

ABSTRACT
Leishmaniasis is a worldwide public health problem caused by protozoan parasites of the genus Leishmania. Leishmania braziliensis is the most important species responsible for tegumentary leishmaniases in Brazil. An understanding of the molecular mechanisms underlying the success of this parasite is urgently needed. An in-depth study on the modulation of gene expression across the life cycle stages of L. braziliensis covering coding and noncoding RNAs (ncRNAs) was missing and is presented herein. Analyses of differentially expressed (DE) genes revealed that most prominent differences were observed between the transcriptomes of insect and mammalian proliferative forms (6,576 genes). Gene ontology (GO) analysis indicated stage-specific enriched biological processes. A computational pipeline and 5 ncRNA predictors allowed the identification of 11,372 putative ncRNAs. Most of the DE ncRNAs were found between the transcriptomes of insect and mammalian proliferative stages (38%). Of the DE ncRNAs, 295 were DE in all three stages and displayed a wide range of lengths, chromosomal distributions and locations; many of them had a distinct expression profile compared to that of their protein-coding neighbors. Thirty-five putative ncRNAs were submitted to northern blotting analysis, and one or more hybridization-positive signals were observed in 22 of these ncRNAs. This work presents an overview of the L. braziliensis transcriptome and its adjustments throughout development. In addition to determining the general features of the transcriptome at each life stage and the profile of protein-coding transcripts, we identified and characterized a variety of noncoding transcripts. The novel putative ncRNAs uncovered in L. braziliensis might be regulatory elements to be further investigated.
KEYWORDS: Noncoding RNAs, Leishmania braziliensis, gene expression, comparative transcriptomics, differential gene expression
Introduction
The protozoan parasites Leishmania spp. are trypanosomatids with a dimorphic life cycle completed between the sandfly digestive tract and mammalian hosts [1]. These parasites are the etiological agents of leishmaniases, a group of diseases with highly diverse clinical presentations. These presentations include a tegumentary form that ranges from cutaneous localized (LCL) or self-healing to a disfiguring and morbid disease affecting the nasopharyngeal mucosae (MCL) and a visceral form (VL) in which hematopoietic organs are affected and that is fatal if not treated. The disease is a serious global public health problem in more than 90 countries [1,2], and the outcome of infection depends mainly on the parasite species and host genetics and immune response [1]. These parasites are classified into two subgenera, and species of the subgenus Viannia are associated with tegumentary leishmaniasis in Central and South America [3,4]. Leishmania (Viannia) braziliensis is a predominant species in Brazil. It is broadly distributed in endemic areas across the country and is the main causative agent of MCL, a highly morbid clinical form affecting 5 to 10% of infected individuals. In MCL, a strong cellular immune response corroborates tissue destruction in the metastatic and pauciparasitary affected loci [5–7].
Leishmania parasites are ingested by the female sandfly vector during blood feeding. Amastigote (AMA) forms, which are mammalian intracellular proliferative forms, are released from ingested macrophages into the digestive tract of the insect and differentiate into the replicative form, procyclic (PRO) promastigotes. After a few days, these promastigotes undergo metacyclogenesis, a process of differentiation that leads to the infective metacyclic (META) promastigote stage. They are then released from the peritrophic membrane and migrate to the stomodeal valve, which is situated between the midgut and the esophagus of the insect [6–8]. Once inside the mammalian host, the META are internalized by phagocytic cells present in the dermis and will proliferate within the phagolysosomes as AMA. The successful adaptation of the parasite to different hostile environments involves important and rapid modifications in its gene expression profile [9–12].
Given the genetic organization of Leishmania, its gene expression is regulated mainly at the posttranscriptional level; RNA polymerase II-transcribed genes lack canonical promoters and are organized as large polycistronic transcription units (PTUs) that may comprise more than a hundred genes with no functional relationships [13,14]. It has been demonstrated that posttranscriptional regulation of gene expression occurs at several levels, from the control of RNA abundance to the translation rate and posttranslational modification or degradation regulation [15–17]. The last two levels may involve the control of the translation process (initiation, elongation and termination), posttranslational modifications and directed protein degradation routes [17–19]. Control of the abundance and stability of mRNAs may involve factors and pathways including cis- and trans-elements, such as elements of the 3ʹ untranslated regions (3ʹUTR), which were suggested in a previous study to function in the posttranscriptional regulation of differentially expressed (DE) genes in L. major and L. infantum [20]. We previously identified a large group of conserved elements present outside the coding sequences (CICS – conserved intercoding sequences) of three different Leishmania species and demonstrated that these elements may act as regulatory elements [21,22]. In addition to cis-elements acting as binding sites for proteins, previous studies on Trypanosoma brucei [23,24] and other eukaryotes [25] and our own study on Leishmania major and Leishmania donovani identified putative noncoding RNAs (ncRNAs) arising from mRNA UTRs; these ncRNAs may act as regulatory elements [22,26].
Genome and transcriptome data from various Leishmania species and isolates suggest complex adaptive traits in these parasites. Despite differences in clinical output, the genomes of different species of Leishmania are syntenic, and protein-coding genes are highly conserved [27]. However, comparative analyses of genomes and transcriptomes of different Leishmania species and isolates indicate that gene dosage variation is an important adaptive trait conferring phenotypic plasticity to Leishmania populations. Gene dosage variation may be a result of individual copy number variation or chromosomal somy changes [28]. The correlation of gene copy dosage with corresponding transcript levels may explain the similarities and differences between isolates and species [29–31]. Furthermore, the regulation of gene expression at the posttranscriptional level and the corresponding regulatory elements and pathways may partially explain species differences. In this scenario, the whole transcriptome of Leishmania species is important for the initial approximation of genetic activity and gene structural features. Comparative transcriptomic analysis identifies the modulation of gene expression differences among species or among developmental stages within species [32,33].
In light of literature findings indicating that ncRNAs act as gene expression regulatory elements in other eukaryotes and the observation that ncRNAs are found in trypanosomatids, we used RNA-seq and in silico analysis to investigate the whole transcriptome and modulation of gene expression profile throughout development in this parasite and explored mRNA content and putative ncRNA transcripts. This study used L. braziliensis parasites in axenic culture to investigate the three main developmental stages. We present incremental data on the genome structure of L. braziliensis, the main characteristics of gene structure and the DE protein-coding genes throughout development. Moreover, we investigated the presence, distribution and characteristics of putative regulatory ncRNAs using an ad hoc computational pipeline. Comparative transcriptome analysis at the three stages revealed the presence of a variety of DE ncRNAs and candidates for functional and regulatory roles in distinct biological processes.
Results and discussion
RNA-seq of L. braziliensis procyclic and metacyclic promastigotes and axenic amastigotes
Biological material
To compare gene expression patterns throughout the life cycle of a Leishmania (Viannia) subgenus, we conducted axenic culturing of Leishmania (Viannia) braziliensis (MHOM/BR/75/M2903, now named L. braziliensis 2903), which allows the in vitro reproduction of different life cycle stages: procyclic promastigotes (PRO), metacyclic promastigotes (META) and amastigotes (AMA) [34,35].
The PRO forms were obtained from the logarithmic phase (day 2), and culture-derived META forms were rescued from a mixed population of promastigotes in the stationary phase (day 5) of culture. The META were extracted based on density differences using the Ficoll enrichment method [36] (Fig. 1A). Promastigotes in the stationary phase (day 5) were incubated in fetal bovine serum (FBS, 100%) at 33°C for 3 days for differentiation into culture-derived AMA [34]. After 10 passages in culture, the AMA were obtained on day 3 (Fig. 1A). Subsequently, the typical PRO, META and AMA morphologies were verified by scanning electron microscopy (Fig. 1B). In addition, we analyzed the mRNA levels of two δ-amastins previously described as markers of the L. braziliensis AMA stage [37]. The δ-amastin transcripts were upregulated at the AMA stage by ~5-fold and ~14-fold relative to those at the META and PRO stages, respectively (Additional file 1: Fig. S1 and Additional file 3: Table S1).
Figure 1.
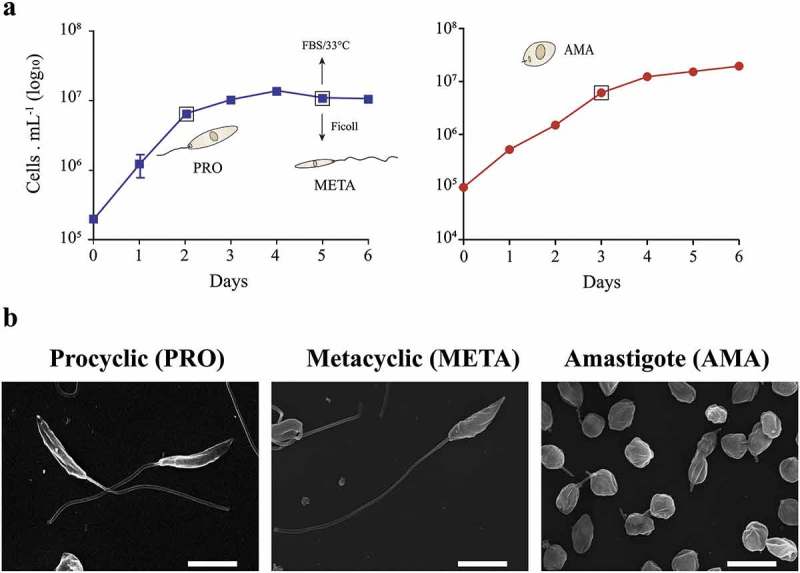
Time course for selection of life cycle stages and morphology of cells used for RNA extraction. (a) Growth curve illustrative of the study design and time points. Growth curves of promastigote forms of L. braziliensis. Culture-derived procyclic forms (PRO) were obtained on day 2, metacyclic forms (META) were purified from a stationary-phase culture (day 5), and amastigotes (AMA) were obtained from differentiated cultures maintained in fetal bovine serum (FBS) at 33°C in a 1% CO2 atmosphere on day 3. Data are shown as the means ± SD, n = 3. (b) Morphology of the different L. braziliensis developmental stages, as indicated above each panel, under scanning electron microscopy. White bars: 5 µm.
Experimental design and correlation between samples
Transcriptome profiling of L. braziliensis throughout its life cycle was performed using RNA-seq technology. The total RNA of three biological replicates from the three main life stages of L. braziliensis was sequenced, resulting in nine cDNA libraries. These libraries were constructed and sequenced using Illumina technology, and ~677 million paired-end reads were generated (Additional file 1: Fig. S2A, Table 1).
Table 1.
RNA-seq results for each Leishmania developmental stage.
| Bowtie2 – LbrM2903 |
|||||
|---|---|---|---|---|---|
| Library | Replicate | Total paired-end reads | % of reads per library* | % of mapped reads | % of reads on chromosome 6a |
| Procyclic | 1 | 69,458,528 | 10.25 | 98.5 | 28.46 |
| Procyclic | 2 | 84,539,426 | 12.47 | 98.54 | 30.48 |
| Procyclic | 3 | 90,215,594 | 13.31 | 98.66 | 25.41 |
| Metacyclic | 1 | 72,520,332 | 10.70 | 98.83 | 22.81 |
| Metacyclic | 2 | 84,021,672 | 12.40 | 98.84 | 23.01 |
| Metacyclic | 3 | 66,830,070 | 9.86 | 99.01 | 23.62 |
| Amastigote | 1 | 54,228,698 | 8.00 | 98.95 | 20.40 |
| Amastigote | 2 | 81,757,314 | 12.06 | 98.76 | 22.21 |
| Amastigote | 3 | 74,281,704 | 10.96 | 99.18 | 32.29 |
a The ribosomal locus (18S and 28S) of L. braziliensis is annotated on chromosome 6. Reads mapped on chromosome 6 were referenced to estimate the representation of the rRNA in the samples.
* total reads (9 libraries): ~677 million reads
Analysis of sequence quality metrics revealed that most of the reads obtained base calling accuracy as determined by a Phred quality score (Q score) above 30 (91% of reads 1 and 83% of reads 2). In addition, more than 98% of the reads were mapped to the reference genome, L. braziliensis 2903-TriTrypDB version 30 [38] (Table 1).
Ribosomal RNAs (rRNAs) were estimated based on the number of reads mapped on chromosome 6 of L. braziliensis 2903 because the largest locus of rRNA is annotated on this chromosome as a result of a misassembly. The representation of this chromosome varied between 20% and 32% per library (Table 1).
To evaluate the consistency of reads distribution per replicate at each stage and compare the homogeneity of reads distribution across stages, we conducted a count distribution of raw reads in annotated CDSs (protein-coding sequences) per library (Additional file 1: Fig. S2B). Furthermore, to analyze the pattern of replicate grouping and the distance between the compared samples (PRO, META, AMA), we used the biological coefficient variation (BCV) distance in a multidimensional scaling (MDS) chart. Analysis revealed the proximity of replicates at each developmental stage and a consistent distance between life cycle stages (Additional file 1: Fig. S2C). This result indicates that the quality of generated data allows differential expression analysis.
Transcriptome profiling
L. braziliensis transcript element structure
Because gene expression regulation in trypanosomatids occurs mainly at the posttranscriptional level [39], RNA stability, storage, degradation and translation rates are important points of control [40,41]. Determining the UTR boundaries is a useful tool for future analysis of possible cis- and trans-acting elements involved in the regulation of gene expression [42,43]. Transcript boundaries have been previously estimated for some Leishmania species [11,23] but not for L. braziliensis.
To estimate the boundaries of annotated L. braziliensis transcripts, we used the genome of L. braziliensis 2903 (version 30 – TriTrypDB) associated with SL-based transcriptome data (unpublished data). PolyA sites were predicted as previously described by Dillon and cols [11], and it allowed us to estimate the terminus of the 3ʹUTR for 38% (3,568) of the CDSs and that of the 5ʹUTR for 81% (7,494) of the annotated CDSs (Additional file 2). These analyses indicated an average length of 1,621 nucleotides (nt) for the L. braziliensis annotated CDSs, ranging from 49 to 19,893 nt (Fig. 2A); the estimated 5ʹUTR mean length was 582 nt (Fig. 2B), and that of the 3ʹUTR was 1,254 nt (Fig. 2C). The average lengths of CDSs and UTRs found for L. braziliensis are comparable to those previously predicted for L. major [11].
Figure 2.
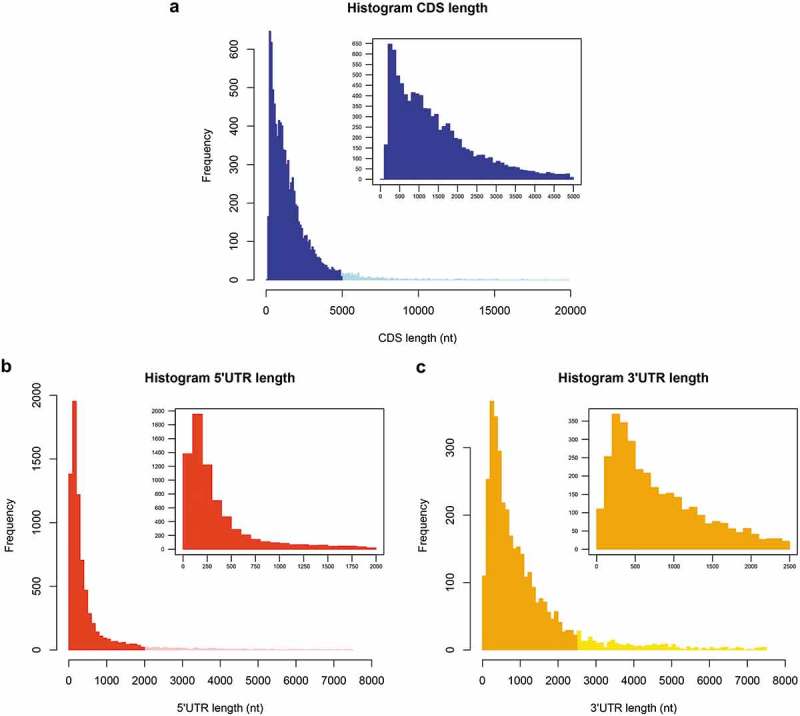
Length distribution of gene elements in Leishmania braziliensis. (a) CDSs (Leishmania braziliensis 2903-TriTrypDB version 30). (b) 5ʹUTRs. (c) 3ʹUTR. For the methodology used to define the main SL acceptor and polyA sites, refer to Methods. The size of each bin is 100 nt.
