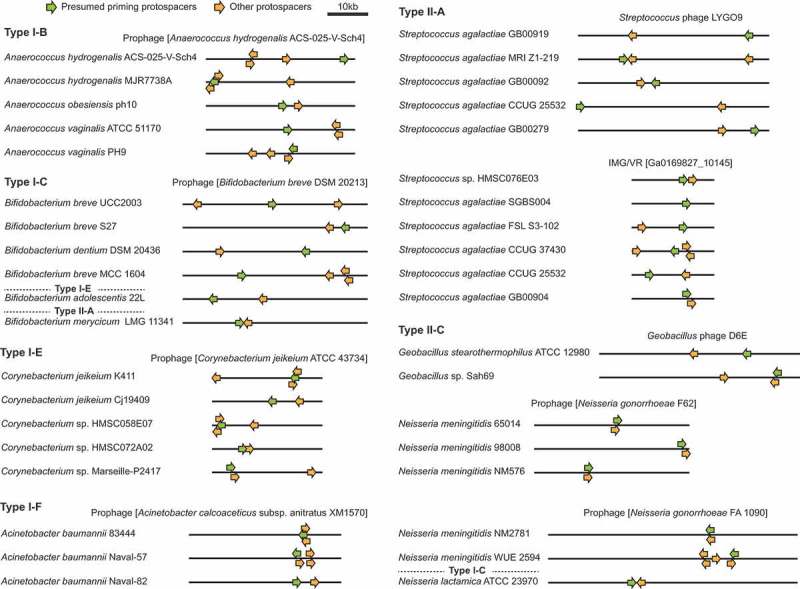
Protospacer clustering on shared target genomes reveals primed CRISPR adaptation. Protospacer clustering for sets of closely-related host species targeting the same phages or prophages. Note that protospacers toward either end of the displayed linear phage genome are considered clustered because most phage genomes undergo circularised states during replication. The IMG/VR accessions represent viral contigs assembled from metagenomic data [59]. In some cases the displayed host species are representative of several related hosts that possess the same spacer sets matching the specified phage. Examples of single hosts with spacers matching multiple targets are provided in Fig. S5.
For type II-C systems we observed some cases of protospacer clustering, within but not between, related bacteria, which supports priming in these hosts (Figure 5). The examples presented are representative of different patterns with multiple prokaryotes targeting the same phage or archaeal virus with different spacer sets. However, in our dataset most phages were targeted by only single hosts (Fig. S4). Differences in target genome injection (e.g. cos-type vs circularly permuted genomes) may also result in some host-target pairs displaying evidence of naïve hotspots [20], whereas other host-target pairs might reveal priming-like adaptation. We also identified examples of single hosts with spacers matching multiple targets that often formed clusters (Fig. S5). Overall, these analyses of individual host-target pairs support the widespread occurrence of priming in type I CRISPR-Cas systems and also the presence of a priming-like pathway in at least some type II systems.
Discussion
Despite the evidence of primed CRISPR adaptation in select type I systems and model organisms in laboratory experiments, it was unknown whether priming was more widespread in other prokaryotes and for other CRISPR-Cas types. In this study, we used a bioinformatic approach to generate and analyse a large dataset of host-target spacer-protospacer pairs to address this question. We found that for type I-B, I-C, I-E, I-F, II-A and II-C systems, spacers are not randomly acquired from target genomes. Instead, their locations are often influenced by older spacers present in the same host. The involvement of these pre-existing spacers and targeting by the Cas-crRNA complex is a hallmark of primed, rather than naïve adaptation.
Our bioinformatics-derived type I-F protospacer mapping distribution closely matches our previously determined experimental spacer acquisition patterns [19,39], thereby confirming that the bioinformatic approach is robust and can identify distributions indicative of primed adaptation. Our bioinformatic evidence for priming in type I-B, I-C, I-E and I-F systems corroborates experimental data that is derived from either single, or just a few, model systems. Moreover, we observed priming-type clustering and asymmetric protospacer mapping distributions in a broad range of bacteria that have not previously been shown to prime (Figures 2(c), 5,& S1).
