

ABSTRACT
Alternative polyadenylation (APA) has been discovered to play regulatory roles in the development of many cancer cells through preferential addition of a poly(A) tail at specific sites of pre-mRNA. A recent study found that APA was involved in the mediation of acute myeloid leukaemia (AML). However, unlike gene expression heterogeneity, little attention has been directed toward variations in single-cell APA for different cell types during AML development. Here, we used single-cell RNA-seq data of a massive population of 16,843 bone marrow mononuclear cells (BMMCs) from healthy and AML patient samples to investigate dynamic APA usage in different cell types. Abnormalities of APA dynamics in the BMMCs from AML patient samples were uncovered compared to the stable APA dynamics in samples from healthy individuals, as well as lower APA diversity between eight cell types in AML patients. Genes with APA dynamics specific to the AML samples were significantly enriched in cellular signal transduction pathways that contribute to AML development. Moreover, many leukaemic cell marker genes such as NF-κB, GATA2 and IAP-Family genes exhibited APA dynamics that specifically affected abnormal proliferation and differentiation of leukemic BMMCs. Additionally, mature erythroid cells displayed greater APA dynamics and global 3′ UTR shortening compared with other cell types. Our results revealed extensive involvement of APA regulation in leukemia development and erythropoiesis at the single-cell level, providing a high-resolution atlas to navigate cellular mRNA processing landscapes of differentiated cells in AML.
KEYWORDS: Alternative polyadenylation, acute myeloid leukemia, erythropoiesis, RNA processing, single-cell RNA-seq
Introduction
Alternative polyadenylation (APA) is a crucial strategy to regulate gene expression, mRNA stability, translation efficiency and localization in eukaryotes through preferential use of distinct polyadenylation (poly(A)) sites in pre-mRNAs [1,2]. APA is prevalent in human genes, >50% of which contain multiple poly(A) sites [3,4]. The involvement of APA regulation in many biological processes in humans, such as cell activation, differentiation and proliferation, and immune defences has been widely observed [5–9]. The contributions of APA to tumorigenesis, cancer development and phenotypes have been increasingly revealed, mainly including a global truncation of 3ʹ untranslated regions (UTRs) resulting in loss of 3′ UTR repressive elements of oncogenes with subsequent activation of oncogenes without genetic alterations [10–13].
Acute myeloid leukaemia (AML) is the most common malignant myeloid cancer and is characterized by increased proliferation of progenitor myeloid cells (‘blasts’ or leukaemic cells) in bone marrow, blood, and other tissues due to blocked differentiation of haematopoietic precursors into mature myeloid cells [14]. In the peripheral blood or bone marrow, 20% or more of cells have been found to be myeloid blasts in AML patients [14]. The 3′ UTR shortening of the oncogenic AML1-ETO (AE) fusion gene due to a proximal poly(A) site preference modulated by APA was reported to enhance AE stability and then promote the growth of leukaemic cells in t(8;21) AML [15]. APA regulation has already been discovered to be highly tissue-specific and cell type-specific by genomic-scale profiling [2,16,17]. However, the genome-wide APA dynamics among different leukaemic cell types and their biological roles in AML development are still unclear.
Single-cell RNA-seq (scRNA-seq) is an ideal method for parallel investigations of large cell populations with mixed cell types, particularly for cells that are difficult to fractionate simultaneously. scRNA-seq is emerging as a key method to study transcriptome varieties at the cellular level, significantly differing from traditional RNA-seq technology in which massive numbers of cells are pooled together, and transcriptome diversities are averaged among many cells [18,19]. By exploring the heterogeneity of cells in terms of the expression levels of genes, scRNA-seq has mainly been used to differentiate cell subpopulations, identify novel cell types, and detect enriched expressed gene sets in specific cell types [20–22].
However, specific protocols for capturing the poly(A) sites of expressed genes of massive cell populations at the single-cell level are not yet available [23]. Many different scRNA-seq protocols have been developed to dissect the heterogeneous transcriptome at the single-cell level by adopting different strategies for individual cell isolation and cDNA library preparation [18,24]. One category of these protocols utilizes a 3ʹ end selection strategy for library construction, resulting in a 3ʹ-most fragment library exhibiting a positional bias in the 3ʹ UTR [18,24]. scRNA-seq data generated through these 3ʹ end selection strategies may reflect genes’ APA preferences, which may be a valuable resource for profiling APA dynamics among cells [23].
In this study, we used a scRNA-seq dataset of bone marrow mononuclear cells (BMMCs, ~17k) from AML patient and healthy samples generated by 3ʹ mRNA counting [24] to evaluate the reliability and validity of scRNA-seq data in representing the poly(A) site usage, investigate the APA dynamics of AML at sample and cell type resolutions, and explore the potential of APA regulation in AML development. Our results showed that scRNA-seq (using a 3ʹ end selection strategy) can represent poly(A) site preferences of genes and be used to study APA dynamics among different cell types. Aberrant APA dynamics in the AML patient samples and an extremely low level of APA diversity among different cell types in the same AML patient sample were both observed in comparison to healthy individuals. Moreover, the APA dynamics specific to the AML patient samples were significantly enriched in pathways involved in AML development, suggesting an important role of APA in leukaemia development. Furthermore, we found many leukemic cell marker genes such as GATA2 and IAP-Family genes exhibited APA dynamics in blasts and immature erythroids of AML patient samples that may be associated with AML cell development. These new findings broaden the application scope of scRNA-seq, and expand our knowledge of APA regulation in AML development.
Materials and methods
Data source
The scRNA-seq datasets were retrieved from the single-cell data website of 10x Genomics (https://support.10xgenomics.com/single-cell/datasets) [24], including 16,843 (~17k) sequenced single cells from two healthy controls and two AML patients (AML027 and AML035) before and after transplant treatment (allogeneic haematopoietic stem cell transplantation, HSCT). As stated by Zheng et al. [24] where these datasets were originally described, scRNA-seq libraries were obtained from the cryopreserved BMMCs of the AML patients and healthy subjects, and constructed using the reagents in the GemCode Single-Cell 3ʹ Library Kit. Through genotype assignment of cells based on single nucleotide variant (SNV) detection, the post-transplant AML027 sample was divided into two sub-samples (86.2% of host cells and 13.8% of donor cells) and the post-transplant AML035 sample was found all donor-derived, which were consistent with the clinical chimerism assays [24]. Therefore, seven samples were analysed in this study, including healthy control 1 and 2, AML035 pre-transplant (host), AML035 post-transplant (donor), AML027 pre-transplant (host), AML027 post-transplant (donor), and AML027 post-transplant (host). The two patients have undergone chemotherapy and erythroleukaemia diagnosis before transplant conditioning [24]. For more details of the diagnoses, treatment protocol, and data collection processes of the patients please refer to [24].
Classification and identification of cell subpopulations
Cells in these samples were classified into distinct subpopulations via k-means clustering on the transcriptomes of single cells. Based on the specific genes identified in each cluster and the well-known markers of immune cell types, the cells were finally assigned to 8 cell types (‘Blasts and Immature Ery 1ʹ, Immature Ery 2ʹ, Immature Ery 3ʹ, ‘Mature Ery’, ‘Immature Granulocytes’, ‘Monocytes’, ‘B’, and ‘T’) as previously described [24]. The cell classification results of BMM single-cell data were obtained from Zheng et al. [24]. The t-distributed stochastic neighbour embedding projection [25] of clustering results and cell type compositions of different samples are shown in Figures S1 and S2 (reproductions of results from Zheng et al. [24]).
The characteristics of each category of cell types are described as follows [24]. ‘Blasts and Immature Ery 1ʹ includes cells expressing CD34, a marker of haematopoietic progenitors, and GATA2, a marker for early erythroid cells. ‘Immature Ery 2ʹ contains cells showing expression of GATA1, a transcription factor essential for erythropoiesis, but not CD71, which is often found in more committed erythroid cells. ‘Immature Ery 3ʹ refers to a cell group showing expression of CD71. ‘Mature Ery’ represents cells exhibiting HBA1 expression, which is a marker of mature erythroid cells. Cells in ‘Immature Granulocytes’ show expression of early granulocyte markers such as AZU1 and IL8 and lack expression of CD16. ‘Monocytes’ refers to cell subpopulations expressing CD14 and FCN1. ‘B’ contains cells showing expression of CD19 and CD79A, which are markers of B cells. ‘T’ includes cells showing expression of T cell markers such as CD3D and CD8A. A list of the cell types and cell counts in each sample are shown in Table S1.