Highly abundant transcripts
The most abundant transcripts were identified by calculating the FPKM (fragments per kilobases of transcript per million mapped reads) for each annotated CDS, and the first FPKM percentile was examined (see Methods). The Venn diagram shows the number of highly expressed genes distributed between the PRO, META and AMA stages (Additional file 1: Fig. S3). Fifty percent (32/63 genes) of the genes common to all stages code for ribosomal proteins (Additional file 3: Table S2); similar results have been obtained for other Leishmania species [44]. In the population of highly abundant genes common to PRO and META (11 genes), 9 are ribosomal protein transcripts. META and AMA shared 6 highly abundant genes; among them was one amastin (LBRM2903_080005000.1) that was previously reported to express in META as well as AMA [45]. AMA and PRO shared only a ribosomal protein gene (LBRM2903_300045100.1) as a highly abundant transcript. Among the 10 most abundant transcripts detected only in PRO, 6 were ribosomal protein-coding genes, and among 5 transcripts exclusively abundant in META, 2 were ribosomal protein-coding genes. Interestingly, for 9 of the ribosomal protein transcripts that were either highly abundant in both META and PRO or exclusively highly abundant in META, an extraribosomal function had been previously described in homologues in other organisms [46–51]. Relevant extraribosomal regulatory roles for some of these proteins classes have been thoroughly investigated and are dependent on posttranslational modifications [46,52]. In AMA, 5 of the 11 highly abundant transcripts were amastin-coding genes.
Differentially expressed (DE) protein-coding genes throughout development
It is important to keep in mind that analysis of DE genes is based on relative abundance of transcripts and that transcript levels do not necessarily have strong correlations with the corresponding protein levels [53]. In addition, this study used axenic conditions; it may have overestimated or underestimated transcript abundance and may have failed to detect differences that only occur in the parasite’s natural niches [54]. Another important consideration is that small statistically significant differences between transcript abundance in different conditions are not necessarily less important or have less impact than larger differences.
In the deposited genome of L. braziliensis 2903 (TryTripDB – version 30), there are 9,269 predicted and annotated protein-coding genes. Previous comparative analysis of transcript levels in Leishmania indicated that fold change (FC) differences are frequently modest [11]. Therefore, we first analyzed the DE genes (Additional file 4) considering only the statistical significance (adjusted p value ≤ 0.05); under this condition, 5,689 transcripts (61%) were DE between PRO and META promastigotes (PROvsMETA). When applying a FC cutoff of 1.5, 1,229 (13%) genes were DE, and with a FC cutoff of 2,256 genes (2.8%) were DE. In the comparative analysis of PROvsMETA, the FCs in the population of downregulated genes reached 3-fold, and those in the population of upregulated genes reached 5.2-fold. Between META and AMA (METAvsAMA) using the same parameters, we detected 4,856 genes (52%), 1,084 (12%) and 264 (2.8%) DE genes, respectively. The FC differences in the group of META-downregulated genes reached 15.1-fold, and those in the group of META-upregulated genes reached 5.7-fold. The comparison between AMA and PRO (AMAvsPRO) using the same cutoff values revealed 6,576 (71%), 1,309 (14%) and 813 (8.8%) DE genes, respectively. In the comparative analysis of AMAvsPRO, the FCs in the population of AMA-downregulated genes reached 3.9-fold, and those in the population of AMA-upregulated genes reached 43.7-fold.
Up- and downregulated genes were visualized using MA plots comparing the developmental stages of the parasite (Additional file 1: Fig. S4 – red dots). Subtle differences (low FCs) were found in PROvsMETA (Additional file 1: Fig. S4A), and large differences (high FCs) were found in AMAvsPRO (Additional file 1: Fig. S4C). Closer proximity between PRO and META than between either and AMA was also observed in the hierarchical clustering (Additional file 1: Fig. S4D) and principal component analyses (Additional file 1: Fig. S4E).
Functional enrichment analysis of DE coding genes
Gene ontology (GO) enrichment analyses of DE protein-coding genes across the development of L. braziliensis were performed. These analyses identified significantly enriched processes based on the up- and downregulated genes at each pairwise developmental stage comparison (Additional file 3: Tables S3-S8). The upregulated biological processes in the PROvsMETA, METAvsAMA and AMAvsPRO comparisons were selected and are presented graphically (Fig. 3).
GO terms related to cellular proliferation (GO:0006412, GO:0006457, GO:0006414 and GO:0008284) were enriched in PRO forms (PROvsMETA – Fig. 3, Additional file 3: Tables S3 and S4), consistent with a proliferative life stage [9,11,55]. GO terms related to cellular motility (GO:0007018 and GO:0007017) were also enriched in PRO when compared to AMA (Additional file 3: Table S4); similar results were found in L. donovani and L. major [9,12]. Protein metabolism- and energy-related GO terms (GO:0015986, GO:0006183 and GO:0006228) were enriched in PRO (PROvsMETA – Fig. 3 and Additional file 3: Table S3) as seen before [11]. The category ATP synthesis (GO:0015986) was enriched in the PRO form in both comparisons (PROvsMETA – Fig. 3 and AMAvsPRO – Additional file 3: Table S4), suggesting increased mitochondrial activity in the PRO. GO terms related to signalization (GO:0044081 and GO:0075130) were enriched in META when compared to PRO (Additional file 3: Table S5) and enriched in AMA when compared to META (Additional file 3: Table S6) and PRO (AMAvsPRO – Fig. 3 and Additional file 3: Table S7), suggesting the relevance of signalization during the differentiation of promastigote to AMA forms, as already described in L. major [10,11]. Phosphorylation-related GO terms (GO:0006468 and GO:0008160) were enriched at infective and mammalian stages (META and AMA) when compared to PRO (Fig. 3 – AMAvsPRO and Additional file 3: Tables S5 and S7). Consistently, protein phosphorylation is a process correlated with differentiation and virulence [9,10,56]. GO terms related to transport (GO:0008160) were enriched in META (Additional file 3: Table S5) and AMA (GO:0008160, GO:0006817 and GO:0005315) in both comparisons (Fig. 3 – AMAvsPRO and Additional file 3: Table S6 and S7). Amastins act as membrane transporters [37] and are AMA-specific or AMA-preferential proteins (as some isoforms are also found in META). The enrichment of transport-related genes in META and AMA might be associated with the upregulation of genes coding for surface proteins involved in signaling and transport or due to the number and abundance of amastins [57]. A similar profile of GO enrichment in META was reported in L. major [9].
Figure 3.
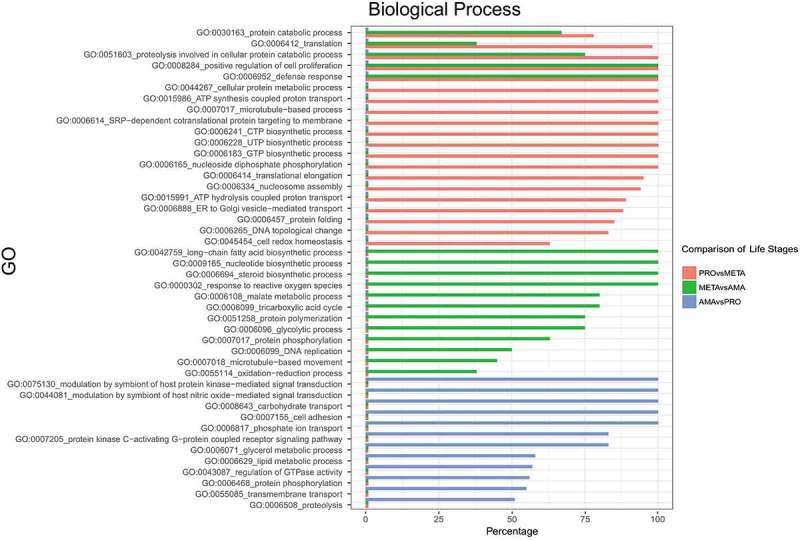
Gene ontology enrichment analysis. Biological processes enriched in genes upregulated in the life cycle stages of L. braziliensis (adjusted p value ≤ 0.05) are shown. Red bars: biological processes upregulated in PRO compared to META. Green bars: biological processes upregulated in META compared to AMA, and blue bars: biological processes upregulated in AMA compared to PRO. The x-axis percentage represents the fraction (%) of genes upregulated within the set of all genes from Leishmania assigned to the corresponding biological process.
In general, GO terms related to proteolysis activities (GO:0051603, GO:0030163, GO:0005839, and GO:0019773), although present at all stages, were enriched in promastigotes (Fig. 3 – PROvsMETA and METAvsAMA and Additional file 3: Tables S3, S4 and S8). Protein catabolism (GO:0030163 and GO:0051603) was enriched in promastigotes in both comparisons (Fig. 3 – METAvsAMA and AMAvsPRO and Additional file 3: Tables S4 and S8), while proteolysis activities related to protein processing (GO:0006508 and GO:0006470) were enriched in the infective and mammalian forms in both comparisons (Fig. 3 – AMAvsPRO and Additional file 3: Tables S5 and S7). Proteolysis activities play key roles in cellular remodeling that occurs through autophagy, a process important to parasite differentiation, which is well described during META to AMA differentiation [19,58,59].
The amastin transcripts are among those with marked FC differences in both comparisons METAvsAMA (Additional file 1: Fig. S4B and Additional file 3: Table S9 – METAvsAMA – downregulated) and AMAvsPRO (Additional file 1: Fig. S4C and Additional file 3: Table S9 – AMAvsPRO – upregulated), reaching a 39.2-fold increase in AMA. These small surface proteins, unique to kinetoplastids, are composed of approximately 45 members [45,60] and are considered essential virulence factors for AMA multiplication into mammalian host cells [37,61]. Although a clear function for amastins remains unknown, it is believed that these proteins interact with the host cell molecules or act as membrane transporters [37]. Some authors have suggested that amastins may be involved in ion or proton traffic through the membrane to adjust the cytoplasmic pH of the parasite [45,60]. Although upregulation of amastins in AMA has been previously reported in other species [12,61], the FCs did not reach the levels of differential expression reported here for L. braziliensis.
We must bear in mind that the high number of genes coding for hypothetical proteins represents an extra obstacle for conducting computational function analysis in these parasites. A large number of hypothetical protein-coding genes (3,466 genes) were observed among DE genes (Additional file 4). Similar results were previously observed in Leishmania and other trypanosomatids [9,11]. We also observed that three of these genes were among those with the highest levels of FC, being upregulated ~44-fold in AMA (Additional file 3: Table S9 – AMAvsPRO – upregulated).
Profiles of DE protein-coding genes
To investigate the most common modulation of gene expression profiles throughout parasite development, DE genes that were either up- or downregulated by a FC ≥ 1.5 (FC cutoff used for reliability) and were DE in all three comparisons (PROvsMETA, METAvsAMA and AMAvsPRO) were selected; 216 such genes were found (Additional file 5). To evaluate the modulation profile throughout development, we used the raw counts to calculate CPM (counts per million); this strategy led to the classification of DE genes into 6 different groups based on their expression profile (Fig. 4). Interestingly, most DE transcripts were present at lower levels in promastigotes, with incremental levels in META and AMA; 176 such genes were detected (Fig. 4 – Group 1). The second largest group (21 genes) included those transcripts with the opposite expression pattern, with the highest levels in promastigotes and the lowest levels in AMA (Fig. 4 – Group 4). The third group contained 17 DE genes with the highest levels in META (Fig. 4 – Group 2). Three other groups with different modulation profiles encompassed either one or no DE genes (Fig. 4 – Groups 3, 5 and 6). Among the 176 genes in Group 1, 26% (46) were amastin or amastin-like transcripts and ~40% (70) coded for hypothetical proteins. The remaining genes (13.8%) were heterogeneously distributed into different classes. Curiously, among the 17 genes in Group 2 (with higher levels in META), 6 were classified as hypothetical protein-coding transcripts and 6 (35%) as RNA or nucleic acid/nucleotide binding proteins. In Group 4, among 21 genes, 6 were hypothetical, and the remaining genes were distributed heterogeneously into several classes.
Figure 4.
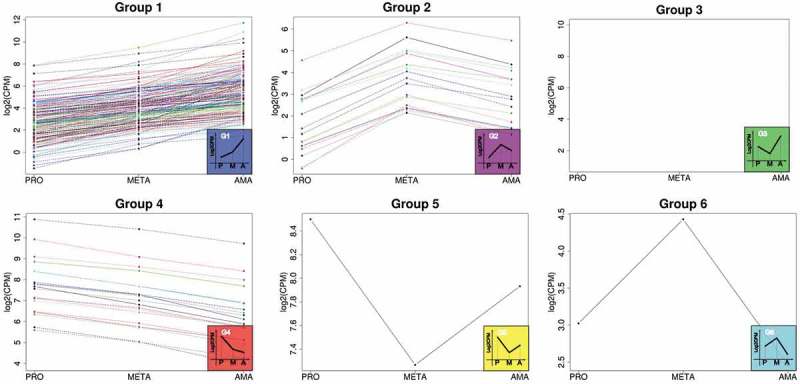
Profiles of differentially expressed protein-coding genes during life cycle progression. The set of genes encompasses only those DE in all three comparisons analyzed (PROvsMETA, METAvsAMA and AMAvsPRO). Total number of DE protein-coding genes = 216, distributed into 6 groups. Group 1: 176 genes, group 2: 17 genes, group 3: no representative, group 4: 21 genes, group 5: 1 gene, group 6: 1 gene. Reads per counts per million (CPM), values in log2.
We also analyzed the group of genes that could be classified as stage-preferential genes. Genes presenting a FC ≥ 2 were compiled, and those presenting a FC ≥ 3 were considered possible stage-specific markers in L. braziliensis (Additional file 3: Table S10). We consistently confirmed the expression profile of some genes that were previously identified as PRO, META or AMA markers in other species. A chaperonin HSP60 (LBRM2903_350028900) was identified as a possible marker for PRO (FC ~3.0, Additional file 3: Table S10) [62,63]. One of the stage-preferential genes detected in META that might be a stage marker was a putative autophagy protein, ATG8 ubiquitin like (LBRM2903_190018000) (FC ~3.6, Additional file 3: Table S10) [19,64]. Similar results have been reported in other Leishmania species, in which ATG8 was upregulated in the motile forms derived from sand fly, termed nectomonads [9]. As mentioned above, several amastin and amastin-like genes were highly prominent transcripts in AMA forms.