Our data also revealed that two main patterns of primed CRISPR adaptation dominate within the type I systems. Type I-B, I-C and I-E displayed protospacer mapping biases towards the target strand 3ʹ of the priming protospacer. Directional biases in type I spacer acquisition are likely due to the activity of Cas3 proteins, which are typically recruited to the non-target strand following Cas-crRNA target recognition and use their helicase activity to unwind DNA in a 3ʹ to 5ʹ direction [41–45]. During strand unwinding driven by the helicase, the HD nuclease of Cas3 generates short fragments of DNA that can form substrates for spacer acquisition [24,46]. In the E. coli type I-E system, Cas3 preferentially cleaves the target strand near PAM-like sequences [24] and these are recognised by a PAM-sensing domain in the Cas1-Cas2 complex [8,47], resulting in the acquisition of spacers that also target this strand. This means that their corresponding protospacers map to the target strand 3ʹ of the PPS. Our data shows that the process of PAM selection on the target strand is likely also conserved for type I-B and I-C systems, as previously observed in some experimental systems. For the type I-E Cas3, bi-directional activity relative to the PPS was observed in vitro in the presence of imperfect targets and Cas1-Cas2 [48], resulting from loading on either the target or non-target strands. Similar bi-directionality has been observed for most characterised type I systems in vivo [19,26,39,40]. This suggests that although there is a bias toward Cas3 loading onto the non-target strand, it can also be recruited to the other strand. This would lead to protospacers mapping in the opposite direction to the PPS.
The protospacer mapping distributions of type I-F systems differed from the other analysed type I systems, as protospacers typically mapped to the non-target strand 5ʹ of the PPS. Type I-F systems are characterised by a fusion of Cas2 and Cas3, which form a Cas1‒Cas2-3 complex that loads onto the non-target strand and exhibits 3ʹ-5ʹ helicase activity [45,49,50]. However, protospacers also map to the non-target strand, implying that PAM selection is opposite to the type I-B, I-C and I-E systems. The mechanistic explanation for this difference is unknown, but since the PAM recognition by the Cas1‒Cas2-3 complex is on the same strand as type I-E (the protospacer strand) [8,50], it is likely that the type I-F Cas2-3 nuclease PAM specificity occurs on the non-target strand.
Here, we observed that for type II-A and II-C systems spacers are typically acquired from phage genomes in a clustered, non-random fashion (Figure 3(c)). Similar clustering of new spacers near the sites of existing target sites has been observed experimentally in the Streptococcus thermophilus type II-A systems [51]. Clustering could potentially result from naïve acquisition hotspots (Figure 1) or from host fitness advantages for spacers directing CRISPR-Cas activity to specific strands and/or locations on phage genomes [51]. In both of these cases, the null hypothesis, expected in the absence of primed adaptation, is for symmetric protospacer mapping distributions (Figure 1(a-c)). For type II-C systems we observed a significant preference for spacers that target the same strand as the PPS, with a bias toward the 5ʹ direction (Figure 4). Therefore, we propose the existence of a priming-like pathway in hosts with type II-C systems, whereby existing spacers can influence and/or enhance the acquisition of new spacers from regions surrounding the existing target sites (priming protospacers).
Mechanistically, priming in type II systems would not be the same as type I systems, because type II systems lack Cas3 to generate substrates for Cas1-Cas2. In type II hosts, the RecBCD or AddAB DNA repair machinery may be recruited and act in prespacer generation at sites where Cas9-mediated dsDNA breaks occur [1]. This is conceptually similar to the role of RecBCD in naïve prespacer generation that occurs in the E. coli type I-E system [18]. However, because existing spacers and Cas-crRNA targeting are involved, this pathway would constitute a priming-like process for the type II, and analogously for type V, systems. Validation of priming in class 2 DNA-targeting systems will require laboratory-based CRISPR adaptation experiments. For example, an extension of previous type II CRISPR adaptation studies [12,13,51] to look at the relative locations of successively acquired spacers would be highly informative.
