

ABSTRACT
SLC47A2 encodes MATE 2-K in the kidney, which mediates the secretion of certain endogenous and exogenous compounds. SLC47A2 was dramatically repressed in patients with renal cell carcinoma (RCC), and a lower level of SLC47A2 might act as a negative prognostic marker, although the mechanism is not well understood. In this study, we aimed to investigate the mechanism via which SLC47A2 is downregulated in RCC. Based on the annotation information of the SLC47A2 locus available in the UCSC genome browser database, we identified a novel lncRNA, which is transcribed from the SLC47A2 locus and named it SANT1. Overexpression and knock-down assays were performed to investigate the effects of SANT1 on cis-regulation of SLC47A2. We verified the direct binding between SANT1 and SFPQ/E2F1/HDAC1 using the cross-linking and immunoprecipitation (CLIP) assay. Chromatin immunoprecipitation was performed to confirm the molecular mechanism via which SANT1 activates the transcription of the SLC47A2 coding region. We observed that SANT1 can cis-regulate its own genetic locus. In tumour-adjacent tissues, the SLC47A2 locus highly expresses SANT1, which can remove the regulatory SFPQ/E2F1/HDAC1 suppressor complex from the promoter region, thereby significantly increasing the levels of the H3K27ac modification and RNAPII binding. Owing to a low SANT1 level, the binding of this inhibitory complex in the promoter region is upregulated in RCC, which results in silencing of the SLC47A2 coding region. In conclusion, we identified a novel lncRNA and elucidated the mechanism via which it regulates SLC47A2 expression in RCC.
KEYWORDS: SANT1, SLC47A2, lncRNA, renal cell carcinoma, epigenetics
Introduction
Renal cell carcinoma (RCC) represents 2–3% cases among all cancers, and is the most common form of kidney cancer [1]. More than 300,000 new patients are diagnosed with RCC worldwide every year [2]. Therefore, understanding the molecular biology of RCC formation and progression is critical for facilitating early diagnosis and developing new therapeutics.
Reports show that RCC is associated with epigenetic modifications of drug transporters. The human organic cation transporter 2 (OCT2/SLC22A2) is downregulated in the proximal tubules of RCC tumour tissues in humans because of methylation of CpG islands around the transcription start sites (TSS) of SLC22A2 [3,4]. In addition, MATE 2-K/SLC47A2 is also strongly repressed in RCC [5]. MATE 2-K, encoded by SLC47A2, is highly expressed in human kidney and mediates the secretion of both endogenous and exogenous compounds. Carnitine and creatinine are the endogenous substrates, and cimetidine, procaine, cisplatin, and oxaliplatin are exogenous substrates of MATE 2-K [6–8]. The balance between H3K4me3 and H3K27me3, and HDAC10 action are responsible for SLC47A2 repression in RCC. H3K4me3 and H3K27me3 provide bivalent regulation of SLC47A2 expression, whereas HDAC10 prevents H3K27ac enrichment at the SLC47A2 promoter, which forms a link between histone deacetylation and methylation [5].
In addition to DNA methylation and histone modifications, non-coding RNAs (ncRNAs) constitute another epigenetic mechanism via which gene expression is regulated. ncRNAs comprise several groups such as miRNA, siRNA, cirRNA, and lncRNA. LncRNAs are more than 200 nucleotide-long endogenous ncRNAs that can regulate gene expression, and are closely correlated with the occurrence and development of diseases [9–11]. LncRNAs perform various regulatory roles, for example, they act as transcriptional regulators, microRNA sponges, scaffolds for protein complexes, and molecular baits in gene regulation [12]. A growing body of evidence indicates that lncRNAs can regulate gene expression at the transcriptional level. For instance, the lncRNA ANCR promotes the enrichment of EZH2 at the Runx2 promoter region and inhibits gene expression by reducing H3K27me3 modification [13].
In preliminary studies, we have observed that MATE 2-K is silenced in RCC, and histone H3K27ac, H3K4me3, and H3K27me3 modifications are involved in regulating gene expression [5]. However, the role of other epigenetic mechanisms involving ncRNAs in the regulation of SLC47A2 expression is still unclear. In this study, we identified a novel lncRNA, SANT1, which can cis-regulate MATE 2-K expression, and identified the regulatory factors involved in forming the RNA-protein (RNP) complex. We further assessed the binding of the regulatory complex at the SLC47A2 promoter by altering SANT1 expression, and finally demonstrated the molecular mechanism via which SANT1 cis-regulates SLC47A2 expression.
Results
Expression pattern of SLC47A2 mRNA and SANT1 in kidney cancer
To determine the changes in the expression of SLC47A2 in RCC and paired adjacent normal tissues, its mRNA and protein levels were detected using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blotting, respectively. SLC47A2 mRNA and protein levels were dramatically lower in RCC tissues than in adjacent normal tissues (Figure 1(a,b)).
Figure 1.