3ʹ end extraction and annotation
After cell classification, valid reads from the mapping result file were extracted and divided into different files (SAM format) according to the cell barcodes and cell type classification results using customized shell scripts. Then, the 3ʹ ends (coordinates of the terminal mapping sites of short reads) were extracted from the SAM files and stored into files in BED format, respectively. Finally, 3ʹ ends in each cell type were annotated into different genes using the map tool of bedtools-v2.26.0. The Ensembl genome annotation of Homo sapiens (Assembly GRCh37/hg19) was adopted for site annotation.
APA dynamics identification
Genes with significant differential APA usage under different conditions are defined as DE-APA (deferentially expressed APA) genes or APA dynamics. DE-APA gene identification included the following procedures. First, the coordinates of 3ʹ ends of all valid short reads mapped to a specific gene in two different conditions were extracted and subjected to the Wilcoxon rank sum test (p < 0.05 for the significance cutoff). Second, the distribution region of 3ʹ ends was divided into uniform bins with a specific window size (default: 100 bp), and corresponding histogram distributions of sites under different conditions were calculated. Then, site distribution differences (SDDs) of genes between conditions were calculated as follows,
where n indicates the number of bins that the 3ʹ end distribution region was divided into; a and b denote two samples to be compared; and represents the percentage of reads with 3ʹ ends located in the ith bin of a specific gene in sample a. Lastly, genes with SDDs ≥ 0.2 and p-values <0.05 in pairwise comparisons were recognized as DE-APA genes. A false discovery rate (FDR) <0.05 was applied to control the false discovery rate in multiple statistical tests. Identification of APA dynamics between different samples of the same cell type or between different cell types in the same sample was performed in this study.
Functional pathway enrichment analysis
To investigate whether DE-APA genes were enriched in specific functional pathways, the DE-APA genes identified from pairwise comparisons were subjected to KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis using clusterProfiler [26] (Fisher’s exact test, p-value <0.01). Pathway annotations and gene function annotations of H. sapiens were retrieved from the KEGG database [27] and the Bioconductor package – org.Hs.eg.db, respectively.
Results
Evaluation of poly(A) profiles extracted from the scRNA-seq data
To evaluate the reliability and validity of scRNA-seq data in representing the poly(A) site preferences of mRNAs, the coordinates of 3ʹ ends extracted from valid mapped reads of each scRNA-seq sample were compared with the poly(A) site annotations obtained from the latest human poly(A) database – PolyA_DB 3 [4], where poly(A) sites were captured by a 3ʹ end sequencing method 3ʹ READS [3]. As shown in Figure 1(a), ~80% of 3ʹ ends obtained from scRNA-seq data overlapped with the authenticated poly(A) sites within 100 bp. An overview of the cumulative distribution of the overlaps also showed that the 3ʹ ends of the scRNA-seq data were not randomly distributed (Figure 1(a)). Additionally, analysis of the poly(A) site annotations showed that only ~20% of the authenticated poly(A) sites overlapped with each other within 100 bp (Figure 1(b)), suggesting that most authenticated poly(A) sites are highly dispersed. Together, these results indicated that the 3ʹ ends from these scRNA-seq data correspond to authenticated poly(A) sites and can be used to differentiate the preferences of poly(A) site usage.
Figure 1.
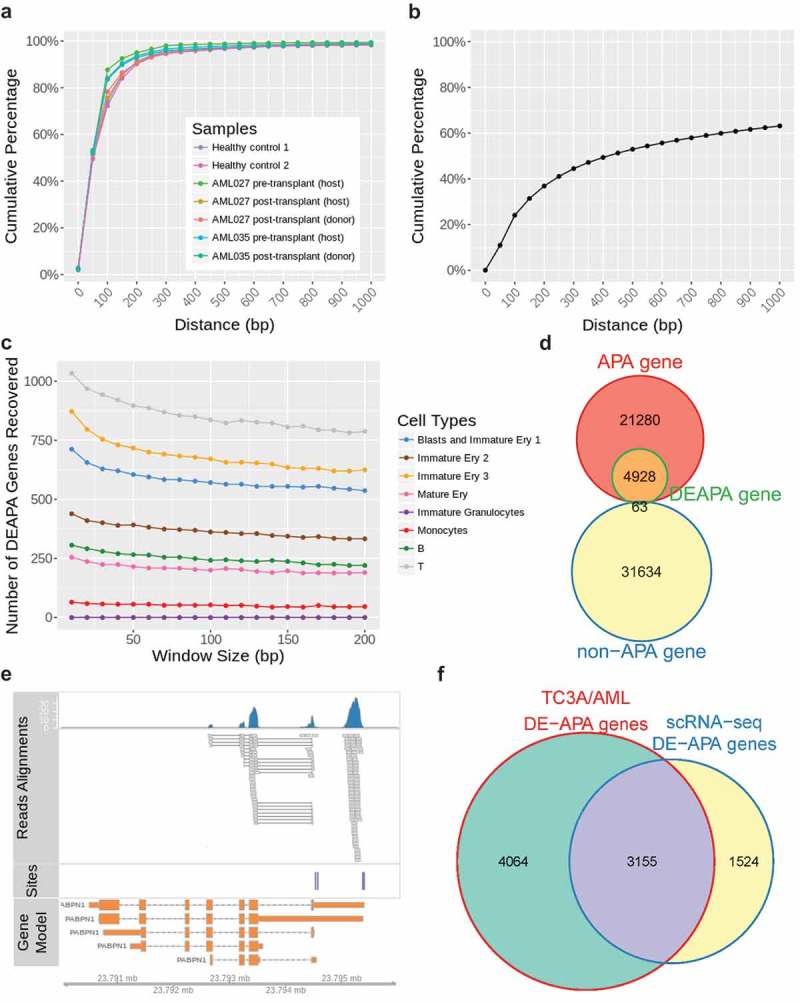
The distance distributions of 3ʹ ends of aligned scRNA-seq reads and the annotated poly(A) sites, and the number of DE-APA genes identified from scRNA-seq data. (a) The cumulative distribution of distances between 3ʹ ends of aligned scRNA-seq reads and their nearest poly(A) annotations; (b) the cumulative distribution of distances between two adjacent poly(A) sites; (c) the numbers of DE-APA genes identified from comparisons of the two healthy samples using different window sizes; (d) the number of non-redundant DE-APA genes belonging to APA gene or non-APA gene category. An APA gene is a gene with ≥2 poly(A) sites, and a non-APA gene is a gene with only one poly(A) site referring to the human poly(A) annotation from PolyA_DB 3/Ensembl; (e) an example of alignment between scRNA-seq reads and annotations from PolyA_DB 3/Ensembl. Top panel, the reads alignments; middle panel, the poly(A) site annotation by PolyA_DB 3; bottom panel, isoforms of gene PABPN1 from Ensembl; (f) the number of non-redundant DE-APA genes identified from scRNA-seq data of AML patient samples overlaps with non-redundant DE-APA genes retrieved from an AML dataset of the TC3A database.
In this study, APA dynamics between different conditions were quantified by the SDD index, and different window sizes could be adopted in the SDD calculation. Here, we first surveyed the impact of window size in DE-APA gene identification by noting the numbers of DE-APA genes in the same cell types between healthy controls 1 and 2 using different window sizes (10 to 200 bp). As shown in Figure 1(c), the smaller the window size, the more DE-APA genes will be recognized. However, no significant decrease in the DE-APA gene number was observed when the window size was ≥100 bp, indicating a minor effect of window size when it approximates to 100 bp. Considering the distance distributions of the 3ʹ ends of the aligned scRNA-seq reads and the annotated poly(A) sites (Figure 1(a,b)), we selected a 100-bp window size for DE-APA gene identification in this study. Furthermore, we divided human genes into two categories, APA and non-APA genes, using the PolyA_DB 3 data and the Ensembl gene annotations. An APA gene is a gene with ≥2 poly(A) sites, and a non-APA gene is a gene with only one poly(A) site in the current annotations. The non-redundant DE-APA genes identified in this study were compared to these two categories of genes, and we found that ~99% of the DE-APA genes belonged to the APA gene group (Fisher’s exact test p < 2.2 × e−16, Figure 1(d)). This result further confirmed the reliability of these scRNA-seq data in representing APA preferences and reliability of our method in APA dynamics identification. An example of alignment between scRNA-seq reads and annotations from PolyA_DB 3/Ensembl is shown in Figure 1(e), two reads clusters were adjacent to the poly(A) site annotations of PolyA_DB 3, and one reads cluster was near to the terminal annotation of an isoform of the gene encoding Nuclear Polyadenylation Binding Protein (PABPN1).
