

ABSTRACT
Trans-activating CRISPR (tracr) RNA is a distinct RNA species that interacts with the CRISPR (cr) RNA to form the dual guide (g) RNA in type II and subtype V-B CRISPR-Cas systems. The tracrRNA-crRNA interaction is essential for pre-crRNA processing as well as target recognition and cleavage. The tracrRNA consists of an antirepeat, which forms an imperfect hybrid with the repeat in the crRNA, and a distal region containing a Rho-independent terminator. Exhaustive comparative analysis of the sequences and predicted structures of the Class 2 CRISPR guide RNAs shows that all these guide RNAs share distinct structural features, in particular, the nexus stem-loop that separates the repeat-antirepeat hybrid from the distal portion of the tracrRNA and the conserved GU pair at that end of the hybrid. These structural constraints might ensure full exposure of the spacer for target recognition. Reconstruction of tracrRNA evolution for 4 tight bacterial groups demonstrates random drift of repeat-antirepeat complementarity within a window of hybrid stability that is, apparently, maintained by selection. An evolutionary scenario is proposed whereby tracrRNAs evolved on multiple occasions, via rearrangement of a CRISPR array to form the antirepeat in different locations with respect to the array. A functional tracrRNA would form if, in the new location, the antirepeat is flanked by sequences that meet the minimal requirements for a promoter and a Rho-independent terminator. Alternatively, or additionally, the antirepeat sequence could be occasionally ‘reset’ by recombination with a repeat, restoring the functionality of tracrRNAs that drift beyond the required minimal hybrid stability.
Introduction
The CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated proteins) system is an adaptive, heritable immune system that is present in nearly all archaea and about one third of bacteria [1–6]. The defense function of the CRISPR-Cas systems involves three stages, namely, (i) adaptation whereby DNA segments (called protospacer) from foreign genetic elements (such as viruses and plasmids) are integrated into a CRISPR array, (ii) CRISPR (cr) RNA maturation whereby a CRISPR array is expressed as a pre-crRNA (long RNA molecule containing multiple spacers inserted between repeats) and cleaved into mature crRNAs that contains the spacer flanked by a portion of an adjacent repeat (cleaved by a complex of Cas proteins and occasionally by an non-Cas RNase), and (iii) interference during which effector Cas nucleases complexed with the crRNA target and cleave cognate DNA molecules. The CRISPR-Cas systems are divided into 2 classes based on the composition of the effector modules: Class 1 systems possess effector complexes that consist of multiple Cas proteins whereas Class 2 effectors consist of a single, multi-domain Cas protein [4,7].
Class 2 systems comprise 3 distinct types with multiple subtypes that are characterized by the domain architectures of their single effector proteins, namely, Cas9 in type II, Cas12 (Cpf1 and C2c1) in type V, and Cas13 (C2c2) in type VI, the only group of CRISPR-Cas systems that exclusively target RNA [4,8]. Thanks to the relative simplicity of the effector module organization, Class 2 CRISPR-Cas systems, initially, of type II and subsequently, of types V and VI as well, have been harnessed for a multitude of genome editing, transcriptome regulation, and other applications [9–11].
In type II, during maturation, the repeat regions of the pre-crRNA interact with a small, non-coding RNA, known as trans-activating CRISPR (tracr) RNA which is encoded in the vicinity of the CRISPR array and cas genes [12–15]. Each of the multiple repeats in a pre-crRNA is bound to a tracrRNA, loaded onto Cas9 protein, and cleaved by a non-Cas RNA endoribonuclease, RNase III (that is, however, dispensable in some variants of subtype II-C [16]), resulting in a repeat-spacer-repeat crRNA that is eventually trimmed into a mature, spacer-repeat crRNA [16–18]. The mature crRNA remains complexed with the tracrRNA, and the two RNAs jointly form the dual guide (g) RNA that, as part of the effector complex, is employed by the type II CRISPR-Cas systems to specifically recognize the cognate target DNA for cleavage by the effector nucleases [19–22]. Additionally, the tracrRNA seems to be involved, along with Cas9, in spacer acquisition [23] although the mechanistic underpinning of this function remains elusive.
The tracrRNA contains an antirepeat, i.e. a region that is (partially) complementary to the repeat, and interacts with the repeat regions of the pre-crRNA to form a hybrid region in the dual-guide RNA (dual-gRNA) that is typically organized into the upper stem, the bulge(s) and the lower stem [15] (Figure 1(A)). In the type II effector complex, the lower stem is buried inside the Cas9 protein molecule and appears to be the functionally most important part of the hybrid region [24]. In engineered single guide (single-g)RNAs, the tracrRNA and crRNA are fused and the hybrid region is abridged to the lower stem, the bulge and a short segment of the upper stem (a large part of the upper stem and additional bulges can be removed) [25]. Nonetheless, the structure of the bulge next to the lower stem appears to be essential for the tracrRNA functionality in S. pyogenes although its specific role remains unknown [14,15]. Additionally, the stability of the hybrid region can affect the function of the effector complex such that, in the gRNA, extension of the upper stem [26] tends to increase cleavage efficiency [27]. Regions of the tracrRNA located downstream of the hybrid area include the nexus, i.e. the first stem-loop of the non-hybrid region, and the Rho-independent terminator which contains a GC-rich hairpin(s) and a 3ʹ poly-U motif [14,15]. Functional dissection of the non-hybrid region of the tracrRNA of Streptococcus pyogenes has shown that the nexus and the hybrid-nexus junction are required for the effector complex interference activity (cleavage of the cognate dsDNA) whereas the terminator hairpin seems to be important for determining orthogonal boundaries between systems. In S. pyogenes, removal of the terminal hairpins only slightly affected the cleavage efficiency [15], whereas in S. aureus, elimination of these hairpins almost completely abolished the cleavage [26]. However, swaps of the terminator regions between tracrRNA from different CRISPR loci (in S. thermophilus) abrogates interference function [15,28,29]. In contrast, swaps of the hybrid regions presenting a similar structure did not impact the effector cleavage function suggesting that nucleotide composition in the hybrid region does not affect the dual-gRNA recognition by Cas9 or interference [15]. Finally, it has been observed that interchanging nexus disrupts effector cleavage function whereas mutations that keep the complementarity of the nexus stem did not affect the effector function [15]. These findings reveal the importance of the nexus structure for the tracrRNA function and the specificity of the nexus with respect to Cas9. In agreement with these results, it has been shown that dual-gRNAs and Cas9 proteins from different species are interchangeable only between closely related organisms which implies tight coevolution between Cas9, repeats and tracrRNAs [29,30].
Figure 1.
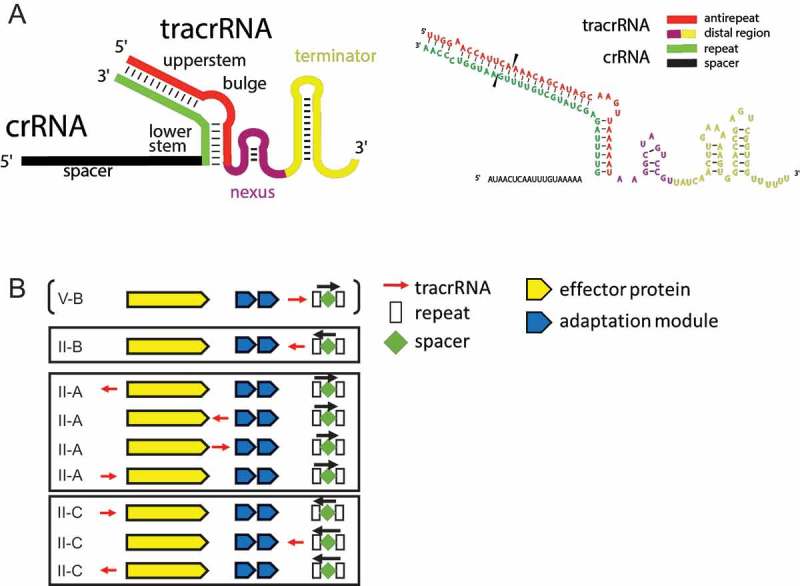
Architectures of the gRNAs and genomic organizations of the tracrRNA-coding CRISPR-cas loci.
