

ABSTRACT
Type II CRISPR-Cas9 systems require a small RNA called the trans-activating CRISPR RNA (tracrRNA) in order to function. The prediction of these non-coding RNAs in prokaryotic genomes is challenging because they have dissimilar structures, having short stems (3–6 bp) and non-canonical base-pairs e.g. G-A. Much of the tracrRNA is involved in base-pairing interactions with the CRISPR RNA, or itself, or in RNA-protein interactions with Cas9. Here we develop a new bioinformatic tool to predict tracrRNAs. On an experimentally verified test set the algorithm achieved a high sensitivity and specificity, and a low false discovery rate (FDR) on genome analysis. Analysis of representative RefSeq genomes (5462) detected 275 tracrRNAs from 165 genera. These tracrRNAs could be grouped into 15 clusters which were used to build covariance models. These clusters included Streptococci and Staphylococci tracrRNAs from the CRISPR-Cas9 systems which are currently used for gene editing. Compensating base changes observed in the models were consistent with the experimental structures of single guide RNAs (sgRNAs). Other clusters, for which there are not yet structures available, were predicted to form novel tracrRNA folds. These clusters included a large and divergent tracrRNA set from Bacteroidetes. These computational models contribute to the understanding of CRISPR-Cas biology, and will assist in the design of further engineered CRISPR-Cas9 systems. The tracrRNA prediction software is available through a galaxy web server.
KEYWORDS: TracrRNA, CM model, small RNA, CRISPR-Cas
Introduction
The CRISPR-Cas (Clustered Regularly Interspaced Palindromic Repeats, CRISPR-associated) system is an RNA mediated adaptive immune system. It is used by many bacterial and archaeal species to protect themselves from incoming foreign nucleic acids [1,2]. The simplest CRISPR-Cas systems consist of a CRISPR array and a set of CRISPR associated (Cas) proteins. The array encodes a non-coding RNA consisting of near identical ‘repeat’ sequences, with unique ‘spacer’ sequences between the repeats (Fig. 1). The spacers may be acquired from foreign nucleic acid sequences, notably viral DNA sequences. During subsequent viral infections, the CRISPR array will be transcribed as a non-coding RNA (precursor CRISPR RNA, pre-crRNA) and the Cas proteins expressed (Fig. 1). The pre-crRNA will then be processed into crRNA, which program the Cas proteins to target foreign nucleic acids. Specific Cas proteins or complexes then cleave the viral DNA.
Figure 1.
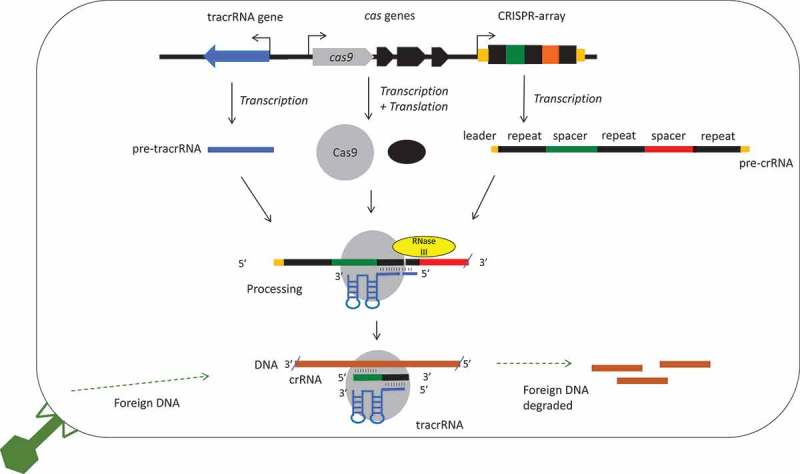
A schematic diagram of a type II CRISPR-Cas9 system focussing on the roles of tracrRNAs in processing and interference. A CRISPR array is shown with repeats in black and spacers in colour, cas genes and the tracrRNA gene are usually nearby. Cotranscribed genes are shown with an arrow. The distances, order and direction of transcription of the genes differ between species, a common arrangement with divergent promoters for cas9 and tracrRNA is shown (see Results). The non-coding RNAs are transcribed into pre-crRNA and pre-tracrRNA, and the proteins transcribed and translated. A 5ʹ leader and 3ʹ trailer is shown for the crRNA (orange)[9]. The RNAs and Cas9 (grey circle) form a complex, specific base pairing occurs between the crRNA repeat (black) and the tracrRNA anti-repeat (blue). Both the crRNAs and tracrRNAs are processed, the crRNA at both ends and tracrRNA at the 5ʹ end. This involves a host factor RNase III (yellow). The complex targets and degrades foreign DNA, though a specific crRNA spacer-DNA protospacer interaction.
There are multiple distinct types of CRISPR-Cas systems that are classified into two classes [3]. Class 1 types, namely type I and III CRISPR-Cas systems, consist of only Cas proteins and CRISPR arrays, and the pre-crRNAs can be processed directly into crRNAs.
Several Class 2 systems, notably type II A, B and C CRISPR-Cas systems also require a small non-coding RNA, the tracrRNA (trans-activating CRISPR RNA) in addition to Cas proteins and crRNAs (Fig. 1) [1,2,4,5]. These have been best studied in Streptococcus pyogenes (type II-A). In S. pyogenes the tracrRNA is transcribed from two promoters into a precursor (pre-tracrRNA, 89 or 171 nt) that is processed into a mature tracrRNA of 75 nt [5]. In engineered systems this has been further shortened to a 67 nt synthetic tracrRNA [6].
The tracrRNAs have an ~ 25 nt ‘anti-repeat’, that is partly complementary to the CRISPR repeat sequence, followed by the ‘nexus’ [7], then a partially folded ‘tail’ [5,8] (Figs. 1 and 2). The pre-crRNAs will form a duplex with tracrRNAs by repeat-anti-repeat base-pairing, and then the RNAs are processed by the nuclease activity of Cas9 and RNase III (Fig. 1) [5,9,10]. After processing, the tracrRNA remains paired with the crRNA, interacting with the Cas9 protein through RNA-protein interactions. This facilitates the targeting and cleavage of target DNA [10], although some systems may also target RNA [11] (Fig. 1).
Figure 2.
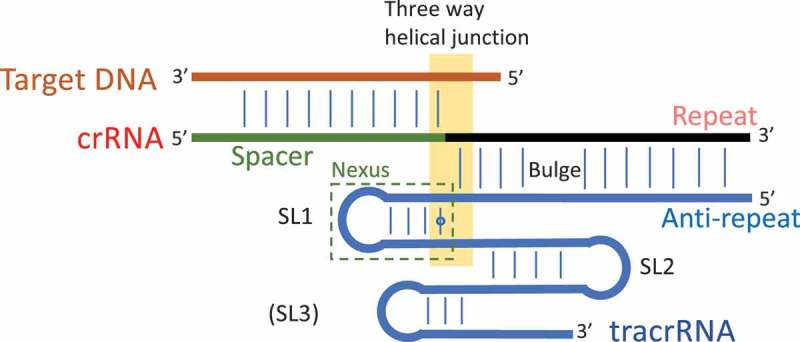
Features of type II interference complexes, based mainly on sgRNA-Cas9 complexes and mutagenesis studies [7,32,56]. The part derived from the crRNA is in red and the tracrRNA in blue, target DNA in brown. The major features (or modules) defined previously in the tracrRNAs are from 5ʹ to 3ʹ. (i) The anti-repeat- this region shows partial complementarity to the repeat in the crRNA, that in some sgRNA have an asymmetric bulge near the junction, separating it into upper and lower stems [15,56]. In sgRNA structures, and synthetic RNA [6] it may be truncated to ~ 11–13 base-pairs but may be 15–25 in the native complex [4,5]. (ii) The nexus- consisting of unpaired bases, stem-loop 1 (SL1), non-canonical pairs (e.g. A-G) and part of the linker between SL1 and SL2 [4]. This region may form more complex structures e.g. a triple helix in C. jejuni [15] (iii) SL2- A second stem loop that may also include non-canonical pairs (A-G) [8]. SL3 – not predicted in all tracrRNAs nor found in all gRNA structures [18]. In gRNA complexes there are backbone and base interactions with the Cas9 protein and the three way helical junction, SL1 and the proximal part of SL2 [32] and with the bulged bases of the repeat-antirepeat hybrid [32]. The loops of the SLs are near the surface of the Cas9 complex.