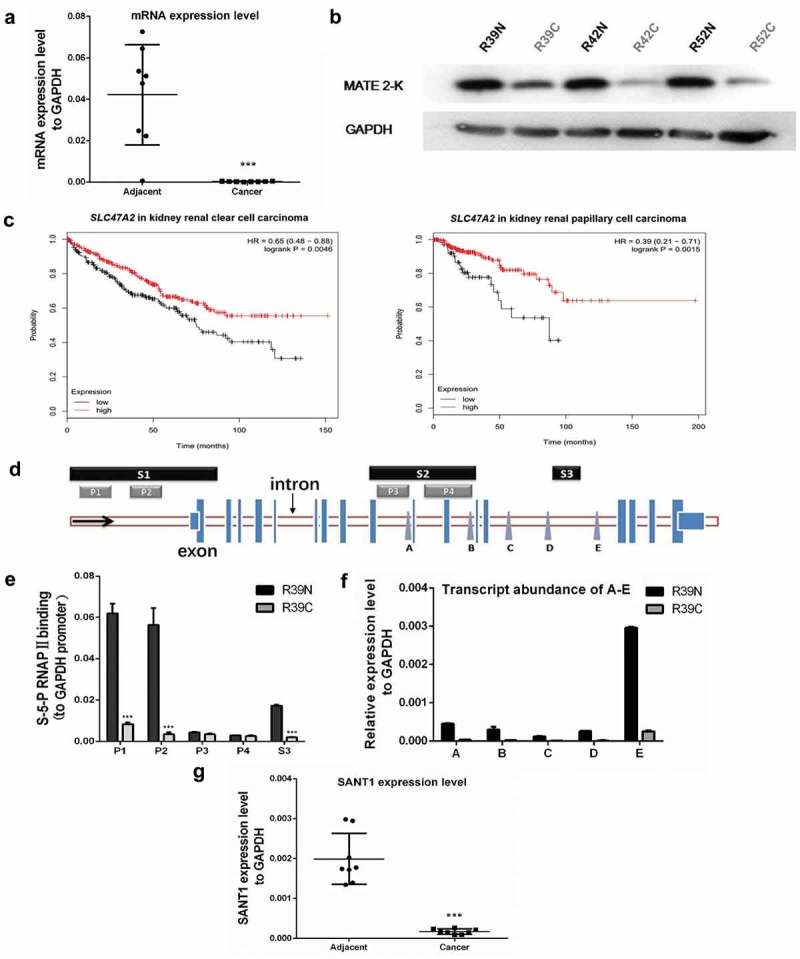
The expression levels of SLC47A2 and SANT1 in RCC tissues were significantly lower than in paired adjacent tissues. (a) The abundance of the SLC47A2 mRNA was strongly reduced in tumour tissues and the expression levels were normalized to GAPDH level (n = 8). (b) The protein levels of MATE 2-K in three RCC tissues (RC) and paired adjacent non-tumour (RN) tissues; the expression levels were normalized to GAPDH level. (c) Kaplan–Meier curves were used to determine the survival probability. Low SLC47A2 expression indicated poor prognosis in kidney renal clear cell carcinoma (p = 0.0046) and kidney renal papillary cell carcinoma (p = 0.0046). (d) The UCSC genome browser revealed the locations of the promoters (P1-P4) and transcription factor (TF) binding sites (S1-S3) in the SLC47A2 locus. The expression levels of non-mRNA transcripts (site A-E) were detected using qPCR. (e) Higher binding levels of S-5-P RNAPII on sections 1 and 3 indicated higher expression levels of mRNA and SANT in adjacent tissue (R39N) than in RCC tissue (R39C). All the data were normalized to the RNAP II binding values in GAPDH promoter. (f) Transcript abundance in the downstream regulatory regions of the SLC47A2 locus. (g) The transcript abundances of SANT1 was strongly reduced in tumor tissues and the expression levels were normalized to that of GAPDH (n = 8).
To determine the correlation between SLC47A2 and prognosis, the univariate test was performed using the Kaplan Meier (KM) plotter database. For renal clear cell carcinoma and renal papillary cell carcinoma, low SLC47A2 expression was shown to be associated with poor prognosis (p = 0.0046 and p = 0.0046, respectively) (Figure 1(c)).
The molecular mechanism via which SLC47A2 is suppressed in RCC is still not well understood. In this study, we investigated whether ncRNAs regulated SLC47A2 suppression. The gene could be regulated by RNAPII occupancy, transcription factor binding, and histone modifications. According to the annotation information from the UCSC genome browser, the SLC47A2 locus might contains four potential promoters (P1-P4) and three transcription factor-binding regions (section 1–3) (Figure S1). The P1 is next to an upstream TSS; P2 is a promoter from both EPD and GeneHancer database; P3 and P4 exhibit abundant RNAPII and Ribosome binding, and they are transcription factor-binding regions too. Three transcription factor-binding regions could be found in ORegAnno section. As shown in Figure 1(d), section 1, which is located upstream of the coding mRNA’s TSS, regulates the expression of its coding gene. However, sections 2 and 3 located in the intronic regions of the SLC47A2 locus might be involved in expressing the SANTs, which might cis-regulate the expression of its coding gene and is also closely related to the especially lower expression level of MATE 2-K in RCC.
To determine the function of sections 2 and 3 in regulating the expression of SANTs in RCC, we determined the S-5-P RNAPII binding levels in these transcriptions factor-binding regions from tumour (R39C) and adjacent (R39N) tissues. We observed that sections 1 (P1 and P2) and 3 displayed significantly lower binding of S-5-P RNAPII in tumour tissue, which suggested lower transcription levels of downstream regions (Figure 1(e)). According to the location of sections 1–3 in the SLC47A2 locus, lower S-5-P RNAPII binding in section 1 corresponded with the lower expression level of MATE 2-K in RCC; section 3, but not section 2, was essential for differential expression of SANT in RCC. To further confirm the relationship between SANT and two non-mRNA-associated transcription factor-binding regions, we determined the expression levels of non-mRNA sequences located downstream of the regulatory regions using qPCR (Figure 1(f)). The transcript abundance of non-mRNA sequences from another two paired tissues were provided in Figure S2. We observed that although both sites A-D (regulated by section 2) and E (regulated by section 3) showed significantly higher expression level in adjacent tissue than in the paired tumour tissue, and the abundance of transcripts from site E was higher than those from other sites. This indicated that section 3 is the most important non-mRNA-associated transcription factor-binding region regulating the expression of SANT in RCC. SANT controlled by section 3 was named SANT1.
SANT1 expression from site E of eight RCC tissues (RC) and paired adjacent tissues (RN) was detected using qPCR. Similar to the SLC47A2 mRNA level, SANT1 was downregulated in RCC (Figure 1(g)). To confirm the sequence of SANT1, the strand-specific qPCRs were carried out. As a result, SANT1 could only be amplified in the sample that reverse transcribed by the Anti-sense primer of site E, and no coding gene transcript could be detected in the samples that reverse transcribed by the Sense or Anti-sense primer of site E (Figure S2). Furthermore, the section 3 is located in the upstream of site E, it seemed that the transcription direction of SANT1 is same to SLC47A2 mRNA. Considering the expression pattern of SANT1, it might regulate SLC47A2 via cis transcriptional regulation and is closely related to lower expression of MATE 2-K in RCC.
SANT1 up-regulated the expression of the SLC47A2 coding region
To investigate the role of SANT1 in regulating the expression of SLC47A2, we obtained the full-length sequence of SANT1 using 5‘/3‘ rapid amplification of cDNA ends (RACE) (Figure S3) and cloned it in an expression vector. The whole sequence of SANT1 is transcribed from the intron SLC47A2 locus and the SANT1 is one member of intronic lncRNA, which is a common form of lncRNA categories (Figure S3).