(A) Generalized molecular architecture of the gRNA. The antirepeat region of the tracrRNA (red) hybridizes with the repeat in the crRNA (green). The hybrid region contains various bulges and often one main bulge separating the upper stem from the lower stem that is adjacent to the distal part of the tracrRNA. The distal part of the tracrRNA folds into the nexus stem loop (violet) and various stem loops forming the Rho-independent terminator (gold). As an example, the sequence of the gRNA from S. pyogenes annotated using the same colors is shown on the right side. Black triangles indicate the location of the RNase III cleavage site.(B) Location of the tracrRNA genes within the CRISPR-cas loci. The tracrRNAs is denoted by a red arrow indicating the direction of transcription. The CRISPR arrays are denoted by white rectangles (repeats) and green diamonds (spacers). The black arrow indicates the direction of the CRISPR array transcription. The cas genes are shown in yellow (effector module) and in blue (adaptation module).
The discovery of additional Class 2 CRISPR-Cas systems, in particular, type V, in which the effectors share the RuvC-like nuclease domain with Cas9 but otherwise, appear to be unrelated [8,31–33], has shed new light on tracrRNA, demonstrating the plasticity of the CRISPR-Cas functionality. In all type V loci, the pre-crRNA is inverted compared to type II, i.e. is oriented as 5ʹ-repeat-spacer-3ʹ (Figure 1(A) and 2(A) top). Subtype V-A systems lack tracrRNA (Figure 2(A) middle) and instead employ a natural single-gRNA that consists of portions of two repeats and a spacer only [34], whereas subtype V-B systems require tracrRNA (Figure 2(A) bottom) similarly to type II [31]. However, the structures of the guide RNAs in type II and subtype V-B are substantially different in that, in subtype V-B, the hybrid region includes two non-contiguous regions of the tracrRNA, and the nexus seems to be missing [35,36].
Figure 2.
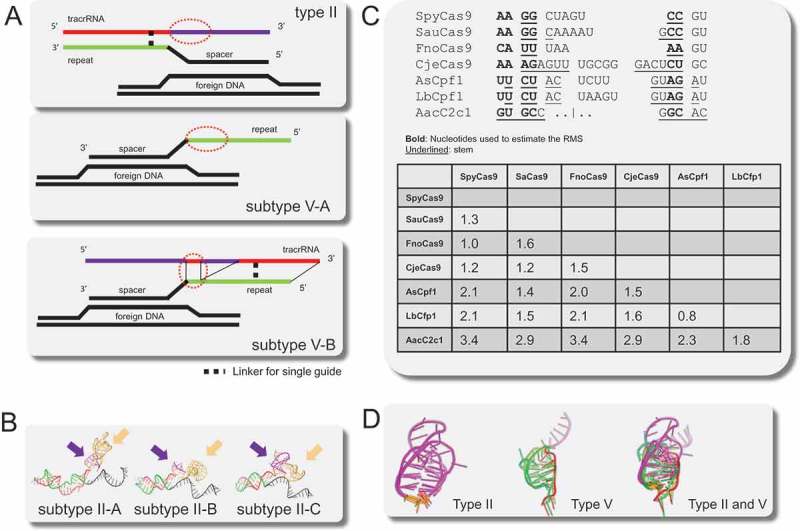
Comparison of the nexus structures in type II and type V gRNAs.
(A) Schematic architectures of the gRNAs of subtype II-A, B, C, subtype V-A, subtype V-B, with the nexus denoted by a red oval. Repeats are shown in green, spacer and DNA target in black, and tracrRNA in red (antirepeat region) and purple (remaining sequence).(B) Comparison of the gRNA orientation in subtypes II-A, II-B and II-C. Violet and gold arrows, respectively, indicate the nexus and the Rho-independent terminator.(C) Nexuses and nexus-like structures extracted from the structure comparison. Paired bases are underlined, bold indicates nucleotides used for estimating the Root Mean Square Deviation (RMSD). The inset table shows RMSD (Angstroms) between the nexuses and nexuses-like structures.(D) Superimpositions of nexuses and nexuses-like structures. The color scheme is the same as in A and B; 5ʹ and 3ʹ flanks of the nexus are colored in orange.
Chylinski and colleagues have undertaken an attempt to decipher the origin and evolution of tracrRNA by identifying and comparing the tracrRNAs from type II loci of diverse bacterial species [13]. They found that the location and orientation of the tracrRNA are not conserved among type II systems, even in closely related species. Furthermore, the sequences of the tracrRNA are short (around 80 nucleotides) and highly divergent which complicates the analysis of tracrRNA evolution.
We took advantage of the recent structural data for guide RNAs (dual and single), the data on tracrRNA expression in various bacteria and the expanding bacterial genomic databases to perform a comprehensive analysis of the evolution of tracrRNA, in search of indications of its ultimate origins. We report evidence of shared structural constraints on the guide RNA architecture among all type II and type V subtypes of CRISPR-Cas systems and of likely multiple origins of the antirepeat regions of tracrRNAs by repeat recruitment and/or recombination.
Results
Structural similarity between guide RNAs
The tracrRNA gene occupies variable positions in the Class 2 CRISPR-cas loci: it can be encoded either upstream of the cas9 gene or between cas9 and the adaptation genes, or between the latter and the CRISPR array, and can be transcribed either codirectionally with the cas genes or in the opposite direction (Figure 1(B)). The type II guide RNA includes, in addition to the spacer, three distinct parts: 1) repeat-antirepeat hybrid that typically contains one or more bulges, with the largest bulge separating the upper and the lower stems, 2) nexus, a small stem-loop structure that separates the proximal portions of the guide RNA containing the hybrid from the distal portion, and 3) the distal region that includes a Rho-independent transcriptional terminator (Figure 1(A)). The subtype V-B guide RNA is organized differently, with the antirepeat comprising the 3ʹ-terminal portion of the tracrRNA and formed by two discontinuous sequences (Figure 2(A)). Subtype V-A lacks tracrRNA so that the single-gRNA is a single molecule consisting of a repeat and a spacer (Figure 2(A)).
We sought to identify structural similarities and differences between the guide RNAs from different types and subtypes of Class 2 CRISPR-Cas systems by comparing the available structures of guide RNAs complexed with Cas proteins. The guide RNA structures were extracted from the crystal structures of interference complexes of type II, subtype A from S. pyogenes (SpyCas9) [37] and S. aureus (SauCas9) [26], subtype B from F. novicida (FnoCas9) [38], and subtype C from C. jejuni (CjeCas9) [39]; and of type V, subtype A from Acidaminococcus sp (AsCpf1) [40] and Lachnospiraceae bacterium (LbCpf1) [41], and subtype B from Alicyclobacillus acidoterrestri (AacC2c1) [35,36]. In the solved structures of type II and subtype V-B, the guide RNA is a single molecule (single-gRNA) in which the tracrRNA is fused to crRNA and the hybrid region is truncated to the lower stem, the bulge and a short part of the upper stem whereas in subtype V-A, it consists of the crRNA alone (Figure 2(A)). Because the single-gRNA guide does not include the complete hybrid region, we focused on comparing the folds and orientations of the single-gRNA regions including the lower stem, the spacer, and the distal region of the tracrRNA, i.e. the nexus and the Rho-independent terminator. First, we extracted type II single-gRNAs and structurally aligned them; examination of the structural alignment shows that the lower stem, the spacer, and the nexus are organized and oriented similarly between the subtypes, whereas the terminal region downstream of the nexus, which contains the terminator, is variable in fold and orientation (Figure 2(B), and Supplementary Figure 1). The lower stem, the spacer and the base of the nexus are embedded within the Cas9 protein structure which constrains their orientations and folding, whereas the nexus loop and the terminator seem to freely fold on the surface of Cas9. Although the nexus regions differ in sequence and the length of the stem (Figure 2(C,D)), the structures could be readily superimposed between subtypes indicating that the junction between the hybrid region and the nexus as well as the base of the nexus stem itself are structurally similar across type II (Root Mean Square Deviation (RMSD: the square root of the mean of the square of the distance between matched atoms) between 1 and 1.6Å; Figure 2(C,D)).
In subtype V-A, the repeat folds into a loop that is located next to the spacer and can be superimposed onto the type II nexus (RMSD between 1.4 and 2.1Å, Figure 2(C,D)). Unlike subtype V-A, subtype V-B loci encode a tracrRNA that hybridizes with the repeat in two discontinuous regions. The hybrid region proximal to the spacer can be superimposed onto both the subtype V-A nexus-like structure (RMSD: 1.8Å) and type II nexus (RMSD between 2.9 and 3.4Å, Figure 2(C,D)). Thus, although the similarity between such small structures should be interpreted cautiously and could emerge by convergence, the fold of the portions of the type V guide RNAs that are located next to the spacer appears to be similar to that in the type II nexus. This nexus-like structure involves the repeat only in subtype V-A, and the first hybrid region between repeat and tracrRNA in subtype V-B (Figure 2(A)).
