

ABSTRACT
Target binding by CRISPR-Cas ribonucleoprotein effectors is initiated by the recognition of double-stranded PAM motifs by the Cas protein moiety followed by destabilization, localized melting, and interrogation of the target by the guide part of CRISPR RNA moiety. The latter process depends on seed sequences, parts of the target that must be strictly complementary to CRISPR RNA guide. Mismatches between the target and CRISPR RNA guide outside the seed have minor effects on target binding, thus contributing to off-target activity of CRISPR-Cas effectors. Here, we define the seed sequence of the Type V Cas12b effector from Bacillus thermoamylovorans. While the Cas12b seed is just five bases long, in contrast to all other effectors characterized to date, the nucleotide base at the site of target cleavage makes a very strong contribution to target binding. The generality of this additional requirement was confirmed during analysis of target recognition by Cas12b effector from Alicyclobacillus acidoterrestris. Thus, while the short seed may contribute to Cas12b promiscuity, the additional specificity determinant at the site of cleavage may have a compensatory effect making Cas12b suitable for specialized genome editing applications.
KEYWORDS: CRISPR-Cas, seed sequence, target binding, off-target activity, genome editing
Introduction
CRISPR-Cas systems provide prokaryotes with adaptive immunity against foreign genetic elements such as phages and plasmids [1–3]. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) loci consist of direct DNA repeats, separated by unique spacer sequences (CRISPR arrays), and cas (CRISPR-associated) genes. While the provenance of the vast majority of spacers remains unknown, some originate from mobile genetic elements such as plasmids and viruses [4]. From the evolutionary standpoint, CRISPR-Cas systems are highly diverse. They can be classified into two classes, several types and multiple subtypes [5]. Despite this diversity, all CRISPR-Cas systems share common principles of function. Transcription of CRISPR array followed by processing of the primary transcript results in production of small CRISPR RNAs (crRNAs), each containing a specific spacer sequence. Individual crRNAs guide Cas ‘effector’ proteins to complementary target sequences (protospacers) causing their endonucleolytic destruction, a process referred to as ‘CRISPR interference’. CRISPR interference thus protects prokaryotic hosts from mobile genetic elements. In DNA-targeting CRISPR-Cas systems, the presence of a protospacer-adjacent motif (PAM) is required for target recognition [6–8], in addition to spacer-protospacer complementarity. Point mutations in a protospacer targeted by CRISPR-Cas or its PAM allow plasmids and viruses to escape CRISPR interference [6,7].
The ability to program target recognition and destruction by short RNAs made it possible to harness CRISPR-Cas effectors for genome editing applications [9]. Partly through historic reasons, the highly active class 2 (single-subunit effectors) Cas9 (Type II) protein from Streptococcus pyogenes (SpCas9) is currently the most widely used programmable nuclease. It recognizes a G-rich PAM downstream of the protospacer and introduces double-stranded break in protospacer DNA that results in blunt ends [10]. Despite its popularity, SpCas9 has some disadvantages caused by its large size, recognition and cleavage of off-target sites partially matching crRNA spacer, and stringent requirement for an NGG PAM, which constrains the choice of targetable sites [11,12]. Considerable efforts have been made to obtain Type II CRISPR-Cas effectors with improved characteristics through rational engineering [13] and bioinformatics searches for new CRISPR-Cas systems and their effectors [14,15]. The latter approach resulted in discovery and validation of new Class 2 CRISPR-Cas systems that formed a new type, Type V [16]. Type V effectors have been grouped into several subtypes, whose distinct effectors independently evolved from unrelated transposases [16,17]. Cpf1 (Cas12a) Type V-A effectors have already found applications in genome editing [18]. Type V-B Cas12b effectors are less studied. Both Cas12a and Cas12b recognize upstream AT-rich PAMs and cleave target DNA in a staggered pattern [14,19–22]. Similarly to Cas9 but unlike Cas12a effectors, Cas12b requires an additional small tracrRNA for processing of CRISPR array transcript to generate mature crRNA [14,15].
Target location is a critical step both during CRISPR interference and defense from mobile genetic elements and in practical applications. Early stages of CRISPR-Cas effectors’ interaction with DNA include PAM recognition, duplex DNA destabilization, and initial pairing of target DNA with crRNA spacer part in the PAM-proximal protospacer segment [23]. The importance of a perfect base pair match in the PAM-proximal sequence (referred to as ‘seed’ [7]) for target binding has been documented in different systems. Interference was abrogated due to mutations in 10-nt seed sequence of the Type I-B H. volcanii system [24], 7-nt seed in the Type I-E E. coli system [7], 8-nt seed in Type I-F P. aeruginosa system [25], and 12-nt seed of the Type II S. pneumoniae system [26]. Conversely, protospacer positions outside the seed can be mismatched with crRNA spacer without affecting target binding.
Molecular aspects of target recognition and cleavage by Cas12b remain to be discovered. Recently, two structures of Cas12b from Alicyclobacillus acidoterrestris (AacCas12b) were determined [20,21]. The structure of AacCas12b bound to single-guide RNA (a hybrid of processed crRNA and tracrRNA) revealed that a short 5-nt segment of the guide was highly structured and adopted a pre-organized nearly A-form conformation [20,21]. The Watson-Crick edges of the bases were exposed toward solvent and available for base pairing with target DNA [20,21]. This ordered segment was on the ‘PAM-side’ of the complex. Other spacer RNA nucleotides were disordered in the binary complex and could not be traced [20,21]. In various RNA-guided systems, the seed sequence of the guide is typically pre-ordered in a helical conformation to reduce the entropy penalty for target binding [27–29]. It therefore appeared that AacCas12b has a short seed. A structure of B. thermoamylovorans Cas12b (BthCas12b) also revealed a preordered 5-nt crRNA segment [22]. A similar arrangement was observed in the Cas12a effector structure [30] and functional analyses indicated that mismatches in the first 5 PAM-proximal base pairs prevent target recognition [14]. It was thus highly surprising that single mismatches introduced in 18 out of 20 positions of the heteroduplex formed by the guide part of sgRNA and the target abolished target cleavage by AacCas12b in vitro, which was interpreted as evidence of a very extensive seed that could lead to exceptional specificity of target recognition [20]. In this work we study sequence specificity determinants of target binding by BthCas12b and AacCas12b. We reveal, consistent with structural expectations, a short 5-nt seed. However, unexpectedly, we also find an additional binding determinant at the site of target cleavage. The data should inform the design of guides and target selection during editing applications by Cas12b effectors.