The SANT1 plasmid was transfected into 786-O, ACHN, 769-P, and Caki-1 cell lines, and the total RNA and protein were harvested 48 h after transfection. Interestingly, SANT1 significantly upregulated the expression of SLC47A2 in all four cell lines, irrespective of cellular heterogeneities (Figure 2(a,c)). The total RNA was also harvested at 72 h after transfection, and similar results were found (Figure S4). The expression of other drug transporters were also determined, which showed that the expression of SLC47A1, ABCB1, ABCC2, and ABCC4 did not change in the presence of high levels of SANT1 (Figure S5), demonstrating the regulatory function of SANT1 in upregulating its own genetic locus. We further constructed a SLC47A2 coding mRNA expression vector and observed that the expression levels of SANT1 in four RCC cells were not induced by high levels of the mRNA (Figure 2(b)). This confirmed that SANT1 is definitely an upstream regulator in the SANT1-mRNA regulatory network. Several targeted siRNAs (Figure S6) were designed to knockdown the expression of the SLC47A2 mRNA (simRNAs) and SANT1 (siSANT1s). The siRNAs were transfected into RCC cells and the expression levels of SLC47A2 were determined using qPCR (Figure 2(d)) and western blot analysis (Figure 2(e)). We observed that SANT1 inhibition significantly reduced the expression of the coding region, which further demonstrated the role of SANT1 in MATE 2-K regulation.
Figure 2.
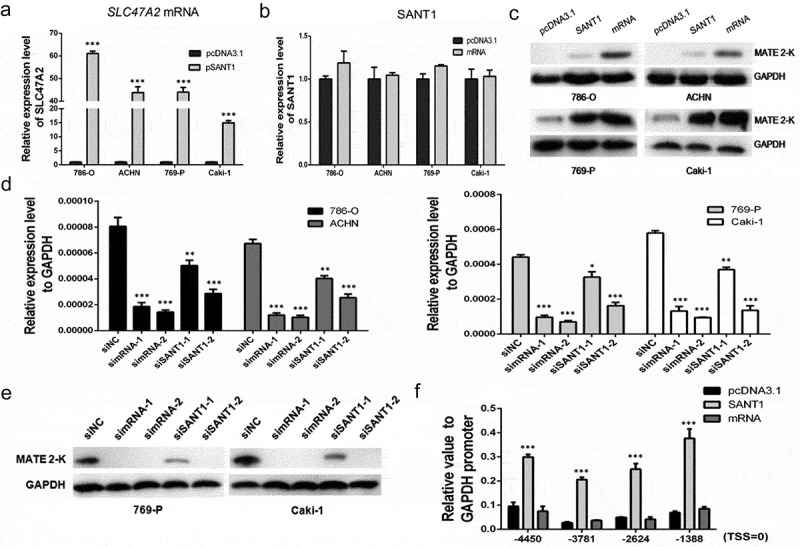
SANT1 overexpression enhanced SLC47A2 expression in RCC cell lines. (a.c) SANT1 upregulated the expression of SLC47A2 mRNA and MATE 2-K. (b) SANT1 expression levels in SLC47A2 mRNA overexpressing cells. (d, e) Four RCC cell lines were transfected with the SLC47A2 mRNA or SANT1 siRNA. SLC47A2 expression was significantly reduced. (f) The coding gene promoter was strongly upregulated by SANT1 in 769-P cells.
We harvested 769-P cells, which highly expressing SANT1 or SLC47A2 mRNA, and evaluated the S-5-P RNA polymerase II (RNAPII) binding rate in section 1, which indicated the transcriptional activity of the coding region. As shown in Figure 2(f), overexpression of the SLC47A2 coding mRNA showed similar S-5-P RNAPII binding level in the negative control, indicating that the coding mRNA was not involved in transcriptional activation in 769-P cells. However, SANT1 strongly promoted activation of the coding gene promoter.
In conclusion, we confirmed that SANT1 enhanced the expression of the coding region by enhancing the activity of the SLC47A2 promoter.
The SANT1 secondary structure is essential for promoting transcription of the SLC47A2 coding region
Previous studies have reported that ncRNAs participate in gene transcriptional regulation by forming numerous RNP complexes [14] or coding micropeptides [15,16]. To further understand the mechanism via which SANT1 regulates gene expression, we analysed the secondary structure of SANT1 and then constructed two types of SANT1 mutants (named SANT1-S1 and SANT1-S2). We amplified a certain sequence from the 3‘ terminus of SANT1 and sub-cloned the amplified products into pcDNA3.1 (+) with two different enzyme digestion schemes (Figure S2). As a result, these two SANT1 mutants exhibited identical fundamental sequence and translation potency but dramatically different secondary structures (Figure 3(a)). To confirm the transcriptional start site of the rescued transcripts, two kinds of forward primers were designed, one is close to the TSS site (S0), and another one is located on the upstream of TSS site (S-57) (Figure S7). As a result, the cDNAs could be amplified by S0/A + 784, and no products were detected by using S-57/A + 784. The plasmids expressed the RNAs with right size as proposed, it is receivable that the plasmids transcribed the SANT1s start from the putative TSS site of the vector. Loops A and B highlighted similarities in the secondary structure between SANT1-S1 and the 3‘ terminal of wild-type SANT1.
Figure 3.