Comparison of Viannia and Leishmania subgenera: species-specific genes and genes upregulated in different life stages
In 2007, Smith and colleagues comparatively analyzed the L. major, L. infantum and L. braziliensis genomes. A total of 49 L. braziliensis-specific genes were found (39 annotated as hypothetical and 10 with predicted function) [27]. We searched for these genes in more recent versions of the L. braziliensis genome and identified 16 genes with predicted function and 33 classified as hypothetical protein-coding genes (Additional file 3: Table S11). Thirty-nine of them were found to be DE between developmental stages, of which 25 were classified as coding for hypothetical proteins and 14 with a predicted function (Table 2). Some of the genes that were over- or underrepresented in META or AMA may be important tools for understanding the peculiar differentiation and infection properties of L. braziliensis compared to other Leishmania species and should be further investigated.
Table 2.
L. braziliensis-specific genes.
| CDS ID | Description | PRO vs META UP | META vs AMA UP | AMA vs PRO UP |
PRO vs META DOWN | META vs AMA DOWN | AMA vs PRO DOWN | Trypanosomatids Orthologous genes with a predicted function |
|---|---|---|---|---|---|---|---|---|
| LBRM2903_110008400 | argonaute-like protein (pseudogene) | 1.2 | 1.23 | - | - | - | 1.48 | L. braziliensis MHOM/BR/75/M2904 – argonaute-like protein (LbrM.11.0360)/L. major – argonaute-like protein (pseudogene) (LmjF.11.0570)* |
| LBRM2903_220013600 | arrestin (or S-antigen), N-terminal domain containing protein, putative | - | 1.38 | - | - | - | 1.3 | - |
| LBRM2903_280015300 | EF hand, putative | - | 1.18 | 1.22 | 1.43 | - | - | - |
| LBRM2903_260012900 | glutathione peroxidase, putative | - | - | 2.99 | 1.85 | 1.58 | - | - |
| LBRM2903_260006200 | phosphoribosyl transferase domain-containing protein, putative | - | - | - | 1.24 | - | - | - |
| LBRM2903_240021600 | protein of unknown function (DUF563), putative | - | - | 1.24 | - | 1.17 | - | - |
| LBRM2903_160010900 | reverse transcriptase (RNA-dependent DNA polymerase), putative | - | - | 3.36 | 1.46 | 2.12 | - | - |
| LBRM2903_110019600 | hypothetical protein | - | 1.18 | - | - | - | 1.28 | - |
| LBRM2903_200006800 | hypothetical protein | - | 1.23 | - | - | - | - | - |
| LBRM2903_230011600 | hypothetical protein | - | 1.22 | - | - | - | 1.23 | - |
| LBRM2903_250014200 | hypothetical protein | 1.32 | - | - | - | - | 1.33 | Crithidia fasciculata strain Cf-Cl – alpha/beta hydrolase family, putative (CFAC1_220014800) |
| LBRM2903_250019400 | hypothetical protein | - | - | 1.59 | 1.33 | 1.18 | - | L. braziliensis MHOM/BR/75/M2904 – ribonuclease 3, putative (LbrM.25.1020) |
| LBRM2903_260005000 | hypothetical protein | - | 1.26 | - | - | - | 1.23 | - |
| LBRM2903_280005300 | hypothetical protein | - | - | 1.37 | 1.2 | 1.14 | - | L. braziliensis MHOM/BR/75/M2904 – TATE DNA transposons (LbrM.11.1160) |
| LBRM2903_280018400 | hypothetical protein | - | - | 2.73 | 2.19 | 1.23 | - | - |
| LBRM2903_290005300 | hypothetical protein | - | - | 1.63 | 1.23 | 1.31 | - | L. braziliensis MHOM/BR/75/M2904 – TATE DNA transposons (LbrM.11.1160) |
| LBRM2903_300032300 | hypothetical protein | - | - | 1.27 | 1.21 | - | - | - |
| LBRM2903_310033200 | hypothetical protein | - | - | 1.78 | 1.91 | - | - | - |
| LBRM2903_320047000 | hypothetical protein | - | - | 1.93 | - | - | - | L. panamensis MHOM/COL/81/L13 – zinc finger-containing protein, putative (LPAL13_320043800) |
| LBRM2903_320047100 | hypothetical protein | - | - | 3.17 | 1.85 | - | - | L. panamensis MHOM/COL/81/L13 – zinc finger-containing protein, putative (LPAL13_320043800) |
| LBRM2903_330006800 | hypothetical protein | 1.34 | - | - | - | - | 1.39 | L. braziliensis MHOM/BR/75/M2904 – RNA Interference Factor 5, putative (LbrM.33.0190) |
| LBRM2903_330043600 | hypothetical protein | - | 1.73 | - | 1.15 | - | 1.5 | T. congolense IL3000 – reverse transcriptase (RNA-dependent DNA polymerase), putative (TcIL3000_0_23600) |
| LBRM2903_340027000 | hypothetical protein | 1.46 | - | - | - | - | 1.32 | - |
| LBRM2903_340045900 | hypothetical protein | - | - | 1.3 | - | - | - | L. braziliensis MHOM/BR/75/M2904 – galactokinase (LbrM.34.3650) |
| LBRM2903_160011000 | hypothetical protein | - | - | 3.24 | - | 2.63 | - | LBRM2903_160010900 (reverse transcriptase) |
| LBRM2903_060019200 | hypothetical protein | - | 1.24 | - | 1.3 | - | - | - |
| LBRM2903_130021900 | hypothetical protein | - | - | 1.14 | 1.14 | - | - | - |
Ontology information from TriTrypDB. The genes were considered specific to the Viannia subgenus. *Although an ortholog was found in L. major (subgenus Leishmania), it has a comment from the annotator clarifying that it is a pseudogene.
In addition to evaluating GO enrichment profiles and species-specific genes, we investigated similarities and differences in transcriptomes between species of the different subgenera by comparing upregulated, DE protein-coding genes. We compared these genes in PRO, META and AMA between L. braziliensis and both L. major and L. mexicana according to available data using orthology as the parameter. We first compared our results from L. braziliensis (PROvsMETA, METAvsAMA, AMAvsPRO) with those of procyclics, metacyclics and lesion-derived amastigotes from an L. major transcriptomic analysis of sand-fly [9]. In L. braziliensis, 3,948 DE genes were upregulated in PRO, 4,465 were upregulated in META, and 3,843 were upregulated in AMA (adjusted p value ≤ 0.05). The L. major orthologous genes were identified using TriTrypDB tools, and the search resulted in 3,913, 4,223 and 3,475 genes in the three stages. These L. major orthologs were then submitted to the list of DE genes identified by Inbar et al [9]. Interestingly, the percentages of orthologous genes that were similarly upregulated in L. major were low: 572 (14.6%) genes in PRO, 513 (12.1%) in META and 437 (12.6%) in AMA (Additional file 4: sheet LbrM–LmjF). Using the same approach, we compared L. braziliensis PRO and META upregulated genes with previously reported upregulated genes of axenic PRO and META of L. major [11]. Our L. braziliensis PROvsMETA comparative analysis revealed 2,888 genes upregulated in PRO and 2,800 in META (adjusted p value ≤ 0.05), and, according to TriTrypDB, the L. major orthologous genes were 2,929 in PRO and 2,575 in META. The number of orthologous genes in the comparison species that were found to be upregulated was 894 (30.5%) in PRO and 900 (34.9%) in META (Additional file 4: sheet Axenic_LbrM–LmjF). We then compared our L. braziliensis axenic forms (PROvsAMA) with available data from L. mexicana axenic forms (PROvsAMA) [44]. In L. braziliensis, there were 3,299 genes upregulated in PRO and 3,276 genes upregulated in AMA (adjusted p value ≤ 0.05), and, according to TriTrypDB, the L. mexicana orthologous genes were 3,230 in PRO and 2,827 in AMA. The orthologous genes found to be upregulated in both species were 741 (23%) in PRO and 422 (15%) in AMA (Additional file 4: sheet Axenic_LbrM–LmxM). These comparative analyses suggest that despite synteny and sequence conservation of protein-coding genes among species, the upregulated-DE genes content differences may contribute to diversity between species and subgenera and may be biologically relevant.
Leishmania braziliensis putative ncRNAs
In general, for most living organisms, there are many gray areas to be investigated concerning the mechanisms and factors involved in the control of gene expression. One route of investigation relies on the discovery and functional characterization of ncRNAs as part of regulatory machineries involved in a diverse number of biological processes in several organisms [65–67]. The plethora of discovered ncRNAs and the several characterized ncRNAs in different organisms contrasts the lack of information on the ncRNA arsenal in Leishmania species; only a few reports mentioning en passant the detection of ncRNAs in different Leishmania species are available [26,41,68–71]. Therefore, we decided to conduct a systematic in-depth transcriptome-wide analysis for the identification of ncRNAs in L. braziliensis, focusing on DE putative ncRNAs.
Because the source of data of the L. braziliensis transcriptomes of PRO, META and AMA was total RNA from each stage, the discovery of ncRNAs presented here, due to the large number of abundant coding RNAs, may be an underestimation. It must be noted that the less abundant transcripts buried in the PTUs may have been missed due to the employed strategy. Complementary approaches that exclude most of the polysomal RNA fraction should be used for rescuing such transcripts [24,72]. It was a challenge to identify individual transcripts (peak of reads), distinguish between a ncRNA candidate and background noise, and exclude those most likely to be non-annotated CDSs in the genome. A read coverage cutoff and a database of known CDSs were used to minimize these difficulties; therefore, the identification of putative ncRNAs began with the definition of transcript strand specificity based on the genome coverage by nucleotide. Two main types of ncRNAs were considered: short (≤ 200 nt) and long (> 200 nt) transcripts. We excluded from the analysis those transcripts found within CDSs when transcribed from the same strand. For the annotation and analysis, a consensus of the putative ncRNAs was generated for all three stages (Additional file 1: Fig. S5).
On the computational pipeline, after the generation of ncRNA consensus sequences, 12,050 ncRNAs were identified (Additional file 1: Fig. S5). From this population of transcripts, those with significant similarity to protein domains (Pfam dataset) were removed from the analysis (659 transcripts). The 11,391 remaining putative ncRNAs were submitted to 5 ncRNA predictors: PORTRAIT, RNAcon, ptRNApred, snoscan and tRNAscan-SE. PORTRAIT predicted 7,085 (62%) ncRNA candidates, RNAcon predicted 7,883 (69%) candidates, and ptRNApred predicted 10,212 (90%) candidates. snoscan identified 242 candidates as possible snoRNAs, and tRNAscan-SE predicted 27 transcripts as tRNAs. From the overall population (12,050), 11,372 transcripts were identified as putative ncRNAs by at least one of the abovementioned predictors (Additional file 6). These putative ncRNAs presented a median size of 281 nt (Additional file 1: Fig. S6). Among the 11,372 putative ncRNAs identified in L. braziliensis, 4,021 were classified as short ncRNAs, and 7,351 were classified as long ncRNAs (lncRNAs; Table 3, Additional File 6).
Table 3.
Genome distribution and general features of putative ncRNAs.
| Chromosome |
Short ncRNAs |
Long ncRNAs |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inter CDS* |
UTR |
Intra CDS |
Inter CDS* |
UTR |
Intra CDS |
|||||||||
| ID | Length | Sense | Antisense | SSR | Sense | Antisense | Antisense | Sense | Antisense | SSR | Sense | Antisense | Antisense | Total |
| LbrM2903_01 | 286,661 | 23 | 8 | 1 | 9 | 1 | 1 | 40 | 2 | 0 | 24 | 3 | 1 | 113 |
| LbrM2903_02 | 352,826 | 21 | 0 | 3 | 13 | 0 | 1 | 45 | 3 | 14 | 27 | 4 | 3 | 134 |
| LbrM2903_03 | 408,717 | 27 | 0 | 0 | 7 | 0 | 7 | 39 | 6 | 1 | 30 | 4 | 1 | 122 |
| LbrM2903_04 | 502,955 | 43 | 4 | 0 | 21 | 2 | 1 | 71 | 2 | 3 | 40 | 1 | 1 | 189 |
| LbrM2903_05 | 505,114 | 29 | 1 | 5 | 19 | 0 | 0 | 70 | 3 | 10 | 35 | 1 | 5 | 178 |
| LbrM2903_06 | 590,413 | 43 | 10 | 2 | 17 | 5 | 3 | 69 | 16 | 6 | 38 | 2 | 1 | 212 |
| LbrM2903_07 | 639,434 | 22 | 1 | 7 | 27 | 3 | 0 | 67 | 4 | 10 | 41 | 3 | 6 | 191 |
| LbrM2903_08 | 537,514 | 37 | 8 | 2 | 18 | 1 | 0 | 70 | 9 | 11 | 40 | 3 | 1 | 200 |
| LbrM2903_09 | 620,965 | 50 | 5 | 1 | 24 | 2 | 1 | 52 | 9 | 2 | 33 | 0 | 0 | 179 |
| LbrM2903_10 | 695,527 | 33 | 6 | 11 | 29 | 5 | 3 | 61 | 4 | 31 | 38 | 10 | 5 | 236 |
| LbrM2903_11 | 603,537 | 28 | 7 | 3 | 19 | 4 | 2 | 46 | 7 | 22 | 49 | 3 | 10 | 200 |
| LbrM2903_12 | 408,254 | 20 | 2 | 1 | 13 | 1 | 0 | 35 | 9 | 7 | 33 | 0 | 0 | 121 |
| LbrM2903_13 | 658,307 | 36 | 0 | 3 | 19 | 1 | 3 | 67 | 9 | 4 | 44 | 1 | 7 | 194 |
| LbrM2903_14 | 650,483 | 59 | 1 | 3 | 34 | 2 | 1 | 61 | 2 | 13 | 63 | 1 | 1 | 241 |
| LbrM2903_15 | 654,844 | 43 | 1 | 3 | 23 | 3 | 1 | 53 | 3 | 3 | 48 | 5 | 4 | 190 |
| LbrM2903_16 | 771,689 | 42 | 2 | 7 | 23 | 3 | 0 | 70 | 5 | 18 | 43 | 5 | 6 | 224 |
| LbrM2903_17 | 740,364 | 28 | 4 | 6 | 30 | 0 | 0 | 65 | 14 | 11 | 52 | 0 | 2 | 212 |
| LbrM2903_18 | 822,729 | 46 | 12 | 1 | 22 | 4 | 1 | 119 | 9 | 8 | 58 | 1 | 4 | 285 |
| LbrM2903_19 | 810,851 | 57 | 9 | 7 | 23 | 3 | 2 | 80 | 5 | 17 | 53 | 5 | 1 | 262 |
| LbrM2903_20.1 | 1,943,446 | 188 | 11 | 14 | 0 | 0 | 2 | 345 | 15 | 32 | 0 | 0 | 5 | 612 |
| LbrM2903_20.2 | 917,802 | 87 | 5 | 6 | 0 | 0 | 1 | 145 | 15 | 10 | 0 | 0 | 3 | 272 |
| LbrM2903_21 | 783,199 | 49 | 1 | 3 | 34 | 2 | 1 | 80 | 7 | 3 | 44 | 2 | 1 | 227 |
| LbrM2903_22 | 728,622 | 40 | 1 | 1 | 19 | 2 | 1 | 88 | 4 | 1 | 59 | 2 | 5 | 223 |
| LbrM2903_23 | 913,624 | 50 | 6 | 6 | 44 | 1 | 0 | 88 | 9 | 11 | 65 | 3 | 5 | 288 |
| LbrM2903_24 | 933,121 | 65 | 3 | 1 | 36 | 2 | 2 | 75 | 5 | 7 | 56 | 7 | 2 | 261 |
| LbrM2903_25 | 968,025 | 56 | 3 | 3 | 39 | 4 | 0 | 91 | 12 | 2 | 76 | 6 | 0 | 292 |
| LbrM2903_26 | 1,030,512 | 66 | 0 | 2 | 48 | 0 | 0 | 100 | 1 | 3 | 75 | 2 | 1 | 298 |
| LbrM2903_27 | 1,281,939 | 68 | 15 | 10 | 55 | 4 | 2 | 103 | 21 | 21 | 101 | 4 | 3 | 407 |
| LbrM2903_28 | 1,239,708 | 66 | 2 | 6 | 47 | 2 | 4 | 147 | 12 | 11 | 83 | 3 | 5 | 388 |
| LbrM2903_29 | 1,257,134 | 70 | 7 | 4 | 51 | 3 | 1 | 115 | 7 | 10 | 104 | 7 | 8 | 387 |
| LbrM2903_30 | 1,460,206 | 119 | 7 | 14 | 52 | 2 | 1 | 149 | 14 | 34 | 108 | 9 | 10 | 519 |
| LbrM2903_31 | 1,738,660 | 103 | 10 | 2 | 55 | 5 | 1 | 248 | 14 | 3 | 134 | 8 | 4 | 587 |
| LbrM2903_32 | 1,666,992 | 126 | 3 | 6 | 55 | 4 | 3 | 121 | 9 | 18 | 122 | 1 | 10 | 478 |
| LbrM2903_33 | 1,581,728 | 99 | 7 | 5 | 68 | 6 | 4 | 125 | 21 | 19 | 135 | 19 | 13 | 521 |
| LbrM2903_34 | 2,192,442 | 122 | 17 | 9 | 95 | 5 | 6 | 207 | 25 | 16 | 152 | 20 | 19 | 693 |
| LbrM2903_35 | 2,890,053 | 156 | 7 | 9 | 106 | 13 | 7 | 221 | 18 | 11 | 230 | 17 | 13 | 808 |
| Scaffolds | - | 4 | 0 | 93 | 0 | 0 | 2 | 8 | 1 | 313 | 1 | 0 | 6 | 428 |
| Total | 2221 | 186 | 260 | 1194 | 95 | 65 | 3636 | 331 | 716 | 2334 | 162 | 172 | ||
| 4021 | 7351 | 11,372 | ||||||||||||
*Inter CDS: term used when the 3ʹUTR border was not determined
In the genome of L. braziliensis, 9,269 protein-coding genes were annotated (TriTrypDB, version 30 [29]). The ratio of protein-coding to noncoding genes in L. braziliensis is supported by similar analyses conducted in different organisms. In Homo sapiens, which has 20,376 protein-coding genes, 22,305 ncRNAs were reported (GRCh38- http://www.ensembl.org/Homo_sapiens/Info/Annotation; accessed September 2018). In Caenorhabditis elegans, 20,222 protein-coding genes have been annotated, whereas 24,765 ncRNAs have been reported (WBcel235 – http://www.ensembl.org/Caenorhabditis_elegans/Info/Annotation; accessed September 2018).