Comparative anatomy of the guide RNAs
To extend our analysis beyond the structurally characterized guide RNAs, we extracted 71 sequences of experimentally validated tracrRNAs from all subtypes of type II and from subtype V-B that have been described previously [13,31,42]. These sequences were supplemented with newly characterized tracrRNAs, primarily, from Lactobacillus (see Materials) resulting in a set of 215 tracrRNAs (Supplementary Data 1). The sequences of these tracrRNAs were used as queries to search for similar sequences in prokaryotic nucleotide sequence databases. These searches yielded 3353 sequences with significant similarity to tracrRNA (see Methods for details). For 2068 of these sequences, a CRISPR array was identified within 10 kbp upstream or downstream of the predicted tracrRNA gene. Subsequently, the cofold structure between tracrRNA and the corresponding consensus repeat was predicted (see Methods).
The subsequent, detailed exploration of the structures and evolution of gRNAs was restricted to type II because the currently available diversity of subtype V-B loci is limited and insufficient for this type of analysis. To analyze in detail the structures of the tracrRNA:crRNA cofolds in the dual-gRNAs, we partitioned the dual-gRNAs into the hybrid region between tracrRNA and the repeat, and the distal portion of the tracrRNA that does not interact with the repeat (Figure 1(A)). The distal regions of the tracrRNAs were identified as the sequences located between the predicted repeat-antirepeat hybrid and the end of the Rho- independent terminators (determined as the end of the BLAST hit), and were clustered at 70% sequence identity, resulting in 134 groups including 49 singletons. From these, 81 groups and 46 singletons, for which a cas9 gene was identified in the vicinity, were selected for further analysis.
Conservation and variability along the tracrRNA molecules
Examination of the tracrRNA alignment for closely related groups of bacteria shows that the highest sequence conservation concentrates in the lower stem of the antirepeat and around the nexus whereas both the upper stem of the antirepeat and the Rho-independent terminator are more variable in size and structure (Figures 3 and 4 and Supplementary Figure 2). We further examined the variation of the hybrid structure among tracrRNAs with similar distal regions. Most of the tracrRNAs within the same group form non-identical hybrid structures with the repeat but the pairing and the position of at least some of the bulges are similar, especially, within the lower stem of the hybrid region and the beginning of the upper stem. For example, the largest tracrRNA group consists of 586 similar tracrRNAs that can fold into 45 distinct (at least one different base pair or a bulge at a different position) hybrid structures of which 41 shared an identical lower stem (gAAAAAg where g and A denote, respectively, GU and AU base pairs) followed by an identical bulge (Supplementary Table 1). The second largest group that consists of 167 similar tracrRNAs included 93 distinct hybrid structures among which 84 shared a lower stem starting with gAAGAG (upper case G denotes a GC pair). The tracrRNA groups showed only limited correspondence to the taxonomy of the corresponding bacteria, probably, due to combined effects of horizontal gene transfer combination and convergent evolution.
Figure 3.

Structure and sequence conservation in tracrRNA: Streptococci.
(A) The gRNA structure. Green shading shows the repeat, red shading shows the antirepeat in the tracrRNA, and white shading shows the distal region of the tracrRNA.(B) Multiple sequence alignment of the tracrRNAs from the ‘branch1’. The bases are colored using Geneious to indicate deviations from the consensus. A phylogenetic tree of the tracrRNAs is shown to the left of the alignment. The first line of the alignment shows the consensus structure of the tracrRNA (in Vienna format) estimated from the alignment of repeat and tracrRNA sequences. Brackets within the antirepeat region indicate pairing with repeat. Blank within the structure line indicate no consensus.
Figure 4.

Structure and sequence conservation in tracrRNA: Bacteroides.
(A) The gRNA structure. Green shading shows the repeat, red shading shows the antirepeat in the tracrRNA, and white shading shows the distal region of the tracrRNA.(B) Multiple sequence alignment of the tracrRNAs from ‘branch4’. The bases are colored using Geneious to indicate deviations from the consensus. A phylogenetic tree of the tracrRNAs is shown to the left of the alignment. The first line shows the consensus structure of the tracrRNA (in Vienna format) estimated from the alignment of repeat and tracrRNA sequences. Brackets within the antirepeat region indicate pairing with repeat. Blank within the structure line indicate no consensus.
Within each tracrRNA group, when a bulge separates the two stems, the upper stem part adjacent to the lower stem displays notable conservation in terms of nucleotide pairing but includes various bulges that seem to appear at random positions (Figures 3 and 4). Thus, tracrRNAs with similar distal regions also form similar hybrid structures with the corresponding repeats, apart from the secondary bulges within the upper stem. These observations are compatible with coevolution of the hybrid and the distal portion of the tracrRNA.
We further compared the hybrid structures across the entire diversity of the type II CRISPR-Cas systems. The II-B hybrids are significantly less stable than those of II-A and II-C whereas the II-A hybrid structures are most stable (the median ∆G values are −47.6 kcal/mol for II-A, −39.4 kcal/mol for II-C, and −28.9 kcal/mol for II-B; see Supplementary Figure 3). In agreement with the structural analysis discussed above, the last nucleotide of the repeat that is adjacent to the spacer is paired with the tracrRNA in 96% of the dual-gRNA. Moreover, in most of the hybrid structures, the lower stem starts with a G:U wobble base pair, which is consistent with the previous report [15]; only in 9 tracrRNA groups, the lower stems start with a conserved G:C pair. Most of these exceptions were observed in those dual-gRNAs where we could not identify the nexus, most likely, due to inaccurate prediction of the co-fold structure. Conservation of the base-pairing in the lower stem suggests an adaptation ensuring that the spacer is fully exposed for the interaction with the target. Indeed, G:U pairs are widespread in various structural RNAs, are often evolutionarily conserved, and can be associated with various functions including recognition sites for proteins (here, most likely, Cas9), other RNA, and ions [43]. Furthermore, G:U pairs have been observed to stabilize a backbone turn in RNA molecules, allowing a sharp turn when GU is located at the end of a helix [44]. This feature is compatible with the structure of the turn at the junction between the lower stem of the dual-gRNA and the nexus.
For each tracrRNA group, we predicted the secondary structure of the distal part (Supplementary Data 2) and mapped these structures onto the Cas9 tree (Supplementary Figure 4). Examination of this mapping (Supplementary Figure 4) shows that, despite the high variability of the tracrRNA sequences and the potential inaccuracy of some of the structure predictions, there are conserved structural patterns in tracrRNAs shared within Cas9 branches. In the cases when the predicted nexus is conserved in a Cas9 branch, it is located 1 to 3 nucleotides downstream of the hybrid region, in agreement with the structural analysis; the rare cases when the nexus was not conserved most likely involved inaccurate prediction due to nexus variability. The junction between the hybrid region and the nexus stem consists of A or AA in II-C tracrRNAs except for R. solanacearum (C), and in II-A except for A. rectalis (CAA), and in most species within the large Lactobacillus Cas9 branch (C) (Supplementary Figure 4, branch2). The size, shape and sequence of the nexus greatly vary within and between the subtypes. Nevertheless, there are clear trends: the base of the stem frequently starts with a G (often GGC), except in subtype II-B, where the stem is AU-rich, and in most species within branch2. In the latter branch, the nexus is large and contains a bulge that separates an AU-rich stem from a GC-rich stem (Supplementary Figure 4). The rest of the stem and the loop of the nexus are highly diverse, e.g. AU-rich in Streptococcus, Listeria, Staphylococcus (Supplementary Figure 4, branch1) and in all II-B systems, but GC-rich in Campylobacter. In the C. jejuni X-ray structure, this GC-rich loop forms a pseudoknot with the 3ʹ end of the tracrRNA [39].
In F. novicida (subtype II-B), the position of the predicted nexus does not correspond to the nexus observed in the X-ray structure. However, a nexus structure similar (although not identical) to that observed in the X-ray structure was predicted using a local folding of 15 nt (Supplementary Figure 5). These observations suggest that, in this bacterium, the native nexus stem-loop configuration is not the most stable fold and is most likely chaperoned by Cas9. In other II-B systems (Supplementary Figure 4), the proximal part of the tracrRNA lacks the features required to form a stable nexus, and one could not be predicted even by comparison with F. novicida tracrRNA, except in 2 groups including Parasutterella excrementihominis and Burkholderiales bacterium where an unstable nexus was identified adjacent to the hybrid region. Thus, in II-B systems, tracrRNAs and repeats seem to co-fold into a dual-gRNA in which both parts, the hybrid and the distal region, are unstable.