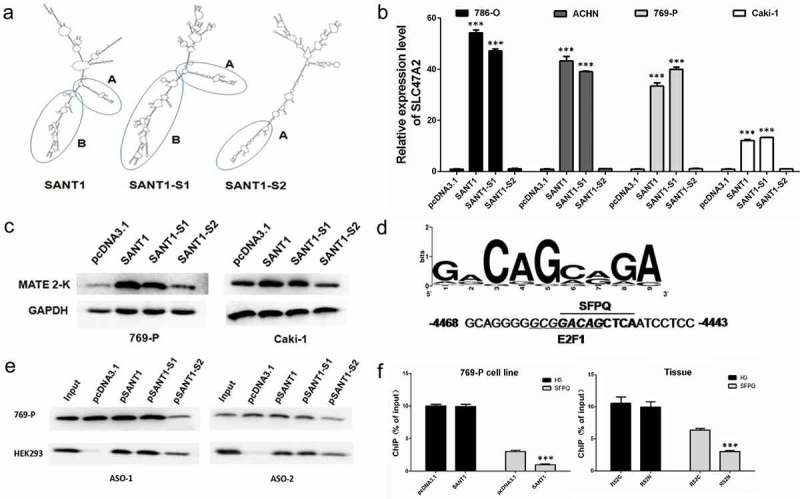
Secondary structure and functions of SANT1, SANT1-S1, and SANT1-S2. (a) The secondary structure of the three SANT1s. (b, c) The perfect secondary structure is the primary requirement for SANT1-mediated promotion of SLC47A2 expression. (d) The binding site of SFPQ in the coding gene promoter. (e) Direct interaction between SANT1s and SFPQ. (f) Overexpression of SANT1 significantly inhibited the binding of SFPQ to the coding gene promoter in 769-P cells and adjacent tissue.
The three types of SANT1 were transfected in RCC cells and the total RNAs and proteins were harvested after 48 KRNB_A_1602436h. SANT1-S1 showed the similar inductive effect as wild type SANT1. As shown in Figure 3(b,c), the expression of the SLC47A2 coding region was significantly increased by overexpressing SANT1 or SANT1-S1, whereas SANT1-S2 could not induce gene expression, irrespective of its sequence similarity with SANT1-S1. This suggested that a perfect secondary structure is the primary requirement for SANT1 function, and that SANT1 might act as a molecular scaffold for constructing RNP complexes.
SANT1 induced off-target binding of E2F1/HDAC1 to the coding gene promoter
As described earlier, SANT1 might be involved in gene regulation via formation of functional lncRNA-protein complexes. To determine the members of this complex, computational analysis was performed to investigate lncRNA-protein interactions. According to the results of the catRAPID omic analysis, the SFPQ protein was forecast to be a strong partner of SANT1 and SANT1-S1 (Table S3). However, SFPQ also exhibited weak interaction with SANT1-S2, irrespective of its differential secondary structure. We further analysed the binding site of SFPQ in the coding gene’s promoter (from −5000 to +300) using the LASAGNA-Search 2.0 software and observed one SFPQ binding site in section 1 (Figure 3(d), in P1 promoter, −4450).
We confirmed a direct interaction between SANT1s and SFPQ using cross-linking and immunoprecipitation (CLIP) (Figure 3(e)). In brief, we transfected pcDNA3.1 (NC), SANT1, SANT1-S1, or SANT1-S2 into 769-P cells and purified the SANT1-protein complex after 48 h using two biotin-labelled antisense oligonucleotides (ASOs). The presence of SFPQ in the purified samples was detected using western blot analysis. As shown in Figure 3(e), SFPQ was detected in SANT1 or SANT1-S1 expressing 769-P using ASO-1 and ASO-2. In the blank plasmid group (NC), the constitutive expression of SANT1 was recognized using ASOs, and the presence of SFPQ was also detected. In contrast, in SANT1-S2-transfected 769-P cells, we observed weak exposure of SFPQ. These results suggested that SANT1-S2 might exhibit a weaker binding activity because of its different structures, which is in agreement with the results of the catRAPID omics report (Table S3). Another possibility was that the defective structure of SANT1-S2 completely impaired the binding of SFPQ; however, the ASOs purified the SFPQ interacted with the constitutively expressed SANT1, and this kind of interaction between ASOs and SANT1 could be competitively inhibited by high levels of SANT1-S2.
To further understand whether SANT1-S2 retained the SFPQ binding ability, we investigated CLIP samples from HEK-293 cells (Figure 3(e)). Constitutive SANT1 expression was low in HEK-293 cells, and no SFPQ was detected in the NC group. SFPQ was detected in the samples purified from three SANT1-expressing cells, and fully functional SANT1s (SANT1 and SANT1-S1) exhibited stronger binding abilities than the activation-deficient one (SANT-S2).
Thus, we concluded that SFPQ is one of the members of the SANT1-associated regulatory complex. SANT1-S2 exhibited weak binding with SFPQ but could not activate gene expression. Considering the significant change in SANT1-S2 secondary structure, we suggested that perfect secondary structure of SANT1 is necessary for forming the intact complex, and that the missing structure in SANT1-S2 might be involved in interacting with other proteins of the regulatory complex.
To confirm the interaction between SANT1 and SFPQ, we determined the binding rates of SFPQ in the coding gene’s promoter (Figure 3(f)). SANT1 and the blank plasmid were transfected into 769-P cells, and as described previously, SANT1 strongly upregulated the expression of the coding gene (Figure 2(f)). In contrast, high SANT1 expression significantly suppressed the binding of SFPQ to the coding gene’s promoter. In addition, the binding level of SFPQ was significantly higher in RCC tissue (R52C) than in paired adjacent tissue (R52N). Owing to higher SANT1 level in adjacent tissues (Figure 1(g)), we suggested that a high level of SANT1 inhibited the binding of SFPQ on the coding gene promoter. It appears that SFPQ binding to the promoter restrained the transcriptional activity by forming a gene repressor complex.
As reported previously, SFPQ frequently acts as a molecular scaffold at target promoters and participates in epigenetic silencing via formation of a corepressor complex by recruiting SIN3A and histone deacetylases (HDAC1 or HDAC3) [17, 18]. Previously, we have shown that E2F1 might be involved in restricting histone acetylation (H3K27ac) of the SLC47A2 promoter by recruiting HDAC10 (5). E2F1/HDAC1 and E2F1/HDAC3 are also well known for inhibiting gene expression by suppressing the H3K27ac modification at gene promoters [5]. Considering the common function of SFPQ and E2F1 in recruiting HDACs, we predicted that the SFPQ/E2F1/HDAC1 complex is instrumental in repressing SLC47A2. Furthermore, the SFPQ binding site on the SLC47A2 promoter is in proximity to an E2F1 binding site, which indicates that these proteins might interact physically (Figure 3(d)).