The sequence conservation of putative ncRNAs, as an indication of selective pressure, was examined to infer or contribute to the proposition of possible functional relevance of the identified ncRNA candidates. The sequences of putative L. braziliensis ncRNAs were compared to the genomic sequences of seven trypanosomatids. The ncRNA conservation percentage was 41% in L. major, 40% in each of L. donovani and L. infantum, and 35% in L. amazonensis. In the more distantly related Leishmania species, L. enriettii and L. tarentolae, ~28% conservation was observed, and in T. brucei, 7% conservation was observed.
Differential expression analysis was performed to compare the transcript levels of 11,372 putative ncRNAs at the three stages of L. braziliensis development (Additional file 6 – sheets PROvsMETA, METAvsAMA and AMAvsPRO). Analogously to the procedure used for the DE protein-coding genes, we analyzed the DE noncoding genes with no filters based only on the statistical significance (adjusted p value ≤ 0.05); in this condition, 3,266 DE ncRNAs were identified in the PROvsMETA comparison (29%, Additional file 1: Fig. S7A). When applying FC cutoffs ≥ 1.5 and ≥ 2, 1,897 (17%) and 625 DE ncRNAs (5.5%) were identified in the same comparison, respectively. In METAvsAMA using the same parameters, we detected 3,058 DE ncRNAs (27%, Additional file 1: Fig. S7B); this number decreased to 2,031 (18%) and 730 (6.4%) DE ncRNAs when applying FC cutoffs of ≥ 1.5 and ≥ 2, respectively. In the AMAvsPRO comparison, 4,380 DE ncRNAs were identified (38%, Additional file 1: Fig. S7C) with no FC cutoff, and 3,332 (29%) and 1,448 DE ncRNAs (12.7%) were identified with FC cutoffs of ≥ 1.5 and ≥ 2, respectively. The ncRNAs with the largest differences (the first 25 putative ncRNAs) in the comparisons between the up- and downregulated ncRNAs are presented in Additional file 3: Tables S12–S14. Because the assembly of the genome of L. braziliensis is not ideal, a large number of sequences are not assigned or are assembled in the chromosomes; these sequences are grouped as ‘Scaffold’. We observed that a large number of the putative ncRNAs identified among the most abundant transcripts in promastigotes were scaffold allotted.
The distribution of ncRNA classes for each ncRNA predictor was analyzed using the list of 11,372 putative ncRNAs. To increase the stringency, we selected those transcripts identified as putative ncRNAs by at least two predictors. With this approach, 9,561 transcripts remained as predicted ncRNAs (Fig. 5A and Additional file 1: Fig. S8). To obtain information on the classification of DE ncRNAs as inferred by the predictors, we selected those ncRNAs presenting FC ≥ 1.5 in at least one of the comparisons (PROvsMETA, METAvsAMA or AMAvsPRO). Within the population of 9,561 ncRNAs identified by at least two predictors, 3,602 such transcripts were identified (Fig. 5B). After exclusion of mRNAs and different rRNAs present in the transcript population, internal ribosome entry site (IRES) and introns were the classes most represented; transcripts classified as introns (group I and group II) represented 25% of the population of putative ncRNAs. In the population of DE transcripts, those classified as introns represented 24% of the population; when the DE ncRNAs were filtered to include only those DE transcripts with an FC ≥ 2, the introns (groups I and II) increased to 32% of the population. It is curious to observe this large number of transcripts classified as introns in an organism virtually lacking cis-splicing and introns [13].
Figure 5.
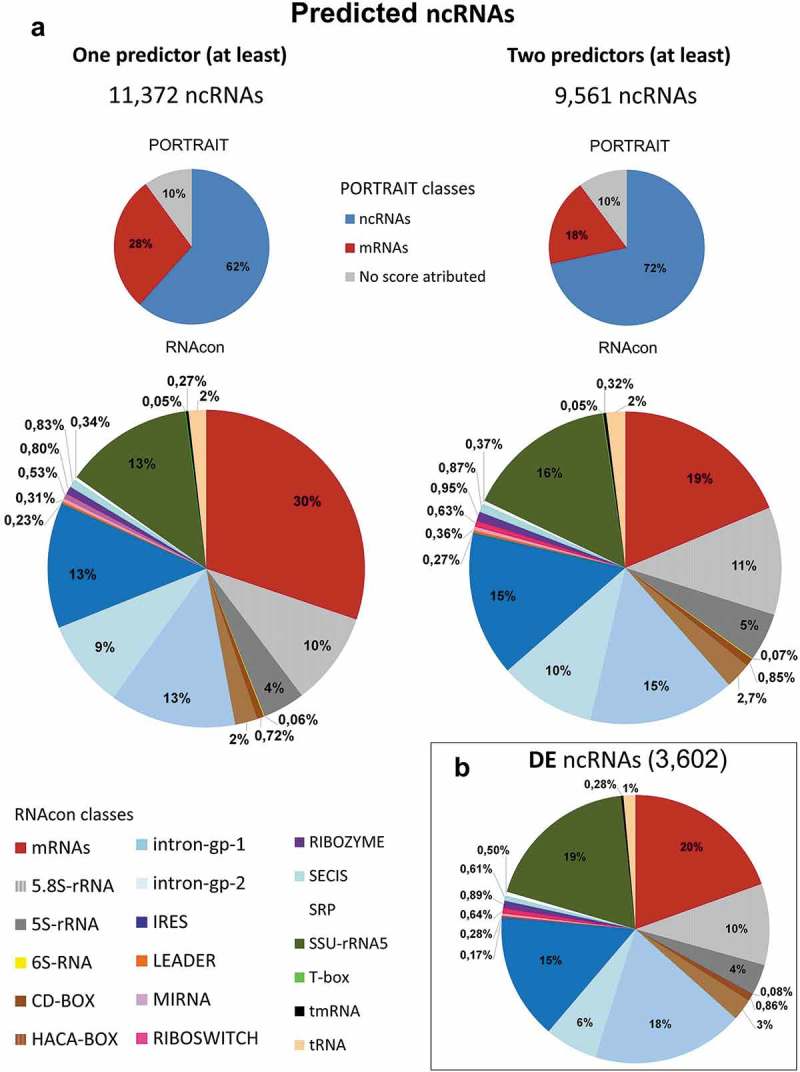
Graphic representation of predicted putative ncRNAs. The in silico identified putative ncRNAs were submitted to five ncRNAs predictors (see Methods). Results from Portrait, which estimates coding and noncoding potential of the transcript, and RNAcon, which discriminates between coding and noncoding RNAs and classifies the ncRNAs under 18 different ncRNAs classes, are presented. (a) On the left, the 11,372 putative ncRNAs that had been predicted by at least one of the 5 predictors are distributed in PORTRAIT classes (pie on the top) and RNAcon classes (pie on the bottom). On the right, in a similar organization, the 9,561ncRNAs predicted by at least two of the 5 programs are depicted and distributed among the different classes. (b) Those differentially expressed (DE) putative ncRNAs (3,602, FC ≥ 1.5) predicted at least by two of the 5 programs are depicted under the RNAcon 18 classes. Portrait and RNAcon classes are color-coded and presented in the figure. The gray ‘no score attributed’ indicates that the transcript is neither ncRNA nor mRNA according to Portrait classification.
Each ncRNA predictor uses different algorithms (see Methods), and combining at least two algorithms to predict a ncRNA may improve stringency and decrease the odds of false-positive ncRNAs. When we used the filter of at least two positive predictions, 1,809 putative ncRNAs were excluded from the original list (11,372).
In addition, to evaluate possible biases in the profile of differential expression throughout development, we selected from the list of ncRNAs that were DE in all comparisons analyzed (PROvsMETA, METAvsAMA and AMAvsPRO) with a cutoff of FC ≥ 1.5. With this analysis, 295 putative ncRNAs were identified (Additional file 7), and they were distributed into 6 groups with different expression profiles (Fig. 6). Interestingly, as observed for CDSs, most DE ncRNAs meeting this criterion were either present at lower levels in PRO than in META and AMA (Group 1–180 ncRNAs) or exhibited the highest levels in PRO and the lowest levels in AMA (Group 4–67 ncRNAs).
Figure 6.
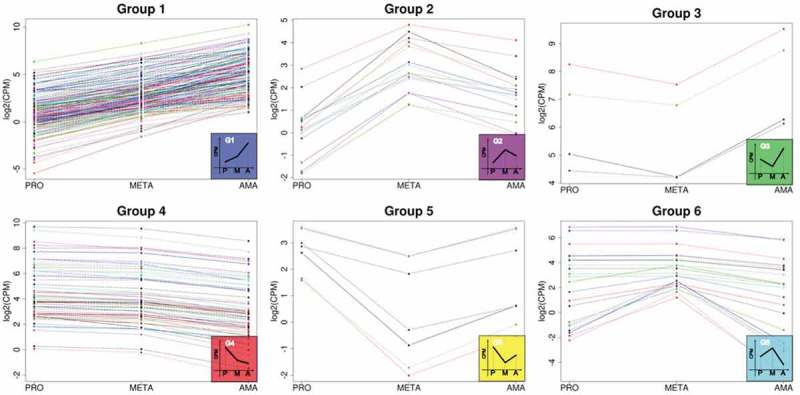
Profiles of differentially expressed putative ncRNAs during life cycle progression. The set of transcripts encompasses only those DE in all three comparisons analyzed (PROvsMETA, METAvsAMA and AMAvsPRO). Total number of DE ncRNAs = 295, distributed into 6 groups. Group 1: 180 ncRNAs, Group 2: 15 ncRNAs, Group 3: 4 ncRNAs, Group 4: 67 ncRNAs, Group 5: 7 ncRNAs, Group 6: 22 ncRNAs. Reads per counts per million (CPM), values in log2.
The total putative ncRNAs (11,372) with the main parameters analyzed were plotted in a single graphical presentation (Additional file 8). This output allows us to discern (i) the profile of modulation throughout development, (ii) the length, (iii) the chromosomal distribution and location relative to the annotated genes and (iv) the sense of transcription compared to annotated PTUs. This overview was helpful for searching for biases of ncRNA categories and their genome distribution. We also used a circos plot to plot only those abovementioned DE protein-coding and putative ncRNAs, totaling 216 and 295 transcripts, respectively. In this representation of DE transcripts only, it is possible to discern the length, chromosomal distribution, location and direction of transcripts. In addition, this circos plot highlights that many ncRNAs present a different expression profile than that of neighboring protein-coding genes. This profile reinforces the hypothesis that the observed DE putative ncRNAs are not mere artifacts or by-products of the polycistronic mode of transcription (Fig. 7).
Figure 7.