To confirm the DE-APA genes in this study, we also compared the 4679 non-redundant DE-APA genes identified from AML patients with those identified from another AML dataset in the TC3A (The Cancer 3ʹ UTR Atlas) database [28]. This dataset contains a compilation of APA events inferred from 172 AML cancer/normal paired standard RNA-seq data, which includes 7219 non-redundant DE-APA genes (5785 DE-APA genes/sample on an average). Consequently, ~67% (3155 out of 4679) of non-redundant DE-APA genes identified from scRNA-seq data of AML patients overlapped with APA events from the AML dataset of TC3A database (Fisher’s exact test p < 2.2 × e−16, Figure 1(f)). This result indicated a high validity of DE-APA genes identified from the scRNA-seq data of AML patients.
Abundant APA dynamics identified from scRNA-seq data
To explore differences in APA site usage between patient and healthy samples, DE-APA genes in each individual cell type between the AML patient sample (AML035 or AML027 before or after undergoing HSCT) and the healthy sample (healthy control 1 or 2) were identified. The DE-APA genes in each individual cell type between the two healthy samples were also surveyed as a control. We found considerable DE-APA genes even between the two healthy samples, especially in the cell types ‘Immature Ery 2ʹ, ‘B’ and ‘T’ (grey points and line in Figure 2(a,c)), because cells of these three types accounted for relatively large portions in the healthy samples (Figure S2), resulting in more genes captured. Considering the distinct cell type compositions and varied gene capture rates of different samples/cell types, the percentages (the number of DE-APA genes/the number of common sequenced genes in the compared conditions) of DE-APA genes were also calculated for comparison (Figure 2(b,d)). After removal of the influence of composition differences among cell types, 10%~13% of the common sequenced genes across seven cell types of the healthy samples were observed with APA dynamics, indicating that diverse APA usage is a natural, widespread phenomenon in healthy BMMCs.
Figure 2.
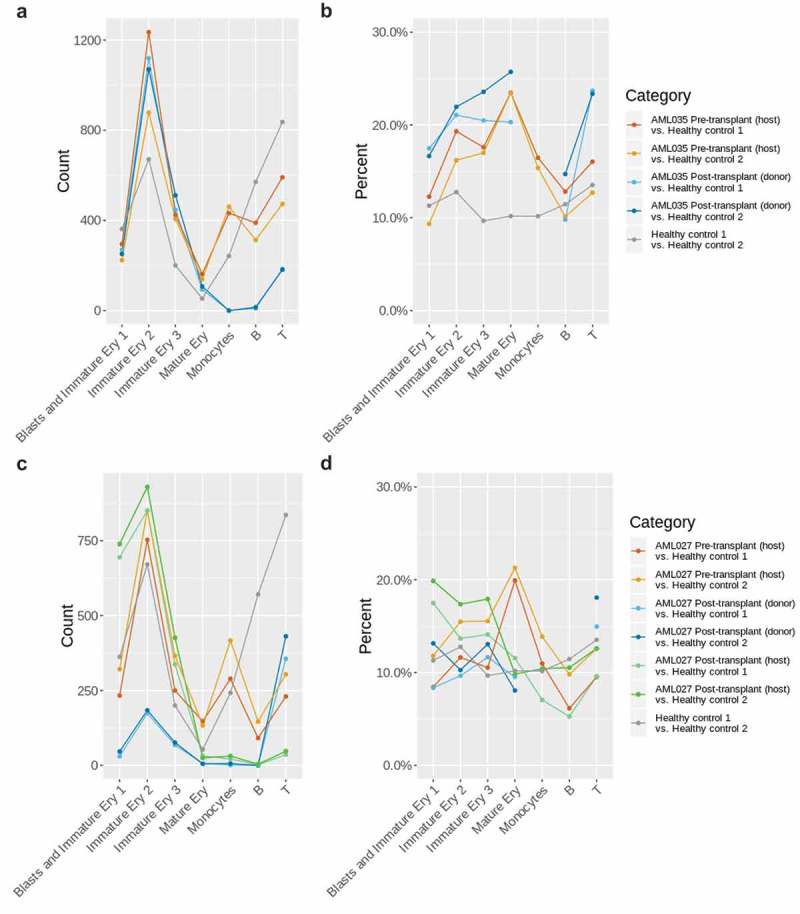
Overview of the DE-APA genes identified between the AML035 or AML027 and healthy samples in the same cell type. (a) The numbers of DE-APA genes identified in each cell type of AML035; (b) the percentages of DE-APA genes identified in each cell type of AML035; (c) the numbers of DE-APA genes identified in each cell type of AML027; (d) the percentages of DE-APA genes identified in each cell type of AML027. The missing points indicate that no or extremely low number of genes were captured in corresponding cell type and sample.
Relative to the percentages of DE-APA genes in healthy BMMCs, higher percentages of DE-APA genes were identified in the cell types ‘Immature Ery 2ʹ, “Immature Ery 3ʹ,”Mature Ery’ and ‘Monocytes’ in the pre-transplant AML035 (host) sample and in the cell types ‘Blasts and Immature Ery 1ʹ,‘Immature Ery 2ʹ, ‘Immature Ery 3ʹ and ‘T’ in the post-transplant AML035 (donor) sample (Figure 2(b)). The increased levels of APA dynamics in AML patient samples suggest a high activity of APA in leukaemic BMMCs. Generally, higher percentages of DE-APA genes were identified in the AML035 post-transplant (donor) sample compared to those in the AML035 pre-transplant (host) sample, especially for the cell type ‘T’, reflecting a difference in APA usage between host and donor cells (Figure 2(b)).
Similar to the AML035 samples, AML027 samples displayed aberrant levels of APA dynamics (Figure 2(c,d)). High levels of APA dynamics were observed in the cell type ‘Mature Ery’ in the pre-transplant AML027 (host) sample, in the cell types ‘Blasts and Immature Ery 1ʹ and ‘Immature Ery 3ʹ in the post-transplant AML027 (host) sample, and in the cell type ‘T’ of the post-transplant AML027 (donor) sample (Figure 2(d)). The percentages of DE-APA genes in the cell types ‘Blasts and Immature Ery’ and ‘Mature Ery’ showed a large difference between pre- and post-transplant AML027 (host) (Figure 2(d)), indicating that HSCT treatment not only affects the cell type compositions of samples (Figure S2) but also the APA usage of genes in cells. An overview of the percentages of DE-APA genes identified in the AML027 and AML035 samples also implied that the impacts of disease or transplant treatment on individuals were different at the level of APA usage.
Specific APA dynamics in AML samples were primarily enriched in disease signal transduction modules
Since APA dynamics commonly exist under normal conditions (Figure 2), we explored how APA dynamics uniquely change in patient samples. Non-redundant DE-APA genes in each sample were obtained by pooling the DE-APA genes identified in different cell types, and then sample-to-sample comparisons were performed to identify APA dynamics specific to each sample (Figure 3(a,b)). Among the 2100 ~ 2700 non-redundant DE-APA genes obtained from the AML035 and healthy samples, ~27% of the DE-APA genes were unique to each sample (Figure 3(a)). In the AML027 sample, 2500 ~ 2600 non-redundant DE-APA genes were monitored; however, the AML027 post-transplant (donor) sample had only 808 DE-APA genes due to a low proportion of donor cells in the AML027 sample after transplant treatment and a low number of transcripts captured (Figure 3(b)). Similar to AML035, 17%~26% of the DE-APA genes were unique to each AML027 sample. The AML035 pre-transplant (host), AML027 pre-transplant (host) and AML027 post-transplant (host) were identified with 724, 680 and 605 sample-specific DE-APA genes, respectively (Figure 3(a,b)), most of which were contributed by blasts, immature erythroids and monocytes (Figure S3). Differently, B and T cells accounted for most of the sample-specific DE-APA genes in healthy samples and AML027 post-transplant (donor) (Figure S3).
Figure 3.