Results
A library-based approach determines protospacer positions important for interference by BthCas12b
The experimental system used in this work is schematically shown in Figure 1. Escherichia coli cells lacking a functional CRISPR-Cas system of their own are transformed with the pCRISPR_Bth plasmid carrying the tracrRNA gene and a miniature single-spacer CRISPR array of the B. thermoamylovorans Type V-B system, and a compatible pCas_Bth plasmid carrying the full set of B. thermoamylovorans cas genes. The components of the B. thermoamylovorans CRISPR-Cas system are placed under the control of inducible promoters. Upon induction, the efficiency of transformation with a third compatible plasmid carrying a sequence matching the CRISPR array spacer and adjacent functional ATTN PAM [15] is decreased several orders of magnitude compared to empty plasmid control due to CRISPR interference.
Figure 1.
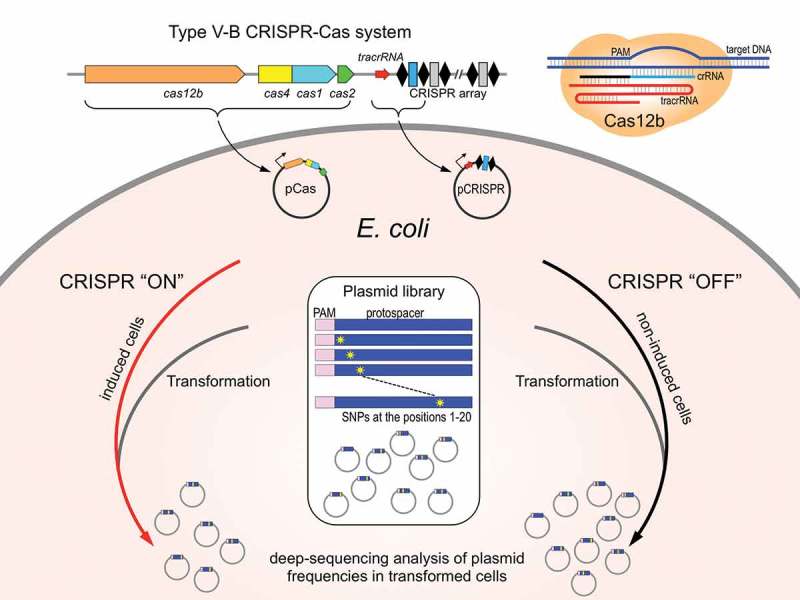
Experimental system to study interference by Type V Cas12b effectors in E. coli.
Type V-B cas genes and a miniature cognate CRISPR array carrying a single spacer cloned on plasmids under control of inducible promoters are introduced in E. coli. Cells are grown in inducing (CRISPR ‘ON’) or uninducing (CRISPR ‘OFF’) conditions and transformed with a library of plasmids carrying mutated protospacer matching the CRISPR array spacer. Plasmids recovered from CRISPR ‘ON’ and CRISPR ‘OFF’ transformants are analyzed by deep sequencing.
The PAM requirements of the BthCas12b effector were earlier deduced using a library-based approach by revealing sequences that were selectively lost from the initial library of protospacer plasmids with randomized PAM positions upon transformation into cells with B. thermoamylovorans CRISPR-Cas system [15]. Here, we used a similar approach to determine the sensitivity of interference by the B. thermoamylovorans CRISPR-Cas system to mismatches between individual positions of crRNA spacer and plasmid protospacer. The PAM in all library members was intact and matched the ATTN consensus. The library was created by pooling plasmids carrying three possible substitutions at protospacer positions 1 through 20 (here and below, protospacer position 1 refers to the base closest to PAM). Note that while the length of the B. thermoamylovorans spacer is 35 base pairs, after processing, only 19 bases of spacer remain in crRNA [15]. Thus, our library interrogated every position of the area involved in crRNA-target protospacer heteroduplex formation. Substitutions at position 20 are outside of heteroduplex and served as control, as they were not expected to have any effect on target recognition/CRISPR interference. Together with wild-type, fully matching, sequence, the library contained 3 × 20 + 1 = 61 members. The library was transformed into induced and control, uninduced, E. coli cells carrying plasmid-borne B. thermoamylovorans CRISPR-Cas system. Pooled transformants were collected. DNA fragments containing the targeted protospacer were amplified and subjected to Illumina sequencing. Interference against individual library members was detected by determining the difference in relative abundance of normalized reads in induced and uninduced cells (see Methods). As can be seen from Figure 2(a), introduction of any mismatch at protospacer positions 1–3 and at position 5 strongly decreased interference. Introduction of T or C instead of G at the fourth position strongly (50-fold or more) decreased interference, while the presence of A at this position had no effect. Mismatches at positions 6 and 7 had much weaker effects on the interference; mismatches at other positions of the protospacer had no effect. The only exception was a G to T mismatch at position 15 that led to a clear, more than 10-fold, decrease in interference.
Figure 2.