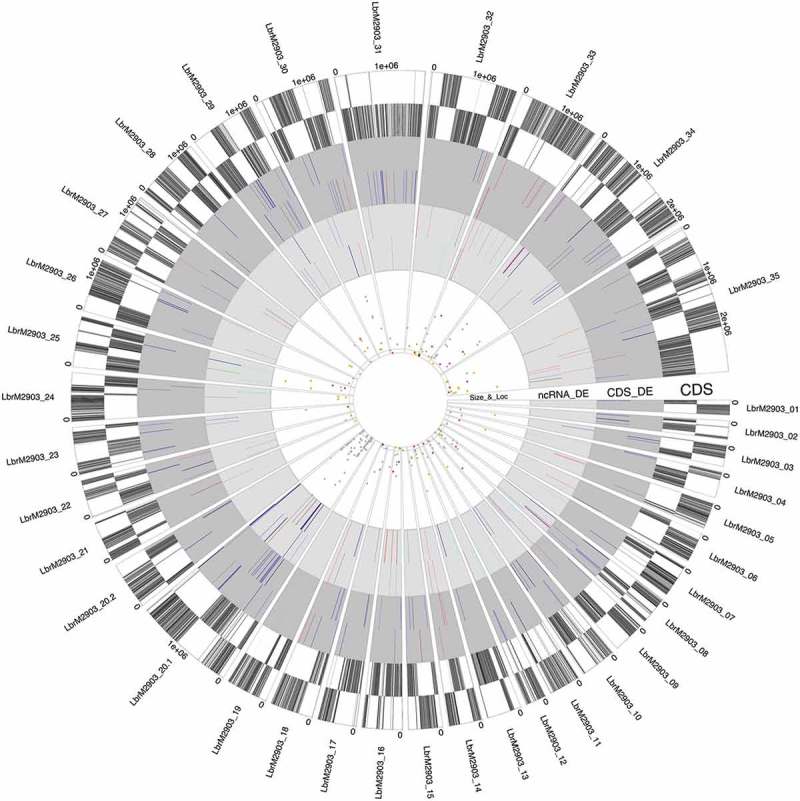
Genomic distribution of DE ncRNA identified in Leishmania braziliensis, represented in a circos plot. Sectors are equivalent to parasite chromosomes. Each sector contains 4 tracks, three of which are divided into two subtracks. From outside to inside, the tracks are named CDS, CDS_DE, ncRNA_DE and Size_&_Loc. The subtracks correspond to the positive and negative strands, transcription oriented clockwise or counterclockwise, respectively. Track 1 (‘CDS’) shows all CDS throughout the parasite chromosomes. Each CDS is represented by a vertical line painted with alternation of 3 grayscale colors to help discriminate individual CDSs and to highlight the annotation and orientation of the polycistronic transcription units (PTUs). Tracks 2 and 3 show the DE protein-coding and ncRNA transcripts, respectively. All of them are DE in all three comparisons. The color of each transcript line depicts the expression profile of each group, as shown in the graphical representation in figure 6 (P, M and A are procyclic, metacyclic and amastigote, respectively). Track 4 shows the DE ncRNAs smaller than 2000 bp. The subtrack delimits the ncRNA length (below or above 200 nucleotides long). The colors are related to the transcript location (orange – 5´UTR, gray – undetermined, yellow – 3´UTR, dark brown – antisense-CDS).
From the list of predicted putative DE ncRNAs, 35 were selected for confirmation by Northern blotting. This selection was based on sequence conservation in other Leishmania species, upregulation in infective (META) and/or mammalian (AMA) proliferative forms and a minimal of two positive predictions by ncRNA prediction programs. Hybridization was conducted with total RNA fractionated in polyacrylamide gels and allowed the identification of at least one transcript (more frequently, more than one) for 22 ncRNA candidates (Table 4). A perfect match between in silico and experimental prediction/results for both parameters analyzed, transcript length and DE pattern, was observed for LbrM2903_33_lncRNA177. For most of the putative ncRNAs, one of the parameters, length or DE pattern, agreed with the in silico prediction and northern blot results (Fig. 8 and Additional file 1: Fig. S9). We have no explanation for the presence of multiple bands or the 100–200-nt transcript common to several of the examined putative ncRNAs. However, we have not applied higher stringency hybridization conditions to exclude non-specific binding of the probe. The pattern of differential expression for six putative ncRNAs (Table 4 – bold) was confirmed, reinforcing a possible regulatory role for ncRNAs in L. braziliensis (Fig. 8 and Additional file 1: Fig. S9).
Table 4.
Putative ncRNAs evaluated by northern blotting.
| ncRNA ID* | Reference genome coordinate | ncRNA length (nt) | Location | Direction | Positive in silico prediction | Conservation | UP | DOWN | UP | DOWN | UP | DOWN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LbrM2903_03_lncRNA44 | 183,923 | 1644 | IC | sense | 2 | 7 | - | - | - | 2.1 | 2.4 | - |
| LbrM2903_08_lncRNA44 | 204,347 | 301 | 3ʹUTR | sense | 3 | 7 | - | - | - | 2.2 | 2.5 | - |
| LbrM2903_08_lncRNA78 | 336,906 | 476 | IC | sense | 2 | 6 | - | - | - | 11.7 | 16.5 | - |
| LbrM2903_08_lncRNA78 | 336,906 | 476 | IC | sense | 2 | 6 | - | - | - | 11.7 | 16.5 | - |
| LbrM2903_08_lncRNA80 | 339,993 | 294 | IC | sense | 2 | 0 | - | - | - | 11.9 | 14.9 | - |
| LbrM2903_10_lncRNA197 | 683,729 | 358 | IC | SSR | 2 | 3 | - | - | - | 2.2 | 2.4 | - |
| LbrM2903_13_lncRNA115 | 541,130 | 1829 | IC | sense | 2 | 3 | - | 3.8 | - | 5.2 | 20.0 | - |
| LbrM2903_16_lncRNA87 | 398,646 | 401 | IC | sense | 3 | 6 | - | - | - | 2.6 | 3.3 | - |
| LbrM2903_16_lncRNA88 | 399,069 | 328 | IC | sense | 3 | 6 | - | - | - | 2.5 | 3.0 | - |
| LbrM2903_16_lncRNA92 | 401,004 | 218 | 3ʹUTR | antisense | 3 | 5 | - | - | - | 3.1 | 3.8 | - |
| LbrM2903_20.1_lncRNA157 | 559,675 | 493 | IC | sense | 2 | 0 | - | 3.1 | - | 10.1 | 32.2 | - |
| LbrM2903_20.1_lncRNA164 | 574,425 | 235 | IC | SSR | 3 | 6 | - | 2.5 | - | 2.5 | 6.8 | - |
| LbrM2903_20.1_lncRNA319 | 1,429,674 | 367 | IC | sense | 3 | 4 | - | 2.7 | - | 4.8 | 12.9 | - |
| LbrM2903_20.1_lncRNA393 | 1,883,386 | 574 | IC | sense | 3 | 3 | - | 2 | - | 4.2 | 8.7 | - |
| LbrM2903_20.1_lncRNA395 | 1,887,223 | 911 | IC | sense | 3 | 5 | - | 2.4 | - | 3.7 | 8.8 | - |
| LbrM2903_22_lncRNA152 | 673,011 | 317 | 3ʹUTR | sense | 3 | 5 | - | 2.2 | 2.1 | - | - | - |
| LbrM2903_23_lncRNA173 | 780,429 | 596 | IC | sense | 2 | 2 | - | - | - | 3.4 | 4.6 | - |
| LbrM2903_25_lncRNA140 | 668,547 | 546 | 3ʹUTR | sense | 2 | 6 | - | 2.1 | 2.3 | - | - | - |
| LbrM2903_29_lncRNA116 | 606,054 | 590 | IC | sense | 3 | 6 | - | - | - | 3.3 | 3.5 | - |
| LbrM2903_29_lncRNA122 | 622,374 | 1185 | 3ʹUTR | sense | 2 | 6 | - | - | - | 2.4 | 2.9 | |
| LbrM2903_30_lncRNA54 | 238,337 | 558 | IC | sense | 3 | 4 | - | 3.1 | 2.6 | - | - | - |
| LbrM2903_31_lncRNA203 | 924,474 | 443 | IC | sense | 3 | 0 | - | 4.1 | 3.7 | - | - | - |
| LbrM2903_32_lncRNA242 | 1,374,540 | 739 | IC | sense | 2 | 5 | - | 2.7 | 2.4 | - | - | - |
| LbrM2903_32_lncRNA243 | 1,376,864 | 428 | IC | sense | 2 | 4 | - | 3.7 | 3.8 | - | - | - |
| LbrM2903_32_lncRNA285 | 1,522,614 | 1253 | IC | sense | 4 | 5 | - | - | - | 2.1 | 2.9 | - |
| LbrM2903_33_lncRNA177 | 692,839 | 300 | IC | sense | 2 | 4 | - | 3.1 | 4.8 | - | - | - |
| LbrM2903_33_lncRNA271 | 1,249,774 | 675 | 5ʹUTR | sense | 3 | 0 | - | 5.4 | 4.4 | - | - | - |
| LbrM2903_33_lncRNA289 | 1,340,368 | 232 | 3ʹUTR | sense | 2 | 0 | - | - | 6.4 | - | - | 5.4 |
| LbrM2903_34_lncRNA377 | 1,635,186 | 310 | 5ʹUTR | sense | 2 | 6 | - | 4.0 | 5.0 | - | - | - |
| LbrM2903_34_lncRNA379 | 1,643,460 | 337 | IC | sense | 3 | 0 | - | 3.4 | 3.3 | - | - | - |
| LbrM2903_34_lncRNA380 | 1,643,833 | 1674 | IC | sense | 3 | 6 | - | 2.1 | 2.2 | - | - | - |
| LbrM2903_34_lncRNA46 | 169,075 | 1347 | 5ʹUTR | sense | 2 | 7 | - | 6.3 | 4.9 | - | - | - |
| LbrM2903_34_lncRNA58 | 199,824 | 468 | IC | sense | 2 | 5 | - | 7.4 | 3.0 | - | 2.3 | - |
| LbrM2903_34_lncRNA62 | 210,928 | 242 | IC | sense | 2 | 5 | - | 4.1 | 3.3 | - | - | - |
| LbrM2903_34_ncRNA23 | 211,281 | 104 | IC | sense | 3 | 6 | - | 5 | 2.9 | - | - | - |
*ncRNA ID: LbrM2903_chromosome_ncRNA (> 200 nt = lncRNA) followed by the given ncRNA number.
Bold denotes ncRNAs confirmed by northern blotting. Positive prediction: the number of positive in silico ncRNA predictions. UP: upregulated transcript in the first cited stage in the comparison; DOWN: downregulated transcript in the first cited stage in the comparison. The columns of the analyzed comparisons depict values in fold change. IC: inter CDS; SSR: strand switch region; (-): transcript not differentially expressed (adjusted p value ≤ 0.05); nt: nucleotides
Figure 8.
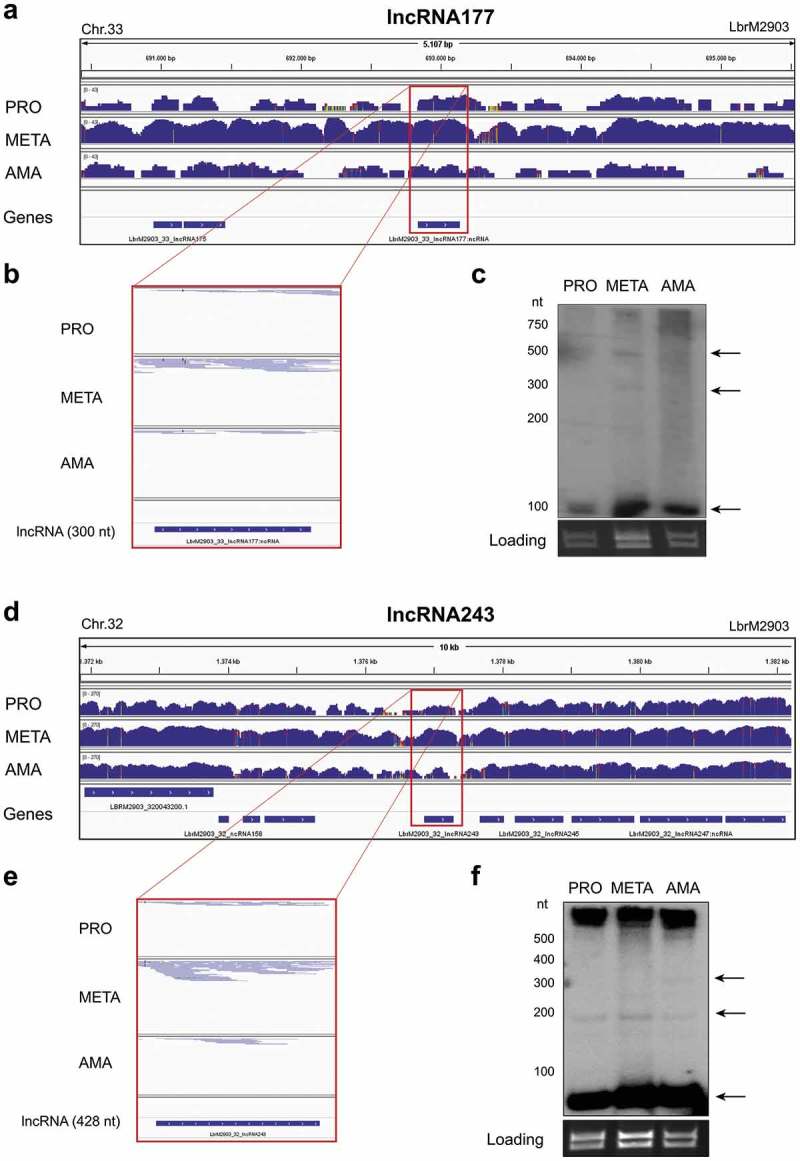
Identification and validation of 2 putative ncRNAs in L. braziliensis. (a) and (d): Regions of chromosomes 33 and 32, enclosing the putative ncRNAs LbrM2903_33_lncRNA177 and LbrM2903_32_lncRNA243, respectively. (b) and (e): extracted and zoomed areas for the corresponding lncRNAs depicting the number of reads in L. braziliensis procyclic, metacyclic and amastigote stages. (c) and (f): Northern blot of L. braziliensis total RNA using specific antisense oligonucleotides to each putative ncRNA. Arrows show multiple bands with approximate sizes to those predicted for the putative ncRNA. Ama: amastigote; Meta: metacyclic; Pro: procyclic; Gene: annotated genes; ncRNA: noncoding RNAs. Thin colorful regions (in a and b) represent divergence between reads and the reference genome.
Conclusion
This study comprises the first in-depth comparative transcriptomic analysis of the three main life cycle stages of the parasite L. braziliensis. The comparison of the transcriptomes of the proliferative (PRO) and infective (META) stages of the insect and of the mammalian proliferative form (AMA) provides information on gene expression variation throughout the life cycle, highlighting stage-specific (or stage-preferential) genes, pathways and biological processes modulation. More importantly, we present the first global scenario of putative ncRNA, apart from the housekeeping ncRNAs, that might play roles as regulatory ncRNAs.
This study contributes to the information on L. braziliensis genome organization and content by defining gene structure features, estimating boundaries of annotated genes and identifying novel putative protein-coding genes. The 3ʹUTR terminus was estimated for 38% of the annotated protein-coding genes, and the 5ʹUTR main start sites were estimated for 81%.
Comparative analyses conducted on the protein-coding gene content at each stage revealed that 61%, 52% and 71% of these genes (adjusted p value ≤ 0.05) were DE in the PROvsMETA, METAvsAMA and AMAvsPRO comparisons, respectively, and GO enrichment analyses revealed no novelties compared to previous similar studies carried out in other species of Leishmania [11,12,20,61]. However, the investigation of the contents of differentially-expressed upregulated genes in procyclics, metacyclics and amastigotes of L. braziliensis and comparisons with the corresponding stages in L. major and L. mexicana yielded interesting results and revealed an apparently not-so-conserved content of upregulated genes in each stage. Data from both Leishmania species from the subgenus Leishmania were used to compare L. braziliensis culture-derived PRO and META with L. major forms obtained with similar protocols. In addition, comparisons of L. braziliensis PRO, META and AMA with L. major forms derived from their natural niches (vector and mice lesions) were performed. Regarding the conservation of the pool of orthologous genes, the comparison of cultured PRO and META from L. braziliensis and L. major revealed that 30.5% (PRO) and 34.9% (META) are orthologous genes. After rescuing the L. major orthologous genes upregulated in PRO, META and AMA obtained from their natural niches, the percentages of orthologous genes represented in both species were lower: 14.6% (PRO), 12.1% (META) and 12.6% (AMA). Comparison between L. braziliensis PROvsAMA and L. mexicana PROvsAMA (both culture-derived parasites) revealed that 23% (PRO) and 15% (AMA) of the upregulated genes were orthologs.