In addition to type II, the type V-B system (c2c1/cas12a) also includes a tracrRNA. However, the basepairing is at the 3ʹ rather the 5ʹ end [12,13]. In this paper, we focus on type II tracrRNAs.
There have been relatively few studies of tracrRNAs in their natural bacterial systems, but considerable characterisation in heterologous systems. The type II system has been modified for use in genetic engineering by deleting much of the crRNA repeat-tracrRNA anti-repeat region and fusing the two RNAs into a single guide RNA (sgRNA) [8,14] or shortening in synthetic tracrRNA [6]. There has been a considerable amount of work done on the structure and functions of these sgRNAs in diverse organisms [1,15].
There have been a few studies that have systematically searched for tracrRNAs, but no software package available to detect them, to date. The broadest study was that of Chylinski et al. (2013) who predicted tracrRNAs in bacterial genomes, near a cas9 gene, and then analysed conserved motifs within the tracrRNAs [4]. They noted that the tracrRNA sequences are highly diverse, and no conserved secondary structures were found apart from a 3ʹ conserved stem-loop structure. This stem-loop may contribute to an intrinsic transcriptional termination of tracrRNA genes [4,5]. In addition, tracrRNAs were not found in some genomes in which they were expected. Briner et al also presented an algorithm to find native tracrRNAs [16] that they used to compare tracrRNAs in several clades [7,17] but did not do a broad survey.
More recently, X-ray crystallographic structures of complexes that include partial repeat-tracrRNA duplexes, as gRNAs, have been published for four bacterial species S. pyogenes [8], Staphylococcus aureus [18], Francisella novicida [19,20] and Campylobacter jejuni [15]. In each of these the repeat forms a ‘hybrid’ by base-pairing with the tracrRNA anti-repeat. This hybrid may contain non-canonical base-pairs, and commonly has a bulge close to the three-way helical junction, just before the start of the tail. These structures are crucial for activity [15]. The tail begins with a stem-loop (SL1) commonly closed with a non-canonical base-pair (A-G) (Fig. 2).
Following the nexus region, the remainder of the tail has one or two stem-loops (SL2, SL3, Fig. 2). Non-canonical base-pairs are found in all four structures. In C. jejuni, the tail stem-loops also interact with other parts of the tail, giving a complex tertiary fold [15]. Mutations in the first stem-loop of S. pyogenes (in the sgRNA) can eliminate cleavage activity completely, whereas the mutations tested in the other two stem-loops have no significant effect or only partially reduce activity [8,16]. In sgRNA constructs additional RNA loops or sequences have be added at the 3ʹ end of the tracrRNA [21,22] or by extending SL2 of S. pyogenes gRNA [23,24] or SL2 of S. aureus gRNA [18] without impairing function.
In this study, we developed a tracrRNA predictor based on the elements of known tracrRNAs, and identified and analysed a large set of novel tracrRNA genes.
Results
Characteristics of known tracrRNAs
The anti-repeats of tracrRNAs have a partial complementarity to repeats in type II CRISPR-Cas9 systems, these repeats can be used for detection of anti-repeats in tracrRNAs. However, there are challenges in applying this matching on a genome-wide scale. Firstly small, partial, diverged, or degenerated arrays including repeats may be missed by stringent array prediction algorithms [25]. These repeats resemble anti-repeats on the reverse complement strand. Secondly, genomes may contain multiple types of arrays [26] e.g. arrays and repeats from type II and other types, but only the type II repeats will have corresponding anti-repeats in tracrRNAs. These first two challenges result in false positives. Thirdly, the repeat-anti-repeat matches may only have short regions of identity (< 10 nt). Fourthly, novel tracrRNAs may be dissimilar to known systems, due to divergence or possibly independent origins. These last two challenges require flexible algorithms to avoid false negatives.
To develop a method for identifying tracrRNAs with high confidence, the features of a set of known tracrRNAs (n = 55) were determined [4]. We then developed an automated system based on a support vector machine, that puts weights on these known features, but does not absolutely require them (Fig. 3).
Figure 3.
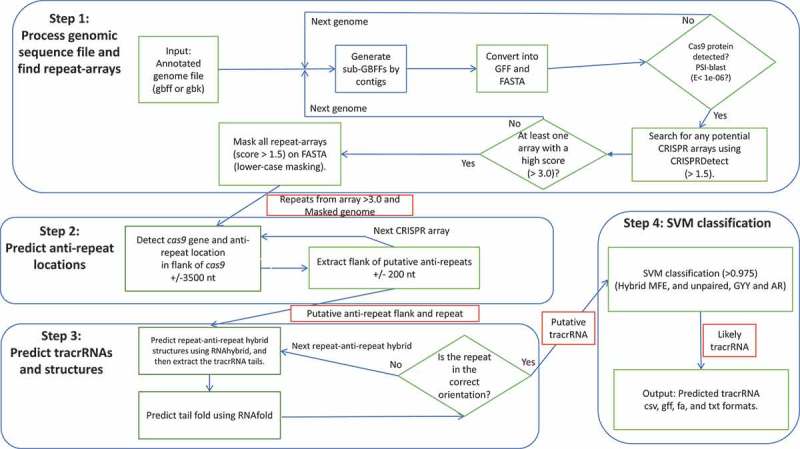
A flow diagram showing the process of tracrRNA prediction. (Results and Discussion and Methods). Step 1. Genomic files are processed to identify CRISPR arrays and cas9 genes Step 2. Anti-repeats from putative tracrRNAs are detected and refined. Step 3. TracrRNA structures and features are predicted. Step 4. The putative tracrRNA are classified by an SVM, and predictions generated in multiple formats.
We observed that these tracrRNAs are commonly located within 500 nt of cas9, although there are exceptions [4,16]. In addition, the crRNA repeat commonly starts with a GYY (G-pyrimidine-pyrimidine) and the tracrRNA tail commonly starts with AR (A-purine, Fig. 3, Fig. S1A-C).
Free energies for the formation of repeat-anti-repeat hybrids are typically below – 40 kcal/mol (Fig. S1D), however, there are frequently mismatches, including a bulge near the 5ʹ end of the repeat (Fig. 2). The longest helices in the repeat-anti-repeat hybrids of known tracrRNAs are typically over 9 nt, and the proportion of non-pairing/mismatch nucleotides are typically below 20%. However, only one of the tracrRNAs has continuous pairing (34/36 nt, Mycoplasma synoviae). The data for all the known and predicted tracrRNAs are available in Supplement 3.