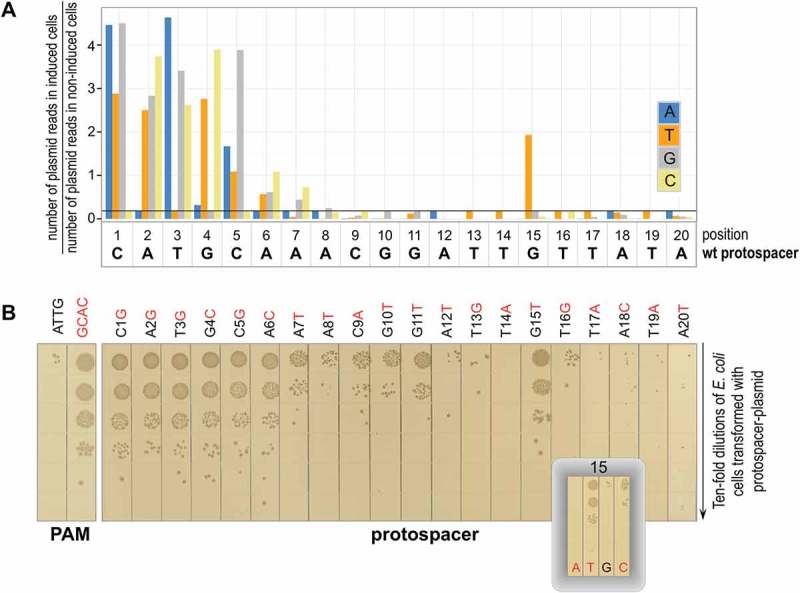
Determining the effects of substitutions introducing single-nucleotide mismatches between crRNA spacer and plasmid protospacer on interference by B. thermoamylovorans CRISPR-Cas system.
(a) Results of analysis of library experiment outlined in Figure 1 are presented. Each bar shows a ratio of normalized abundances of reads containing a nucleotide substitution (color code shown on the right) at every position of the 20-nt protospacer sequence in CRISPR ‘ON’ and CRISPR ‘OFF’ cells. Black horizontal line represents the wild-type protospacer ratio (indicative of CRISPR interference). High ratios indicate that a corresponding sequence is not subject to interference. A representative result of experiment performed in triplicate is presented. Results of the other two experiments are shown in the Supplementary Figure S1(b) Transformation efficiencies of plasmids carrying indicated single nucleotide substitutions at the protospacer. As controls, plasmids carrying fully matching protospacers with a functional ATTG and a nonfunctional GCAC PAM were tested (left). After transformation, cells were serially diluted, and aliquots were deposited on the surface of agar plates containing appropriate antibiotic. Results of overnight growth at 37°C are shown (a composite of several plates). Results obtained with plasmids carrying each of the three mutants in position 15 are shown in an inset.
The results obtained with protospacer plasmid library were confirmed by determining transformation efficiencies of individual plasmids carrying substitutions throughout the protospacer length (Figure 2(b)). For position 15, each of the three possible plasmids were tested (Figure 3(b), inset). In agreement with results of library analysis, a strong decrease of interference by the G to T substitution at this position was observed. The G to C substitution had a weak effect on transformation efficiency while the G to A substitution behaved as wild-type (no transformants/strong interference).
Figure 3.
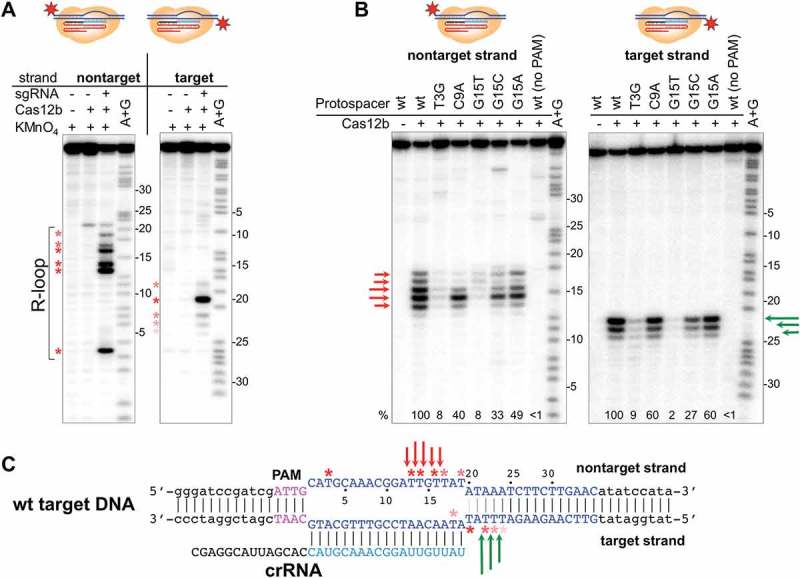
R-loop formation and protospacer cleavage by the B. thermoamylovorans Cas12b effector.
(a) KMnO4 probing of R-loop complex formed by BthdCas12b on 5ʹ-radioactively labeled DNA fragment containing fully matching protospacer. Numbers on the right side of each gel indicate protospacer positions for each DNA strand (a position closest to PAM is numbered as 1). Asterisks show thymine positions in the melted regions with various red color intensities reflecting extent of KMnO4 modification.(b) BthCas12b complexes were formed on 5ʹ-radioactively labeled DNA fragments containing fully matching (wt) or indicated mutant versions of protospacer at conditions that allow target cleavage. Reaction products were separated by denaturing PAGE and revealed by autoradioagraphy. Arrows of different colors represent cleavage sites in the target or nontarget strands. Cumulative relative cleavage efficiencies (amount of radioactivity in cleavage products bands compared to radioactivity in all cleaved and uncleaved DNA bands) are indicated below the gels; cleavage efficiency of fully-matching target is taken as 100%. (c) The positions of KMnO4 modification and cleavage sites in the fully matching protospacer R-loop complex are schematically shown.
In vitro analysis of target cleavage by BthCas12b
Purified recombinant BthCas12b was combined with in vitro transcribed sgRNA whose 19-nt guide part matched the spacer of mature crRNA produced from miniature CRISPR array. The in vitro assembled effector complex was next combined with a DNA fragment containing fully matching protospacer and consensus PAM and localized melting of DNA in the R-loop complex was revealed by KMnO4 probing (Figure 3(a)). The experiment was conducted using a catalytically inactive BthdCas12b to prevent target cleavage. Every thymine in the protospacer non-target strand (positions 3, 13, 14, 16, 17, and 19) was sensitive to KMnO4 in the presence of effector, indicating that a fully open R-loop complex is formed. Curiously, a thymine at position 22 was sensitive to KMnO4 in the presence of BthCas12b with or without sgRNA, possibly indicating a specific interaction of the BthCas12b protein with an AT rich region outside of PAM-distal end of protospacer. Protospacer target strand thymines were resistant to KMnO4 modification, an expected result as they shall be present in the RNA-DNA heteroduplex. In contrast, target strand thymine 20, just outside the heteroduplex was highly sensitive to modification. Thymines at positions 18 and 22, 23, and 24 were also sensitive, though their level of reactivity was weaker. The sensitivity of these residues to modification depended on the presence of sgRNA.