Interestingly, among the orthologs upregulated in AMA or META in all three species, we detected genes involved in transport across the membrane (ABC transporters; ion, amino-acid and sugar transporters), LPG biosynthesis, amastin and amastin-like proteins, different classes of protein kinases and proteases, genes related to DNA repair, autophagy-related genes, signaling and vesicular transport genes, pore-forming protein transcripts, and proteasome and ubiquitin machinery-related proteins, among others. Approximately 40% of the transcripts of orthologous genes in each comparison are predicted to code for hypothetical proteins. The finding of orthologous genes similarly upregulated in a given stage among the three species suggests that their sequence, levels and functional conservation might be central to parasite survival and success. The lack of a high proportion of orthologs in the subpopulation of upregulated genes (in any stage) may be partially explained by gene duplication events and somy changes; the genome plasticity of the Leishmania genus facilitates the generation of paralogous genes with potentially similar functions or alternative ones. These results reveal differences between species and subgenera that may have biological significance. Nevertheless, the observed differences may derive from a diversity of factors, including the distinct computational tools and parameters used in the different studies, study variation in the origins of the parasite forms (natural niche vs culture-derived parasites), and differences among subgenera. These similarities and differences should be explored, and their study may improve our understanding of the clinical and biological features of different Leishmania species.
Investigation of common gene expression profiles (DE genes through all comparisons with FC ≥ 1.5) during parasite development revealed that most DE transcripts presented increased levels of expression from promastigotes to AMA (Group 1), whereas the second largest group of transcripts depicted the opposite profile (Group 4). The majority of genes in group 1 were annotated as hypothetical proteins (40%) or amastin/amastin-like transcripts (26%). Investigation and analysis of cis-elements common to transcripts presenting a similar expression profile must be performed to unravel possible posttranscriptional regulons.
To identify putative ncRNAs based on total RNA sequencing, we used the coverage of reads for positive and negative strands. The identified transcripts were considered candidates for short RNAs or lncRNAs when i) they were outside the CDS region or within them, transcribed from the opposite strand; ii) they presented no similarity to known protein domains (Pfam database); and iii) they received at least one positive prediction for known ncRNA characteristics. Thus, 11,372 putative ncRNAs were identified in L. braziliensis (4,021 as short ncRNAs and 7,351 as lncRNAs), with a median size of 281 nt and between 27% and 41% conserved in other Leishmania species. The identified ncRNAs represent 15% of the L. braziliensis genome (predicted ncRNAs are contained in 5,295,295 nt, and the genome length is 35.210.471 nt, including scaffolds). Analysis of DE ncRNAs (adjusted p value ≤ 0.05) identified 29%, 27% and 38% DE ncRNAs in the PROvsMETA, METAvsAMA and AMAvsPRO comparisons, respectively. The expression profile of DE ncRNAs in all comparisons (FC ≥ 1.5–295 putative ncRNAs) showed a similar distribution among different expression profiles throughout the life cycle compared to the protein-coding genes; most of the noncoding transcripts presented a Group 1 expression profile, followed by transcripts in Group 4. Interestingly, and supportive of the hypothesis that ncRNAs may be functional in the parasite, we observed that many of ncRNAs presented an expression profile that differs from the neighboring protein-coding genes in several genomic regions, as witnessed in the circos plot (Fig. 7).
A frequent finding for many of the ncRNAs analyzed by northern blotting was the presence of multiple hybridization-positive signals in the polyacrylamide fractionated RNA. As mentioned above, these signals could be the result of RNA processing or degradation or technical artifacts. For those ncRNAs to be further investigated as putative regulatory factors, experimental conditions for northern blotting analysis should be modified to exclude non-specific binding of the probe. If they are confirmed as products of RNA degradation, they might nonetheless have functional roles. Much has been discussed regarding RNA degradomes, and it is clear that they are reservoirs for RNA degradation products that may act as signaling molecules or participate in mechanisms that control gene expression. Studies of RNA degradomes indicate that RNA degradation is an underestimated source of regulatory molecules and that it has relevance for cellular homeostasis [73].
Functional analyses of these transcripts must be conducted to confirm and identify possible biological roles in the parasite. Nevertheless, we presented evidence that suggests that the identified transcripts might be functional regulatory ncRNAs. The in silico detection of the ncRNA transcripts revealed the differential expression of many of these transcripts throughout the parasite life cycle, some of which were confirmed as small transcripts by northern blotting. Additionally, different tools and algorithms were used to predict transcripts as ncRNAs. Corroborating the hypothesis that ncRNA transcripts are functional elements and not the result of polycistronic transcription promiscuity, many ncRNA sequences were found to be conserved across species, which might suggest selective pressure.
This work comprises the first comparative analysis of the transcriptomes of the PRO, META and AMA forms of L. braziliensis, revealing the expression profile of protein-coding genes and modulation of biological processes throughout the life cycle. Moreover, for the first time, a panorama of the putative ncRNAs of the parasite is presented. The putative ncRNAs might act as signaling molecules or in gene expression regulation, and investigation of their role may improve our understanding of the biology of this parasite and contribute to a better understanding of the processes of gene expression regulation in Leishmania.
Methods
Leishmania life stage obtainment: procyclic, metacyclic and axenic amastigote
L. braziliensis (M2903 – MHOM/BR/75/M2903) promastigotes were routinely cultured in M199 medium supplemented as previously described [35]. To maintain virulence and infectivity, BALB/c mice were inoculated with fresh parasites. After 6 weeks of infection, the mice were euthanized, and their lymph nodes were extracted and transferred to promastigote culture medium for the recovery of parasites. Every 2 days, the parasites were subcultured 0.1:10 in fresh medium, remained in culture until the sixth passage, and then frozen in freezing medium. Prior to the experiments, the parasites were thawed and kept in culture until the fourth passage (tenth passage after the immune system of BALB/c).
We established culture conditions to obtain META promastigotes and axenic AMA from promastigote cultures in the stationary phase of growth. To obtain the META forms, i.e., the infective forms of Leishmania, promastigotes on the fifth day of the stationary growth phase were submitted to centrifugation in Ficoll solution (10%) [36] to obtain an enriched fraction of the META stage. The culture of parasites on the fifth day of the stationary growth phase was also used for differentiation of the promastigote form in axenic AMA from culture in FBS at 33ºC and 5% CO2 [34]. After the differentiation process, the parasites were subcultured 0.1:10 in fresh serum and remained in culture until the tenth passage, when the experiments were carried out. To visualize the morphology of each life stage, samples from PRO, Ficoll-purified META and axenic AMA were fixed in two solutions, 2% glutaraldehyde, 2% paraformaldehyde, 0.05% CaCl2, cacodylate 0.1 M and osmium 2% and 0.2 M cacodylate, and then subsequently transferred to ethanol (30% to 100%). Samples were visualized using scanning electron microscopy (Jeol JSM-6610 LV; Multiuser Laboratory, Ribeirão Preto Medical School/University of São Paulo-FMRP/USP).
Rt-qPCR
The cells were lysed using TRIzol reagent (Invitrogen). Direct-Zol RNA Miniprep (Zymo Research) was used for RNA purification. The extracted RNA was treated with DNase Turbo (Thermo Fisher Scientific), and an RT-qPCR assay was performed according to Freitas Castro and cols [26]. Data were analyzed according to the ΔΔCt method [74] using the geometric mean of two selected housekeeping genes (G6PD and rRNA45) for normalization according to a previously described strategy [75]. RT-qPCR data were analyzed using GraphPad Prism 5 (Prism). Where shown, the data correspond to the means and standard deviations (± SD) from 3 independent experiments. Statistics were performed by Student’s t-test (two-tailed), and asterisks indicate statistically significant differences between samples, p ≤ 0.002 (**) and p ≤ 0.0001 (***).
RNA isolation and cDNA library preparation
The total RNA of 108 cells of L. braziliensis was extracted by using the Direct-zol™ extraction kit (Zymo Research) from cultured PRO and AMA. The experiment was performed in biological triplicate (a culture flask for each replicate). RNA samples were evaluated for quantity and quality by fluorometric quantitation (Qubit RNA BR Assay Kit – Thermo Fisher Scientific) and an Agilent Bioanalyzer system (with the RNA 6000 Nano Kit – Agilent, Waldbronn, Germany) according to the manufacturers’ instructions. Subsequently, RNA samples were submitted to treatment with a Ribo-Zero Epidemiology kit (Illumina) for rRNA depletion. Libraries were constructed for the 9 samples using the TruSeq® Stranded Total RNA kit following the manufacturer’s instructions.
RNA-seq data generation
Paired-end reads (150 bp) were obtained in the NextSeq® Illumina platform using a NextSeq 500/550 Kit v2 high output with 300 cycles. RNA sequencing data can be accessed from the Sequence Read Archive (SRA) database using accession number SRP162992.
Library processing
Cutadapt version 1.4.1 (http://journal.embnet.org/index.php/embnetjournal/article/view/200) was used for adapter cleaning. The quality metrics, size and number of reads were obtained with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped by Bowtie2 version 2.1.0 [76] with the reference genome of L. braziliensis 2903 (version 30 – TriTrypDB [38]). Alignment parameters were – N 1, to allow only 1 mismatch, and – local, to dismiss the requirement of end-to-end read alignment. Picard (https://broadinstitute.github.io/picard/) version 2.0.1 was used to convert sam to bam files and to group, sort and index these files. Mapping of the reads was visualized and analyzed with IGV (Integrative Genomic Viewer) version 2.3.91 [77].
CDSs and UTR length definition
CDS length was obtained from TriTrypDB L. braziliensis 2903 (version 30). The UTRs were defined by combining the coordinates of CDS, SL (spliced-leader) and polyadenylation (polyA) sites. The SL sites were obtained with a genomic library constructed to identify these sites [78] in L. braziliensis PRO promastigotes (unpublished data – Peter Myler). The sequenced reads were mapped with Bowtie2 allowing 1 mismatch in the alignment (–N 1). Picard software was used to manipulate the mapping files. The coverage of each nucleotide was obtained with genomecoverageBed (BEDTools package version 2.26 [79]), and a minimum coverage of 10 reads was considered to define regions with possible SL sites. A Perl script connected each CDS to the nearest upstream SL site with deeper coverage (main site), defining the 5ʹUTR region. For the 3ʹUTR definition, the sequenced total RNA libraries were remapped to rescue those reads with polyA tracks, with no match in the genome. For that, we excluded the – local parameter. Nonmapped reads were considered candidates to have polyA tails. Sequences with at least four continuous ‘As’ were selected and trimmed using the cutadapt program. After remapping these reads with the reference genome, a minimum coverage of 10 reads was considered to define regions with possible polyA sites. Each annotated CDS was associated with the nearest downstream polyA site with deeper coverage (main site). This methodology was based on similar work by Dillon and cols on L. major [11].
Transcript frequency
The most abundant transcripts were identified using FPKM calculation for each annotated CDS. The first FPKM percentile for all replicates of PRO, META and AMA was analyzed. The CDSs found in all replicates at each developmental stage were considered the most abundant. The VennDiagram [80] package was used to produce the Venn diagram in R.
Differential gene expression analysis
The number of reads in CDSs per library was obtained with featureCounts [81]. An R script [82] was developed to first analyze the correlation between the replicates and to perform DE analysis. The correlation between replicates included a comparison of the raw reads count distribution and the similarities/differences between the replicates with an MDS chart and BCV distance. For differential expression analysis, genes with a read count equal to zero were removed. DESeq was used (Bioconductor package) [83,84] to conduct the DE analysis. Genes with an adjusted p value < 0.05 were considered DE. This methodology was used for differential expression analysis of CDSs and putative ncRNAs.
Functional enrichment analyses
The GOseq [85] package of R was used for GO term enrichment analysis. Up- and downregulated DE genes were analyzed separately and compared between life cycle stages, and a p value cutoff ≤ 0.05 was used.
L. braziliensis species-specific genes
L. braziliensis species-specific genes were identified in 2007 in comparison to L. major and L. infantum. To identify these genes among those DE at each of the developmental stages, it was necessary to update the gene IDs provided by the authors. With the list of IDs, the sequences were searched in the NCBI (National Center for Biotechnology Information). These nucleotide sequences formed the database for similarity search (BLAST). The hits of the 49 species-specific genes with the annotated CDSs of L. braziliensis (2903 – TriTrypDB – version 30) were manually analyzed.
Comparisons of the contents of DE genes: L. braziliensis, L. major and L. mexicana
To compare the contents of DE genes that were upregulated in each stage of L. braziliensis with corresponding data from L. major and L. mexicana, we first rescued the orthologous genes available in TriTrypDB for each species (using the ‘transform by orthology’ tool). The number of upregulated L. braziliensis DE genes unique to each of the different life stages was 3,948 in PRO, 4,465 in META and 3,843 in AMA. The L. major orthologs were then submitted to the list of upregulated DE genes in procyclics and metacyclics obtained from the vector gut and mice lesion-derived amastigotes (PROvsMETA, METAvsAMA and AMAvsPRO comparisons) from Inbar et al [9]. The L. braziliensis procyclic and metacyclic upregulated genes from the PROvsMETA contrast were also compared with L. major axenic procyclic and metacyclic upregulated genes from Dillon et al [11]. The upregulated genes of L. braziliensis procyclics and amastigotes from the PROvsAMA contrast were compared with those of L. mexicana culture-derived procyclics and axenic amastigotes from Fiebig et al [44].