The formation of the SFPQ/E2F1/HDAC1 complex was confirmed in 769-P using the co-immunoprecipitation (co-IP) assay. We selected SFPQ, E2F1, or HDAC1 as the target protein, and precipitated the entire complex from the total cell lysate using anti-SFPQ, anti-E2F1, or anti-HDAC1 antibodies, respectively. As shown in Figure 4(a), we successfully determined the existence of SFPQ, E2F1, and HDAC1 in each co-IP sample, which strongly suggested the existence of the SFPQ/E2F1/HDAC1 complex. To further verify the relationship between SANT1 and the SFPQ/E2F1/HDAC1 complex, we performed the co-IP assay in HEK293 cells, which expresses extremely low levels of SANT1, and confirmed the formation of the SFPQ/E2F1/HDAC1 complex in the absence of SANT1. Therefore, we proposed two potential mechanisms: 1) SANT1 is involved in target binding of the SFPQ/E2F1/HDAC1 complex at the SLC47A2 promoter but is not necessary for its formation; 2) SANT1 participates in the formation of the SLC47A2-regulatory complex, but does not affect the formation of other similar regulatory complexes.
Figure 4.
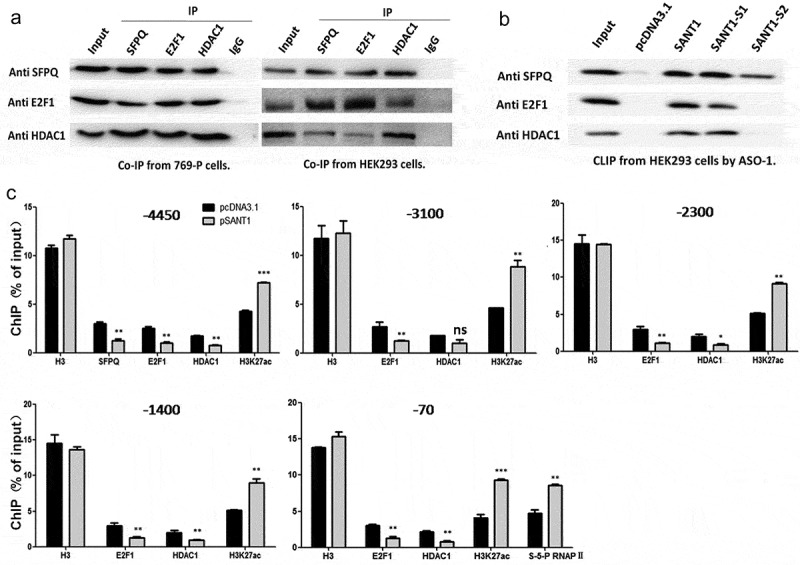
SANT1 altered the binding of SFPQ/E2F1/HDAC1 to the coding gene promoter. (a) Formation of the transcription suppressor SFPQ/E2F1/HDAC1 complex. (b) The perfect secondary structure of SANT1 is necessary for the formation of a functional RNP complex. (c) High expression of SANT1 promotes H3K27ac modification and S-5-P RNAPII binding to the SLC47A2 promoter by inducing off-target binding of E2F1/HDAC1 to the coding gene promoter.
To confirm the role of SANT1 in the formation of the SFPQ/E2F1/HDAC1 complex, we transfected the blank vector, SANT1, SANT1-S1, or SANT1-S2 into HEK293 cells and then purified RNA-binding proteins via CLIP using ASO-1 as the probe. As shown in Figure 4(a), the members of the SFPQ/E2F1/HDAC1 complex were detected in each precipitated sample. All members were detected in cells expressing SANT1 and SANT1-S1, but only SFPQ was detected in cells expressing SANT1-S2 (Figure 4(b)). Considering the extreme differences in secondary structure between SANT1/SANT1-S1 and SANT1-S2, we concluded that the perfect secondary structure of SANT1 is necessary for forming a functional RNP complex.
To further understand the function of SANT1 in regulating the binding of the SFPQ/E2F1/HDAC1 complex to the promoter, we overexpressed SANT1 in 769-P cells and detected the binding rate of the SFPQ/E2F1/HDAC1 complex to the entire SLC47A2 promoter (Figure 4(c)). According to the computational analysis of binding of transcription factors, the coding gene’s promoter contained only one SFPQ (−4450) binding site, which was in proximity to an E2F1 binding site (Figure 3(d)). We also detected abundant E2F1 binding sites from −4000 to +1 in the coding gene’s promoter. Therefore, the other four E2F1 biding sites (−3100, −2300, −1400, and −70) were also selected for determining the E2F1/HDAC1 binding rate after overexpressing SANT1 in 769-P cells.
As shown in Figure 4(c), overexpression of SANT1 reduced the binding rate of SFPQ/E2F1/HDAC1 to the SFPQ (also E2F1) binding site in the promoter region. On the contrary, the level of H3K27ac, which is regulated by HDAC1, increased significantly. In addition, the extent of E2F1/HDAC1 binding was also significantly reduced at the other E2F1 promoter binding sites. These results showed that the level of the H3K27ac modification increased over the entire SLC47A2 promoter and that the binding of S-5-P RNAPII was strongly upregulated in the region proximal to the TSS. This suggested that SANT1 overexpression resulted in higher level of H3K27ac modification and S-5-P RNAPII binding in the SLC47A2 promoter by inducing off-target binding of E2F1/HDAC1.
Discussion
SLC47A2 was identified as a promising diagnostic biomarker and is considered to be tightly associated with risk of developing RCC as its transcription is significantly repressed in all patients tested [5]. Furthermore, univariate analyses indicated that SLC47A2 expression is a prognostic risk factor for patients with RCC, and lower level of SLC47A2 might act as a negative prognostic marker (Figure 1(c)). SLC47A2 expression is strongly suppressed in RCC (Figure 1(a,b)), which might significantly change the function of kidney cells in excreting anti-cancer drugs and endogenous substances [5]. However, evidence regarding the role of SLC47A2 in RCC aetiology or progression is absent.
With the development of large-scale sequencing technology, the aberrant transcriptional profiles and transcription regulatory mechanisms in kidney cancer were investigated, and many ncRNAs were found to be involved in the progression of kidney cancer [19,20]. The abnormal expression of ncRNAs plays important roles in tumour genesis, growth, or metastasis through their interactions with other cellular macromolecules [21]. Accumulating evidence suggests that lncRNAs are important regulators of almost all gene expression networks and are involved in regulatory processes, ranging from epigenetic, transcriptional, and post-transcriptional processes [22].