The differences observed here between type II systems are intriguing (Figure 4) because the major difference between II-A and II-C, is only the additional Csn2 protein in type II-A systems. Csn2 contributes to spacer acquisition [12,13,52] and might contribute to the adaptation differences we observed between the subtypes. However, for type II-A systems our data cannot exclude the presence of a priming-like pathway, that results in a symmetric distribution (Figure 4), perhaps mediated by Csn2. There was insufficient data to analyse type II-B systems. However, in the future, a comparison including type II-B would be informative, given that the cas1 and cas2 genes of II-B systems are more closely related to the type I systems [53] where priming occurs. Cas4 is present in II-B systems and in the type I-B system it is required for priming [26]. Therefore, if II-B systems undergo primed adaptation, Cas4 is likely to be involved.
In conclusion, we have revealed that primed adaptation occurs widely in nature in four type I CRISPR-Cas systems, and provide evidence of naïve hotspots and/or a priming-like pathway in some type II systems. The patterns of primed adaptation differ between the CRISPR-Cas systems, which likely results from biochemical and mechanistic differences between the distinct Cas proteins [1]. Although we had insufficient data to draw conclusions about priming in other type I systems (e.g. types I-A and I-D), we predict that it is likely to occur. However, this will require further genomic data, or experimental approaches, for verification. At present, the main limitation of the power of our bioinformatics approach is the number of diverse host and target genome sequences available. Thus, future analyses of other systems will require sequencing of more relevant genomes. Overall, our study shows that priming in type I systems is widespread and that priming-like CRISPR adaptation also occurs in some type II systems.
Methods
Identification of Cas proteins and typing of CRISPR-Cas systems
Two approaches were used to assign CRISPR-Cas systems to genomes. Makarova et al., (2015) established a set of Hidden Markov Models (HMM) to describe all known Cas proteins, and defined a CRISPR-Cas subtype classification scheme that is based on the constituent cas genes. Using these models, we searched all of the proteins in Refseq Archaea and Bacteria (Version 83; 21/07/2017) using HMMER (hmmer.org). The proteins that matched to one of the models were then compared to the conserved domain database (CDD, NCBI) and the proteins that matched better to a non-Cas model were removed. Using this method 448,741 cas genes were identified. The second approach was to use the Refseq gene annotations, which included 419,645 cas genes. Of these genes, 2,037 (0.5%) were not found by the HMM search, but the HMM search found an additional 29,096 (6.4%). When both methods found a cas gene the annotations were consistent. The subtypes of CRISPR-Cas systems present in each genome were then assigned based on the presence of signature genes [3]. The CRISPR repeats of some subtypes also fall within highly-conserved CRISPR repeat families, which we used to verify the assigned system subtypes [54].
Identification of CRISPR loci, spacers and target protospacers
The Refseq version 83 archaea and bacteria genomes were searched for CRISPR loci using CRISPRDetect v2.2 [55]. CRISPRDetect uses CRISPRDirection to call the orientation of the array and identify the leader [56]. The orientations of arrays were checked by comparison to known conserved repeat families within subtypes (as above). For type II systems, we also used tracrRNA sequences to increase the accuracy of CRISPR orientation calls [57]. Overall, 59.2% of Archaea and 35.9% of Bacteria possessed both CRISPR arrays and cas genes and were used for further analysis. Spacer sequences from the arrays were searched for protospacer matches in the ‘Phage’ division of Genbank (which includes both bacteriophages and archaeal viruses) and the PHAST prophage database (2/2017), which contains data from both bacterial and archaeal genomes [58] using SWIPE [54]. To increase the size of the target dataset for type I-C and type II systems we also searched their spacers against the IMG/VR_2018 database, which contains >700,000 viral contigs including diverse metagenomic samples [59]. The SWIPE bit score was calculated as +1 match, −1 mismatch, −10 gap open and −2 gap extension. A SWIPE search of a dinucleotide-shuffled PHAST dataset was used to determine appropriate thresholds for the bit score cut-off; set at ≥20 for the PPS and ≥23 for all other protospacers (Fig. S6). These thresholds allowed a 32 nt spacer with 6 or fewer mismatches for the priming protospacer. We deem this conservative because priming has been shown to occur with 10 or more mismatches in a 32 nt spacer [23]. The SWIPE results were grouped into host-target pairs, with each pair representing all of the spacers and corresponding protospacer matches (hits) for a specific host and specific target. Most host-target pairs consisted of just a single spacer matching the target (Fig. S7), whereas two or more matches are required to investigate the presence of primed CRISPR adaptation. Therefore, only host-target pairs with more than one spacer matching the target were retained. Some hosts were represented in more than one host-target pairing (Figure 3(a)). To exclude potential clustering biases from short or incomplete target genomes, only host-target pairs with a target genome length ≥10 kb were analysed.