Putative ncRNA identification
The process of identification of putative ncRNAs began with the definition a set of transcripts obtained by total RNA sequencing of three L. braziliensis life stages: PRO and META promastigotes and AMA. Because transcription in Leishmania occurs in polycistronic blocks, followed by post transcriptional processing (splicing and polyadenylation) leading to the generation of mature mRNAs, the use of programs that identify transcripts appropriate for organisms that perform monocistronic transcription are unsuitable. To minimize this problem, we identified the transcripts in an alternative way using the parameters of minimum coverage (number of mapped reads), size and location of a transcript for the prediction of putative ncRNAs (Additional file 1: Fig. S5). The definition of the transcripts began by grouping triplicates to obtain the largest number of reads, increasing the chance that low-expressed ncRNAs were identified. These libraries were merged with the Picard program (MergeSamFiles). To define the boundaries of the transcripts, we obtained the reads coverage by genome position and used the program igvtools count [77] with the parameters – strand first (considering the original strand of the transcript) and – windowSize 1 of coverage for each position in the genome. An in-house Perl script was developed to split the coverage file by chromosome. The coverage files were analyzed by in-house Perl scripts that defined the boundaries of the transcripts on the positive (+) and negative (-) strands of the chromosomes. Transcripts of 50 to 200 nt with minimal coverage (per position) of 100 reads were grouped as short ncRNAs, and those > 200 with minimal coverage of 50 reads were grouped as lncRNAs. Those transcripts within annotated CDSs were discarded, except when transcribed from the complementary strand. An in-house Perl script was developed to apply these filters to the identified transcripts. At the end of this step, short and long putative ncRNAs were identified for PRO, META and AMA, generating tabular and gff files. The consensus of the ncRNA regions between PRO, META and AMA was generated considering the largest overlapping region between the three life cycle stages. The BEDTools merge program [79] was used to generate consensus, with the parameters -s (to group only regions on the same strand) and -o distinct (nonduplicated regions) applied.
ncRNA characterization
Once the putative ncRNAs were identified, known protein domains in these regions were searched for, thus avoiding the possibility that non-annotated CDSs in the genome were considered putative ncRNAs. A search for sequence similarity in the 6 reading frames (blastx) was performed against the Pfam database, which contains a large set of protein families [86,87]. Blastx was executed (e-value parameter less than 10−6) in the cluster of the LCCA-USP (Laboratory of Advanced Scientific Computing). Putative ncRNAs with a hit in Pfam were discarded. To enhance the reliability of the sequences identified as ncRNA candidates, predictors of specific characteristics of ncRNAs were used: PORTRAIT [88], RNAcon [89], snoscan [90], tRNAscan program-SE [91], and ptRNApred [92].
ncRNA conservation between Leishmania species
For conservation analysis of the identified ncRNAs, a sequence similarity search was performed with the genomes of L. major Friedlin, L. infantum JPCM5, L. amazonensis MHOMBR71973M2269, L. donovani BPK282A1, L. tarentolae ParrotTarII, L. enriettii LEM3045, and T. brucei TREU927 (TriTrypDB – version 29). One hit was considered positive with an e-value ≤ 10−5.
ncRNAs Northern blots
A group of 35 putative ncRNAs was submitted to Northern blotting to confirm the transcript, checking its length. They were chosen by the following criteria: transcript length (≥100 nt); ncRNA prediction (minimum of 2 positive predictions); and DE with FC ≥ 2, either upregulated in META and/or AMA forms and conserved in at least three Leishmania species or, in opposition, L. braziliensis specific. Northern blot experiments were performed using total RNA extracted from L. braziliensis PRO, META and AMA. RNA samples were fractionated in 15% polyacrylamide 8 M urea gel at 160 V (1.5 hours), stained with ethidium bromide and electrotransferred to the Hybond-N+ membrane (GE Lifesciences). RNA hybridization was carried out in Amersham rapid hybridization buffer (GE Healthcare) overnight at 42°C. Random primed probes were produced in the presence of [α-32P] dCTP, as described previously [93], and 3ʹ end-labeled probes were produced in the presence of [γ −32P] ATP [26]. The primers used for probe generation are listed in Additional file 3: Table S1.
Funding Statement
This work was supported by FAPESP [2013/50219-9] and CNPq [305775/2013-8]. PCR was supported by a CAPES and FAPESP TT5 fellowship [2016/16429-4]. NMMT was supported by a FAPESP fellowship [2015/16684-1]. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Finance Code 001
Acknowledgments
We thank the research support staff of the Center for Medical Genomics (CMG) at the Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto, HCFMRP/USP, particularly Kamila Chagas Peroni Zueli, for library construction and RNA sequencing. We thank Viviane Ambrosio and the Laboratório Multiusuário de Microscopia Eletrônica (LMME) for technical assistance.
Additional files
File: Additional file 1; file format: PDF (.pdf); title: Supplementary figures; description: Supplementary figures with legends.
File: Additional file 2; file format: .xlsx; title: CDSs and UTRs; description: protein-coding genes for which 5ʹUTRs and/or 3ʹUTRs have been predicted in L. braziliensis 2903. Column A: CDSs are identified according to genome annotation; column B, chromosome identification; columns C and D, coordinates of the start and end of either 5ʹUTRs or 3ʹUTRs, respectively; column E, UTR length; Column F, SL ID or polyA ID (see methodology); column G, SL or polyA coverage.
File: Additional file 3; file format: PDF (.pdf); title: Supplementary tables; description: Supplementary tables with legends.
File: Additional file 4; file format: .xlsx; title: Differentially expressed protein-coding genes; description: Annotated protein-coding genes found as differentially expressed with an adjusted p value ≤ 0.05. In the last 3 sheets, the orthologous genes upregulated in L. major and L. mexicana (LbrM—LmjF, Axenic_LbrM—LmjF and Axenic_LbrM—LxmM) are depicted.
File: Additional file 5; file format: .xlsx; title: Differentially expressed protein-coding genes during life cycle progression; description: DE Protein-coding genes in all three comparisons analyzed and clustered by level of expression throughout life cycle stages; groups 1 to 6. The spreadsheet is organized in sheets 1 to 6 representing the corresponding groups (G1 to G6). In each sheet, column A contains the list of DE protein-coding genes, and columns B to D depict the CPM (mean of triplicates) for PRO, META and AMA, respectively.
File: Additional file 6; file format: .xlsx; title: putative ncRNAs information; description: The sheet ‘ncRNA General Info’ contains generated information on the 11,372 putative ncRNAs identified, including the results of the ncRNA differential expression analysis. In columns A to L, the following information is presented: ncRNA ID (A), chromosome (B), (C and D) lower and higher chromosomal coordinates, (E) strand transcribed, (F) length, (G) location (within UTRs, antisense- within CDSs, undetermined, strand switch region), (H) direction (with respect to PTU), (I) PORTRAIT prediction, (J and K) ptRNApred prediction, (L) snoscan prediction, (M and N) RNAcon prediction, (O) tRNAscan-SE prediction (P) prediction score, (Q) upregulated in META, (R) upregulated in AMA, and (S) conservation score. Subsequent sheets named ‘PROvsMETA’, ‘METAvsAMA’ and ‘AMAvsPRO’ for DE ncRNAs are listed (up- and downregulated) with corresponding FC values for all comparisons.
File: Additional file 7; file format: .xlsx; title: Differentially expressed putative noncoding genes (ncRNAs) during life cycle progression; description: DE ncRNAs in all comparisons analyzed (three main Leishmania life cycle stages) and clustered by levels of expression at each life cycle stage.
File: Additional file 8; file format: .pdf; title: Genomic distribution of ncRNAs identified in Leishmania braziliensis, represented in a circos plot; description: The 36 sectors are equivalent to the parasite chromosomes as annotated and deposited in TriTrypDB. The 5 tracks named CDS, P/M, M/A, A/P, 3xDE are subdivided into two subtracks, which correspond to the plus and minus strands; transcription oriented clockwise or anticlockwise, respectively. Track 1 (CDS) shows all annotated CDSs throughout the parasite chromosomes. Each CDS is represented by vertical lines in 3 grayscale colors to help discriminate individual CDSs (Legend track 1). The observation of both subtracks allows the prediction of the location and orientation of the polycistronic transcription units (PTUs). Tracks 2, 3, and 4 show the DE ncRNA found in each contrast (PROvsMETA ‘P/M’, METAvsAMA ‘M/A’, AMAvsPRO ‘A/P’; p value ≤ 0.05, FC ≥ 1.5; 4301 records). The color of each ncRNA line corresponds to the Log2FC value. The range of Log2FC values is 5.48 (red) to −3.83 (green, legend tracks 2, 3, and 4); 0 is represented by white. Track 5 shows the DE ncRNA in all three contrasts (3xDE). The color coding is equivalent to that in Fig. 7 (G1 – blue, G2 – magenta, G3 – green, G4 – red, G5 – yellow, G6 – cyan; Legend track 4). Track 6 shows DE ncRNA smaller than 2000 bp (4172 of 4301 ncRNA DE located in chromosomes). The subtrack limit is defined by a line separating ≤ 200 nt transcripts. The colors indicate the localization of the ncRNA (orange – 5´UTR, gray – undetermined, yellow – 3´UTR, dark brown – CDS; track 5). Features predicted in scaffolds are not represented in this figure (47 CDS and 221 ncRNA).
Authors’ contributions
PCR, NMMT and AKC conceived, designed and wrote the manuscript. NMMT prepared the samples of the parasite life cycle stages. PCR conducted the bioinformatics analysis. FFC and RDMM designed some figures and contributed to fragments of the manuscript text. RDMM clustered the RNA-seq results into six groups of differentially expressed genes in the three parasite life stages. NMMT, LD and TPAD conducted the northern blotting analysis. FFC and TPAD conducted the RT-qPCR experiments. EJRV supported the design of the ncRNA identification pipeline. PJM provided the SL libraries and reviewed the manuscript. All authors read and approved the final manuscript.
Availability of data and material
The Leishmania braziliensis total RNA sequencing data can be accessed from the SRA (accession SRP162992). The ncRNA identification pipeline is available from the corresponding author, AKC, on reasonable request.
Competing interests
The authors declare no competing interests.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
The use of mice was approved by the Ethical Commission of Ethics in Animal Research from Ribeirão Preto Medical School of the University of São Paulo. They certified that Protocol 153/2016 (‘Investigation of noncoding RNAs in Leishmania (Viannia) braziliensis’) was in accordance with Ethical Principles in Animal Research adopted by the National Council for the Control of Animal Experimentation (CONCEA) in 09/26/2016.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Grimaldi G, Tesh RB.. Leishmaniases of the new world: current concepts and implications for future research. Clin Microbiol Rev. 1993;6(3):230–250. PubMed PMID: 8358705; PubMed Central PMCID: PMCPMC358284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27(5):305–318. PubMed PMID: 15225981. [DOI] [PubMed] [Google Scholar]
- [3].Llanes A, Restrepo CM, Del Vecchio G, et al. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci Rep. 2015;5:8550 PubMed PMID: 25707621; PubMed Central PMCID: PMC4338418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cuervo P, Cupolillo E, Nehme N, et al. Leishmania (Viannia): genetic analysis of cutaneous and mucosal strains isolated from the same patient. Exp Parasitol. 2004;108(1–2):59–66. PubMed PMID: 15491550. [DOI] [PubMed] [Google Scholar]
- [5].Azulay RD, Azulay Junior DR. Immune-clinical-pathologic spectrum of leishmaniasis. Int J Dermatol. 1995;34(5):303–307. PubMed PMID: 7607788. [DOI] [PubMed] [Google Scholar]
- [6].Queiroz A, Sousa R, Heine C, et al. Association between an emerging disseminated form of leishmaniasis and Leishmania (Viannia) braziliensis strain polymorphisms. J Clin Microbiol. 2012;50(12):4028–4034. PubMed PMID: 23035200; PubMed Central PMCID: PMC3503016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Silva-Almeida M, Pereira BA, Ribeiro-Guimaraes ML, et al. Proteinases as virulence factors in Leishmania spp. infection in mammals. Parasit Vectors. 2012;5:160 PubMed PMID: 22871236; PubMed Central PMCID: PMC3436776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bates PA. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol. 2007;37(10):1097–1106. Epub 2007/04/18 PubMed PMID: 17517415; PubMed Central PMCID: PMCPMC2675784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Inbar E, Hughitt VK, Dillon LA, et al. The transcriptome of Leishmania major developmental stages in their natural sand fly vector. MBio. 2017;8(2). Epub 2017/04/04 DOI: 10.1128/mBio.00029-17 PubMed PMID: 28377524; PubMed Central PMCID: PMCPMC5380837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dillon LA, Suresh R, Okrah K, et al. Simultaneous transcriptional profiling of Leishmania major and its murine macrophage host cell reveals insights into host-pathogen interactions. BMC Genomics. 2015;16:1108 Epub 2015/12/29 PubMed PMID: 26715493; PubMed Central PMCID: PMCPMC4696162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dillon LA, Okrah K, Hughitt VK, et al. Transcriptomic profiling of gene expression and RNA processing during Leishmania major differentiation. Nucleic Acids Res. 2015;43(14):6799–6813. PubMed PMID: 26150419; PubMed Central PMCID: PMCPMC4538839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Saxena A, Lahav T, Holland N, et al. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol Biochem Parasitol. 2007;152(1):53–65. PubMed PMID: 17204342; PubMed Central PMCID: PMC1904838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ivens AC, Peacock CS, Worthey EA, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309(5733):436–442. PubMed PMID: 16020728; PubMed Central PMCID: PMCPMC1470643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Beverley SM. Protozomics: trypanosomatid parasite genetics comes of age. Nat Rev Genet. 2003;4(1):11–19. PubMed PMID: 12509749. [DOI] [PubMed] [Google Scholar]
- [15].McKee AE, Silver PA. Systems perspectives on mRNA processing. Cell Res. 2007;17(7):581–590. PubMed PMID: 17621309. [DOI] [PubMed] [Google Scholar]
- [16].Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416(6880):499–506. PubMed PMID: 11932736. [DOI] [PubMed] [Google Scholar]
- [17].Hershey JW, Sonenberg N, Mathews MB. Principles of translational control: an overview. Cold Spring Harb Perspect Biol. 2012;4(12). DOI: 10.1101/cshperspect.a011528 PubMed PMID: 23209153; PubMed Central PMCID: PMCPMC3504442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kumar P, Sundar S, Singh N. Degradation of pteridine reductase 1 (PTR1) enzyme during growth phase in the protozoan parasite Leishmania donovani. Exp Parasitol. 2007;116(2):182–189. Epub 2006/ 12/30 PubMed PMID: 17275814. [DOI] [PubMed] [Google Scholar]
- [19].Williams RA, Smith TK, Cull B, et al. ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 2012;8(5):e1002695 Epub 2012/05/17 PubMed PMID: 22615560; PubMed Central PMCID: PMCPMC3355087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rochette A, Raymond F, Ubeda JM, et al. Genome-wide gene expression profiling analysis of Leishmania major and Leishmania infantum developmental stages reveals substantial differences between the two species. BMC Genomics. 2008;9:255 PubMed PMID: 18510761; PubMed Central PMCID: PMCPMC2453527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vasconcelos EJ, Terrão MC, Ruiz JC, et al. In silico identification of conserved intercoding sequences in Leishmania genomes: unraveling putative cis-regulatory elements. Mol Biochem Parasitol. 2012;183(2):140–150. PubMed PMID: 22387760. [DOI] [PubMed] [Google Scholar]