Determining the parameters for the support vector machine (SVM)
A number of parameters were predicted from the training set, six were found to be most useful for the Support Vector Machine (SVM) classification. The ‘length of the longest helix’, and the ‘proportion of non-pairing nucleotides’ in the hybrid are used in the final SVM because they were good discriminators (triangles and circles in Fig. S2). In addition, the minimum free energy of the hybrid, the cas9-tracrRNA intergenic distance, the presence of a ‘GYY repeat-start’ and the presence of an ‘AR tail-start’, were parameters used by the SVM (Fig. 3).
The SVM was tested by leave-one-out cross-validation using the positive and the negative training dataset, and the receiver operating characteristic (ROC) curve demonstrated the robustness of the SVM (Fig. S2). From the distribution of SVM true-positive probabilities of known tracrRNAs in the positive training dataset, the probability cutoff for true positives (TP) was chosen to be 0.975. One reported tracrRNA from Legionella pneumophila has a score lower than this (0.970), which may due to the presence of a 10 nt bulge on the anti-repeat side of the repeat-anti-repeat hybrid, that increases the proportion of non-pairing nucleotides.
Genome wide prediction
We first identified genomes with both cas9 genes and CRISPR arrays, by searching genomes for cas9 genes using PSI-BLAST and Hidden Markov Models (HMMs), and CRISPR arrays were predicted using CRISPRDetect (Fig. 3). The CRISPR repeats from confidently predicted arrays (CRISPRDetect score > 3.0) were used to locate putative tracrRNA anti-repeats within 3500 bases of the cas9 gene (see Methods). Annotated coding sequences and CRISPR arrays (score > 1.5) were not included in the search. In particular, imperfect arrays (scores 1.5–3.0) were excluded as they may have repeats that would generate false positives.
After locating the putative anti-repeats, the flank of the anti-repeat was extracted (± 200). Repeat-anti-repeat hybrid structures and start position of the tracrRNA tails were predicted within this region using RNAhybrid [27], with constraints (Methods). The secondary structures of the tracrRNA tails were predicted using RNAfold [28]. The separate RNAhybrid (anti-repeat) and RNAfold (tail) predictions were combined. The direction of the anti-repeat depends on accurate prediction of the repeat direction. Therefore, we corrected the initial CRISPRDetect direction to increase accuracy for type II arrays (Methods), and eliminated matches from erroneous directions.
We used the prediction pipeline on all RefSeq 84 reference and representative bacterial genomes (n = 5462, Sept 2017) to predict tracrRNAs. We then selected all tracrRNA predictions with a single SVM TP probability of ≥ 0.975. The dataset has 275 predicted tracrRNAs, including the 55 tracrRNAs in the SVM positive-training dataset from 165 genera (S3). The Streptococcus pyogenes M1 476 and the Staphylococcus aureus M0408 tracrRNAs was missed in this multi-genome screen, as both organisms are not representative bacterial species in RefSeq84 genomes. Our predictions also differ in lengths and start-end coordinates (Supplementary Figure S3). At the 5ʹ end, we determined the likely end by using the repeat and locating the anti-repeat (using RNAhybrid). This will tend to give shorter predictions than the previous method that attempted to identify promoters. The median difference was 6 bases and interquartile range 2–16 bases (Supplementary Figure S2). In C. jejuni, our prediction is 11 bases longer, extending the repeat-antirepeat base-pairing [4]. Such a longer precursor was reported by Dugar et al. 2013, although the major processed RNA detected was shorter [4,11,29]. Experimentally determined tracrRNAs are processed/shortened at the 5ʹ end to within the repeat-antirepeat hybrid (Fig. 1).
At the 3ʹ end compared to the previous analysis, the median difference was 3 bases and interquartile range 2–20 bases (Supplementary file S2). They predicted terminators using TranstermHP whereas we used a simpler method (TTTT) to estimate the end. For comparison we also used the best performing rho-independent terminator prediction software RNIE [30] to find rho-independent transcription terminators, but signals were only found in 17 of the 55 training-set tracrRNAs. These were all concordant with our predictions.
The characteristics of all the tracrRNA can be found in Supplementary file S4, their genomic contexts in S5, and mapping to a taxonomic tree in S6. In addition, there were 85 genomes with more than one tracrRNA prediction. It is possible that some genomes have more than one tracrRNA, but alternatively that the second prediction is a false positive. Our stringent SVM cutoff detects one or more tracrRNAs within 3500 bases in 360 of 482 cas9 and CRISPR-array containing genomes. After excluding genomes with a truncated and potentially non-functioning cas9 gene (< 2000 bases long), 299 out of 362 CRISPR have at least one tracrRNA, a sensitivity of 0.83. These 85 multiple predictions are provided in the supplements, but not used further for this initial conservative analysis. In the galaxy version of the tracrRNA predictor, cas9 flanking distance, SVM cutoff, and other parameters can be varied by the user.
As an alternative measure of the FDR (false discovery rate), a region +/-17,500 bases flanking the cas9 was examined, along with a range of SVM cutoffs (0.850 – 0.975). Most known tracrRNA lie within 3500 bases of the cas9 (Fig S1), so additional predicted tracrRNAs in this longer sequence are likely false positives. At high stringency (0.975), after excluding genomes with a truncated cas9 gene 302 genomes have at least one tracrRNA within 17,500 bases, of these there were 7 genomes with one or more potential ‘false positive’ tracrRNA predictions within 3,501–17,500 bases. This gives an FDR of 0.023 (7/302) at this cutoff. This data is shown in S5, and the best prediction within 3,500 bases for each gene in S3. A lower SVM cutoff of 0.95 gave an estimated FDR of 0.066 and sensitivity of 0.94 within 3500 nt of the cas9 (Table S6). Such looser settings may be suitable for analysis of a few genomes of interest using the online tracrRNA predictor.
The direction of transcription of each of the three components of the type II systems were analysed. The most common arrangement of the three, found in 60% of those on a single contig, was that the cas9 genes and tracrRNA were adjacent and transcribed in opposite directions followed by the CRISPR array (140/236 as sketched in Fig. 1). The CRISPR array was slightly more commonly (59%) transcribed in the same direction as the cas genes (83 vs 57) (Fig. 1).
Clustering of tracrRNA sequences
We separately clustered the sequences of CRISPR repeats, anti-repeats, tracrRNA tails, and full length tracrRNAs of all 275 tracrRNAs. We further analysed those clusters with 3 or more sequences. Anti-repeats formed 19 clusters and tracrRNA tails formed 15 clusters. Repeats formed 23 clusters and full-length tracrRNAs formed 19 clusters. Cluster assignments, including two-member clusters, are shown on a taxonomic tree in S3. Sequences in clusters were aligned using CLUSTALW, and RNAalifold and covariance models built using CMbuild.
Clusters were compared, as covariance models, using CMcompare (Methods) to calculate the similarity between each pair of clusters, giving a ‘link score’. A high link score indicates strong similarity. When the folds of the tail clusters were compared to themselves they had link scores 80–120, diagonal in Fig. 4A. In addition, some clusters generated similar models in addition what had been detected by the initial similarity (red boxes, notably 17 and 36, see below). However, in general, tails form distinct folds, with link scores below 40 (Fig. 4A).
Figure 4.