Assignment of spacer acquisition order
The spacers in each host-spacer pair were assigned a chronological order based their positions within the host CRISPR loci. Spacers are typically inserted into the leader end of CRISPR arrays, thus the order of spacer acquisition can be implied (Figure 3(a)). However, for hosts with multiple arrays, the order of acquisition for spacers in different arrays is sometimes ambiguous. In our final dataset, 93.7% of host-target pairs contained target-matching spacers originating from a single array. For the other 6.3% of the data, we reduced potential bias from incorrectly assigned spacer acquisition orders by only including host-target pairs where the putative oldest spacer (i.e. furthest from the corresponding leader by spacer number) was in the shortest array (i.e. we assume, based on our past experimental data, that shorter arrays have lower spacer integration rates [19,39]).
Reducing host and target redundancy
We initially found matches (hits) for 96,384 spacers to 877,897 phage, archaeal virus or prophage sequences (target protospacers), representing 790,722 different host-target pairs. Of the host-target pairs, 54,882 contained at least two spacers. The high number of hits relative to total spacers was due to redundancy, caused by multiple cases of near identical host or target sequences. To resolve this, and other potential ambiguities, we used an objective filtering process. Firstly, host redundancy was reduced by merging host-target pairs that shared target genomes with identical protospacer patterns (based on the host spacer order and target strand and position). For non-identical cases of shared targets, if none of the protospacers overlapped then all host-target pairs were kept. If a single protospacer overlapped then the host-target pairs were kept if the ambiguity was only due to the PPS; i.e. these represent multiple independent priming events. If the ambiguity was due to the PPS and PS+1, then the host-target sets were merged, and only the PPS and PS+1 were analysed. Secondly, target redundancy was removed. This was often caused by a single set of host spacers matching multiple different phage genomes. This was removed using a similar scheme, except that both host spacers and the target strand, direction and distance relative to the PPS were considered as common features. Although our stringent approach excluded many host-target pairs from the final dataset, it ensured that the data were of high quality. The final dataset was examined manually and representative sets shown in Figure 5 & S5.
Simulation of random distributions and clustering analyses
To generate the Monte Carlo simulated random distributions we randomised the strand and protospacer mapping positions for every host-target pair in the final dataset. We then assigned the PPS strand and relative protospacer mapping distributions using the same approach that we used for the observed data (Figure 3(a,b)). For each subtype, we generated 1,000 random distributions and report the mean and 95th percentile for the relative protospacer mapping density. This approach takes into consideration both the number of observed target matching spacers and the size of target genomes within each subtype dataset.
Funding Statement
This work was supported by the Marsden Fund [14-UOO-009];Rutherford Discovery Fellowship [11-UOO-016]; Division of Health Sciences Career Development Post-Doctoral Fellowship [Not applicable].
Acknowledgments
This work was supported by the Marsden Fund (PCF, CMB) and a Rutherford Discovery Fellowship (PCF) both from the Royal Society of New Zealand. RHJS was funded by University of Otago Division of Health Sciences Career Development Post-Doctoral Fellowship.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
supplemental data can be accessed here.