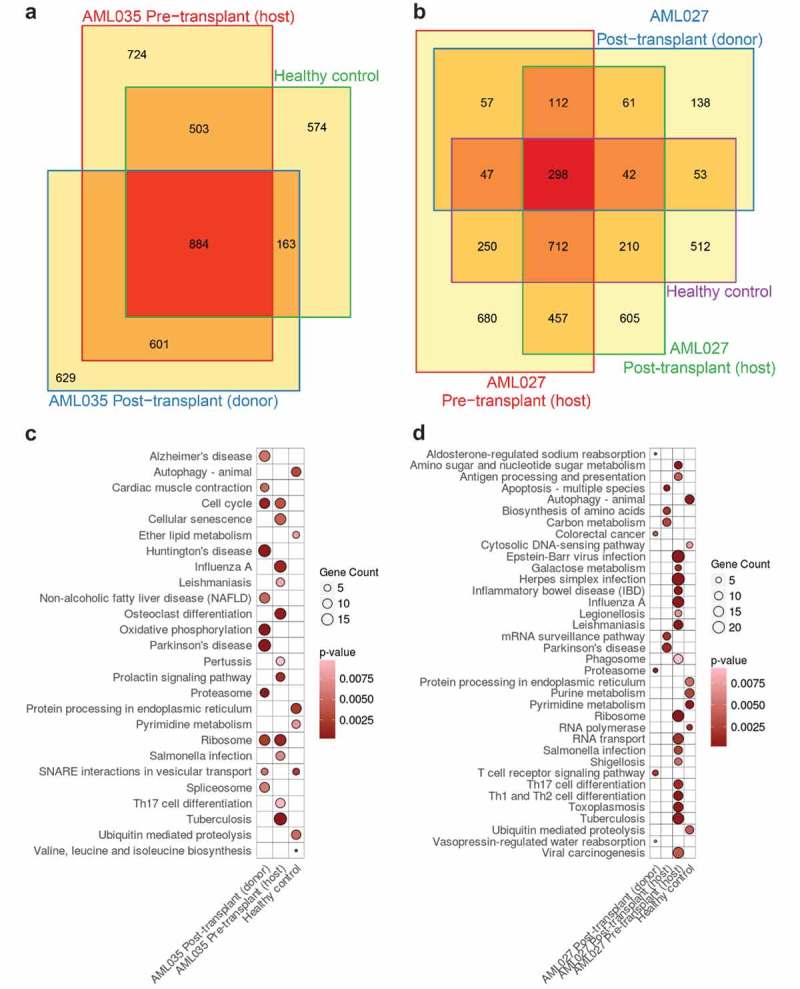
Sample-specific DE-APA genes identified from different samples. (a) Overlaps among DE-APA genes identified from the AML035 and healthy samples; (b) overlaps among DE-APA genes identified from the AML027 and healthy samples; (c) the enriched pathways of DE-APA genes specific to AML035 and healthy samples; (d) the enriched pathways of DE-APA genes specific to AML027 and healthy samples. Specific DE-APA genes identified from comparisons of healthy samples 1 and 2 are shown as Healthy control.
To determine the effect of unique APA dynamics on BMMCs of AML patient samples, each group of sample-specific DE-APA genes was subjected to functional pathway enrichment analysis. The DE-APA genes specific to the AML035 and AML027 patient samples were clearly enriched in different pathways compared to those in the healthy samples (Figure 3(c,d)). The DE-APA genes specific to the healthy samples were enriched in the pathways of autophagy-animal, ubiquitin-mediated proteolysis, protein processing in the endoplasmic reticulum, etc. (Figure 3(c,d)). In contrast, the DE-APA genes specific to the AML patient samples were significantly enriched in many pathways relevant to disease signalling transduction (Figure 3(c,d)). In particular, in the AML027 pre-transplant (host) and AML035 pre-transplant (host) samples, specific DE-APA genes were both extremely overrepresented in pathways involved in disease progression (Figure 3(c,d)). The specific DE-APA genes of the AML035 pre-transplant (host) sample were also significantly enriched in pathways involved in cell proliferation, cell survival and apoptosis, such as the pathways of cell cycle, cellular senescence, prolactin signalling, and osteoclast differentiation (Figure 3(c)). For the enriched pathways in both AML027 and AML035 pre-transplant (host) samples (Figure 4), two modules including the NF-κB and JAK-STAT signalling pathways were extremely overrepresented. The modules of the MAPK, phosphoinositide3-kinase (PI3K)/Akt, and Raf/ERK signalling pathways were found among the enriched pathways of the AML035 samples. Thus, we found that specific APA dynamics in pre-transplant AML patient samples function at the cellular signalling transduction and cell fate processes of their BMMCs.
Figure 4.
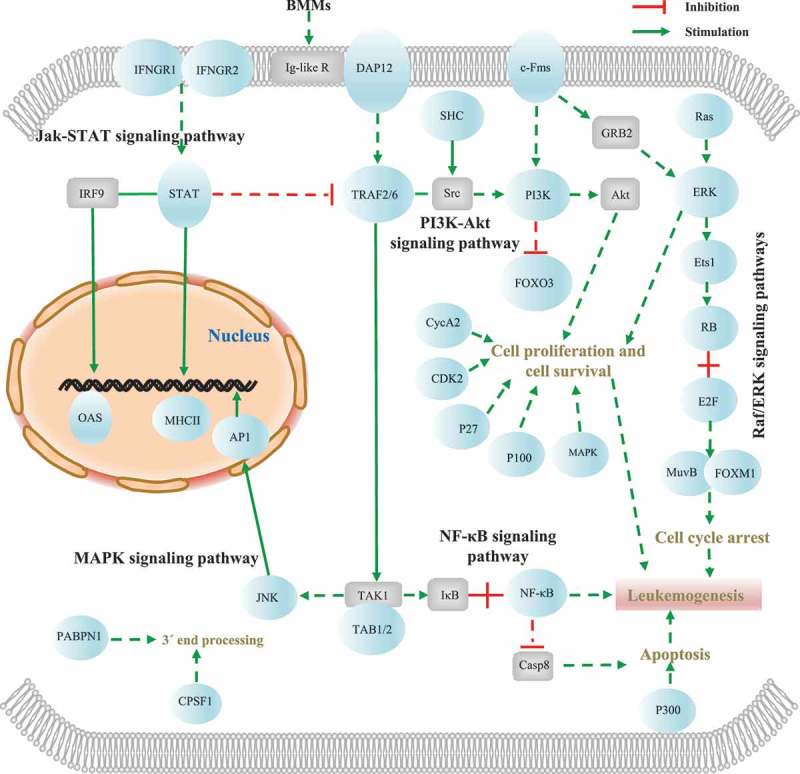
The key modules and genes related to AML development among the enriched disease signal transduction pathways in the AML035 and AML027 pre-transplant (host) samples. Blue ellipse represents the specific DE-APA genes, and grey rectangle represents key metabolic protein without APA dynamics. Dashed line represents multiple enzyme reactions which were not concerned here, and solid line represents a direct enzyme reaction between two subjects. Details of these DE-APA genes are listed in Table S3.
After HSCT treatment, the specific DE-APA genes of the AML027 post-transplant (host) were enriched in pathways of apoptosis-multiple species, Parkinson’s disease, mRNA surveillance pathway, biosynthesis of amino acids, and carbon metabolism (Figure 3(d)). In the pathway of apoptosis, we found FADD encoding Fas associated via death domain preferred using proximal poly(A) site in ‘Immature Ery 2ʹ (Table S2), while BIRC2 and BIRC3 encoding Baculoviral IAP repeat-containing protein (IAPs) preferred using distal poly(A) site in ‘Blasts and Immature Ery 1ʹ (Figure 5(a); Table S2), and XIAP and BIRC5 encoding IAPs showed APA dynamics in ‘Immature Ery 2ʹ and ‘Immature Ery 3ʹ (Figure 5(b); Table S2). The specific DE-APA genes in the AML035 post-transplant (donor) sample were also enriched in many disease-related pathways, such as Parkinson’s disease, Huntington’s disease and Alzheimer’s disease, while the specific DE-APA genes in the AML027 post-transplant (donor) sample were enriched in other pathways, such as T cell receptor signalling, aldosterone-regulated sodium reabsorption, and vasopressin-regulated water reabsorption.
Figure 5.