- [22].Terrão MC, Rosas de Vasconcelos EJ, Defina TA, et al. Disclosing 3ʹ UTR cis-elements and putative partners involved in gene expression regulation in Leishmania spp. PLoS One. 2017;12(8):e0183401 Epub 2017/08/31 PubMed PMID: 28859096; PubMed Central PMCID: PMCPMC5578504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kolev NG, Franklin JB, Carmi S, et al. The transcriptome of the human pathogen Trypanosoma brucei at single-nucleotide resolution. PLoS Pathog. 2010;6(9):e1001090 PubMed PMID: 20838601; PubMed Central PMCID: PMC2936537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Michaeli S, Doniger T, Gupta SK, et al. RNA-seq analysis of small RNPs in Trypanosoma brucei reveals a rich repertoire of non-coding RNAs. Nucleic Acids Res. 2012;40(3):1282–1298. Epub 2011/ 10/05 PubMed PMID: 21976736; PubMed Central PMCID: PMCPMC3273796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mercer TR, Wilhelm D, Dinger ME, et al. Expression of distinct RNAs from 3ʹ untranslated regions. Nucleic Acids Res. 2011;39(6):2393–2403. Epub 2010/ 11/12 PubMed PMID: 21075793; PubMed Central PMCID: PMCPMC3064787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Freitas Castro F, Ruy PC, Nogueira Zeviani K, et al. Evidence of putative non-coding RNAs from Leishmania untranslated regions. Mol Biochem Parasitol. 2017;214:69–74. Epub 2017/04/03 PubMed PMID: 28385563. [DOI] [PubMed] [Google Scholar]
- [27].Smith DF, Peacock CS, Cruz AK. Comparative genomics: from genotype to disease phenotype in the leishmaniases. Int J Parasitol. 2007;37(11):1173–1186. PubMed PMID: 17645880; PubMed Central PMCID: PMCPMC2696322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Iantorno SA, Durrant C, Khan A, et al. Gene expression in Leishmania is regulated predominantly by gene dosage. MBio. 2017;8(5). Epub 2017/09/12 DOI: 10.1128/mBio.01393-17 PubMed PMID: 28900023; PubMed Central PMCID: PMCPMC5596349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bussotti G, Gouzelou E, Côrtes Boité M, et al. Genome dynamics during environmental adaptation reveal strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification. MBio. 2018;9(6). Epub 2018/11/06 DOI: 10.1128/mBio.01399-18 PubMed PMID: 30401775; PubMed Central PMCID: PMCPMC6222132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dumetz F, Imamura H, Sanders M, et al. Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio. 2017;8(3). Epub 2017/05/23 DOI: 10.1128/mBio.00599-17 PubMed PMID: 28536289; PubMed Central PMCID: PMCPMC5442457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rogers MB, Hilley JD, Dickens NJ, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21(12):2129–2142. Epub 2011/10/28 PubMed PMID: 22038252; PubMed Central PMCID: PMCPMC3227102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. .PubMed PMID: 19015660; PubMed Central PMCID: PMCPMC2949280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. PubMed PMID: 18516045. [DOI] [PubMed] [Google Scholar]
- [34].Doyle PS, Engel JC, Pimenta PF, et al. Leishmania donovani: long-term culture of axenic amastigotes at 37 degrees C. Exp Parasitol. 1991;73(3):326–334. PubMed PMID: 1915747. [DOI] [PubMed] [Google Scholar]
- [35].Kapler GM, Coburn CM, Beverley SM. Stable transfection of the human parasite Leishmania major delineates a 30-kilobase region sufficient for extrachromosomal replication and expression. Mol Cell Biol. 1990;10(3):1084–1094. PubMed PMID: 2304458; PubMed Central PMCID: PMC360971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Almeida Marques-Da-Silva E, de Oliveira JC, Figueiredo AB, et al. Extracellular nucleotide metabolism in Leishmania: influence of adenosine in the establishment of infection. Microbes Infect. 2008;10(8):850–857. .PubMed PMID: 18656412 [DOI] [PubMed] [Google Scholar]
- [37].de Paiva RM, Grazielle-Silva V, Cardoso MS, et al. Amastin knockdown in Leishmania braziliensis affects parasite-macrophage interaction and results in impaired viability of intracellular amastigotes. PLoS Pathog. 2015;11(12):e1005296 PubMed PMID: 26641088; PubMed Central PMCID: PMC4671664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Aslett M, Aurrecoechea C, Berriman M, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38(Databaseissue):D457–62. PubMed PMID: 19843604; PubMed Central PMCID: PMCPMC2808979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mayer MG, Floeter-Winter LM. Pre-mRNA trans-splicing: from kinetoplastids to mammals, an easy language for life diversity. Mem Inst Oswaldo Cruz. 2005;100(5):501–513. PubMed PMID: 16184228. [DOI] [PubMed] [Google Scholar]
- [40].Kramer S. Developmental regulation of gene expression in the absence of transcriptional control: the case of kinetoplastids. Mol Biochem Parasitol. 2012;181(2):61–72. PubMed PMID: 22019385. [DOI] [PubMed] [Google Scholar]
- [41].Rastrojo A, Carrasco-Ramiro F, Martin D, et al. The transcriptome of Leishmania major in the axenic promastigote stage: transcript annotation and relative expression levels by RNA-seq. BMC Genomics. 2013;14:223 PubMed PMID: 23557257; PubMed Central PMCID: PMC3637525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].De Gaudenzi JG, Noé G, Campo VA, et al. Gene expression regulation in trypanosomatids. Essays Biochem. 2011;51:31–46. .PubMed PMID: 22023440. [DOI] [PubMed] [Google Scholar]
- [43].Clayton C, Shapira M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol Biochem Parasitol. 2007;156(2):93–101. Epub 2007/07/19 PubMed PMID: 17765983. [DOI] [PubMed] [Google Scholar]
- [44].Fiebig M, Kelly S, Gluenz E. Comparative life cycle transcriptomics revises Leishmania mexicana genome annotation and links a chromosome duplication with parasitism of vertebrates. PLoS Pathog. 2015;11(10):e1005186 Epub 2015/10/09 PubMed PMID: 26452044; PubMed Central PMCID: PMCPMC4599935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rochette A, McNicoll F, Girard J, et al. Characterization and developmental gene regulation of a large gene family encoding amastin surface proteins in Leishmania spp. Mol Biochem Parasitol. 2005;140(2):205–220. PubMed PMID: 15760660. [DOI] [PubMed] [Google Scholar]
- [46].Zhou X, Liao WJ, Liao JM, et al. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015;7(2):92–104. Epub 2015/ 03/03 PubMed PMID: 25735597; PubMed Central PMCID: PMCPMC4481666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mans BJ, Pienaar R, Latif AA, et al. Diversity in the 18S SSU rRNA V4 hyper-variable region of Theileria spp. in Cape buffalo (Syncerus caffer) and cattle from southern Africa. Parasitology. 2011;138(6):766–779. Epub 2011/ 02/25 PubMed PMID: 21349232. [DOI] [PubMed] [Google Scholar]
- [48].Llanos S, Serrano M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle. 2010;9(19):4005–4012. Epub 2010/10/09 PubMed PMID: 20935493; PubMed Central PMCID: PMCPMC3615335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Eng FJ, Warner JR. Structural basis for the regulation of splicing of a yeast messenger RNA. Cell. 1991;65(5):797–804. PubMed PMID: 2040015. [DOI] [PubMed] [Google Scholar]
- [50].Badjatia N, Park SH, Ambrósio DL, et al. Cyclin-dependent kinase CRK9, required for spliced leader trans splicing of pre-mRNA in trypanosomes, functions in a complex with a new L-type cyclin and a kinetoplastid-specific protein. PLoS Pathog. 2016;12(3):e1005498 Epub 2016/03/08 PubMed PMID: 26954683; PubMed Central PMCID: PMCPMC4783070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Beyer AR, Bann DV, Rice B, et al. Nucleolar trafficking of the mouse mammary tumor virus gag protein induced by interaction with ribosomal protein L9. J Virol. 2013;87(2):1069–1082. Epub 2012/ 11/07 PubMed PMID: 23135726; PubMed Central PMCID: PMCPMC3554096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jia J, Arif A, Willard B, et al. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol Cell. 2012;47(4):656–663. Epub 2012/07/05 PubMed PMID: 22771119; PubMed Central PMCID: PMCPMC3635105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].de Almeida-Bizzo JH, Alves LR, Castro FF, et al. Characterization of the pattern of ribosomal protein L19 production during the lifecycle of Leishmania spp. Exp Parasitol. 2014;147:60–66. Epub 2014/10/05 PubMed PMID: 25290356. [DOI] [PubMed] [Google Scholar]
- [54].Holzer TR, McMaster WR, Forney JD. Expression profiling by whole-genome interspecies microarray hybridization reveals differential gene expression in procyclic promastigotes, lesion-derived amastigotes, and axenic amastigotes in Leishmania mexicana. Mol Biochem Parasitol. 2006;146(2):198–218. Epub 2006/01/06 PubMed PMID: 16430978. [DOI] [PubMed] [Google Scholar]
- [55].Soto M, Iborra S, Quijada L, et al. Cell-cycle-dependent translation of histone mRNAs is the key control point for regulation of histone biosynthesis in Leishmania infantum. Biochem J. 2004;379(Pt 3):617–625. PubMed PMID: 14766017; PubMed Central PMCID: PMCPMC1224130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Daba S, Mansour NS, Youssef FG, et al. Vector-host-parasite inter-relationships in leishmaniasis. IV. Electrophoretic studies on proteins of four vertebrate bloods with and without Leishmania infantum or L. major. J Egypt Soc Parasitol. 1997;27(3):795–804. PubMed PMID: 9425823. [PubMed] [Google Scholar]
- [57].Maclean LM, O’Toole PJ, Stark M, et al. Trafficking and release of Leishmania metacyclic HASPB on macrophage invasion. Cell Microbiol. 2012;14(5):740–761. Epub 2012/02/24 PubMed PMID: 22256896; PubMed Central PMCID: PMCPMC3491706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Williams RA, Tetley L, Mottram JC, et al. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol Microbiol. 2006;61(3):655–674. Epub 2006/06/27 PubMed PMID: 16803590. [DOI] [PubMed] [Google Scholar]
- [59].Besteiro S, Williams RA, Coombs GH, et al. Protein turnover and differentiation in Leishmania. Int J Parasitol. 2007;37(10):1063–1075. Epub 2007/03/31 PubMed PMID: 17493624; PubMed Central PMCID: PMCPMC2244715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rafati S, Hassani N, Taslimi Y, et al. Amastin peptide-binding antibodies as biomarkers of active human visceral leishmaniasis. Clin Vaccine Immunol. 2006;13(10):1104–1110. PubMed PMID: 17028214; PubMed Central PMCID: PMC1595312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rochette A, Raymond F, Corbeil J, et al. Whole-genome comparative RNA expression profiling of axenic and intracellular amastigote forms of Leishmania infantum. Mol Biochem Parasitol. 2009;165(1):32–47. PubMed PMID: 19393160. [DOI] [PubMed] [Google Scholar]
- [62].Requena JM, Montalvo AM, Fraga J. Molecular chaperones of Leishmania: central players in many stress-related and -unrelated physiological processes. Biomed Res Int. 2015;2015:301326 PubMed PMID: 26167482; PubMed Central PMCID: PMC4488524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schluter A, Wiesgigl M, Hoyer C, et al. Expression and subcellular localization of cpn60 protein family members in Leishmania donovani. Biochim Biophys Acta. 2000;1491(1–3):65–74. PubMed PMID: 10760571. [DOI] [PubMed] [Google Scholar]
- [64].Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–178. PubMed PMID: 17632063. [DOI] [PubMed] [Google Scholar]
- [65].Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(1):R17–29. PubMed PMID: 16651366. [DOI] [PubMed] [Google Scholar]
- [66].Mattick JS. The functional genomics of noncoding RNA. Science. 2005;309(5740):1527–1528. PubMed PMID: 16141063. [DOI] [PubMed] [Google Scholar]
- [67].Mattick JS. RNA regulation: a new genetics? Nat Rev Genet. 2004;5(4):316–323. PubMed PMID: 15131654. [DOI] [PubMed] [Google Scholar]
- [68].Dumas C, Chow C, Müller M, et al. A novel class of developmentally regulated noncoding RNAs in Leishmania. Eukaryot Cell. 2006;5(12):2033–2046. Epub 2006/10/27 PubMed PMID: 17071827; PubMed Central PMCID: PMCPMC1694821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Atayde VD, Shi H, Franklin JB, et al. The structure and repertoire of small interfering RNAs in Leishmania (Viannia) braziliensis reveal diversification in the trypanosomatid RNAi pathway. Mol Microbiol. 2013;87(3):580–593. Epub 2012/ 12/26 PubMed PMID: 23217017; PubMed Central PMCID: PMCPMC3556230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lambertz U, Oviedo Ovando ME, Vasconcelos EJ, et al. Small RNAs derived from tRNAs and rRNAs are highly enriched in exosomes from both old and new world Leishmania providing evidence for conserved exosomal RNA Packaging. BMC Genomics. 2015;16:151 PubMed PMID: 25764986; PubMed Central PMCID: PMCPMC4352550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Eliaz D, Doniger T, Tkacz ID, et al. Genome-wide analysis of small nucleolar RNAs of Leishmania major reveals a rich repertoire of RNAs involved in modification and processing of rRNA. RNA Biol. 2015;12(11):1222–1255. Epub 2015/05/13 PubMed PMID: 25970223; PubMed Central PMCID: PMCPMC4829279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Rederstorff M, Bernhart SH, Tanzer A, et al. RNPomics: defining the ncRNA transcriptome by cDNA library generation from ribonucleo-protein particles. Nucleic Acids Res. 2010;38(10):e113 PubMed PMID: 20150415; PubMed Central PMCID: PMCPMC2879528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jackowiak P, Nowacka M, Strozycki PM, et al. RNA degradome–its biogenesis and functions. Nucleic Acids Res. 2011;39(17):7361–7370. Epub 2011/06/07 PubMed PMID: 21653558; PubMed Central PMCID: PMCPMC3177198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45 PubMed PMID: 11328886; PubMed Central PMCID: PMCPMC55695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034 PubMed PMID: 12184808; PubMed Central PMCID: PMCPMC126239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. PubMed PMID: 22388286; PubMed Central PMCID: PMCPMC3322381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. Epub 2012/ 04/19 PubMed PMID: 22517427; PubMed Central PMCID: PMCPMC3603213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jensen BC, Ramasamy G, Vasconcelos EJ, et al. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. BMC Genomics. 2014;15:911 PubMed PMID: 25331479; PubMed Central PMCID: PMCPMC4210626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. PubMed PMID: 20110278; PubMed Central PMCID: PMCPMC2832824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35 Epub 2011/ 01/26 PubMed PMID: 21269502; PubMed Central PMCID: PMCPMC3041657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. PubMed PMID: 24227677. [DOI] [PubMed] [Google Scholar]
- [82].R Core Team R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. [Google Scholar]
- [83].Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80 PubMed PMID: 15461798; PubMed Central PMCID: PMCPMC545600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106 PubMed PMID: 20979621; PubMed Central PMCID: PMCPMC3218662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Young MD, Wakefield MJ, Smyth GK, et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14 Epub 2010/ 02/04 PubMed PMID: 20132535; PubMed Central PMCID: PMCPMC2872874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Finn RD, Mistry J, Tate J, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38(Databaseissue):D211–22. PubMed PMID: 19920124; PubMed Central PMCID: PMCPMC2808889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Finn RD, Coggill P, Eberhardt RY, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44(D1):D279–85. Epub 2015/ 12/15 PubMed PMID: 26673716; PubMed Central PMCID: PMCPMC4702930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Arrial RT, Togawa RC, Brigido M. Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: case study of the pathogenic fungus Paracoccidioides brasiliensis. BMC Bioinformatics. 2009;10:239 PubMed PMID: 19653905; PubMed Central PMCID: PMCPMC2731755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Panwar B, Arora A, Raghava GP. Prediction and classification of ncRNAs using structural information. BMC Genomics. 2014;15:127 PubMed PMID: 24521294; PubMed Central PMCID: PMCPMC3925371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lowe TM, Eddy SR. A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283(5405):1168–1171. PubMed PMID: 10024243. [DOI] [PubMed] [Google Scholar]
- [91].Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. PubMed PMID: 9023104; PubMed Central PMCID: PMCPMC146525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Gupta Y, Witte M, Möller S, et al. ptRNApred: computational identification and classification of post-transcriptional RNA. Nucleic Acids Res. 2014;42(22):e167 Epub 2014/ 10/10 PubMed PMID: 25303994; PubMed Central PMCID: PMCPMC4267668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132(1):6–13. PubMed PMID: 6312838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Leishmania braziliensis total RNA sequencing data can be accessed from the SRA (accession SRP162992). The ncRNA identification pipeline is available from the corresponding author, AKC, on reasonable request.