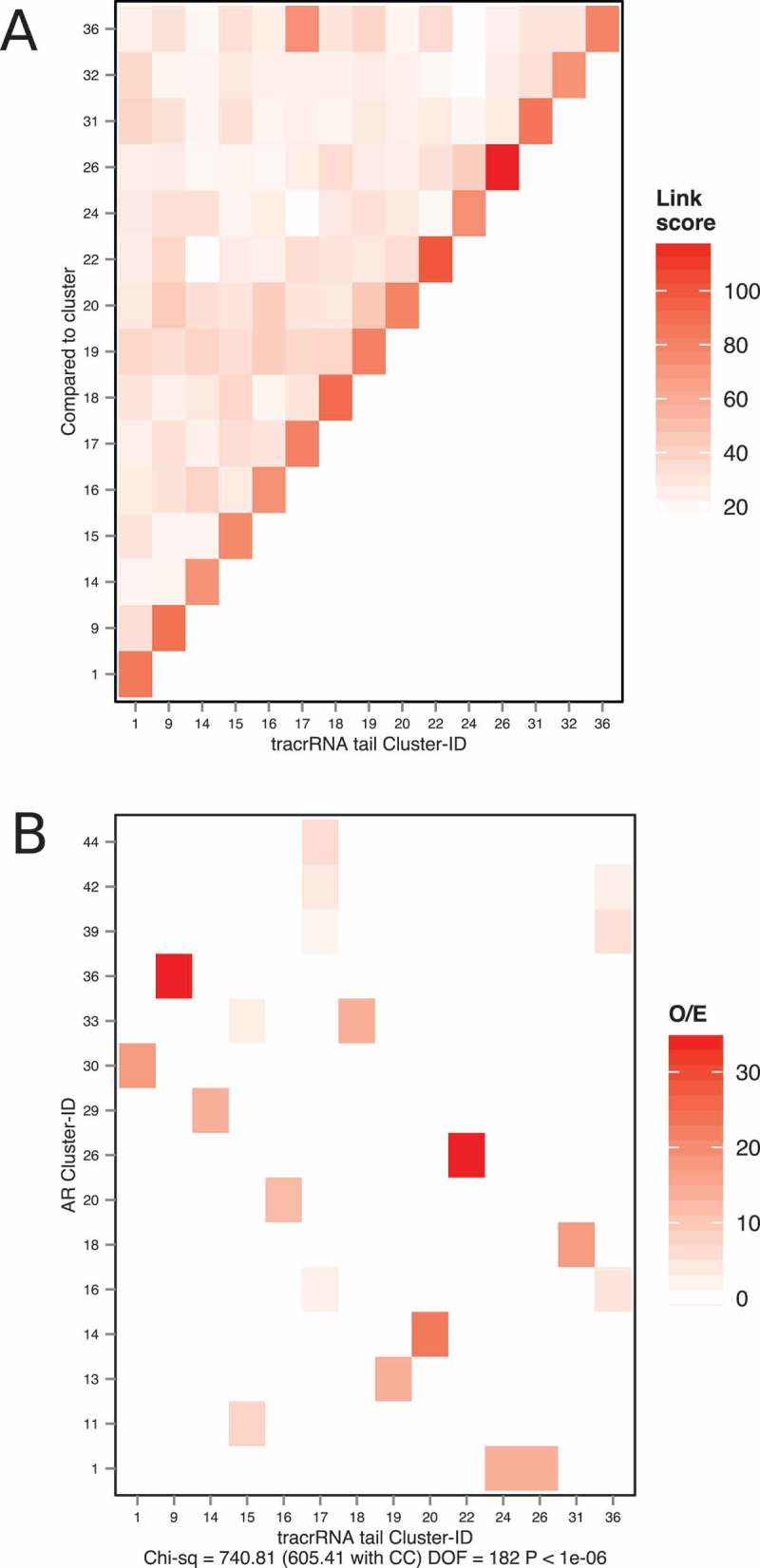
(A) Comparisons between tail-clusters. Similarities between clusters are calculated as link scores using CMCompare. A high link score indicates strong similarity (red). Self matches are on the diagonal. Only those clusters with 3 or more members were analysed so cluster IDS are discontinuous (e.g. 1, 9, 14). (B) the association between anti-repeats (AR) and tracrRNA tails. Most tail clusters correspond to one anti-repeat cluster (e.g. tail 1 and AR 30). To test for the association between anti-repeats and tracrRNA tails, we take tracrRNAs where the tail and the anti-repeat both belong to clusters with 3 or more sequences. The observed frequencies of tail and anti-repeat cluster assignments were divided by the expected frequencies of cluster assignments assuming that the tails and the anti-repeats are clustered independently.
Repeat and anti-repeat clusters correlate strongly (Chi-square = 1048.65, df 221, p-value < 1e-06, details not shown), as expected [5]. In addition, the relatively well conserved anti-repeat portion largely determines the tracrRNA cluster. Therefore, we further examined the most independent tracrRNA components – the anti-repeat and tail sequences and their common folds (Fig. 4B).
Association between anti-repeats and tracrRNA-tails
A possible path by which tracrRNAs evolved is from antisense transcription of the repeat [5]. In which case the initial tail would be any sequence following the anti-repeat. This could likely be a variable spacer sequence, or possibly a 3ʹ trailer (Fig. 1). Depending on whether this happened once or independently in different clades, the sequences of tails and anti-repeats may, or may not, be associated.
We tested the correlation between anti-repeats and tracrRNA-tail members (Chi-square = 605.41, df 182, p-value < 1e-06, Fig. 4B). In most cases a single tracrRNA tail cluster correlates with an anti-repeat (AR) cluster, for example, tail cluster 1 and anti-repeat cluster 30, 9 and 36, 14 and 29 etc. In two cases anti-repeat clusters are split between two tail clusters. Particularly, tail cluster 15 is associated with AR clusters 11 and 33 and tail cluster 17 and 36 weakly associated with three AR clusters. This correlation supports the association between the repeat and the tracrRNA tail, rather than between tails across genera.
Common folds in tracrRNA sequences
We wanted to examine the similarities and differences between the folds in the tracrRNA tails. Fig. 6 shows the taxonomic relationship (NCBI) of the 165 representative genera that had tracrRNAs. Only those clades with tracrRNA tail clusters are shown in full (the full tree is in S5). For each genus, the assignments to tail clusters are shown, clades with no tracrRNA tail clusters are shown collapsed (Fig. 5). As noted in previous studies, tracrRNA tail sequences are highly diverse across species. In support of this diversity, those predicted in 124 of the 165 genera do not cluster. However, where they do cluster it is notable that tracrRNA tails of related genera tend to fall within the same clusters (e.g. clusters 17, 19 and 36, Fig. 5.)
Figure 6.
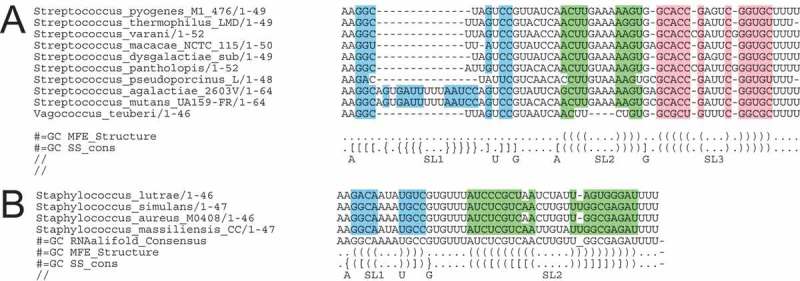
Conserved sequences and structures in tracrRNA tails. Bases that match to the structure in the ‘SS_cons’ line are coloured. The structures in the GC lines are shown in dot-bracket notation. With curved brackets representing common base pairs predicted by RNAalifold and curly or square brackets added manually based on gRNA complexes crystal structures. (A) Alignment of S. pyogenes like tracrRNA tails (Cluster 15). (B) Alignment of S. aureus like tracrRNA tails (Cluster 14).