References
- [1].Jackson SA, McKenzie RE, Fagerlund RD, et al. CRISPR-Cas: adapting to change. Science. 2017;356. [DOI] [PubMed] [Google Scholar]
- [2].Barrangou R, Horvath P, New CRISPR.. Horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3:143–162. [DOI] [PubMed] [Google Scholar]
- [3].Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR–Cas systems. Nat Rev Microbiol. 2015;13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nature. 2017;15:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Westra ER, Swarts DC, Staals RH, et al. The CRISPRs, they are a-changin’: how prokaryotes generate adaptive immunity. Annu Rev Genet. 2012;46:311–339. [DOI] [PubMed] [Google Scholar]
- [6].Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. [DOI] [PubMed] [Google Scholar]
- [7].Deveau H, Barrangou R, Garneau JE, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang J, Li J, Zhao H, et al. Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR-Cas systems. Cell. 2015;163:840–853. [DOI] [PubMed] [Google Scholar]
- [9].Rollie C, Graham S, Rouillon C, et al. Prespacer processing and specific integration in a Type I-A CRISPR system. Nucleic Acids Res. 2018;46:1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lee H, Zhou Y, Taylor DW, et al. Cas4-dependent prespacer processing ensures high-fidelity programming of CRISPR arrays. Mol Cell. 2018;70:48–59 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kieper SN, Almendros C, Behler J, et al. Cas4 facilitates PAM-compatible spacer selection during CRISPR adaptation. Cell Rep. 2018;22:3377–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Heler R, Samai P, Modell JW, et al. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015;519:199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wei Y, Terns RM, Terns MP, et al. Cas9 function and host genome sampling in type II-A CRISPR–cas adaptation. Genes Dev. 2015;29:356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Silas S, Mohr G, Sidote DJ, et al. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016;351:aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yosef I, Goren MG, Proteins QU. DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fineran PC, Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–209. [DOI] [PubMed] [Google Scholar]
- [17].Ivancic-Bace I, Cass SD, Wearne SJ, et al. Different genome stability proteins underpin primed and naive adaptation in E. Coli CRISPR-Cas Immunity. Nucleic Acids Res. 2015;43:10821–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Levy A, Goren MG, Yosef I, et al. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Staals RH, Jackson SA, Biswas A, et al. Interference-driven spacer acquisition is dominant over naive and primed adaptation in a native CRISPR-Cas system. Nat Commun. 2016;7:12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Modell JW, Jiang W, La M. CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature. 2017;544:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Datsenko KA, Pougach K, Tikhonov A, et al. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat Commun. 2012;3:945–947. [DOI] [PubMed] [Google Scholar]
- [22].Swarts DC, Mosterd C, van Passel MW, et al. CRISPR interference directs strand specific spacer acquisition. PLoS One. 2012;7:e35888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fineran PC, Gerritzen MJ, Suarez-Diez M, et al. Degenerate target sites mediate rapid primed CRISPR adaptation. Proc Natl Acad Sci USA. 2014;111:E1629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kunne T, Kieper SN, Bannenberg JW, et al. Cas3-derived target DNA degradation fragments fuel primed CRISPR adaptation. Mol Cell. 2016;63:852–864. [DOI] [PubMed] [Google Scholar]
- [25].Semenova E, Savitskaya E, Musharova O, et al. Highly efficient primed spacer acquisition from targets destroyed by the Escherichia coli type I-E CRISPR-Cas interfering complex. Proc Natl Acad Sci USA. 2016;113:7626–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li M, Wang R, Zhao D, et al. Adaptation of the Haloarcula hispanica CRISPR-Cas system to a purified virus strictly requires a priming process. Nucleic Acids Res. 2014;42:2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rao C, Chin D, Ensminger AW. Priming in a permissive type I-C CRISPR-Cas system reveals distinct dynamics of spacer acquisition and loss. RNA. 2017;23:1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Savitskaya E, Semenova E, Dedkov V, et al. High-throughput analysis of type I-E CRISPR/Cas spacer acquisition in E. coli. RNA Biol. 2013;10:716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Grissa I, Vergnaud G, The PC. CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol. 2011;9:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Staals RH, Brouns SJ. Distribution and mechanism of the type I CRISPR-Cas systems In Barrangou R and Van der Oost J CRISPR-Cas systems. Springer; 2013. p. 145–169. [Google Scholar]