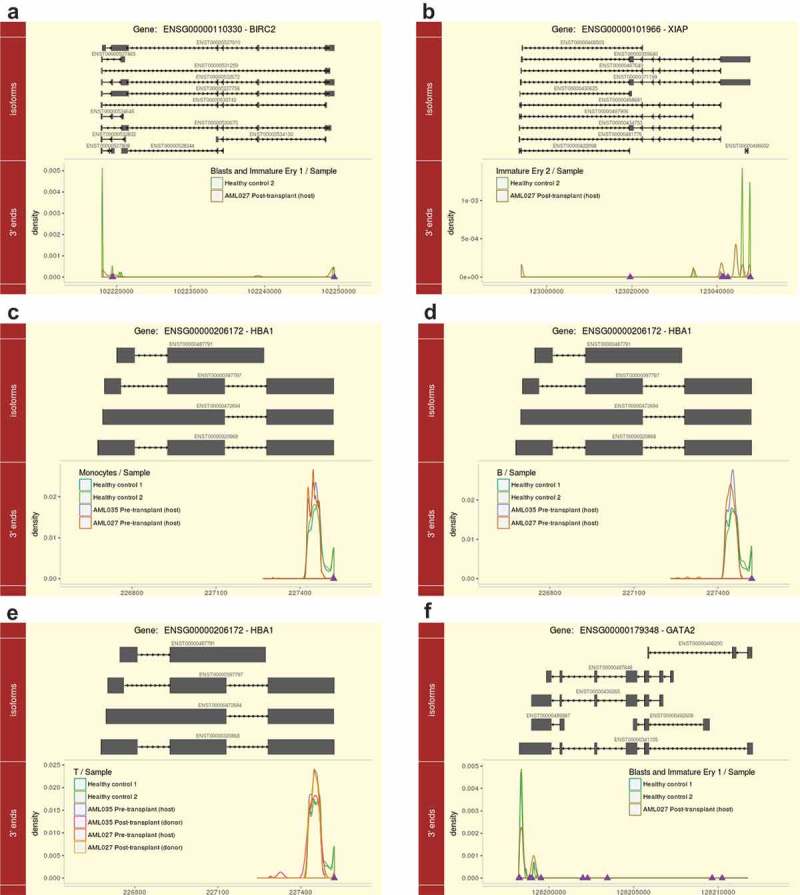
APA dynamics of IAP-Family genes and cell marker genes in specific samples. (a) BIRC2 in the cell type ‘Blasts and Immature Ery 1ʹ; (b) XIAP in the cell type ‘Immature Ery 2ʹ; (c) HBA1 in the cell type ‘Monocytes’; (d) HBA1 in the cell type ‘B’; (e) HBA1 in the cell type ‘T’; (f) GATA2 in the cell type ‘Blasts and Immature Ery 1ʹ. Lines in different colours represent different samples, and samples with no or extremely low expression of the corresponding gene are not shown. Purple triangles represent the annotated poly(A) sites from PolyA_DB 3.
Specific DE-APA genes in patient samples were highly related to AML development
The enriched modules including the NF-κB, JAK/STAT, Raf/ERK, PI3K/Akt, and MAPK signalling pathways were reported to be aberrantly regulated in AML [29–31]. To further investigate the functional consequences of the specific APA dynamics in these enriched modules, we focused on the specific DE-APA genes in the two AML pre-transplant patient samples (Figure 4 and Table S3). In the NF-κB signalling pathway, the gene NFKB1 encoding nuclear factor kappa B subunit 1 (NF-κB) tended to use a distal poly(A) site in the cell type ‘Monocytes’, and displayed down-regulated expression in both AML pre-transplant samples (Table S3). NF-κB is a crucial regulator of multiple cellular processes, including cell differentiation, proliferation, apoptosis, inflammation, and leukaemogenesis [32,33]. Increased activation of NF-κB has been reported to be associated with leukemic cells, and inhibition of NF-κB leads to elimination of leukemic cells with minimal effects on normal host cells [31]. Interestingly, several members of the NF-κB inhibitor family (NFKBIA, NFKBIB, and NFKBIZ) encoding IκB, which contain ankyrin-repeat domains and inhibit NF-κB complexes by cytoplasmic trapping [34], also showed APA dynamics in these AML patient samples. Meanwhile, in its downstream module MAPK signalling pathway, the gene TAB1 encoding TAB1/2 and the gene MAPK8 encoding JNK both preferred using distal poly(A) sites and were down-regulated in ‘Immature Ery 2ʹ in the AML035 sample compared to the expression in the healthy samples (Table S3).
In the JAK-STAT signalling pathway, signal transducer and activator of transcription (STAT) protein-coding gene STAT1 showed a preference of distal poly(A) sites and was down-regulated in ‘Monocytes’ in the AML027 pre-transplant sample. STAT has been shown to play important roles in myeloid differentiation, oncogenesis, and leukaemia [35]. In the Raf/ERK signalling pathway of the AML035 sample before transplant treatment, the NRAS gene encoding Ras showed preferential use of proximal poly(A) sites in ‘Immature Ery 2ʹ, which has been detected to show a point mutation in approximately 15 ~ 25% of AML cases and can activate the PI3K/Akt signalling pathway in AML [36]. In T cells of the AML pre-transplant samples, MAPK1 encoding ERK with a preference for a distal poly(A) site and ETS1 encoding ETS-1 transcription factor with a preference for a proximal poly(A) site both showed increased gene expression levels. ETS1 expression was found to be decreased in the presence of AML blasts [37]. In the PI3K/Akt signalling pathway, PIK3CB encoding PI3K tended to use a proximal site and was highly down-regulated in ‘Immature Ery 2ʹ of the AML035 sample (Table S3 and Figure 4). The genes in the PI3K/Akt signalling pathway were confirmed to contribute to the proliferation, survival and drug resistance of AML blasts [29]. These results suggest that APA is widely associated with the occurrence of AML by regulating key genes in the NF-κB, JAK/STAT, Raf/ERK, PI3K/Akt, and MAPK signalling pathways.
Additionally, we identified many DE-APA genes related to cell proliferation, survival, and apoptosis (Figure 4 and Table S3). For instance, gene CDKN1B encoding P27 preferred to use distal poly(A) sites and showed down-regulation, while gene CREBBP encoding P100 with a proximal poly(A) site preference was down-regulated in ‘Monocytes’ in the AML027 pre-transplant (host) sample. APA dynamics were also observed in key subunits or factors involved in 3′ end cleavage and polyadenylation of mRNA precursors (Figure 4). The gene encoding Cleavage and Polyadenylation Specificity Factor subunit 1 (CPSF1) tended to use proximal poly(A) sites and exhibited higher expression levels in ‘Immature Ery 2ʹ in the AML035 pre-transplant (host) sample than in the healthy sample. Another gene encoding PABPN1 exhibiting preferential use of distal poly(A) sites was mainly observed in the AML samples before transplant treatment (Figures S4 and S5).
APA dynamics of cell type markers
The APA usage of the marker genes of the distinct cell types in this study was examined, and only three marker genes, HBA1, GATA2, and GATA1, were found to have significantly different APA usage patterns (Figure 5(c-f) and Table S4). The marker gene HBA1 of cell type ‘Mature Ery’ preferred using more proximal poly(A) sites in ‘Monocytes’ and ‘B’ cells of the AML027 pre-transplant (host) (Figure 5(c,d)), and ‘T’ cells of the AML035 pre-transplant (host) and AML027 post-transplant (donor) samples (Figure 5(e)). Correspondingly, the percentage of B cells decreased, but the percentage of mature erythroid cells dramatically increased in AML027 pre-transplant (host) sample compared to those in the healthy samples (Figure S2). We also detected a decreased portion of T cells and an increased portion of mature erythroid cells in the AML035 pre-transplant (host) sample compared with those in the healthy samples (Figure S2). Additionally, the marker gene GATA2 of early erythroid cells showed unique APA dynamics in the cell type ‘Blasts and Immature Ery 1ʹ in the AML027 post-transplant (host) sample (Figure 5(f)). GATA2 preferred using more proximal sites and displayed higher expression levels in ‘Blasts and Immature Ery1ʹ in the AML027 post-transplant (host) sample compared to that in the healthy samples (Figure 5(f)). Remarkably, the AML027 post-transplant (host) sample exhibited an abnormally large portion of cells of the cell type ‘Blasts and Immature Ery 1ʹ (Figure S2). These observations suggested that the APA dynamics of marker genes may contribute to the cell type composition changes in the corresponding sample.