Figure 5.
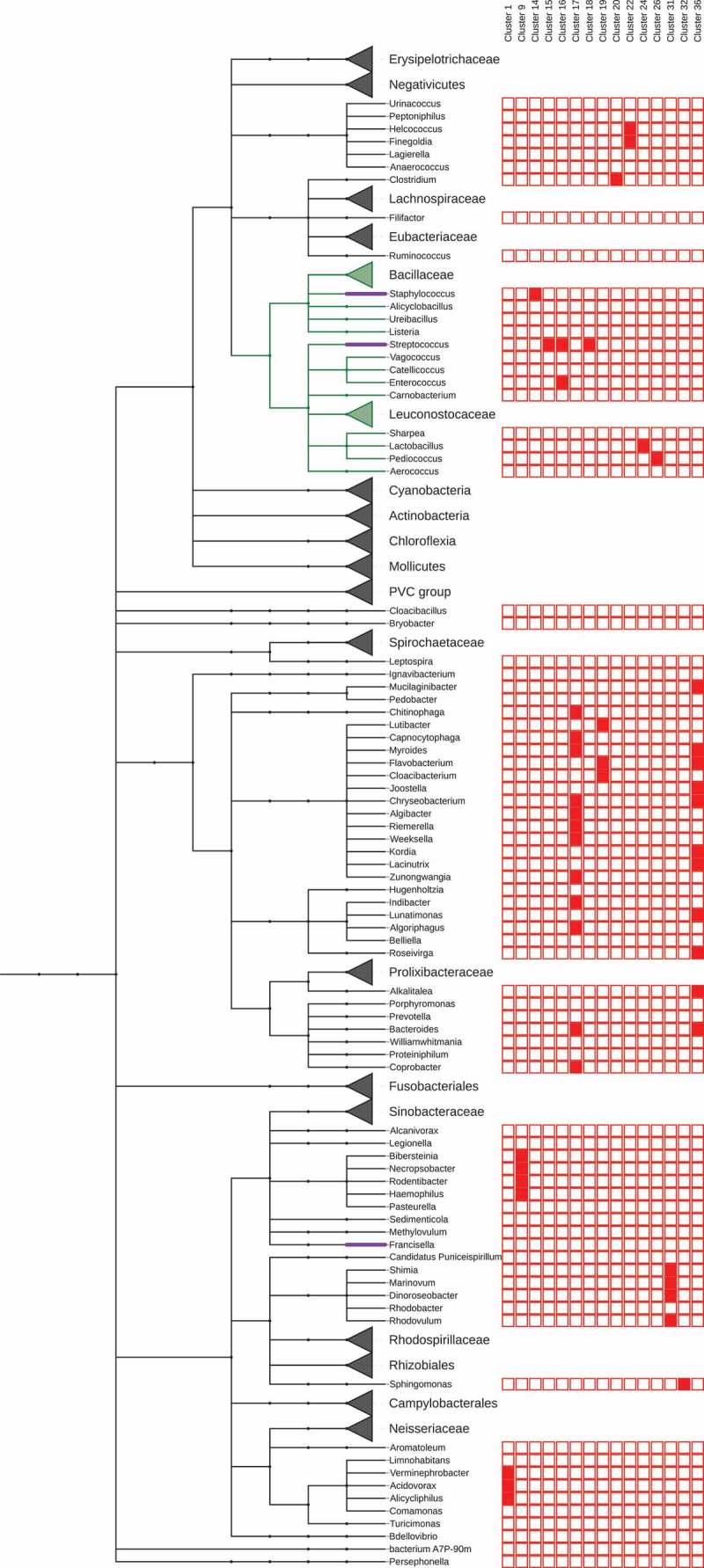
tracrRNA tail clusters distribution across genera. The relationships between the genera shown are based on NCBI’s taxonomic database. Clades with no clusters are collapsed (triangles). Supplementary figure S3 is an extended version of this figure with 165 genera, tail, repeat, ant-repeat and tracrRNA clusters shown.
Streptococci tail clusters
The best characterised tracrRNA are those of Streptococci, particularly S. pyogenes and S. thermophilus, although much of their function has been inferred from studies done on gRNAs in vitro [8,31] or in heterologous environments (e.g. mammalian cells [7,16]). When interacting with cas9, the tail of the S. pyogenes gRNA in complexes forms three short stem-loops that include non-canonical base pairs [8]. Streptococcal tracrRNA tails are part of three clusters 15, 16 and 18, and tracrRNAs from related Bacillales (e.g. Lactobacillus) form two similar clusters 24, 26. Clusters 15, 16 and 24 are broadly similar to the three groups described by Briner et al 2014, for whole Streptococcus and Lactobacillus tracrRNA. These are similar and could be combined into one alignment (Fig. 6, initial separate clusters are in S6).
The alignment of the streptococcal supercluster is shown in Fig. 6A. The common structure predicted by RNAalifold is shown below the sequence alignment in dot-bracket format, it predicts only SL2 and SL3. The structure predicted by RNAalifold was refined manually and presented in dot-bracket format (Fig. 6A), compensating base-pair changes also support the predicted stem-loops. Additional non-canonical base pairs observed in the gRNA structure that cannot be predicted by RNAalifold, for example an A-G base pair, have been added manually at the ends of SL1 and SL2. These bases are invariant in the alignment and thus their function is supported by this analysis. Mutation of these ‘nexus’ bases disrupts interference with gRNA [7] and the interaction with spCas9 in the gRNA structure [32].
Interestingly, two of the stems show evidence of different length forms. SL1 in several Streptococci have a 13 base additional sequence that could form a longer stem (e.g. S. mutans). This could form an extended stem with potential folding indicated Fig. 6A, and can occur without disrupting the U that is pinching out and interacting with Cas9 in an S. pyogenes derived gRNA [32]. Such extended SL1a have been engineered by adding the MS2 stem-loop in gRNA to add a protein binding site [32]. In the S. pyogenes derived gRNA there are no interactions with spCas9 beyond the first non-canonical pair of SL2 [8]. SL2 could be shorter in the diverged Vagococcus teuberi sequence and structure (Fig. 6A).
The database of RNA families (Rfam) has a single tracrRNA model (RF02348) that was built mainly from reported streptococcal tracrRNA sequences [5,14]. Its membership currently corresponds most closely to this supercluster, but the alignment did not predict the conserved stem-loops SL1 and SL2. Only one stem-loop, SL3 that was modelled by Rfam, is predicted because only this common stem-loop had been predicted when the Rfam model was built [5,14]. This loop stem-loop 3 (SL3 in Figure 7) may form part of a rho-independent terminator.
Figure 7.