Depending on their functional models, lncRNAs can regulate adjacent or own loci via cis regulation, or regulate distal genes via trans regulation [23]. In this study, we focused on detecting the cis regulatory model of SLC47A2. Initiation and elongation of most lncRNAs require the same transcriptional machinery as other mRNAs, and lncRNAs are regulated by RNAPII occupancy, transcription factor binding, and histone modifications [23]. We obtained the full-length sequence of lncRNA SANT1 by evaluating the binding rate of S-5-P RNAPII at the potential transcriptional regions in the SLC47A2 locus, and finally confirmed the cis regulatory model of SANT1 in four RCC cell lines.
Many lncRNAs display cis regulatory functions by altering transcription factor binding to gene promoter regions [24]. However, recent findings showed that certain functional micropeptides can be encoded by lncRNAs [15,16]. It appears that lncRNAs act as gene regulators by forming RNP complexes or translating functional micropeptides. To further understand the regulatory mechanism of SANT1, we analysed its secondary structure and constructed two SANT1 mutants (Figure 3(a)). Figure 3(b,c) show that SANT1 does not translate potential micropeptides but acts as a modular scaffold in activating the performance of the SLC47A2 coding gene’s promoter; additionally, SANT1 must have a perfect secondary structure for forming a regulatory RNP complex (Figures 3, 4). Furthermore, lncRNAs act as miRNA sponge, thereby regulating miRNA expression, which in turn suppresses the targeted binding between specific genes and miRNAs [25]. It is also possible that SANT1 regulates the expression of SLC47A2 through lncRNA–mRNA/miRNA interaction in RCC, which will be extensively studied in future.
The SFPQ protein was forecast to be a strong partner of SANT1 and SANT1-S1 (Table S3), and we confirmed the direct binding between SANT1/SANT1-S1 and SFPQ using the CLIP assay. The binding rate of SFPQ to the SLC47A2 promoter was significantly suppressed by high SANT1 level (Figure 3(f)), which is indicative of the inhibitory ability of the SFPQ-associated functional complex. SFPQ/HDACs [17,18] and E2F1/HDACs [5] are well-known to repress gene expression by decreasing H3K27ac modification in the gene promoter. Considering that HDAC1 is the common member in SFPQ/HDAC and E2F1/HDAC complexes, we predicted that the SFPQ/E2F1/HDAC1 complex was a transcriptional repressor of SLC47A2. The existence of the SFPQ/E2F1/HDAC1 complex was verified using co-IP, and SANT1 was involved in relocating the regulatory repressor from the promoter region. However, in tumour tissues, lower expression of SANT1 and higher expression of E2F1 [5] resulted in significantly higher binding of E2F1/HDAC1 to the SLC47A2 promoter, which repressed MATE 2-K by reducing H3K27ac modification and S-5-P RNAPII binding.
In this study, we observed that repression of SLC47A2, which was identified as a promising diagnostic biomarker, correlated positively with overall survival in 818 patients with RCC cancer (Figure 1(c)). While investigating the molecular mechanism underlying SLC47A2 repression, we identified a novel human lncRNA, SANT1, which is transcribed from the SLC47A2 locus. We identified a new mechanism of SANT1-mediated SLC47A2 regulation, which involved the relocation of an inhibitory SFPQ/E2F1/HDAC1 complex from the promoter and increase in H3K27ac modification and S-5-P RNAPII binding. In RCC tissue, low SANT1 expression increased E2F1/HDAC1 binding to the entire coding gene promoter, which induced a significantly lower level of histone H3K27ac modification (Figure 5). The SANT1-SLC47A2 regulatory network promoted abnormal gene expression in RCC. Considering the tight correlation between SANT1 and SLC47A2 expression, SANT1 expression level, as well as SLC47A2 expression, can be used as a prognostic marker for RCC. However, the reason behind the extremely low SANT1 level is still unclear, and the mechanism underlying the association of SLC47A2 repression with poor prognosis in patients with renal cancer requires further investigation.
Figure 5.
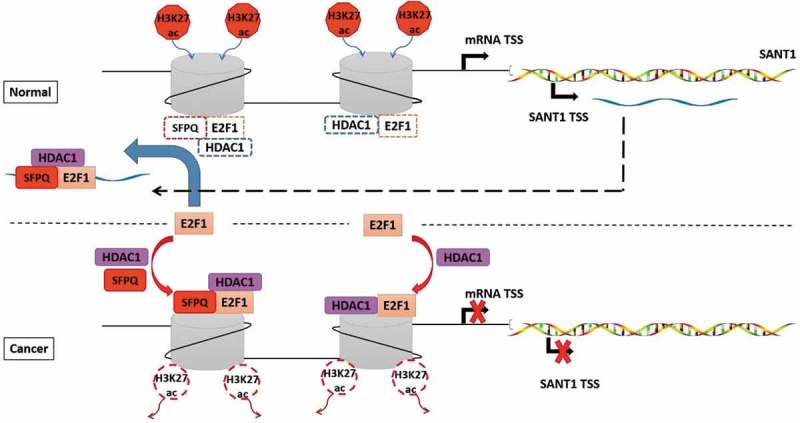
SANT1 induced off-target binding of E2F1/HDAC1 to the entire coding gene promoter.
Materials and methods
Tissues and cell culture
The paired RCC samples were provided by the Specimen Bank of Zhejiang Cancer Hospital (Hangzhou) and were approved for use by the Zhejiang Cancer Hospital Ethics Committee ([2014]-08-76). Patients‘ clinical information is shown in Supplementary Table S1.
HEK293 and human RCC cell lines 786-O, 769-P, ACHN, and Caki-1 were purchased from the Chinese Academy of Science Committee on type culture collection cell libraries. HEK293 was cultured in Dulbecco’s modified Eagle’s medium (DMEM), 786-O and 769-P were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium, ACHN was cultured in minimal essential medium (MEM), and Caki-1 was cultured in McCoy’s 5 A medium. All media contained 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin.