Healthy samples showed higher APA diversity among different cell types than AML patient samples
To investigate APA usage preferences in different cell types in each sample, pairwise comparisons of APA usage between cell types in each AML patient and healthy sample were performed (Figures 6 and S6). More than 10% of genes exhibited APA dynamics according to pairwise cell type comparisons of the healthy samples (Figure 6(a,b)), suggesting that APA diversity at the cell type level is common under normal conditions. However, significantly low percentages of DE-APA genes (<10% of the common sequenced genes) were identified in the pairwise cell type comparisons in the five AML patient samples (Figures 6(c-e) and S6). Particularly, APA diversity of immature granulocytes showed the highest level in comparison to other cell types of the AML027 post-transplant (host) (Figure 6(e)).
Figure 6.
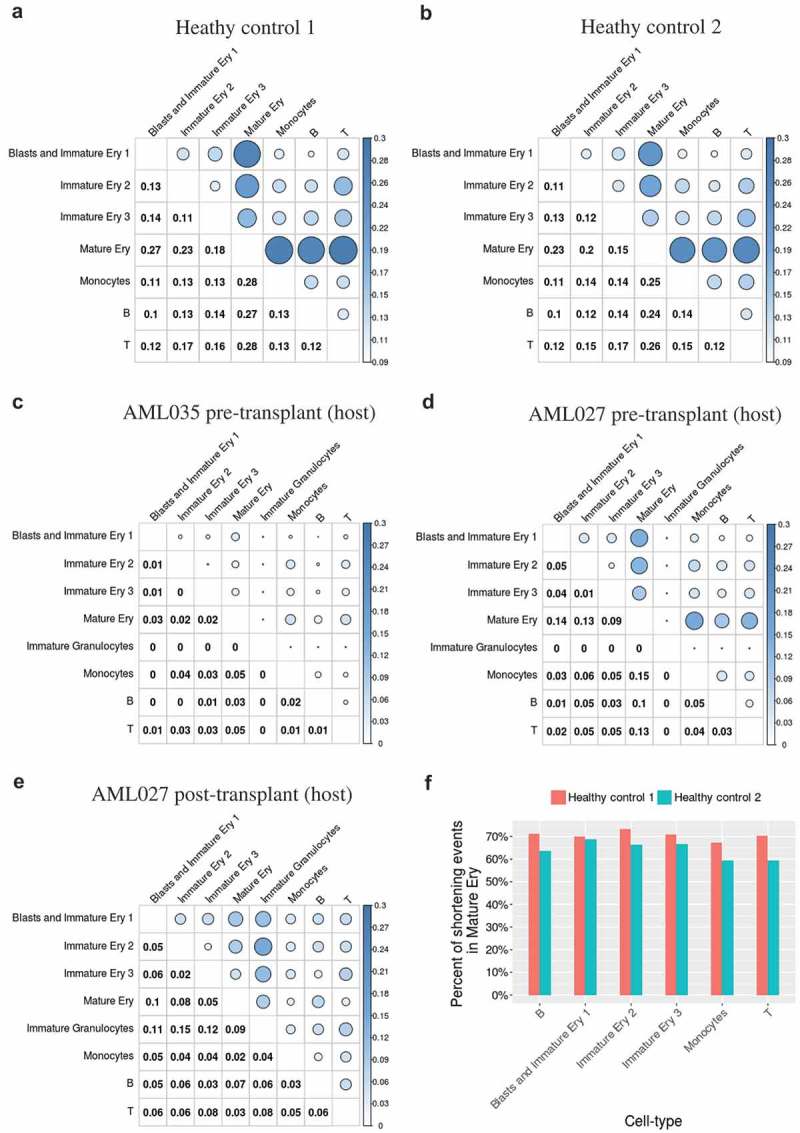
The percentages of DE-APA genes in each paired cell types. The percentages of DE-APA genes identified in each pairwise cell type comparison in healthy control 1 (a), healthy control 2 (b), AML035 pre-transplant (host) (c), AML027 pre-transplant (host) (d), and AML027 post-transplant (host) (e); (f) the percentages of DE-APA genes with shortened 3ʹ UTR in mature erythroid cells compared to other cell types in healthy samples.
Despite the diverse differences discovered between cell types and the difference in APA usage between healthy control samples 1 and 2 (Figure 2(b), 10%~13% of common sequenced genes are DE-APA genes across seven cell types), healthy controls 1 and 2 exhibited an extremely similar atlas of APA dynamics in inner pairwise comparisons of distinct cell types (Pearson’s correlation coefficient r = 0.98, p < 4.6 × e−16), indicating that APA dynamics between cell types are not a random phenomenon. Moreover, the cell type ‘Mature Ery’ showed obviously greater APA dynamics compared with the other cell types in both healthy samples (Figure 6(a,b)), and most of the DE-APA genes in ‘Mature Ery’ displayed shortened 3′ UTRs in both healthy samples (more than 60%, Figure 6(f)), as well as in AML patient samples (Figure S7). The diversity between ‘Mature Ery’ and its predecessors ‘Blasts and Immature Ery1ʹ, ‘Immature Ery2ʹ, or ‘Immature Ery3ʹ decreased gradually, implying a relationship between APA diversity and cell development and/or differentiation during erythropoiesis under normal conditions.
As high levels of APA diversity were found in the cell type ‘Mature Ery’, we next analysed the DE-APA genes in this cell type. In total, 292 and 236 non-redundant DE-APA genes were discovered in the cell type ‘Mature Ery’ in healthy samples 1 and 2, respectively. Among them, 56 and 45 genes were simultaneously observed across comparisons with the other 6 cell types (hereafter referred to as core DE-APA genes), respectively (Figure S8(a)). Most importantly, significantly high numbers of DE-APA gene overlap (106, Fisher’s exact test p < 2.2 × e−16) and core DE-APA gene overlap (18, Fisher’s exact test p < 2.2 × e−06) were observed between the two healthy samples. Details of the 18 core DE-APA genes shared by both healthy samples are shown in Table 1 (details of all core DE-APA genes are shown in Table S5). Among the 18 core DE-APA genes, gene FOS is a member of the Fos family, which dimerize with Jun family to form the AP-1 transcription factor complex (Figure S8(b,c)). In addition, the genes encoding poly(A) binding protein cytoplasmic 1 (PABPC1), eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) and eukaryotic translation elongation factor 2 (EEF2), which are related to poly(A) RNA binding, and PFN1 encoding profiling 1 showed distinct APA usage in mature erythroid cells compared to those in the other cell types (Figure S8(d)).
Table 1.
Details of the 18 core DE-APA genes.
| Ensemble Gene ID | Gene Name | Description |
|---|---|---|
| ENSG00000070756 | PABPC1 | poly(A) binding protein cytoplasmic 1 |
| ENSG00000089157 | RPLP0 | ribosomal protein lateral stalk subunit P0 |
| ENSG00000100316 | RPL3 | ribosomal protein L3 |
| ENSG00000108518 | PFN1 | profilin 1 |
| ENSG00000111640 | GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| ENSG00000123416 | TUBA1B | tubulin alpha 1b |
| ENSG00000140264 | SERF2 | small EDRK-rich factor 2 |
| ENSG00000140988 | RPS2 | ribosomal protein S2 |
| ENSG00000147403 | RPL10 | ribosomal protein L10 |
| ENSG00000156508 | EEF1A1 | eukaryotic translation elongation factor 1 alpha 1 |
| ENSG00000167526 | RPL13 | ribosomal protein L13 |
| ENSG00000167658 | EEF2 | eukaryotic translation elongation factor 2 |
| ENSG00000170345 | FOS | Fos proto-oncogene, AP-1 transcription factor subunit |
| ENSG00000184009 | ACTG1 | actin gamma 1 |
| ENSG00000198034 | RPS4X | ribosomal protein S4, X-linked |
| ENSG00000198886 | MT-ND4 | mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 4 |
| ENSG00000204628 | RACK1 | receptor for activated C kinase 1 |
| ENSG00000273189 | AC010619.2 | NA (encoding a RNA) |
Discussion
APA can be globally regulated in response to changes in cell proliferation and differentiation [2]. Global shortening of 3ʹ UTR length modulated by APA with activating oncogenes without genetic alteration in cancers [11,13], and extensive APA dynamics associated with pathological conditions have also been uncovered [38]. However, these findings were generally based on sequencing methods or protocols requiring a high cell input [3,11,39], and the APA heterogeneity between single cells was not considered. AML is characterized by clonal expansion of abnormally or poorly differentiated myeloid precursors infiltrated into the bone marrow and blood [14]. A previous study reported that APA contributed to leukaemia development by regulating AML1-ETO (AE) fusion gene expression in t(8;21) AML patients [15]. The APA dynamics induced by heterogeneous single leukemic BMMCs still need to be isolated and clarified, especially in samples with mixed and varied rare cell populations. In this study, we profiled the APA usage of eight BMM cell types in both AML patient and healthy samples using scRNA-seq data and provided a high-resolution atlas to depict APA dynamics between AML patients and healthy individuals, or between different cell types in the same sample.