Bacteroidetes tracrRNA tails. Sequences of the tails of the tracrRNAs from clusters 17 and 36 are shown. All are predicted to form common SL1and SL2, with a A-G pair at the beginning of SL1, and a bulged U. SL3 is not predicted in the common MFE structure by RNAAlifold, but most are predicted to form a SL3, representative secondary structure predictions are shown (an extended figure with all predictions is in the supplement).
Staphylococci cluster
Only a small number of Staphylococci species have CRISPR-Cas systems, but the system found in some S. aureus strains has been successfully modified for genetic engineering [18,33,34]. In contrast to the 3 stem-loops in S. pyogenes gRNA, the S. aureus tail (cluster 14) is predicted to form two longer stem-loops, although the second was not visible in the gRNA complex crystal structure [18].
As SL2 had not been resolved in the gRNA structure, we tested to see if it was supported by covariation. Indeed, six of the 14 base-pairs in SL1 and SL2 were supported by compensating base-pair changes shown as square brackets in Fig. 8 (see the SS_cons line). This supports the formation of the two stem-loops. The alignment supports the A-G base-pair seen in the gRNA at the beginning of SL1. The A-A base-pair observed in the loop would not be predicted by the current MFE methods such as RNAAlifold, but possible base pairing of either A-U or A-A is supported in the model (Fig. 6B), saCas9 protein interacts with the A [18]. The underlined base pairs in SL2 are supported by compensating base-pair changes in the model, which supports the predicted structure shown in [18].
Studies have shown the final SL is dispensable or modifiable, without affecting interference, in both the S. aureus and the S. pyogenes gRNA complexes [6,18,33]. This supports a notion that this SL has a different role, perhaps as a transcription terminator during the biogenesis of the tracrRNA in bacteria. It is GC rich (Fig. 6A,B) and followed by a run of U’s where the tracrRNA terminates [5], although the function of this stem-loop has not been tested experimentally. These gRNAs or synthetic tracrRNAs do not normally require a bacterial termination mechanism as they are synthesised in eukaryotes or in vitro [6].
tracrRNAs from phylum bacteroidetes
Some tracrRNAs clustered with many members across genera. A diverse range of 13 genera from the phylum Bacteroidetes formed clusters 17, 19 and 36. These organisms represent a large group of widely distributed Gram-negative bacteria and include animal pathogens and organisms with unusual biology. In some species e.g. Riemerella and Prevotella, the CRISPR systems have been described, but no tracrRNA or gRNA has been experimentally characterised for any of these genera[35,36].
Clusters 17 and 36 were sufficiently similar and can be combined (Fig. 4A). An alignment is shown in Fig. 7, with some individual folds shown. All individual folds are shown in Fig. S7. These sequences are very similar at the 5ʹ end but dissimilar at the 3ʹ end. Our predictions show a three stem-loop structure, with an A-G terminating base pair in SL1 as seen in both S. aureus and S. pyogenes. However, the A-G at the beginning of SL2 is not conserved, this base pair is also lacking in S. aureus, but the SL2 is predicted to be much shorter in these tracrRNAs. These tracrRNAs form a third abundant class of tracrRNA.
Discussion
A diverse set of type II tracrRNAs
We developed a tracrRNA predictor and applied to find a large set of 275 tracrRNAs. Because this set comes from only the representative sequences in RefSeq, usually only one sequence per species and only a few tracrRNAs per genera, this gives a diverse set of tracrRNA for construction of covariance model that does not contain many closely related sequences from a single species (e.g. S. pyogenes). In future work, it should be possible to use our confidently predicted dataset to build more diverse models to detect exceptional tracrRNAs. We have provided sequence alignments in Stockholm format, and covariance models suitable for use with Infernal (CMsearch), that can be used find further tracrRNAs.
In some genomes (85) more than one tracrRNA was predicted. This may be due to the presence of more than one tracrRNA for a single Cas9 and CRISPR type providing redundancy. They may also include some false predictions, from degenerate arrays. Inclusion of a tracrRNA from these 85 genomes did not add many sequences to the clusters, indicating they are not near identical duplicates. The web implementation of the tool allows the user to investigate multiple predictions.
Clustering of and evolution of tracrRNAs
It has previously been observed that the base-pairing patterns in repeat-anti-repeat hybrids are similar amongst closely related bacterial species [4,5]. We observed this in a much larger dataset. Furthermore, in this study, we observed a correlation between tracrRNA tails and the relationship between species. For each genus, the tracrRNAs tails tend to be located within the same cluster, or be split into clusters that are similar. It would be expected that cas9 genes, tracrRNAs, and crRNAs co-evolve within species.
Evolution of tracrRNAs
tracrRNA may have arisen from anti-sense transcription of repeats, then translocation of a repeat or proto-tracrRNA to a nearby part of the genome. Existent repeat sequences are diverse within CRISPR-Cas systems, reflecting likely multiple origins, but better conserved within type II systems. We cluster repeats into 23 clusters, including the three CRISPRMap type II repeat clusters [37]. It is possible that the tracrRNA may have evolved once, or multiple times, then co-evolved with CRISPR arrays and cas9 genes. In order to begin to address this we looked for, and observed, association between the CRISPR anti-repeats and the tails of the corresponding tracrRNAs. CRISPR repeats of tracrRNAs from the same tail-sequence cluster tend to fall within specific repeat-sequence clusters, instead of spreading across all of them. This observation also supports co-evolution of repeat and tracrRNA. Regardless of whether the tracrRNA originated from a degenerated CRISPR array and/or the translocation of a degenerated CRISPR repeat, the tail of the tracrRNA co-evolves with the CRISPR array nearby.
Genomic locations of tracrRNAs
We observed that the majority of tracrRNAs are transcribed from genes in the opposite direction to cas9 but located nearby. The CRISPR arrays were commonly located after the cas genes in either orientation. This arrangement suggests possible co-regulation of the tracrRNAs and cas9, possibly being transcribed from a bidirectional promoter. On the other hand, the CRISPR array would have a separate promoter. If proto-tracrRNAs arose by antisense transcription of arrays then this may require subsequent translocation of the tracrRNA gene.
Most (45/55 training) tracrRNAs are located within 500 bases of the cas9 gene. Notable exceptions are the Type II-C systems where some are adjacent and some have the cas1 and cas2 genes between the tracrRNA and cas9. This is similar to some of the II-A and II-B arrangements (e.g. F. novicida and N. lactamica c.f. C. jejuni [38]). This arrangement is only seen in C. jejuni in our analysis, not for example in the related C. fetus genome. This may indicate that the C. jejuni arrangement is exceptional.
Other small CRISPR associated RNAs
Here, we have focussed on the tracrRNA of type II systems. Type V systems also have tracrRNAs, but they do not have a folded tail joining to the 3ʹ end of the anti-repeat, instead, they have a folded leader joining to the 5ʹ end of the anti-repeat. Our predictor can, by design, detect the anti-repeat but did not extract the leader (data not shown). We propose to extend our predictor to extract tracrRNAs in Type V systems in the future. Furthermore, there is one other class of small RNAs that can also interact with CRISPRs. In the type II-B system of Francisella novicida there is an additional unique small CRISPR/Cas-associated RNA (scaRNA) [39,40]. This RNA, and the tracrRNA have an additional role: to repress an endogenous bacterial lipoprotein (blp) mRNA[39,41,42]. In this process the 5ʹ end of the tracrRNA (anti-repeat) base-pairs to the mRNA, rather than crRNA [39–42]. In our analysis this scaRNA overlaps the CRISPR array and is masked.