To explore in greater detail the variability of the distal part of the tracrRNAs, we selected 4 groups of tracrRNAs associated with closely related Cas9 proteins which were similar enough to to be confidently aligned but sufficiently variable for informative analysis. We obtained 3 alignments of II-A tracrRNAs that cover the ‘branch1’ (contains some Streptococci, Listeria and Lactobacilli tracrRNAs) (Figure 3), the ‘branch2’ (contains Lactobacilli tracrRNAs folding into a large double nexus) (Supplementary Figure 6), and the ‘branch3’ (contains Streptococcus, Enterococcus, Staphylococcus tracrRNAs) (Supplementary Figure 7), and one alignment of II-C tracrRNAs from multiple Bacteroides species (‘branch4’) (Figure 4). In the branch1 (Figure 3), the tracrRNAs are well conserved within the antirepeat region, with only a few scattered substitutions. Thus, it seems likely that, in these bacteria, the tracrRNAs are exchangeable between CRISPR-Cas systems, even among different species and genera [15]. In the other groups of tracrRNAs, the antirepeat regions are only conserved within species and in some cases, within genera. However, the lower stem as well as the junction and the stem of the nexus comprise the best conserved region in each of the respective alignments (Figures 3 and 4). The conservation of this portion of the tracrRNA is particularly notable in the branch1 alignment where the region of tracrRNA involved in the lower stem GUUAAAU, the junction AA and the adjacent region forming the base of the nexus (GG), are identical in all species. However, the nexus loops are variable and, in several Streptococcus species, contain a large, 18 nt insert. Because this insertion does not seem to disrupt the base of the nexus, it most likely does not affect tracrRNA functions. The large loop of the nexus in C. jejuni has been shown to interact with the distal region of the tracrRNA [39]. The 18 nt insertion in Streptococci might interact with a distinct partner but at present, there are no indications of its identity.
In the other tracrRNA groups, the lower stem, junction and nexus also are the regions of the most pronounced conservation albeit not to the extent they are conserved in the branch1. In addition, in the branch4 alignment, the junction between the nexus and the terminator is highly conserved, with only sparse mutations compared to the terminator region and the antirepeat region (except for the lower stem), whereas in the other groups, no strong conservation was detected of this region. Although, in the available X-ray structures of the guide RNA, this junction does not show specific, tight interactions with Cas9, its conservation in Bacteroides could indicate that such interactions exist in the respective effector complexes. Because the location (downstream of the lower stem, separated by AA) and the stem of the nexus in Bacteroides (stem base: GG) are compatible with the general patterns of the nexus organization, the predicted structure is likely to be correct. In the branch2, the large double nexus stem separated by a conserved AA-AA bulge. (Supplementary Figure 6). Additionally, the bulge seems to delimit the nexus stem (upper stem of the nexus) that is composed of an AA junction (the bulge) and a GG in the beginning of the stem, both observed as a general trend in the nexuses of other tracrRNAs. Thus, the Lactobacillus tracrRNAs could have evolved its nexus from a AA-G nexus stem. Generally, the nexus seems to be a hot spot for insertion resulting in broad variability among different groups of tracrRNAs (Supplementary Figure 2). The conservation of the overall structure combined with the wide diversity of the nexus between tracrRNA groups is best compatible with a structural role, i.e. preventing the distal part of the tracrRNAs from interacting with the spacer.
Finally, we compared the ∆G estimates for the hybrid formation and for folding of the distal part of the tracrRNA. A significant, positive correlation between the ∆G estimates for the two portions of the dual-gRNA was observed for all subtypes of type II although the ranking correlation coefficient was notably lower for subtype II-A than it was for subtypes II-B and II-C (Figure 5). These observations suggest that evolution of the two unrelated parts of tracrRNA occurs under similar constraints that are likely to be determined by the structure of the respective Cas9 proteins.
Figure 5.
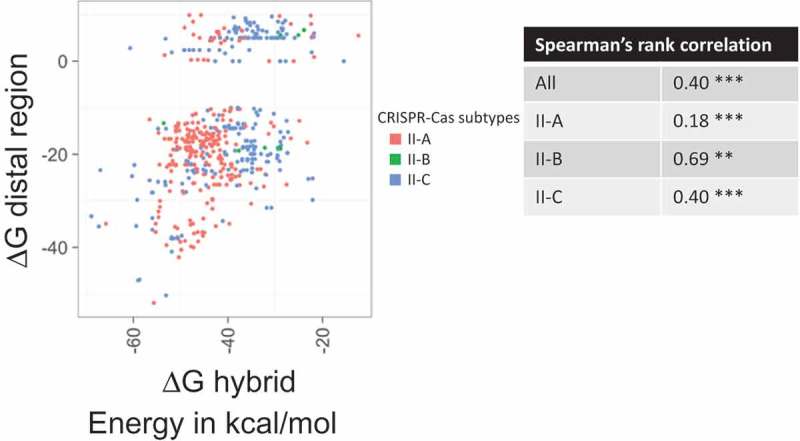
Comparison of the estimated free energies of the repeat-antirepeat hybrid and of the tracrRNA distal region fold. Each point indicates free energies (∆G, kcal/mol) for gRNAs of subtypes II-A (coral), II-B (green), II-C (light blue). The inset table shows Spearman’s rank correlation coefficients. The significance is indicated by *** for p-value< 0.005 and by ** for p-value< 0.05.
Coevolution of tracrRNA and Cas9
Because Cas9 intimately interacts with tracrRNA, it could be expected that the two coevolve. Given that the tracrRNA alignments lack sufficient information to construct reliable phylogenetic trees, we compared the clustering of tracrRNAs by sequence similarity with clusters and trees of Cas9 proteins. Notably, within each group, the tracrRNA genes occupied the same position in the corresponding CRISPR-cas loci and are transcribed in the same direction (Supplementary Figure 4). For 41 tracrRNA groups (Supplementary Table 1), we found that the respective Cas9 proteins belonged to the same Cas9 cluster (30% sequence identify, see Methods) suggesting that similar tracrRNAs interact with similar Cas9 proteins. However, for 26 tracrRNA groups, we found 2 different associated Cas9 clusters, and for 14 tracrRNA groups, there were 3 or more Cas9 clusters (Supplementary Table 1). Furthermore, one large group of tracrRNAs that consists of 167 similar tracrRNAs was found to be associated with 6 Cas9 clusters. A more detailed examination shows that the respective Cas9 proteins indeed belonged to distinct branches of the phylogenetic tree which is compatible with coevolution between tracrRNA and Cas9 (Supplementary Figure 4). Because Cas9 is a large, fast-evolving protein [29,45], it appears likely that, in the cases when multiple Cas9 clusters are associated with the same tracrRNA group, a small subset of tracrRNA-interacting residues is shared by the respective Cas9 proteins whereas the other parts of the proteins vary, resulting in limited overall similarity.
Origin of the tracrRNA antirepeat: recruitment of repeat with subsequent divergence
An obvious evolutionary scenario for the antirepeat portion of the tracrRNA is recruitment of a repeat followed by subsequent limited divergence resulting in the accumulation of the characteristic bulges. Given that the repeat sequences between different subtypes of type II and even within each subtype are dissimilar, this scenario implies that tracrRNA independently evolved in numerous CRISPR-Cas variants. To explore the routes of tracrRNA evolution, we estimated the ∆G values for co-folds of all type II tracrRNAs with the native repeats from the same CRISPR-cas locus and with the repeats from other type II loci. The comparison of the ∆G values shows that, in the great majority of cases, the native hybrid is more stable than any of the heterologous hybrids (Figure 6). Equal stability of native and non-native hybrids was observed only within groups of closely related CRISPR-cas loci that correspond to short branches in the Cas9 phylogenetic tree (Figure 6). The rare cases where non-native hybrids had lower ∆G than native ones (Figure 6) likely reflect horizontal transfer of tracrRNA genes. This pattern is compatible with the evolution of the antirepeat by repeat recruitment, with subsequent divergence. However, it does not appear possible to infer the depth of tracrRNA evolution.
Figure 6.