Bioinformatic analysis
Potential promoter analysis of the SLC47A2 locus was performed using the UCSC genome browser (GRCh37/hg19). SANT1 secondary structure prediction was performed on M-Fold. SANT1-protein interaction was predicted using catRAPID. SFPQ and E2F1 binding sites analysis were performed using LASAGNA-Search 2.0 and PROMO, respectively. In the current study, the prognostic values of SLC47A2 in RCC were analysed using KM plotter, which updated gene expression data and survival information from 818 patients with renal cancer.
Chromatin immunoprecipitation assay (ChIP)
ChIP was performed per the protocol of the ChIP assay kit (Beyotime, P2078). In briefly, RCC samples were fully shredded with scissors and ground with a homogenizer, and then were rotational cross-linked with 1% formaldehyde for 30 min, cells were cross-linked with 1% formaldehyde for 10 min at 37°C. Then the samples were neutralized with glycine for 5 min at room temperature, all samples were washed by cold PBS, then collected and stored on ice. Next, the samples were lysed with SDS lysis buffer, and DNA was shredded to fragments of 200–1000 bp by sonication. An equal amount of chromatin was immunoprecipitated at 4°C overnight with 1.5 µg of the following antibodies: anti-H3 (Abcam, ab1791, 1mg/ml), anti-S-5-P RNAPII (Abcam, ab5095, 1mg/ml), anti-SFPQ (GeneTex, GTX114209, 1.36mg/ml), anti-E2F1 (GeneTex, GTX101235, 1mg/ml), anti-HDAC1 (GeneTex, GTX100513, 1mg/ml), and anti-H3K27ac (Abcam, ab4729, 1mg/ml). Antibodies were added to each aliquot of pre-cleared chromatin and incubated overnight. Protein A + G-agarose beads were added and incubated for 2 h at 4°C. After reversing the cross-links, the immunoprecipitated DNA was purified and subjected to qPCR analysis, and the primers used are listed in Supplementary Table S2.
RACE of SANT1
Total RNA (2 μg) from adjacent (R39N) tissue was reverse transcribed using the primer 3-AD, which contains a lock-docking oligo (dT). The three rounds of 3‘ RACE PCR were performed using 3RACE-S1/NUP, 3RACE-S2/NUP, and 3RACE-S3/NUP.
5‘ RACE amplification was performed using the ribonucleotide tailing method. Total RNA (4 μg) was reverse transcribed using the primer 5RACE-A1. The cDNA mixture was incubated with RNase H for 1 h at 37°C, and then the cDNA was processed to generate a ribo-tail using terminal deoxynucleotidyl transferase (TdT) (Takara, 2230A) and rCTP. The first round of PCR was performed using 5-AD/5RACE-A2, and the following amplifications were performed using NUP/5RACE-A3, NUP/5RACE-A4, and NUP/5RACE-A5. All the primers are listed in Supplementary Table S2.
Plasmid construction
Full-length SLC47A2 mRNA, SANT1, and SANT1 mutants (SANT1-S1 and SANT1-S2) were cloned into the pcDNA3.1(+) vector. The SLC47A2 mRNA was amplified using mRNA-S/mRNA-A. Full-length SANT1 and SANT1-S1were amplified using SANT1-S/SANT1-A and SANT1-S1/SANT1-A1, respectively. SANT1-S2 was amplified from SANT1-S1 using SANT1-S2/SANT1-A2. The SLC47A2 mRNA and SANT-S1 were sub-cloned into the EcoR Ⅴ restriction enzyme site. SANT1 was sub-cloned between Sac I/Xho I restriction enzyme sites, and SANT-S2 was sub-cloned into Sac I/Xba I sites. The differences between the three forms of SANT1 are shown in Supplementary Figure S1 and primers are shown in Supplementary Table S2.
To confirm the transcriptional start site of the rescued SANT1s transcripts, total RNAs from the plasmid-rescued cells were extracted as described before. The RNAs were treated by DNase Ⅰ and reverse transcribed using the primer A923. The PCR detections were carried out using S0/A784 or S-57/A784. The location of primers is shown in Supplementary Figure S6, and the sequences could be found in Supplementary Table S2.
Real-time PCR
Total RNA was extracted using the total RNA mini-prep kit (Tiangen) and then reverse transcribed into cDNA with PrimeScript RT master mix (Takara). Real-time PCR was performed using the SYBR Premix EX Taq (Takara); the reaction system included 1 μL cDNA, 1 μL primer, 0.2 μL ROX dye, 4.8 μL SYBR® Premix Ex TaqTM, and 3 μL double distilled H2O. mRNA expression level was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. The primers used are listed in Supplementary Table S2.
Western blot analysis
Cells were cultured in six-well plate, washed with phosphate buffered saline (PBS) after maturation, harvested with 0.25% trypsin, and then lysed using radioimmunoprecipitation assay (RIPA) (Beyotime, P0013B) supplemented with phenylmethylsulfonyl fluoride. The concentrations of the protein samples were quantified using the bicinchoninic acid (BCA) protein assay kit (Beyotime). Equal amounts of protein extracts were separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride (PVDF) membranes. The PVDF membranes were blocked for 2 h in 5% nonfat milk and incubated overnight with primary antibodies at 4°C, followed by incubation with secondary antibodies for 2 h at room temperature. Finally, the PVDF membranes were exposed to colouration using G-box. MATE 2-K antibody (Sigma, HPA062112, 1:200 dilution) was used in western blot analysis.
Co-IP
Cells were cultured in a 10-cm cell culture dish and washed with PBS after maturation. The cells were digested with 0.25% trypsin and collected into 1.5 mL tubes. The cells were washed once with precooled PBS. Whole cell lysates were prepared using Pierce™ IP lysis buffer (Thermo, 87787) supplemented with protease inhibitors. Lysates were incubated with 2 µg of antibodies and 30 µl of protein A + G agarose (Beyotime, P2012). The complexes were collected by centrifugation. Target proteins were detected using western blot analysis. Antibodies used for co-IP included anti-SFPQ (GeneTex, GTX114209, 1.36mg/ml), anti-E2F1 (GeneTex, GTX101235, 1mg/ml), anti-HDAC1 (GeneTex, GTX100513, 1mg/ml), and anti-IgG (Santa Cruz, sc-2027, 1mg/ml).