We next investigated cleavage of both fully matching and mismatched targets in vitro using R-loop complexes formed by the active BthCas12b effector. In the wild-type, fully matching, complex cleavage at positions 12–17 of the non-target strand (with major cleavage site at position 14) and at positions 22, 23, and 24 of the target strand (with major cleavage site at position 22) was observed (Figure 3(b)). A target with the T3G substitution (abolishes interference in vivo) was poorly cleaved at both strands (less than 10% of fully matching target cleavage); the pattern of residual cleavage bands was, however, unchanged. A C9A target (subject to interference in vivo) was cleaved with 60% efficiency in the target strand. In non-target strand, cleavage was also decreased, though the major site, at position 14, was not strongly affected. The G15T mismatch strongly decreased the overall cleavage efficiency to below 10% of the fully matching complex, with the major cleavage site in the non-target strand position 14 practically being abolished. In contrast, the G15C and G15A mismatches had no such effect (though cleavage patterns of mismatched targets differed from the fully matching pattern, particularly at position 16 of non-target strand, where decreased cleavage efficiency was evident). We conclude that overall, the results of in vitro analysis confirm the in vivo data. Observed decreased cleavage efficiency at the target strand without changes in cleavage patterns are consistent with decreased binding of the effector to mismatched targets. Complex changes in the non-target strand cleavage, suggest that the BthCas12b nuclease center involved in cleavage of this strand possesses some sequence specificity.
In vitro analysis of target binding by BthCas12b
To determine whether mismatches also affect target binding a fluorescent beacon assay that was previously employed to study target interactions by Type I E. coli Cascade effector and Type II SpCas9 effector [31,32] was used (Figure 4(a)). The BthCas12b beacon is a double-stranded structure assembled from three oligonucleotides (Figure 4(b)). Oligo 1 is the longest of the three; it is labeled with fluorescein at the 3ʹ end and consists of two functional parts of roughly the same length. The 3ʹ-proximal part is complementary to the sgRNA spacer. The 5ʹ proximal part is complementary to oligo 2. Binding of oligo 2 creates a double-stranded structure upstream of the spacer part with consensus PAM. Oligo 3 is complementary to the spacer part of oligo 1 and contains a quencher that decreases fluorescence of fluorescein. To selectively test BthCas12b-sgRNA binding to DNA targets, a catalytically inactive BthdCas12b derivative that lacks endonuclease activity but binds target DNA when programmed with guide RNA was used [15]. The addition of BthdCas12b charged with sgRNA fully matching the spacer part of oligo 1 led to a rapid increase in beacon fluorescence (Figure 4(c)), presumably due to displacement of oligo 3. When the BthdCas12b-sgRNA complex was combined with double-stranded unlabeled oligonucleotide with fully matching protospacer sequence and consensus PAM before the addition of the beacon (‘target DNA competitor’, Figure 4(a)), a very slow increase in beacon fluorescence was observed due to sequestration of the effector. In contrast, a competitor with non-consensus PAM but a fully matching protospacer had very small effect on fluorescence increase of the beacon (Figure 4(c)). Thus, the beacon competition assay provides a rapid and sensitive way to follow BthCas12b effector interaction with its targets. A consensus PAM competitor with a T3G mismatch had little effect on fluorescence increase, indicating that a mismatch at this position affects target binding, which should thus be the reason for poor cleavage of targets with this mismatch (Figure 3(b)). In contrast, the C9A competitor competed as well as the wild-type, consistent with the in vivo interference and in vitro cleavage data. Competitor with the G15T substitution competed worse than the wild-type but clearly better than the T3G mutant (Figure 4). The effect of substitution in this position was base specific since the G15A competitor was indistinguishable from wild-type. We conclude that a G to T mismatch at position 15 decreases the binding of the BthCas12b-sgRNA complex studied here, while a G to A mismatch has no such effect.
Figure 4.
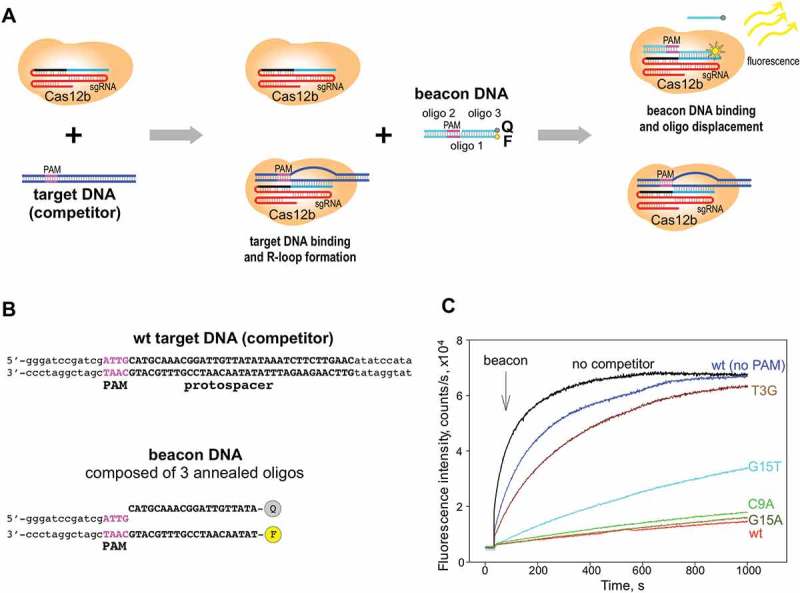
Binding to target DNA studied using the BthCas12b beacon assay.