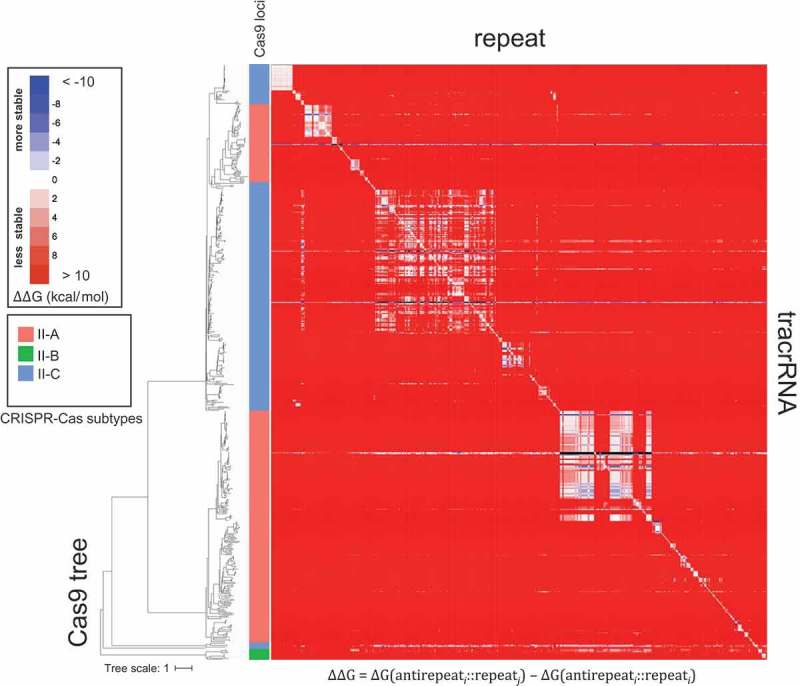
Comparison of the estimated free energies of the repeat-antirepeat hybrids between native and non-native tracrRNA-repeat pairs.
The color code for the free energy differential (∆∆G) is shown in the inset. In the heatmap, the tracrRNAs and repeats are positioned according to the topology of the phylogenetic tree of Cas9 that is shown to the left of the map. Thus, diagonal elements represent native pairs (∆∆G = 0), and the distance from the diagonal reflects the evolutionary distance between the source loci of the tracrRNA and the repeat. Red, blue and white colors, respectively, indicate non-native repeat-antirepeat hybrids with positive (less stable), negative (more stable), and approximately equal similar ∆∆G values compared to the native hybrid. The leaf colors in the tree show the subtypes of the native gRNAs: II-A, coral; II-B, green; II-C, light blue.
The free energy differential between the native and non-native hybrids (∆∆G) is highly significant positive correlation with the evolutionary distance between the respective loci estimated from the Cas9 tree (Figure 7). The correlation disappears in comparisons across large evolutionary distances (between subtypes or branches within the same subtype such as II-A; see middle and upper cloud in Figure 7), conceivably, because at such distances, the tracrRNA have unrelated origins (Supplementary Table 2). These observations are compatible with multiple origins of the antirepeats from the native repeats with subsequent limited divergence. However, the possibility of divergence beyond recognition cannot be ruled out.
Figure 7.
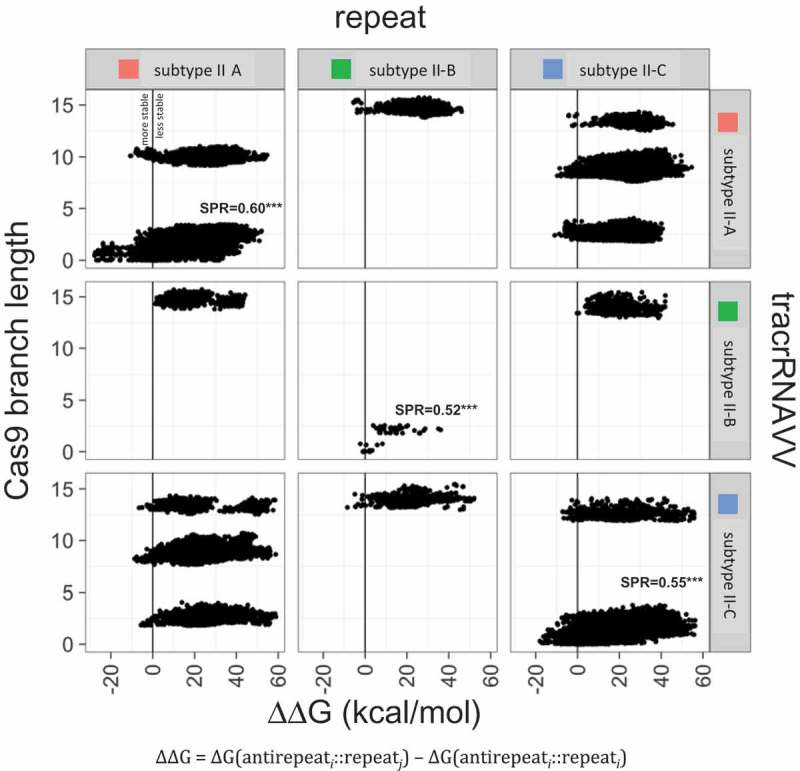
Correlation between the free energy differential between the native and non-native hybrids and the evolutionary distance between CRISPR-cas loci. inferred from the Cas9 phylogenetic tree. The free energy differential (∆∆G, kcal/mol) between native and non-native repeat-antirepeat hybrids is compared with the evolutionary distance between the respective loci estimated from the phylogenetic tree of Cas9. The different point clouds represent different branches of the Cas9 tree. The Spearman’s correlation coefficients for each comparison are given in Supplementary Table 2; *** indicates p-value < 0.005.
We further sought to explore the evolution of the repeat-antirepeat hybrid structure in the branch1, branch2, branch3 and branch4 (see above). For each branch, we reconstructed the ancestral sequences of the repeat and the tracrRNA using the Cas9 subtree and ΔG was estimated for each ancestral dual-gRNA (see Methods). As shown in Figure 8, the ΔG values of ancestral and extant repeat-antirepeat hybrids showed a significant positive correlation with the distance to the root of the Cas9 tree, i.e. an apparent decrease in the hybrid stability, for the branch1. In contrast, significant negative correlation was observed in both the branch2 and the branch3, where the branch4 showed no significant correlation. However, in all 4 groups, the difference in energy between each dual-gRNA and its immediate ancestor (ΔΔG) varied symmetrically between approximately −5 and 5 kcal/mol (Figure 9). This behavior suggests that, within certain energetic constraints, the hybrid structure drifts randomly toward either higher better or lower stability, especially in the upper stem region (Figures 3 and 4).
Figure 8.
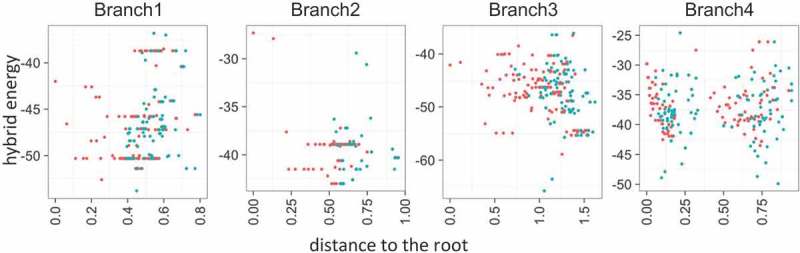
Evolution of the repeat-antirepeat hybrid stability. The free energy of the hybrid energy (∆G, kcal/mol) of ancestral (red) and extant (blue) gRNA is compared to the distance from the root of the corresponding Cas9 subtree.
Figure 9.
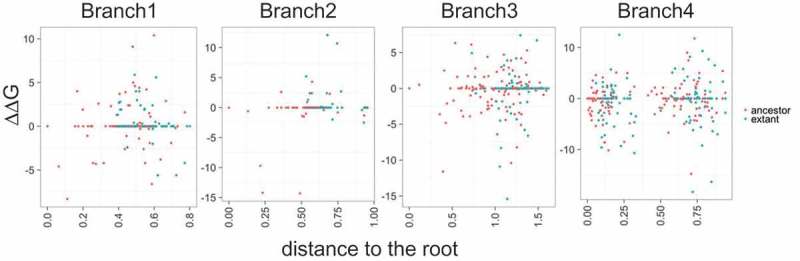
Distribution of the repeat-antirepeat hybrid stability in ancestral and extant gRNAs. Free energy differentials between reconstructed ancestral (red) and extant (blue) gRNAs and their closest ancestors (∆∆G, kcal/mol) are compared to the distance from the root of the corresponding Cas9 subtree.