CLIP
Cells were cultured in a 10-cm cell culture dish. The cells were exposed to UV light to crosslinking the protein-RNA complexes in vivo. Cells were harvested by trypsinization and washed three times with pre-cooled PBS. PBS was removed and 2 ml of lysis buffer (Thermo Fisher, 87788) supplemented with protease, RNase, and DNase inhibitors were added to the pellet to lyse the cells. The lysate was centrifuged at 12,000 g for 10 min at 4°C, and the supernatant collected for RNA isolation. Protein concentration of eluate was determined with the BCA method. ~35 µg protein was transferred to a 1.5 ml tube, then incubated with 200 pmol of antisense oligonucleotides (ASOs), mixed and incubated at 70°C for 5 min. ASOs used in CLIP are listed in Supplementary Table S2. Blocked streptavidin agnetic beads were added, and the mixture was incubated for 30 min at room temperature with constant shaking. The beads were retrieved using a magnet and the supernatant was collected for reference. The beads were then washed three times with 750 µl B&W buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA, pH 8.0) at 55°C. Finally, 50 µl of B&W buffer was added and the sample split: 20 µl for RNA and 30 µl for protein analysis. Removed the B&W buffer and the beads were resuspended by 20 µl of Tris-HCl (10 mM, pH 7.5) and then incubated at 90°C for 10 min until the RNA released from beads. For protein analysis, the beads were resuspended in 20 µl of Laemmli buffer and denatured at 100°C for 5 min.
Statistical analysis
Results are presented as mean ± standard deviations (SD). Statistical comparisons were made using the Mann–Whitney test in GraphPad Prism 5, and differences between groups were considered significant if the P value was <0.05.
Funding Statement
This work was supported by the [National Natural Science Foundation of China] under Grant [number 81773817] and [Key Technologies R&D Program of China] under Grant [number 2017YFC0908600].
Acknowledgments
We thank Ms. Haihong Hu for managing the instruments and helping us perform the experiments.
Complying with ethics of experimentation
The procedures used in this study were approved by the Zhejiang Cancer Hospital Ethics Committee ([2014]-08-76).
Data deposition
The nucleotide sequence of SANT1 has been submitted to the GenBank with the accession number MH971975.
Data availability statement
The data that support the finding of this study are available from the corresponding author (Su Zeng and Lushan Yu), upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Ljungberg B, Bensalah K, Canfield S, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol. 2015;67:913–924. [DOI] [PubMed] [Google Scholar]
- [2].Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2014;136:E359–E386. [DOI] [PubMed] [Google Scholar]
- [3].Liu Y, Zheng X, Yu Q, et al. Epigenetic activation of the drug transporter OCT2 sensitizes renal cell carcinoma to oxaliplatin. Sci Transl Med. 2016;8(348):348ra97. [DOI] [PubMed] [Google Scholar]
- [4].Zheng X, Liu Y, Yu Q, et al. Response to Comment on “Epigenetic activation of the drug transporter OCT2 sensitizes renal cell carcinoma to oxaliplatin”. Sci Transl Med. 2017;9(391):eaam6298. [DOI] [PubMed] [Google Scholar]
- [5].Yu Q, Liu Y, Zheng X, et al. Histone H3 lysine 4 trimethylation, lysine 27 trimethylation, and lysine 27 acetylation contribute to the transcriptional repression of solute carrier family 47 member 2 in renal cell carcinoma. Drug Metab Dispos. 2017;45:109. [DOI] [PubMed] [Google Scholar]
- [6].Omote H, Hiasa M, Matsumoto T, et al. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol Sci. 2006;27:587–593. [DOI] [PubMed] [Google Scholar]
- [7].Damme K, Nies AT, Schaeffeler E, et al. Mammalian MATE (SLC47A) transport proteins: impact on efflux of endogenous substrates and xenobiotics. Drug Metab Rev. 2011;43:499–523. [DOI] [PubMed] [Google Scholar]
- [8].Staud F, Cerveny L, Ahmadimoghaddam D, et al. Multidrug and toxin extrusion proteins (MATE/SLC47); role in pharmacokinetics. Int J Biochem Cell Biol. 2013;45:2007–2011. [DOI] [PubMed] [Google Scholar]
- [9].Kapranov P, Cheng J, Dike S, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484. [DOI] [PubMed] [Google Scholar]
- [10].Fok ET, Scholefield J, Fanucchi S, et al. The emerging molecular biology toolbox for the study of long noncoding RNA biology. Epigenomics. 2017;9:1317–1327. [DOI] [PubMed] [Google Scholar]
- [11].Quinn JJ, Chang HY.. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2015;17:47. [DOI] [PubMed] [Google Scholar]
- [12].Jain S, Thakkar N, Chhatai J, et al. Long non-coding RNA: functional agent for disease traits. RNA Biol. 2017;14(5):522–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhu L, Xu PC. Downregulated LncRNA-ANCR promotes osteoblast differentiation by targeting EZH2 and regulating Runx2 expression. Biochem Biophys Res Commun. 2013;432:612–617. [DOI] [PubMed] [Google Scholar]
- [14].Hafner M, Landthaler M, Burger L, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Anderson Douglas M, Anderson Kelly M, Chang C-L, et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bazzini AA, Johnstone TG, Christiano R, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. Embo J. 2014;33(9):981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Snijders AP, Hautbergue GM, Bloom A, et al. Arginine methylation and citrullination of splicing factor proline- and glutamine-rich (SFPQ/PSF) regulates its association with mRNA. Rna. 2015;21:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Knott GJ, Bond CS, Fox AH. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 2016;44:3989–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yu G, Yao W, Wang J, et al. LncRNAs expression signatures of renal clear cell carcinoma revealed by microarray. PLOS ONE. 2012;7:e42377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Blondeau JJC, Deng M, Syring I, et al. Identification of novel long non-coding RNAs in clear cell renal cell carcinoma. Clin Epigenetics. 2015;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang X, Hamblin MH, Yin K-J. The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. 2017;14(12):1705–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300. [DOI] [PubMed] [Google Scholar]
- [24].Zhang B, Arun G, Mao YS, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2(1):111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Song X, Cao G, Jing L, et al. Analysing the relationship between lncRNA and protein-coding gene and the role of lncRNA as ceRNA in pulmonary fibrosis. J Cell Mol Med. 2014;18:991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the finding of this study are available from the corresponding author (Su Zeng and Lushan Yu), upon reasonable request.