(a) The principle of the competitive beacon assay is schematically shown.(b) The structure of the BthCas12b beacon and of the wild-type, fully matching DNA competitor probe.(c) Effects of indicated competitor DNA fragments on the kinetics of beacon binding to BthdCas12b complex are shown. Concentrations of beacon, BthdCas12b, and dsDNA competitor fragments are 1, 10, and 50 nM, respectively.
Protospacer positions important for interference by AacCas12b
To determine how general is the requirement for spacer-protospacer match outside the seed for Cas12b effectors, limited analysis of target recognition by the Alicyclobacillus acidoterrestris Cas12b was carried out. The in vitro cleavage sites by AacCas12b are located similarly to those observed in the BthCas12b complexes (Figure 5(a)), with non-target strand being cleaved close to protospacer position 15, while the target strand being cleaved downstream of the crRNA-DNA duplex (Figure 5(b)). Using an in vivo interference system similar to the one developed for BthCas12b (Figure 1), we showed that AacCas12b efficiently prevents transformation of plasmids containing a protospacer matching crRNA spacer provided there is a 5ʹ TTA PAM. High transformation efficiency was observed when PAM was substituted for a GGA sequence (Figure 5(c)). A T3G substitution in the protospacer, introducing a mismatch in the seed, also restored transformation efficiency, as expected. The effect of mismatches at protospacer positions 14, 15, and 16 was systematically probed. As can be seen, mismatches at positions 14 and 16 had no effect on interference. In contrast, substitutions at position 15 partially (A15T) or fully (A15C) abolished interference. We thus conclude that a match at an internal position of protospacer at the cleavage site is a general requirement for interference by Cas12b effectors.
Figure 5.
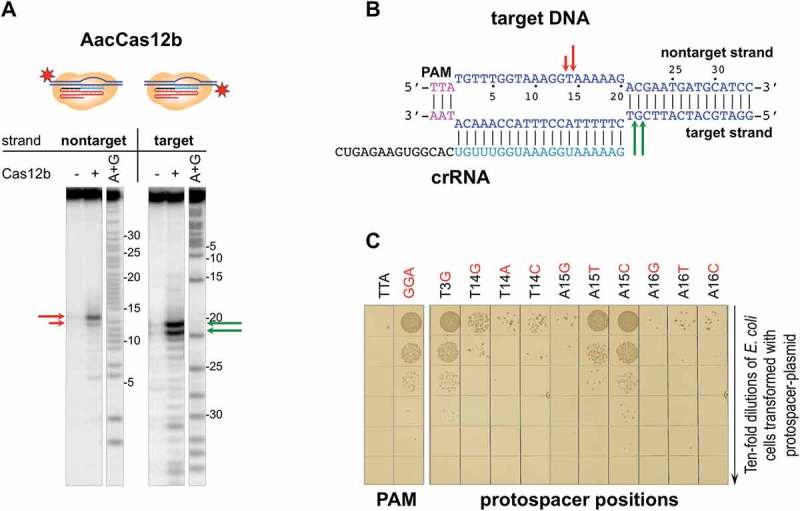
Determining the effects of substitutions introducing single-nucleotide mismatches between crRNA spacer and plasmid protospacer on interference by (A). acidoterrestris CRISPR-Cas system.
(a) AacCas12b complexes were formed on 5ʹ-radioactively labeled DNA fragments containing fully matching protospacer at conditions that allow target cleavage. Reaction products were separated by denaturing PAGE and revealed by autoradioagraphy. Arrows of different colors represent cleavage sites in the target or nontarget strands. (b) Schematic representation of data obtained in panel (a). (c) Transformation efficiencies of plasmids carrying each of the three mutants in position 14, 15, and 16, as well as a mismatch in position 3 (seed) at the protospacer. As controls, plasmids carrying fully matching protospacers with a functional TTA and a nonfunctional GGA PAM were tested (left). After transformation, cells were serially diluted, and aliquots were deposited on the surface of agar plates containing appropriate antibiotic. Results of overnight growth at 37°C are shown.
Discussion
In this work, we experimentally defined a 5-nt seed for a Cas12b effector from B. thermoamylovorans. The size of the seed is fully consistent with structural expectations and matches the seed reported for an unrelated Cas12a effector [14,22]. Yet, our results are different from the reported extensive, 18-nt, seed for Cas12b from Alicyclobacillus acidoterrestris [20], which is evolutionary close to BthCas12b. The nature of this discrepancy remains unknown, but the data for BthCas12b, obtained both in vivo and in vitro, seem unequivocal and are supported by our less extensive analysis of AacCas12b.
Unexpectedly, in vivo data show that interference efficiency by BthCas12b is strongly reduced by a G15T substitution in the protospacer sequence and in vitro data show that this is due to reduced binding that leads to the absence of target cleavage. Other substitutions at position 15 have either minor or no effect. The sequence specific influence of mismatches with crRNA guide at position 15 may be a consequence of destabilization of BthCas12b R-loop complex, distortion of its catalytically-competent conformation or both. In the structure of BthCas12b-sgRNA-DNA complex a base at position 15 of the target strand forms hydrogen bonds with Asn282 and Thr280 of the effector protein. This property distinguishes the position 15 base from other non-seed target strand bases among which only the neighboring base at position 14 forms a contact with the protein while other nucleotides interact only via their backbones [22]. This feature is also conserved in the structure of AacCas12b-sgRNA-DNA complex where among the non-seed target strand protospacer bases only the position 15 base forms hydrogen bonds with the protein [21]. Indeed, we show that mismatches at position 15 abolish interference by AacCas12b. Another, not necessarily excluding possibility is that the active site responsible for non-target site cleavage also possesses sequence specificity. This inference is supported by altered pattern of non-target strand cleavage sites in complexes with substitutions at position 15 revealed in our work. Be as it may, Cas12b interactions with either target or non-target strand position 15 nucleotides seem to either promote R-loop complex formation initiated at the perfect seed or inhibit it. Structural analysis of target opening intermediates that include non-target DNA strand (missing in current structures) will be necessary to address these questions. In the meantime, the importance of position at the cleavage site for target binding by BthCas12b, AacCas12b (and likely other Type V-B effectors) should be considered when designing guides/selecting targets for editing with this effector.