Discussion
TracrRNA is a small RNA molecule that is required for the maturation of the pre-crRNA and interference in most of the Class 2 CRISPR systems, including all subtypes of type II and subtype V-B but not subtype V-A or type VI. The functions of the guide RNAs and the tracrRNAs in particular have been studied in great detail, especially, in connection with the genome editing applications [14,15,17]. In contrast, the origin and evolution of tracrRNA remain poorly understood. Here, we harnessed the available structures and sequences of guide RNAs to analyze evolutionary conservation and variability of tracrRNA, its coevolution with CRISPR effector proteins and its relationship with the repeats, in an attempt to decipher the origins and the routes of evolution of tracrRNAs.
Structural constraints on the guide RNA appear to ensure spacer specificity
Although the sequences of the tracrRNAs are poorly conserved, the structure of the lower stem with the hybrid region between the tracrRNA and the repeat, and the nexus position and structure are highly similar among type II dual-gRNAs. In the effector complexes, these portions of the tracrRNA are deeply buried inside the Cas9 protein structure [37]. The interactions with Cas9 apparently impose the selective pressure on these regions resulting in structural constraints. Moreover, we identified nexus-like structures also in subtype V-B, where they are formed by one of the two discontinuous hybrid regions between the tracrRNA and the repeat, and subtype V-A where they emerge solely from the crRNA (Figure 2). Thus, the nexus structure, which is positioned next to the spacer, seems to be conserved among all Class 2 systems except for the RNA-targeting type VI in which the structures of the single gRNA and the effector protein are completely different from those in types II and V [46–48]. Because of its location, next to the spacer in both the sequence and the structure of the effector complex, it seems likely that the role of the nexus is to prevent interaction between tracrRNA (in type II), repeat (subtype V-A), or both (in subtype V-B) and the spacer. Additionally, in type II, the 5ʹ-terminal base of the repeat almost invariably forms a G:U pair with the 3ʹ-terminal base of the antirepeat, thus preventing interaction between the repeat and both the spacer and the DNA target, and orienting the distal part of tracrRNA with a sharp turn away from the spacer. Both structural features, the nexus and the base-pairing of 5ʹ-end of the repeat with the tracrRNA, appear to ensure full availability of the spacer for specific and complete interaction with the DNA target. These inferences are clearly amenable to experimental validation.
Evolution of tracrRNA: random walk within the stability constraints and likely multiple origins of the antirepeats from repeats
The structure of the tracrRNA is shared across a wide range of CRISPR-Cas systems but its sequence is not, which creates an obvious challenge for the analysis of tracrRNA evolution. Our reconstruction of the evolution of tracrRNAs in 4 groups of type II systems suggests random drift within a certain window of the repeat-antirepeat hybrid. The implication is that selection only maintains the minimal required level of complementarity between the repeat and the antirepeat, with no strong selection for a perfect hybrid, or a particular, optimal stability. This scenario is compatible with the apparent random location of the secondary bulges in the upper stem (Figures 3 and 4). As discussed above, the structural features of the hybrid are likely to optimize the interaction between the guide RNA and the effector protein and maximize the exposure of the spacer. Furthermore, conservation of the upper stem could be important for the pre-crRNA processing by RNase III. However, this conservation seems to be rather permissive. Indeed, it has been shown [12] that mutations in the complementary regions of tracrRNA and pre-crRNA impeded (but did not abrogate) maturation catalyzed by RNase III whereas mutations preserving pairing did not affect maturation. Subsequently, it has been demonstrated that bacterial RNases III enzymes can be interchangeable such that switching RNase III between various type II systems, even with widely different guide RNAs, did not hinder maturation [29]. Considering the various location and extent of bulges within the upper stem region of type II dual-gRNAs, these findings imply that the hybrid region does not undergo a strong selection pressure for recognition by RNase III. At least in part, this relative promiscuity, most likely, owes to the fact that RNase III is a highly conserved protein [49].
The presence of a minimally stable stem seems to be the basic requirement for a functional guide RNA. Notably, in subtype V-A, this requirement seems to be fulfilled by the palindromic structure of the repeat itself so that a tracrRNA is not needed. Further experiments are required to test these possibilities.
A natural explanation for the origin of the antirepeat in tracrRNA is that it evolved from a repeat.
Given that the sequences of the repeats are apparently unrelated between subtypes and even between distant variants within a subtype, the most likely scenario for the antirepeat evolution appears to be one with multiple cases of repeat recruitment, followed by limited divergence of the antirepeat resulting in random accumulation of bulges and mismatches. Under this scenario, evolution of the tracrRNA would require rearrangement of the CRISPR-array such that a repeat is relocated to one of the positions shown in Figure 1(B); clearly, this difference in the positions of the tracrRNA among CRISPR-cas loci is compatible with multiple origins of the tracrRNAs. A functional tracrRNA would emerge if the new position of the (anti)repeat is flanked by sequences that can meet the minimal requirements for, respectively, a promoter and a Rho-independent terminator. Given the limited sequence constraints on both types of elements, emergence of a tracrRNAs could be a relatively frequent event. Indeed, in S. pyogenes, the tracrRNA gene has 2 promoters from which 2 forms of the tracrRNAs can be expressed, a short and a long ones (89 nt and 171 nt, respectively) [12]. Both these tracrRNA variants function with similar efficiencies, and their processed, mature forms are identical. This case illustrates the evolutionary plasticity of CRISPR-cas loci, and more specifically, the relatively high frequency of sequences capable of acting as promoters in microbial genomes resulting in a relatively high likelihood that a relocated antirepeat lands next to a promoter. Alternatively or additionally, evolution of tracrRNAs could include occasional ‘reset’ of the antirepeat by recombination with the repeat. Such reset would rescue tracrRNAs that drift outside the window of stability that is compatible with functionality. A corollary of this evolutionary scenario is the convergent origin of the shared structural features of the guide RNA, such as the nexus. Given the simplicity of these structures and our observation on the similarity of the nexuses in type II, subtype V-B and subtype V-A (in this case, formed by the crRNA alone), convergent origin of these features indeed appears likely. This conclusion is further corroborated by the lack of sequence similarity among the repeat sequences and the different origins of the Cas1 proteins in type II and the two type V subtypes [31].
We found that the tracrRNAs almost always formed more stable hybrids with the native repeats than with heterologous repeats from other organisms (Figure 6). Furthermore, strong positive correlation was observed between the stability differential of the repeat-antirepeat hybrids in dual-gRNAs and the evolutionary distances (Figure 7) between the corresponding Cas9 proteins within tight branches in the phylogenetic tree but this correlation disappeared at longer evolutionary distances. All these observations are compatible with the multiple origins of tracrRNAs but cannot rule out a single origin followed by divergence beyond recognition. Furthermore, although we clearly observed divergent evolution of the tracrRNA in tight groups of type II systems, we cannot assess the actual depth of such vertical evolution, or put another way, the frequency of tracrRNA emergence or the antirepeat reset. Ultimately, the problem of the tracrRNA origin is tightly linked with the origin of the repeats themselves in different CRISPR-Cas variants. There is currently no conclusive evidence of either divergent or convergent origin of the repeats although the lack of sequence similarity between and sometimes even within subtypes and the recently demonstrated ability of the Cas1-Cas2 protein complex to generate CRISPR arrays de novo [50] appear to favor convergence.
An additional class of substantially less frequent events in the evolution of tracrRNA seems to involve recombination between CRISPR-cas loci within the same or different genomes that are sufficiently closely related to allow cross functionality. Such event would result in the rare observation of higher hybrid stability in heterologous compared to native repeat-antirepeat pairs (Figure 6; Supplementary Figure 8). Further evolutionary reconstructions for multiple groups of bacteria with identified tracrRNAs will provide for more direct testing of this evolutionary scenario, in particular, by observation of newborn tracrRNAs and recombination events.
Materials and methods
Structural analysis of the guide RNA
The structures of RNA guides were extracted from the available PDB structures of interference complexes in subtype II-A: S. pyogenes (SpyCas9, pdb code: 5FW3 [37]), S. aureus (SauCas9, pdb code: 5B2T [26]); subtype II-B: F. novicida (FnoCas9, pdb code: 5B2O [38]); subtype II-C: C. jejuni (CjeCas9, pdb code: 5X2G [39]; subtype V-A: Acidaminococcus sp. (AsCpf1, pdb code: 5B43 [40] and Lachnospiraceae bacterium; LbCpf1, pdb code: 5XUS [41]); and subtype V-B: Alicyclobacillus acidoterrestris (AaC2c1, pdb code: 5U34 [35]). In all structures, the crRNA is linked to the tracrRNA in a single RNA guide. Using PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC), we structurally aligned tracrRNA nexuses, by fitting the backbones of 6 nucleotides, including 2 nucleotides upstream of the nexus and 4 nucleotides that form the base of the stem (2::2). These positions were selected because they are shared among all type II structures. These positions were mapped onto the type V structures after manual examination of potential structural superposition of the type II and type V structures. All RMSD values were estimated from these positions using PyMoL.