To the best of our knowledge, this is the first work to investigate genome-wide APA dynamics using scRNA-seq data that was not based on the poly(A) site mapping protocol and to study single-cell APA dynamics in a massive population of BMMCs of various cell types. Our study revealed the validity and applicability of scRNA-seq data in reflecting poly(A) site preferences of genes using a 3ʹ end selection strategy for library construction (Figure 1), and successfully utilized the scRNA-seq data to investigate APA dynamics among individual samples and cell types. These findings extend the application scope of scRNA-seq data, which were previously mainly used to quantify cell-to-cell heterogeneity across gene expression levels and to classify cell subpopulations [18,19]. To overcome the low coverage of scRNA-seq in differentiating the APA preferences between different cells, we proposed an approach adopting a strategy of combining repertoires of cells, which were classified into same cell groups by scRNA-seq, to monitor APA diversity between different cell types/conditions. This approach can be applied to similar datasets and invoke more innovative ideas or methods to mine APA dynamics from scRNA-seq data. It is important to note that, in terms of detecting real de novo poly(A) sites or differentiating adjacent poly(A) sites, the approach of utilizing 3ʹ-based scRNA-seq data to delineate APA dynamics is not as accurate or stable as methods based on experimental poly(A) site mapping protocols. However, most of the poly(A) site mapping protocols are bulk RNA-seq based which cannot be used to dissect APA at single resolution, such as 3ʹ READS [3], 3ʹ seq [38], PAS-seq [40], A seq [41], and PAT-seq [42]. The only available single cell poly(A) site mapping method BATSeq comes with low sensitivity and is hard to be quantitative [17,23]. Thus, our method is one way of quantifying APA dynamics at single-cell resolution when efficient single-cell poly(A) site mapping protocols are not available.
Compared with the APA levels in healthy samples, aberrant levels of APA diversity were detected in the AML patient samples in sample-to-sample comparisons in the same cell types and in cell type-to-cell type comparisons in the same samples (Figures 2, 6, and S6). High levels of APA diversity were found at the sample scale in the AML patient samples (the percentages of DE-APA genes were generally distributed between 15% and 25%, Figure 2). Consistently, the median number of unique molecular identifier (UMI) counts per cell of BMMCs from AML patient samples were found to be higher than those in the two healthy samples, reflecting the abnormal transcriptional process of leukemic BMMCs [24]. In contrast, low levels of APA diversity were observed between cell types in the AML patient samples (the percentages of DE-APA genes were generally distributed less than 10%, Figures 6(c-e) and S6). APA is correlated with different cell proliferation and differentiation statuses in many kinds of cancers [11,13]. The abnormal APA profiles in BMMCs of AML patient samples may be associated with aberrant proliferation and/or poor differentiation of leukemic BMMCs [14].
We found the most sample-specific DE-APA genes were from the BMMCs of two AML pre-transplant (host) samples (Figure 3(a,b)), and a majority of sample-specific DE-APA genes in AML pre-transplant samples, and AML027 post-transplant (host) were contributed by blasts and immature erythroids (Figure S3). Dissimilarly, most of the sample-specific DE-APA genes in healthy samples and AML027 post-transplant (donor), which exhibited a similar cell type composition to healthy samples, were contributed by B and T cells (Figure S3). The sample-specific DE-APA genes in the two AML pre-transplant (host) samples were significantly enriched in pathways relevant to disease signal transduction (Figure 3(c,d)). Among these enriched pathways, five modules, including the NF-κB, JAK/STAT, Raf/ERK, PI3K/Akt, and MAPK signalling pathways, which govern many biological processes, such as cell proliferation, differentiation, apoptosis, inflammation, oncogenesis, and leukaemia, displayed specific APA dynamics in the BMMCs of AML patient samples (Figure 4). The five modules were reported to be activated and to mediate abnormal haematopoiesis in AML [29–31,35,36]. These results suggest that APA may play an essential role in AML development, especially by regulating genes involved in key signal transduction pathways to modulate haematopoiesis. In addition, many specific DE-APA genes involved in cell proliferation, survival, and apoptosis were also identified in leukemic BMMCs (Figure 4 and Table S3). For instance, the down-regulated CDKN1B gene encoding P27 preferred using distal poly(A) site in ‘Monocytes’ in the AML027 pre-transplant (host) sample. P27 is an inhibitor of cell cycle progression, and FLT3 and FLT3-ITD can phosphorylate tyrosine residue 88 of P27, which impairs the function of cell proliferation prevention through inactivation of cyclin-dependent kinases in AML cells [43,44]. These findings are consistent with the main characteristic of AML that haematopoietic precursors are blocked from differentiating into mature myeloid cells, and further confirm the high involvement of APA in leukemic cells.
Furthermore, APA was found involved in disease relapse of the AML027 patient sample. The AML027 post-transplant (host) were detected with large populations of atypical blast and granulocyte precursors (Figure S2), which was diagnosed with disease relapse and return of the malignant host AML [24]. At the sample scale, post-transplant AML027 (host) had the highest level of APA dynamics in the cell type ‘Blasts and Immature Ery 1ʹ compared to other samples (Figure 2(d)), and showed a higher level of APA diversity in the immature granulocytes than other seven cell types (Figure 6(e)), indicating a high activity of APA in blast and granulocyte precursors of AML027 post-transplant (host). In the ‘Blasts and Immature Ery1ʹ of the AML027 post-transplant (host) sample, we found that the early erythroid cells marker gene GATA2 exhibited a preferential usage of proximal sites (Figure 5(f)), and a higher expression level than that in the healthy samples (Figure 5(f)). GATA2 is involved in the control of proliferation and differentiation of myeloid progenitors, and has been reported to be an AML-predisposing gene [45]. The proximal poly(A) site usage of GATA2 may result in its up-regulated expression to escape targeting by cis-elements such as miRNA in cancer [2]. Besides, in the enriched pathway of apoptosis in the AML027 post-transplant (host) sample, IAP-Family genes such as XIAP, BIRC2, and BIRC3 were found to change the usage of APA sites (Figure 5(a,b); Table S2), which are responsible for adverse prognostic of patients with AML, and exist posttranscriptional regulation in their gene expression [46]. These results indicate that APA may be involved in disease relapse of the AML027 post-transplant patient.
APA is regulated by the 3′ end processing machinery composed of more than 20 core polyadenylation factors [2]. Here, many specific DE-APA genes in the AML patient samples encode poly(A) factors such as PABPN1 and CPSF1. The gene encoding PABPN1, which has a regulatory role in APA usage [5], preferred using a distal poly(A) site in the AML patient samples before transplant treatment (Figures S4 and S5). Dysfunctional or down-regulated PABPN1 could induce widespread enhancement of polyadenylation shift from distal sites to proximal sites [38,47]. Moreover, enhancement of CPSF1 with proximal poly(A) site usage was observed in ‘Immature Ery 2ʹ in the AML patient samples (Figure 4), which could promote 3ʹ UTR shortening of the AML1-ETO (AE) fusion gene and increase the AE stability and growth rate of t(8;21) AML cells [15]. As dysregulation or mutation of poly(A) cis-elements or factors has already been discovered to cause or contribute to human diseases [48], variations of these poly(A) factors in AML patients may participate in regulating abnormal APA dynamics of leukemic BMMCs.
By investigating the scRNA-seq dataset generated from parallel transcriptional profiling of a massive population of BMMCs, we found that APA dynamics readily occurred between different cell subpopulations in the normal BMMCs; 10%~13% of genes across seven cell types in the two healthy samples were involved in APA dynamics (Figure 2), and 10%~28% of genes between distinct cell types in the same healthy sample exhibited APA dynamics, which was significantly higher than that in the AML patient samples (Figures 6 and S6). Furthermore, the APA dynamics between cell types in the two healthy samples exhibited high similarity (Figure 6(a,b)). These results reflected high and stable activity of APA regulation in routine cellular process of BMMC development, suggesting that APA may regulate myeloid cell development, which is consistent with previous findings that many cell developmental processes, such as embryonic development [49] and reprogramming of differentiated cells into induced pluripotent stem cells [50], were regulated by APA.