- [32].Staals RHJ, Agari Y, Maki-Yonekura S, et al. Structure and activity of the RNA-targeting type III-B CRISPR-Cas complex of Thermus thermophilus. Mol Cell. 2013;52:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Elmore J, Deighan T, Westpheling J, et al. DNA targeting by the type I-G and type I-A CRISPR-Cas systems of Pyrococcus furiosus. Nucleic Acids Res. 2015;43:10353–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Majumdar S, Zhao P, Pfister NT, et al. Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furiosus. RNA. 2015;21:1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Silas S, Lucas-Elio P, Jackson SA, et al. Type III CRISPR-Cas systems can provide redundancy to counteract viral escape from type I systems. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Deng L, Garrett RA, Shah SA, et al. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol. 2013;87:1088–1099. [DOI] [PubMed] [Google Scholar]
- [37].Westra ER, van Houte S, Oyesiku-Blakemore S, et al. Parasite exposure drives selective evolution of constitutive versus inducible defense. Curr Biol. 2015;25:1043–1049. [DOI] [PubMed] [Google Scholar]
- [38].Almendros C, Guzmán NM, García-Martínez J, et al. Anti-cas spacers in orphan CRISPR4 arrays prevent uptake of active CRISPR–Cas I-F systems. Nat Microbiol. 2016;16081. [DOI] [PubMed] [Google Scholar]
- [39].Richter C, Dy RL, McKenzie RE, et al. Priming in the type I-F CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014;42:8516–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Strotskaya A, Savitskaya E, Metlitskaya A, et al. The action of Escherichia coli CRISPR-Cas system on lytic bacteriophages with different lifestyles and development strategies. Nucleic Acids Res. 2017;45:1946–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sinkunas T, Gasiunas G, Fremaux C, et al. Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system. EMBO J. 2011;30:1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Westra ER, Pbg VE, Künne T, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell. 2012;46:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mulepati S, Bailey S. In vitro reconstitution of an Escherichia coli RNA-guided immune system reveals unidirectional, ATP-dependent degradation of DNA target. J Biol Chem. 2013;288:22184–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sinkunas T, Gasiunas G, Waghmare SP, et al. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013;32:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rollins MF, Chowdhury S, Carter J, et al. Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proc Natl Acad Sci. 2017;114:E5113–E5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mulepati S, Bailey S. Structural and biochemical analysis of nuclease domain of clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 3 (Cas3). J Biol Chem. 2011;286:31896–31903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shipman SL, Nivala J, Macklis JD, et al. Molecular recordings by directed CRISPR spacer acquisition. Science. 2016;353:aaf1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Redding S, Sternberg SH, Marshall M, et al. Surveillance and processing of foreign DNA by the Escherichia coli CRISPR-Cas system. Cell. 2015;163:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Richter C, Gristwood T, Clulow JS, et al. In vivo protein interactions and complex formation in the Pectobacterium atrosepticum subtype I-F CRISPR/Cas system. PLoS One. 2012;7:e49549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fagerlund RD, Wilkinson ME, Klykov O, et al. Spacer capture and integration by a type I-F Cas1-Cas2-3 CRISPR adaptation complex. Proc Natl Acad Sci. 2017;201618421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Paez-Espino D, Morovic W, Sun CL, et al. Strong bias in the bacterial CRISPR elements that confer immunity to phage. Nat Commun. 2013;4:1430. [DOI] [PubMed] [Google Scholar]
- [52].Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science (New York, NY). 2007;315:1709–1712. [DOI] [PubMed] [Google Scholar]
- [53].Chylinski K, Makarova KS, Charpentier E, et al. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:6091–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rognes T. Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation. BMC Bioinformatics. 2011;12:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Biswas A, Staals RHJ, Morales SE, et al. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Biswas A, Fineran PC, Brown CM. Accurate computational prediction of the transcribed strand of CRISPR non-coding RNAs. Bioinformatics. 2014;30:1805–1813. [DOI] [PubMed] [Google Scholar]
- [57].Chyou T, Brown CM. Prediction and diversity of tracrRNAs from type II CRISPR-Cas systems. RNA Biol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhou Y, Liang Y, Lynch KH, et al. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Paez-Espino D, Chen IA, Palaniappan K, et al. IMG/VR: a database of cultured and uncultured DNA viruses and retroviruses. Nucleic Acids Res. 2017;45:D457–D465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.