Experimental characterization of tracrRNAs in various subtype II-A CRISPR loci
Previous predictions of tracrRNAs in Lactobacilli [15] were validated by RNA sequencing and boundary mapping as described in detail elsewhere [14]. The antirepeat portion of the tracrRNA was identified by BLAST nucleotide alignment [51] between the consensus CRISPR repeat and the putative tracrRNA sequence. The tracrRNA sequence was then extended at the 3ʹ end until a Rho-independent transcription terminator was identified, typically, as a GC-rich hairpin followed by a string of Ts/Us. For the validation of tracrRNA boundaries, short RNA molecules (less than 200 nt) were sequenced by HiSeq Illumina 2500 sequencing with single-end 150 nt read length of TruSeq Small RNA samples. Samples were de-multiplexed, reads trimmed and filtered to remove adapters and low quality bases (Phred 20). Then, sequences shorter than 15 nt were removed, and reads were mapped to the reference genome using Bowtie2 [52].
TracrRNA sequences
We initially used 216 tracrRNAs that have been previously predicted [13,31,42] and cover all types II subtypes. To extend this tracrRNA data set, we searched for similar tracrRNA using BLASTN (parameters – task blastn-short – word_size 8) [51] in an in-house database containing prokaryotic nucleotide sequences encompassing 4,961 complete genomes and 43,599 partial genomes, assembled from NCBI FTP database (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/) in March 2016. The hits with e-values below 0.005 and covering more than 50 percent of the tracrRNA query were selected as significant (Supplementary Data 3). The significant hits were then filtered to retain potential tracrRNAs within a 10 kb distance to a CRISPR array. CRISPR arrays were identified using CRISPRFinder [53]. Sequence and locus alignment were performed within the Geneious framework v.11.0.5 [54].
Prediction of the RNA guide structure
To predict the secondary structure of the dual-gRNA, the tracrRNA sequence was first hybridized with the corresponding repeat sequence (tracrRNA::repeat) using the RNAhybrid software [55]. The non-hybrid portions of the tracrRNAs were clustered (the region downstream of the hybrid) at 70% sequence identity using uclust [56]. The structure of each singleton was predicted using RNAfold [57]. For each cluster, the sequence were aligned using MUSCLE [58], and the structures were predicted using RNAaliFold [57]. Specific cases including repeats that did not engage the 5ʹ base or tracrRNAs that lacked an identifiable nexus structure within the non-hybrid region of tracrRNA were further investigated using local structure prediction (fold of short region) with Mfold [59,60] and by inferring the structure using the experimentally solved guide RNA structures (see above). The phylogenetic trees of tracrRNAs were used to order tracrRNA sequences in the multiple sequence alignments (Figures 3 and 4 and Supplementary Figures 6 and 7). The trees were built using the alignments shown in Figures 3 and 4 and Supplementary Figures 6 and 7, and reconstructed using RAxML with the GTR gamma nucleotide model [61].
Classification and annotation of the CRISPR-Cas systems
The CRISPR-cas loci were identified and annotated essentially as described previously [7,8,62]. Briefly, the sequences of the previously identified effector Cas proteins [7,8] were used as queries to search the in-house prokaryotic database (see above) using PSIBLAST [51] (evalue< 10e-4 and dbsize = 2*10e+ 7). The significant hits and the protein sequences encoded by the adjacent genes (10 kb upstream and downstream) were and annotated using COG [63], pfam [64], CDD [65] databases, and custom Cas protein profiles [62]. All these protein sequences (the original hits and the protein products of adjacent genes) were clustered at 30% sequence identity using uclust [56], and the redundant sequences (more than 90% sequence identity) were discarded. The resulting sets of non-redundant sequences from each cluster were aligned using MAFFT [66], and alignment columns containing gaps in more than 75% of the sequences were removed. To further refine the alignments, a PSSM (Position-Specific Scoring Matrix) was constructed for each cluster. All protein sequences and smalls clusters that failed to show significant similarity to the respective PSSMs using PSI-BLAST [51] were discarded as likely false positives, and the remaining sequences were clustered again. Three iterations of this procedure were performed. The remaining proteins were finally clustered at 50% sequence identify using uclust. Clusters containing Cas9, Cas1, Cas2, Cas4 or Cns2 proteins were extracted and used to identify type II CRISPR-cas system loci and annotate the subtypes. CRISPR arrays located within 10 kb of the clusters of cas genes were identified using CRISPRFinder [53].
Phylogenetic analysis of cas9
To construct the Cas9 tree, the previously described hierarchical approach was employed [67]. Briefly, the Cas9 sequences were clustered by sequence similarity (30% of sequence identity), and for each cluster, a multiple alignment was constructed using MUSCLE [58]. Then, the Cas9 alignments were combined using the HHsearch suite [68] if the resulting score between the two alignments was higher than the default HHalign-Kbest threshold; otherwise, the HHalign-Kbest scores were recorded in a similarity matrix from which a UPGMA dendrogram was produced [69]. For each cluster, the alignment positions with gaps in more than 50% of the sequences and homogeneity values less than 0.1 were discarded [70]. The remaining positions were used to reconstruct a phylogenetic tree using FastTree [71] with the WAG evolutionary model and the discrete gamma model with 20 rate categories. The same program was used to compute SH (Shimodaira-Hasegawa)-like node support values. The Cas9 tree analysis was performed using ete toolkit version 3 [72] and Biopython version 1.70 [73]. The dual-gRNA features and locus configuration were mapped on the Cas9 tree using iTOL [74].
Cross-hybridization of dual-gRNAs
To explore the tracrRNA-repeat interchangeability, we used only the antirepeat region of the tracrRNA and selected unique repeat-antirepeat pairs. The final data set included 535 unique repeat-antirepeat pairs. In this dataset, the free energy (∆G) of the hybrid structure was calculated for all 286,225 repeat-antirepeat combinations. The ∆G values for all these hybrids were used to construct a heatmap using iTOL [74]; the heatmap was ordered using the phylogenetic tree of Cas9.
CRISPR array orientation
To determine the orientation of CRISPR arrays, it was assumed that the orientation of the tracrRNA, namely, 5ʹ anti-repeat and 3ʹ non-hybrid region is conserved in type II. Using RNAhybrid [55], we hybridized the tracrRNA sequence (correctly oriented) to both possible repeat orientations (plus and minus strand), estimated the free energy for both hybrid structures and selected the repeat orientation that yielded the the more stable hybrid.
Reconstruction of ancestral dual-gRNA sequences
Ancestral dual-gRNA sequences were reconstructed for 4 distinct groups of tracrRNAs (branch1, branch2, branch3 and branch4). The 4 groups of tracrRNAs were selected such that the sequences were sufficiently conserved to be confidently aligned but presented enough variability for informative analysis. For each group, an alignment of the repeat sequences was generated and a rooted Cas9 subtree was extracted from the complete Cas9 phylogeny. Each group was analyzed independently. Using the repeat alignment and the Cas9 subtree, the ancestral repeat sequences were reconstructed for each tree node using FastML (parameters: no tree reconstruction, no branch optimization, GTR nucleotide model) [75]. The same procedure was followed to reconstruct the ancestral tracrRNA sequence at each node. Then, for each node of the tree, the ancestral hybrid structure was inferred and the ∆G value was calculated. The hybrid energy of both extant and ancestral dual-gRNAs was compared to the distance to the tree root, and additionally, ΔΔG was calculated as the difference between the ∆G values of a given hybrid structure (extant or ancestral) and is immediate ancestor.
Funding Statement
This work was supported by the U.S. Department of Health and Human Services [Intramural funds, to EVK], and the NC Ag Foundation.
Acknowledgments
We thank Koonin group members for useful discussions. GF, SS, KSM, YIW and EVK are supported by intramural funds of the US Department of Health and Human services (to the National Library of Medicine). ABC and RB are supported by the NC Ag Foundation.