Materials and methods
Plasmids and strains
To generate Bacillus thermoamylovorans cas operon, four gBlocks gene fragments were synthesized with codon optimization for expression in E. coli cells (IDT Inc.) and assembled with a linearized pACYCDuet-1 plasmid using Gibson Assembly kit (NEBuilder HiFi DNA Assembly Cloning Kit from NEB). The resulting plasmid, pCas_Bth, contains Bth cas genes under the control of T7 RNA polymerase promoter. For crRNA expression, the pCRISPR_Bth plasmid was generated by cloning a double-stranded DNA fragment (gBlock form IDT Inc.) containing a minimized Bth CRISPR-array (one spacer flanked by two repeats) and the natural leader region [15] into the NcoI and NotI sites of the pCDF-1b vector. The 35-bp spacer (5ʹ-CATGCAAACGGATTGTTATATAAATCTTCTTGAAC) is identical to the first spacer of the native Bth CRISPR array [15]. The leader region is carrying the coding sequence for the tracrRNA [15].
The construction of pCas_Aac, pCRISPR_Aac plasmids was described elsewhere [15]. Briefly, Alicyclobacillus acidoterrestris cas operon was amplified from the genomic DNA of A. acidoterrestris ATCC 49,025 and PCR products was cloned into NcoI and KpnI sites of pACYCDuet-1 plasmid. Synthetic DNA fragment (gBlock from IDT Inc.) containing the leader region for the tracrRNA expression and minimized Aac CRISPR-array was cloned into the NcoI and NotI sites of the pCDF-1b vector. The resulting pCRISPR_Aac plasmid contained the spacer (5ʹ-TGTTTGGTAAAGGTAAAAAGACGAATGATGCATCC) corresponding to the first spacer of the native A. acidoterrestris CRISPR array [15].
The Bth and Aac target protospacer plasmids were constructed by cloning appropriate double-stranded oligonucleotides containing the ATTG PAM (Bth protospacer plasmid) or the TTA PAM (Aac protospacer plasmid) and wild-type or mutant protospacer sequences into the EcoRV site of the pT7Blue vector. A variant of the target plasmid carrying a GCAC sequence (for Bth plasmid) and a GGA sequence (for Aac plasmid) instead of PAM and a fully matching protospacer was created and used as a negative control.
E. coli BL21-AI cells carrying the T7 RNA polymerase gene under the araP promoter were used as a host for pCas, pCRISPR and protospacer plasmids.
The pET28_BthCas12b and the pET28_AacCas12b expression plasmids were constructed by sub-cloning the cas12b genes from the pCas plasmids into pET28a vector. pET28_BthdCas12b plasmid expressing the dead mutant derivative of the BthCas12b (harboring D952A/N954A substitutions [15]) was created by site-directed mutagenesis of pET28_BthCas12b.
Transformation assay
E. coli BL21-AI cells carrying the pCas and pCRISPR plasmids were grown overnight in liquid LB medium supplemented with 25 µg/ml chloramphenicol, 25 µg/ml streptomycin. The cultures were diluted 1:100 in 15 ml of fresh LB with 25 µg/ml chloramphenicol, 25 µg/ml streptomycin and 1 mM arabinose, and allowed to grow till OD600 reached 0.5. A parallel culture was grown in the absence of arabinose. Cells were collected, washed three times with ice-cold 10% glycerol and transformed, by electroporation, with 10 ng of protospacer plasmids. To monitor CRISPR interference, 10-fold dilutions of transformation mixtures were plated on LB agar supplemented with 25 µg/ml chloramphenicol, 25 µg/ml streptomycin, and 100 µg/ml ampicillin. All transformation experiments were performed in triplicates.
Plasmid library experiment
The library was prepared by combining equal amounts of 60 mutant protospacer plasmids and the wild-type protospacer plasmid. 30 ng of plasmid DNA library was used to transform pre-induced or uninduced competent E. coli cells carrying pCas_Bth and pCRISPR_Bth plasmids. After 1-hour outgrowth at 37°C, 50 µl transformation mixture aliquots were spread on multiple LB agar plates supplemented with 25 µg/ml chloramphenicol, 25 µg/ml streptomycin, and 100 µg/ml ampicillin. For each sample, ~ 8000 colonies were pooled and plasmid DNA was isolated. PCR amplification of target protospacer region was performed with Bth Lib F (5ʹ- GCATGCCTGCAGGTCGACTCTAGAGG) and Bth Lib R (5ʹ- GTTGTAAAACGACGGCCAGTGAATTC) primers. PCR products of 138 bp were gel purified using Thermo Scientific GeneJET Gel Extraction Kit and High Throughput Illumina Sequencing was done at the Waksman Institute Genomics Core Facility. All samples were prepared and processed in triplicates.
Library analysis
For each sample, ca. 1,000,000 reads were obtained after filtering and quality control procedures (Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc; http://dx.doi.org/10.14806/ej.17.1.200). The frequency of each unique sequence within the sample was normalized by calculating a ratio of the number of corresponding reads to the total number of reads in the sample. The relative abundance of individual sequences between the samples was calculated as a ratio of their frequencies in induced and uninduced cells and was visualized using R (v3.3.1) (http://www.R-project.org) and ggplot2 (v2.2.1) package [33].