Among the eight cell types of BMMCs in the healthy samples, mature erythroid cells exhibited outstanding APA dynamics (Figure 6(a,b)), and DE-APA genes had global 3′ UTR shortening compared to other cell types (Figure 6(f)). The APA diversity between erythroid cells in premature stages, including ‘Blasts and Immature Ery1ʹ, ‘Immature Ery2ʹ, and ‘Immature Ery3ʹ, and mature erythroid cells decreased gradually (Figure 6(a,b)), which may reflect developmental processes in erythroid cells, implying a relationship between APA regulation and erythroid cell development. We identified several core DE-APA genes that were unique to mature erythroid cells (Figure S8(a); Table 1). Among them, FOS (Figure S8(b,c)) can dimerize with the Jun family to form the AP-1 transcription factor complex, which participates in erythroid proliferation, differentiation and apoptosis [51–53]. Additionally, Profilin 1 encoded by PFN1 (Figure S8(d)) has an essential role in the development of haematopoietic stem cells in bone marrow in mice [54]. From the results, we deduce that APA regulation plays certain roles in normal erythropoiesis.
Overall, our results uncovered the potential value of large-scale scRNA-seq data in studying APA regulation and the important roles of APA regulation in leukaemia development and erythropoiesis, that indispensable APA dynamics contribute to cell diversity and stable APA dynamics can maintain homeostasis in the host organism.
Funding Statement
This work was supported in part by grants from the National Key R&D Project of China (2016YFE0108800), U.S. NSF (IOS-154173), the Fundamental Research Funds for the Central Universities in China (Xiamen University: 20720170076), the China Postdoctoral Science Foundation (2017M622067), and the National Natural Science Foundation of China (61802323, 31801268, and 61573296).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Mayr C. Evolution and biological roles of alternative 3′UTRs. Trends Cell Biol. 2016;26:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoque M, Ji Z, Zheng D, et al. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods. 2013;10:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang R, Nambiar R, Zheng D, et al. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018;46:D315–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. [DOI] [PubMed] [Google Scholar]
- [6].Huang G, Huang S, Wang R, et al. Dynamic regulation of tandem 3′ untranslated regions in zebrafish spleen cells during immune response. J Immunol. 2016;196:715–725. [DOI] [PubMed] [Google Scholar]
- [7].Hwang H-W, Saito Y, Park CY, et al. cTag-PAPERCLIP reveals alternative polyadenylation promotes cell-type specific protein diversity and shifts araf isoforms with microglia activation. Neuron. 2017;95:1334–1349.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Qiu F, Fu Y, Lu C, et al. Small nuclear ribonucleoprotein polypeptide A–mediated alternative polyadenylation of STAT5B during Th1 cell differentiation. J Immunol. 2017;199:3106–3115. [DOI] [PubMed] [Google Scholar]
- [9].Yoon OK, Hsu TY, Im JH, et al. Genetics and regulatory impact of alternative polyadenylation in human B-Lymphoblastoid cells. PLoS Genet. 2012;8:e1002882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Masamha CP, Wagner EJ. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis. 2018;39:2–10. [DOI] [PubMed] [Google Scholar]
- [11].Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Singh P, Alley TL, Wright SM, et al. Global changes in processing of mRNA 3ʹ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009;69:9422–9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xia Z, Donehower LA, Cooper TA, et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun. 2014;5:5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–1152. [DOI] [PubMed] [Google Scholar]
- [15].Shima T, Davis AG, Miyauchi S, et al. CPSF1 regulates AML1-ETO fusion gene polyadenylation and stability in t(8;21) acute myelogenous leukemia. Blood. 2017;130:2498. [Google Scholar]
- [16].Neve J, Burger K, Li W, et al. Subcellular RNA profiling links splicing and nuclear DICER1 to alternative cleavage and polyadenylation. Genome Res. 2016;26:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Velten L, Anders S, Pekowska A, et al. Single-cell polyadenylation site mapping reveals 3ʹ isoform choice variability. Mol Syst Biol. 2015;11:812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Saliba A-E, Westermann AJ, Gorski SA, et al. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 2014;42:8845–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wen L, Tang F. Single-cell sequencing in stem cell biology. Genome Biol. 2016;17:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu SJ, Nowakowski TJ, Pollen AA, et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016;17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rato S, Rausell A, Muñoz M, et al. Single-cell analysis identifies cellular markers of the HIV permissive cell. PLoS Pathog. 2017;13:e1006678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tirosh I, Venteicher AS, Hebert C, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539:309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen W, Jia Q, Song Y, et al. Alternative polyadenylation: methods, findings, and impacts. Genomics Proteomics Bioinformatics. 2017;15:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- [26].Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS J Integr Biol. 2012;16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Feng X, Li L, Wagner EJ, et al. TC3A: the cancer 3′ UTR atlas. Nucleic Acids Res. 2018;46:D1027–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martelli AM, Nyåkern M, Tabellini G, et al. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911. [DOI] [PubMed] [Google Scholar]
- [30].Sakamoto KM, Grant S, Saleiro D, et al. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol Genet Metab. 2015;114:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Volk A, Li J, Xin J, et al. Co-inhibition of NF-κB and JNK is synergistic in TNF-expressing human AML. J Exp Med. 2014;211:1093–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kuo H-P, Wang Z, Lee D-F, et al. Epigenetic roles of MLL oncoproteins are dependent on NF-κB. Cancer Cell. 2013;24:423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chapman SJ, Khor CC, Vannberg FO, et al. IκB genetic polymorphisms and invasive pneumococcal disease. Am J Respir Crit Care Med. 2007;176:181–187. [DOI] [PubMed] [Google Scholar]
- [35].Benekli M, Baer MR, Baumann H, et al. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. [DOI] [PubMed] [Google Scholar]
- [36].Steelman LS, Pohnert SC, Shelton JG, et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. [DOI] [PubMed] [Google Scholar]
- [37].Venton G, Labiad Y, Colle J, et al. Natural killer cells in acute myeloid leukemia patients: from phenotype to transcriptomic analysis. Immunol Res. 2016;64:1225–1236. [DOI] [PubMed] [Google Scholar]
- [38].Jenal M, Elkon R, Loayza-Puch F, et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012;149:538–553. [DOI] [PubMed] [Google Scholar]
- [39].Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2017;18:35–45. [DOI] [PubMed] [Google Scholar]
- [40].Shepard PJ, Choi E-A, Lu J, et al. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. Rna. 2011;17:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Martin G, Gruber AR, Keller W, et al. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012;1:753–763. [DOI] [PubMed] [Google Scholar]
- [42].Wu X, Liu M, Downie B, et al. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci. 2011;108:12533–12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Peschel I, Podmirseg SR, Taschler M, et al. FLT3 and FLT3-ITD phosphorylate and inactivate the cyclin-dependent kinase inhibitor p27 Kip1 in acute myeloid leukemia. Haematologica. 2017;102:1378–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Uras IZ, Bellutti F, Sexl V. p27 in FLT3-driven acute myeloid leukemia: many roads lead to ruin. Haematologica. 2017;102:1299–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hahn CN, Chong C-E, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tamm I, Kornblau SM, Segall H, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–1803. [PubMed] [Google Scholar]
- [47].de Klerk E, Venema A, Anvar SY, et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res. 2012;40:9089–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Danckwardt S, Hentze MW, Kulozik AE. 3′ end mRNA processing: molecular mechanisms and implications for health and disease. Embo J. 2008;27:482–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ji Z, Lee JY, Pan Z, et al. Progressive lengthening of 3ʹ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci. 2009;106:7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ji Z, Tian B. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE. 2009;4:e8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jacobs-Helber SM, Wickrema A, Birrer MJ, et al. AP1 regulation of proliferation and initiation of apoptosis in erythropoietin-dependent erythroid cells. Mol Cell Biol. 1998;18:3699–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jacobs-Helber SM, Abutin RM, Tian C, et al. Role of JunB in erythroid differentiation. J Biol Chem. 2002;277:4859–4866. [DOI] [PubMed] [Google Scholar]
- [53].Ney PA, Sorrentino BP, McDonagh KT, et al. Tandem AP-1-binding sites within the human beta-globin dominant control region function as an inducible enhancer in erythroid cells. Genes Dev. 1990;4:993–1006. [DOI] [PubMed] [Google Scholar]
- [54].Zheng J, Lu Z, Kocabas F, et al. Profilin 1 is essential for retention and metabolism of mouse hematopoietic stem cells in bone marrow. Blood. 2014;123:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.