Competing interests
The authors declare that they have no competing interests.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.van der Oost J, Westra ER, Jackson RN, et al. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol. 2014;12:479–492. nrmicro3279 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61. nature15386 [pii]. [DOI] [PubMed] [Google Scholar]
- 3.Barrangou R, Horvath P. A decade of discovery: CRISPR functions and applications. Nat Microbiol. 2017;2:17092 nmicrobiol201792 [pii]. [DOI] [PubMed] [Google Scholar]
- 4.Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78. S1369-5274(17)30023-1 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohanraju P, Makarova KS, Zetsche B, et al. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147 aad5147 [pii]; 353/6299/aad5147 [pii]. [DOI] [PubMed] [Google Scholar]
- 6.Hille F, Richter H, Wong SP, et al. The biology of CRISPR-Cas: backward and forward. Cell. 2018;172:1239–1259. S0092-8674(17)31383-1 [pii]. [DOI] [PubMed] [Google Scholar]
- 7.Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. nrmicro3569 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017. nrmicro.2016.184 [pii] DOI: 10.1038/nrmicro.2016.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096 1258096 [pii] 346/6213/1258096 [pii]. [DOI] [PubMed] [Google Scholar]
- 10.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. S0092-8674(14)00604-7 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;169:559 S0092-8674(17)30417-8 [pii]. [DOI] [PubMed] [Google Scholar]
- 12.Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. nature09886 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–737. 24321 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briner AE, Barrangou R. Guide RNAs: a glimpse at the sequences that drive CRISPR-Cas systems. Cold Spring Harb Protoc. 2016;2016:pdb top090902 2016/7/pdb.top090902 [pii]. [DOI] [PubMed] [Google Scholar]
- 15.Briner AE, Donohoue PD, Gomaa AA, et al. Guide RNA functional modules direct Cas9 activity and orthogonality. Mol Cell. 2014;56:333–339. S1097-2765(14)00751-5 [pii]. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Heidrich N, Ampattu BJ, et al. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell. 2013;50:488–503. S1097-2765(13)00364-X [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charpentier E, Richter H, van der Oost J, et al. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev. 2015;39:428–441. fuv023 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hille F, Charpentier E. CRISPR-Cas: biology, mechanisms and relevance. Philos Trans R Soc Lond B Biol Sci. 2016;371 20150496 [pii] rstb.2015.0496 [pii] DOI: 10.1098/rstb.2015.0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. science.1225829 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrangou R. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip Rev RNA. 2013;4:267–278. [DOI] [PubMed] [Google Scholar]
- 21.Karvelis T, Gasiunas G, Miksys A, et al. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013;10:841–851. 24203 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plagens A, Richter H, Charpentier E, et al. DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol Rev. 2015;39:442–463. fuv019 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heler R, Samai P, Modell JW, et al. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015;519:199–202. nature14245 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang F, Doudna JA. CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46:505–529. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. 2016;85:227–264. [DOI] [PubMed] [Google Scholar]
- 26.Nishimasu H, Cong L, Yan WX, et al. Crystal structure of staphylococcus aureus Cas9. Cell. 2015;162:1113–1126. S0092-8674(15)01020-X [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, Lian W, Jia Y, et al. Optimized guide RNA structure for genome editing via Cas9. Oncotarget. 2017;8:94166–94171. 21607 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esvelt KM, Mali P, Braff JL, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. nmeth.2681 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fonfara I, Le Rhun A, Chylinski K, et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:2577–2590. gkt1074 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chylinski K, Makarova KS, Charpentier E, et al. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:6091–6105. gku241 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and functional characterization of diverse Class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–397. S1097-2765(15)00775-3 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stella S, Alcon P, Montoya G. Class 2 CRISPR-Cas RNA-guided endonucleases: Swiss Army knives of genome editing. Nat Struct Mol Biol. 2017;24:882–892. nsmb.3486 [pii]. [DOI] [PubMed] [Google Scholar]
- 33.Murugan K, Babu K, Sundaresan R, et al. The revolution continues: newly discovered systems expand the CRISPR-Cas toolkit. Mol Cell. 2017;68:15–25. S1097-2765(17)30655-X [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell. 2015;163:759–771. S0092-8674(15)01200-3 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Gao P, Rajashankar KR, et al. PAM-dependent target DNA recognition and cleavage by C2c1 CRISPR-Cas endonuclease. Cell. 2016;167:1814–1828 e1812. S0092-8674(16)31665-8 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Chen P, Wang M, et al. C2c1-sgRNA complex structure reveals RNA-guided DNA cleavage mechanism. Mol Cell. 2017;65:310–322. S1097-2765(16)30813-9 [pii]. [DOI] [PubMed] [Google Scholar]
- 37.Nishimasu H, Ran FA, Hsu PD, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. S0092-8674(14)00156-1 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirano H, Gootenberg JS, Horii T, et al. Structure and Engineering of Francisella novicida Cas9. Cell. 2016;164:950–961. S0092-8674(16)30053-8 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamada M, Watanabe Y, Gootenberg JS, et al. Crystal structure of the minimal Cas9 from campylobacter jejuni reveals the molecular diversity in the CRISPR-Cas9 systems. Mol Cell. 2017;65:1109–1121 e1103. S1097-2765(17)30121-1 [pii]. [DOI] [PubMed] [Google Scholar]
- 40.Yamano T, Nishimasu H, Zetsche B, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–962. S0092-8674(16)30394-4 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao P, Yang H, Rajashankar KR, et al. Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res. 2016;26:901–913. cr201688 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burstein D, Harrington LB, Strutt SC, et al. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–241. nature21059 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varani G, McClain WH. The G x U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000;1:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark BFC, Klug A. Structure and function of tRNA with special reference to the three dimensional structure of yeast phenylalanine tRNA. Proc Tenth FEBS Meet. 1975;39:183–206. [Google Scholar]
- 45.Takeuchi N, Wolf YI, Makarova KS, et al. Nature and intensity of selection pressure on CRISPR-associated genes. J Bacteriol. 2012;194:1216–1225. JB.06521-11 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abudayyeh OO, Gootenberg JS, Konermann S, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573 aaf5573 [pii] science.aaf5573 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.East-Seletsky A, O’Connell MR, Burstein D, et al. RNA targeting by functionally orthogonal Type VI-A CRISPR-Cas enzymes. Mol Cell. 2017;66:373–383 e373. S1097-2765(17)30238-1 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.East-Seletsky A, O’Connell MR, Knight SC, et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016. nature19802 [pii] DOI: 10.1038/nature19802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aguado LC, tenOever BR. RNase III nucleases and the evolution of antiviral systems. Bioessays. 2018;40 DOI: 10.1002/bies.201700173 [DOI] [PubMed] [Google Scholar]
- 50.Nivala J, Shipman SL, Church GM. Spontaneous CRISPR loci generation in vivo by non-canonical spacer integration. Nat Microbiol. 2018;3:310–318. 10.1038/s41564-017-0097-z [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. nmeth.1923 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–57. gkm360 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kearse M, Moir R, Wilson A, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. bts199 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–454. 34/suppl_2/W451 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. btq461 [pii]. [DOI] [PubMed] [Google Scholar]
- 57.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26 1748-7188-6-26 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol. 2008;453:3–31. [DOI] [PubMed] [Google Scholar]
- 61.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. bti191 [pii]. [DOI] [PubMed] [Google Scholar]
- 62.Makarova KS, Koonin EV. Annotation and classification of CRISPR-Cas systems. Methods Mol Biol. 2015;1311:47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galperin MY, Makarova KS, Wolf YI, et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015;43:D261–269. gku1223 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Punta M, Coggill PC, Eberhardt RY, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–301. gkr1065 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marchler-Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–226. gku1221 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. mst010 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peters RS, Niehuis O, Gunkel S, et al. Transcriptome sequence-based phylogeny of chalcidoid wasps (Hymenoptera: Chalcidoidea) reveals a history of rapid radiations, convergence, and evolutionary success. Mol Phylogenet Evol. 2017;120:286–296. S1055-7903(17)30336-6 [pii]. [DOI] [PubMed] [Google Scholar]
- 68.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. bti125 [pii]. [DOI] [PubMed] [Google Scholar]
- 69.Sokal RR, Michener CD. A statistical method for evaluating systematic relationships. Univ Kans Sci Bull. 1958;38:1409–1438. [Google Scholar]
- 70.Yutin N, Makarova KS, Mekhedov SL, et al. The deep archaeal roots of eukaryotes. Mol Biol Evol. 2008;25:1619–1630. msn108 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol. 2016;33:1635–1638. msw046 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Talevich E, Invergo BM, Cock PJ, et al. Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinf. 2012;13:209 1471-2105-13-209 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–245. gkw290 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ashkenazy H, Penn O, Doron-Faigenboim A, et al. FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res. 2012;40:W580–584. gks498 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.