Cas12b protein expression and purification
Cas12b proteins were purified from E. coli BL21(DE3) cells transformed with appropriate plasmids. 0.4 l of LB supplemented with 50 μg/ml kanamycin were inoculated with cells from freshly transformed cells. The cultures were grown at 37°C to OD600 0.6–0.9, induced with 0.5 mM IPTG and grown for additional 6–8 hours at room temperature. Cells were harvested and resuspended in buffer A (20 mM Tris-HCl pH, 8.0, 500 mM NaCl, 4 mM imidazole pH 8.0, 5% (v/v) glycerol, 0.2 μg/ml phenylmethylsulfonyl fluoride (PMSF)) supplemented with protease inhibitor cocktail Roche cOmplete, EDTA-free (Sigma) and disrupted by sonication. Cleared lysates were obtained by centrifugation at 15 000 g for 60 minutes, filtered through 0.22 micron filter (Millipore) and applied onto a 1-ml chelating Hi-Trap Sepharose column (GE Healthcare) equilibrated with buffer A. The column was washed with buffer A containing 25 mM imidazole, and the proteins were eluted with buffer A containing 200 mM imidazole. Protein-containing fractions were pooled, diluted 10 times with TGED buffer (20 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 1 mM EDTA, 2 mM μ-mercaptoethanol) and loaded onto a 1-ml Hi-Trap Heparin column (GE Healthcare) equilibrated with TGED. The column was washed with TGED containing 500 mM NaCl, and proteins were eluted with TGED containing 1 M NaCl. Protein fractions were pooled, concentrated using Microsep centrifugal devices 30K (Pall Corp), dialyzed against buffer B (20 mM Tris-HCl, pH 8.0, 200 mM NaCl, 50% (v/v) glycerol, 0.5 mM EDTA, 2 mM β-mercaptoethanol) and stored at −80°C.
sgRNA purification
Cas12b sgRNA was transcribed in vitro from PCR-generated dsDNA template using T7 RNA polymerase (New England Biolabs) according to manufacturer recommendations and was purified after electrophoresis in 10% polyacrylamide 6M urea gels. The template was amplified using a long oligonucleotide Bth_sgRNA and two short oligonucleotides T7prom_mod and Bth_sgRNA_rev (all from Integrated DNA Technologies). Oligonucleotide sequences are listed in Supplementary Table S1.
Cas12b target cleavage and KMnO4 probing
dsDNA target fragments containing protospacer sequence or its mutant derivatives were prepared from 60-nt oligonucleotides (from IDT Inc.), as described in [34]. In each case, either the target or nontarget strand oligonucleotide was labeled with [γ-32P]-ATP at its 5ʹend. The labeled DNA targets were purified on Micro Bio-spin 6 columns (Bio-Rad) and used for the in vitro assays at 50 – 100 nM concentrations.
In vitro target cleavage assays were performed with Cas12b at 37°C in cleavage buffer (20 mM Tris-HCl pH 8.0, 5 mM MnCl2, 100 mM NaCl, 3 mM β-mercaptoethanol). Cas12b-sgRNA complexes were formed by combining, in 10 μl, Cas12b and sgRNA (200 nM each) and incubating at 37°C for 10 minutes. Next, 50 nM of dsDNA target was added. After 60 minutes of incubation at 37°C, the reactions were stopped by the addition of the equal volume of urea-formamide loading buffer, resolved by 10% denaturing PAGE and visualized and quantified using PhosphorImager.
BthdCas12b-sgRNA complexes were formed as above in a buffer containing 20 mM trisHCl pH8.0, 5 mM MgCl2, 100 mM NaCl. After target addition, reactions were incubated for 10 minutes at 37°C and treated with 1 mM KMnO4 at room temperature for 40 seconds. Reactions were terminated by the addition of 300 mM β-mercaptoethanol, followed by ethanol precipitation and 20-min treatment with 10% piperidine at 95°C. Reaction products were treated with chloroform, ethanol precipitated, dissolved in 8 μl of urea-formamide loading buffer, resolved by 10% denaturing PAGE and visualized using PhosphorImager.
Fluorometric measurements
The BthCas12b beacon was formed by mixing oligonucleotide 1 labeled with fluorescein at 3ʹ end, unmodified oligonucleotide 2, and oligonucleotide 3 labeled with Iowa BlackR FQ at 5ʹ end (final oligonucleotide concentrations were within low μM range) in a buffer containing 40 mM Tris, pH 7.9, 100 mM NaCl by heating for 1 min at 90°C and slow cooling to 20°C. Oligonucleotides 2 and 3 were taken in 30% excess of oligonucleotide 1 to avoid the presence of free oligonucleotide 1 in the samples. Control experiments verified that such excess of oligonucleotides 2 and 3 had no effect on beacon binding.
Fluorescence measurements were performed using a QuantaMaster QM4 spectrofluorometer (PTI) in binding buffer (20mM Tris HCl pH 7.9, 100 mM NaCl, 5% glycerol, 0.1 mM DTT and 1 mM MgCl2) containing 0.02% Tween 20 at 25°C. Final assay mixtures (800 μl) contained 10 nM of BthdCas12b protein, 15 nM sgRNA, 1 nM beacon and 50 nM target DNA competitor substrates. The fluorescein fluorescence intensity was recorded with an excitation wavelength of 498 nm and an emission wavelength of 520 nm. Time-dependent fluorescence changes were monitored after the addition of negligible volume of BthdCas12b beacon to a cuvette followed by manual mixing; the mixing dead-time was 15 s. Competition experiments were analyzed as previously described [23].