The CRISPR-Cas system has been modified for genetic engineering, and the role of tracrRNA/gRNA is to facilitate site-specific gene modification at high precision. In addition to the understanding of CRISPR-Cas biology, these computational models of tracrRNAs will assist in the design of engineered CRISPR-Cas9 systems.
Materials and methods
Training datasets for the SVM
From published tracrRNA [4] we built the positive training dataset, adding the tracrRNA of S. aureus, giving a total of 55 tracrRNAs. Precise ends of 10 of the 55 tracrRNAs have been confirmed experimentally. Our SVM classifier was trained based on the attributes of the repeat-anti-repeat hybrid and the 3-way junction parts of the repeat-tracrRNA duplex, as well as the distance between the tracrRNA and the cas9 gene. Attributes of the tracrRNA tail were not considered in the prediction, to allows unbiased tracrRNA-tail sequence analyses.
To build the positive training dataset, genomic sequence files in GBFF format were downloaded from RefSeq 84. For each published tracrRNA, we reproduced it by using CRISPRDetect to generate the CRISPR array at an array-quality score cutoff of 3.0, BLASTN to locate the anti-repeat (word-size 7, mismatch penalty −1, gap-opening penalty −2, gap-extension penalty −1, match-reward 1, and e-value cutoff 20), and RNAhybrid to calculate the repeat-anti-repeat hybrid structure and locate the 3-way junction.
The CRISPR repeat sequence was then used as a query to find the anti-repeat part of the tracrRNA in the BLASTN step. Genomic sequence was then extracted around the anti-repeat. We then applied RNAhybrid to the CRISPR repeat and the extracted genomic sequence with default settings but requiring at least one base-pair from position 1 to 3 relative to the 5ʹ end of the repeat, to calculate the repeat-anti-repeat hybrid structure and the free-energy, and at the same time locate the end of the hybrid. We then extracted the tail of the tracrRNA up to the next U-tetramer, U tetramers were not considered in the first 40 nucleotides.
Because the prediction of repeat direction is not completely accurate [25] we tested both orientations. The initial prediction of the direction of the array was done using CRISPRDetect, and for type II arrays, the direction prediction was further refined based on features of known type-II arrays and tracrRNAs.
Type II CRISPR repeats commonly begin with GYY and this was used as the primary indicator of direction. If both, or none of, the array repeat predictions began with GYY repeat start, we used the CRISPRDetect/CRISPRDirection prediction. All tracrRNAs with a tail starting with the conserved AR, were retained as an alternative (Fig. S4). This algorithm will not eliminate potential tracrRNAs where the CRISPR repeats differ and do not start with GYY or the tails do not start with AR.
After the direction-reconfirmation step, for each tracrRNA prediction we calculated the probability of a true-positive prediction with a support-vector machine (SVM). The SVM has 6 parameters. The presence of the conserved GYY repeat start, and the presence of the conserved AR tail start supported the CRISPR array direction chosen as well as building the SVM. The minimum free-energy (MFE) of the repeat-anti-repeat hybrid, the tracrRNA-Cas9 intergenic distance, the length of the longest helix in the repeat-anti-repeat hybrid, and the proportion of non-pairing nucleotides in the repeat-anti-repeat hybrid, were also included in the SVM.
Additional statistical parameters, such as position of the first bulge in the repeat-anti-repeat hybrid from the three-way junction and the nucleotide composition of the repeat-anti-repeat hybrid, were also considered, but including them in the SVM did not increase prediction accuracy (not shown).
To build negative training dataset, for each CRISPR repeat we randomly shuffled the CRISPR repeat and the corresponding tracrRNA to generate a ‘false CRISPR repeat’ and a ‘false tracrRNA’. These were matched using RNA-hybrid, if a hybrid is not found after shuffling the sequences, the randomisation process was repeated until a hybrid was detected. A randomly selected intergenic distance was assigned assuming that the false tracrRNA can be anywhere in the genome but not within the cas9 gene.
Specific parameters of the SVM were chosen after extracting and analysing the positive and the negative training datasets. In general terms, the characteristics that published tracrRNAs had in common, or are discriminators of true tracrRNAs, were considered as potential SVM parameters. The final SVM was built using the application SVM-train in the LIBSVM suite [43]. We used the two-class support-vector classifier with radial-basis kernel function under the default settings. The SVM was validated by leave-one-out cross-validation using the positive and negative training datasets. The robustness of the SVM was evaluated by plotting and examining the ROC curve.
Genome wide predictions of tracrRNAs
Amino acid sequences in the GBFF files are extracted. PSI-Blast was then used with an e-value cutoff of 1e-06 to search for Cas9 using the multiple-alignments of Cas9-family proteins published by Makarova et al 2015 [44] (PSI-BLAST models mkCas0193, cd09643, COG3513, cd09704 and mkCas0192), and the start-end coordinates of the Cas9 coding sequence were determined. This process was complemented by a hidden Markov model search using HMMsearch with an e-value cutoff of 1e-06, using HMM models of Cas9 published by Burstein et al 2016 [45].
In a similar way to the construction of the positive training dataset, for each genome with a Cas9 prediction, we used CRISPRDetect to predict putative CRISPR arrays (array-quality score cutoff 3.0). Then we masked out all annotated genes and all CRISPR-arrays predicted, and used BLASTN to locate the anti-repeat parts of putative tracrRNAs using the same set of parameters we used to generate the positive training dataset, and the CRISPR repeat sequences as queries. We then extracted genomic flanking sequences around the putative anti-repeats, and applied RNAhybrid to the CRISPR repeats predicted and the genomic extracts, to calculate the repeat-anti-repeat hybrid structures and the free energies, and at the same time locate the 3-way junctions of the repeat-tracrRNA duplexes. RNAhybrid were run using the following constraints: base-pairing in the first three positions of the repeat-anti-repeat hybrid, 3–15 mismatches in the hybrid, and a helix of over 9 base-pairs. RNAhybrid allows such constraints as has been used for the similar task of determining miRNA-mRNA interactions[46]. The region following the anti-repeat (the tracrRNA tail) was extracted, minimum length 40, max 204 up to the first four U’s (a putative terminator). The structure of the repeat-tracrRNA duplex were also calculated using RNAcofold [47] and examined using regular expressions corresponding to known tracrRNA families (shown in S3). The secondary structure of the tracrRNA tails were predicted using RNAfold. RNAfold and RNAcofold were run with default settings, but without allowing for lonely base-pairs.
Because the prediction of array direction or strand was not always congruent with published data, both the predicted CRISPR repeat and the reverse-complement of it were used to predict tracrRNAs. We examined repeat-anti-repeat hybrids of known tracrRNAs to find conserved sequences in the CRISPR repeat (GYY) and the 3-way junction part of the tracrRNAs (AR), and used them to re-determine the array direction.
The SVM was applied to all tracrRNA predictions, to calculate the probability of a TP prediction. This calculation was accomplished by using the application SVM-predict in the LIBSVM suite. To establish a probability cutoff for true predictions, we applied the established prediction pipeline including the SVM to re-predict the known tracrRNAs in the positive training dataset to calculate the TP probabilities and examined their distribution.
Software availability
(2) The software is available through a galaxy server (galaxy.otago.ac.nz). Data is available as supplements, and updates will be available from http://bioanalysis.otago.ac.nz/tracrRNA.
Mapping tracrRNA positions back to genomes
The positions of the cas9 (HMM search on RefSeq predicted proteins, methods above), CRISPR arrays (CRISPRDetect), and tracrRNAs were recorded (csv table in S4). This was used to determine the order and orientation of these genes. The flanking regions of the tracrRNAs (+/- 5000 nt) were extracted and re-annotated using an in house modified version of prokka [48]. The genbank format output (.gbk) was visualised in Geneious [49] (S5).
Clustering of tracrRNA sequences
Clustering of repeats, anti-repeats, tracrRNAs and tails was done using UCLUST [50] at an identity cutoff of 0.65. Alignments were made of the sequences in each cluster [51]. We then made covariance models from the tail cluster alignments using CMbuild [52] using default parameters, and compared them using CMcompare [53]. Consensus fold of each tail-cluster alignment were calculated using RNAAlifold [54], and visualized them using Ralee [55]. Alignments, Stockholm format files, and covariance models are available in S4. Repeats for all CRISPR types have been clustered previously as part of CRISPRMap [37], co-occurrence between the three type II clusters and those generated here is shown in S4.
Comparison of common folds in clusters
Comparisons were made using CMcompare [52]. To determine to significance of the co-clustering of anti-repeats and tracrRNA tails, we selected tracrRNAs where the anti-repeat and tail belong to clusters of 3 or more sequences. The observed frequencies (O) of tail and anti-repeat cluster assignments, and the expected frequencies (E) of tail and anti-repeat cluster assignments assuming statistical independence, were calculated; and the O/E values were visualized on a heatmap in R. A Chi-Squared test was used to test the significance of co-clustering.
Funding Statement
This work was supported by the Marsden Fund Council from Government funding, managed by Royal Society Te Apärangi, and a University of Otago Grant.
Supplementary material
Supplemental data for this article can be accessed here.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
References
- 1.Jiang F, Doudna JA.. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46:505–529. [DOI] [PubMed] [Google Scholar]
- 2.Mir A, Edraki A, Lee J, et al. Type II-C CRISPR-Cas9 biology, mechanism, and application. ACS Chem Biol. 2018;13:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobi AM, Rettig GR, Turk R, et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods. 2017;121–122:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briner AE, Donohoue PD, Gomaa AA, et al. Guide RNA functional modules direct Cas9 activity and orthogonality. Mol Cell. 2014;56:333–339. [DOI] [PubMed] [Google Scholar]
- 8.Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carte J, Christopher RT, Smith JT, et al. The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in streptococcus thermophilus. Mol Microbiol. 2014;93:98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charpentier E, Richter H, van der Oost J, et al. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev. 2015;39:428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dugar G, Leenay RT, Eisenbart SK, et al. CRISPR RNA-Dependent Binding and Cleavage of Endogenous RNAs by the Campylobacter jejuni Cas9. Mol Cell. 2018;69:893–905 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017;15:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada M, Watanabe Y, Gootenberg JS, et al. Crystal structure of the minimal Cas9 from campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol Cell. 2017;65:1109–1121. [DOI] [PubMed] [Google Scholar]
- 16.Briner AE, Henriksen ED, Barrangou R. Prediction and validation of native and engineered Cas9 guide sequences. Cold Spring Harb Protoc. 2016;2016:pdbprot086785. [DOI] [PubMed] [Google Scholar]
- 17.Briner AE, Lugli GA, Milani C, et al. Occurrence and diversity of CRISPR-Cas systems in the genus bifidobacterium. PLoS One. 2015;10:e0133661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimasu H, Cong L, Yan WX, et al. Crystal structure of staphylococcus aureus Cas9. Cell. 2015;162:1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirano H, Gootenberg JS, Horii T, et al. Structure and engineering of Francisella novicida Cas9. Cell. 2016;164:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swarts DC, van der Oost J, Jinek M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol Cell. 2017;66:221–233 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mali P, Aach J, Stranges PB, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31:833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng AW, Jillette N, Lee P, et al. Casilio: a versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling. Cell Res. 2016;26:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konermann S, Brigham MD, Trevino AE, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zalatan JG, Lee ME, Almeida R, et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas A, Staals RH, Morales SE, et al. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shmakov SA, Sitnik V, Makarova KS, et al. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio. 2017;8:e01397–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofacker IL, Fontana W, Stadler PF, et al. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994;125:167–188. [Google Scholar]
- 29.Dugar G, Herbig A, Forstner KU, et al. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple campylobacter jejuni isolates. PLoS Genet. 2013;9:e1003495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner PP, Barquist L, Bateman A, et al. RNIE: genome-wide prediction of bacterial intrinsic terminators. Nucleic Acids Res. 2011;39:5845–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karvelis T, Gasiunas G, Miksys A, et al. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013;10:841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishimasu H, Yamano T, Gao L, et al. Structural Basis for the Altered PAM Recognition by Engineered CRISPR-Cpf1. Mol Cell. 2017;67:139–147 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Najm FJ, Strand C, Donovan KF, et al. Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat Biotechnol. 2017;36:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tajkarimi M, Hm W. CRISPR-Cas systems in bacteroides fragilis, an important pathobiont in the human gut microbiome. Front Microbiol. 2017;8:2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu DK, Yang XQ, He Y, et al. Comparative genomic analysis identifies structural features of CRISPR-Cas systems in Riemerella anatipestifer. BMC Genomics. 2016;17:689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alkhnbashi OS, Costa F, Shah SA, et al. CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformatics. 2014;30:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson BM, Louwen R, van Baarlen P, et al. Differential distribution of type II CRISPR-Cas systems in agricultural and nonagricultural campylobacter coli and campylobacter jejuni isolates correlates with lack of shared environments. Genome Biol Evol. 2015;7:2663–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sampson TR, Weiss DS. Cas9-dependent endogenous gene regulation is required for bacterial virulence. Biochem Soc Trans. 2013;41:1407–1411. [DOI] [PubMed] [Google Scholar]
- 40.Sampson TR, Saroj SD, Llewellyn AC, et al. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013;497:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westra ER, Buckling A, Fineran PC. CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol. 2014;12:317–326. [DOI] [PubMed] [Google Scholar]
- 42.Hille F, Richter H, Wong SP, et al. The Biology of CRISPR-Cas: backward and Forward. Cell. 2018;172:1239–1259. [DOI] [PubMed] [Google Scholar]
- 43.Chang CC, Lin C-J. LIBSVM: a library for support vector machines. ACM Trans Intell Syst Technol. 2011;2:1–27. [Google Scholar]
- 44.Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burstein D, Harrington LB, Strutt SC, et al. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Umu SU, Gardner PP. A comprehensive benchmark of RNA-RNA interaction prediction tools for all domains of life. Bioinformatics. 2017;33:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bernhart SH, Tafer H, Muckstein U, et al. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol Biol. 2006;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069.24642063 [Google Scholar]
- 49.Kearse M, Moir R, Wilson A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. [DOI] [PubMed] [Google Scholar]
- 51.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 52.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honer Zu Siederdissen C, Hofacker IL. Discriminatory power of RNA family models. Bioinformatics. 2010;26:453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernhart SH, Hofacker IL, Will S, et al. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffiths-Jones S. RALEE–RNA ALignment editor in Emacs. Bioinformatics. 2005;21:257–259. [DOI] [PubMed] [Google Scholar]
- 56.Garcia-Doval C, Jinek M. Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases. Curr Opin Struct Biol. 2017;47:157–166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.