Funding Statement
This work was supported by the National Institute of General Medical Sciences [R01 GM10407]; Busch Biomedical Research grant; Ministry of Education and Science of the Russian Federation Subsidy Agreement # 14.606.21.0006 unique project identifier RFMEFI60617X0006; Skolkovo Institute of Science and Technology [Skoltech-MIT Next generation Program Grant];
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007. March 23;315(5819):1709–1712. PubMed PMID: WOS:000245106900039; English. [DOI] [PubMed] [Google Scholar]
- 2.Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008. August 15;321(5891):960–964. PubMed PMID: 18703739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marraffini LA, Sontheimer EJ.. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008. December 19;322(5909):1843–1845. . PubMed PMID: 19095942; PubMed Central PMCID: PMCPMC2695655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shmakov SA, Sitnik V, Makarova KS, et al. The CRISPR Spacer Space Is Dominated by Sequences from Species-Specific Mobilomes. MBio. 2017. September 19;8(5). PubMed PMID: 28928211; PubMed Central PMCID: PMCPMC5605939 DOI: 10.1128/mBio.01397-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015. November;13(11):722–736. PubMed PMID: 26411297; PubMed Central PMCID: PMCPMC5426118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deveau H, Barrangou R, Garneau JE, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008. February;190(4):1390–1400. PubMed PMID: 18065545; PubMed Central PMCID: PMCPMC2238228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semenova E, Jore MM, KA Datsenko, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A. 2011. June 21;108(25):10098–10103. PubMed PMID: 21646539; PubMed Central PMCID: PMCPMC3121866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009. March;155(Pt 3):733–740. PubMed PMID: 19246744. [DOI] [PubMed] [Google Scholar]
- 9.Sander JD, Joung JK.. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014. April;32(4):347–355. PubMed PMID: 24584096; PubMed Central PMCID: PMCPMC4022601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012. August 17;337(6096):816–821. PubMed PMID: 22745249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng R, Lin G, Li J.. Potential pitfalls of CRISPR/Cas9-mediated genome editing. FEBS J. 2016. April;283(7):1218–1231. . PubMed PMID: 26535798. [DOI] [PubMed] [Google Scholar]
- 12.Nakade S, Yamamoto T, Sakuma T.. Cas9, Cpf1 and C2c1/2/3-What’s next? Bioengineered. 2017. May 4;8(3):265–273. . PubMed PMID: 28140746; PubMed Central PMCID: PMCPMC5470521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slaymaker IM, Gao L, Zetsche B, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016. January 1;351(6268):84–88. PubMed PMID: 26628643; PubMed Central PMCID: PMCPMC4714946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015. October 22;163(3):759–771. PubMed PMID: 26422227; PubMed Central PMCID: PMCPMC4638220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell. 2015. November 5;60(3):385–397. PubMed PMID: 26593719; PubMed Central PMCID: PMCPMC4660269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017. March;15(3):169–182. PubMed PMID: 28111461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017. June;37:67–78. PubMed PMID: 28605718; PubMed Central PMCID: PMCPMC5776717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017. January;35(1):31–34. PubMed PMID: 27918548; PubMed Central PMCID: PMCPMC5225075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fonfara I, Richter H, Bratovic M, et al. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016. April 28;532(7600):517–521. PubMed PMID: 27096362. [DOI] [PubMed] [Google Scholar]
- 20.Liu L, Chen P, Wang M, et al. C2c1-sgRNA Complex Structure Reveals RNA-Guided DNA Cleavage Mechanism. Mol Cell. 2017. January 19;65(2):310–322. PubMed PMID: 27989439. [DOI] [PubMed] [Google Scholar]
- 21.Yang H, Gao P, Rajashankar KR, et al. PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell. 2016. December 15;167(7):1814–1828 e12. PubMed PMID: 27984729; PubMed Central PMCID: PMCPMC5278635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu D, Guan X, Zhu Y, et al. Structural basis of stringent PAM recognition by CRISPR-C2c1 in complex with sgRNA. Cell Res. 2017. May;27(5):705–708. PubMed PMID: 28374750; PubMed Central PMCID: PMCPMC5520857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mekler V, Minakhin L, Severinov K. Mechanism of duplex DNA destabilization by RNA-guided Cas9 nuclease during target interrogation. Proc Natl Acad Sci U S A. 2017. May 23;114(21):5443–5448. . PubMed PMID: 28484024; PubMed Central PMCID: PMCPMC5448204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maier LK, Lange SJ, Stoll B, et al. Essential requirements for the detection and degradation of invaders by the Haloferax volcanii CRISPR/Cas system I-B. RNA Biol. 2013. May;10(5):865–874. PubMed PMID: 23594992; PubMed Central PMCID: PMCPMC3737343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiedenheft B, Van Duijn E, Bultema JB, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci U S A. 2011. June 21;108(25):10092–10097. PubMed PMID: 21536913; PubMed Central PMCID: PMCPMC3121849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang W, Bikard D, Cox D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013. March;31(3):233–239. PubMed PMID: 23360965; PubMed Central PMCID: PMCPMC3748948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parker JS, Parizotto EA, Wang M, et al. Enhancement of the seed-target recognition step in RNA silencing by a PIWI/MID domain protein. Mol Cell. 2009. January 30;33(2):204–214. PubMed PMID: 19187762; PubMed Central PMCID: PMCPMC2642989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorski SA, Vogel J, Doudna JA. RNA-based recognition and targeting: sowing the seeds of specificity. Nat Rev Mol Cell Biol. 2017. April;18(4):215–228. . PubMed PMID: 28196981. [DOI] [PubMed] [Google Scholar]
- 29.Kunne T, Swarts DC, Brouns SJ. Planting the seed: target recognition of short guide RNAs. Trends Microbiol. 2014. February;22(2):74–83. . PubMed PMID: 24440013. [DOI] [PubMed] [Google Scholar]
- 30.Yamano T, Nishimasu H, Zetsche B, et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell. 2016. May 5;165(4):949–962. PubMed PMID: 27114038; PubMed Central PMCID: PMCPMC4899970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mekler V, Minakhin L, Semenova E, et al. Kinetics of the CRISPR-Cas9 effector complex assembly and the role of 3ʹ-terminal segment of guide RNA. Nucleic Acids Res. 2016. April 7;44(6):2837–2845. PubMed PMID: 26945042; PubMed Central PMCID: PMCPMC4824121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuznedelov K, Mekler V, Lemak S, et al. Altered stoichiometry Escherichia coli Cascade complexes with shortened CRISPR RNA spacers are capable of interference and primed adaptation. Nucleic Acids Res. 2016. December 15;44(22):10849–10861. PubMed PMID: 27738137; PubMed Central PMCID: PMCPMC5159557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wickham H. ggplot2: elegant Graphics for Data Analysis. New York: Springer-Verlag; 2009. [Google Scholar]
- 34.Mekler V, Pavlova O, Severinov K. Interaction of Escherichia coli RNA polymerase sigma70 subunit with promoter elements in the context of free sigma70, RNA polymerase holoenzyme, and the beta’-sigma70 complex. J Biol Chem. 2011. January 7;286(1):270–279. . PubMed PMID: 20952386; PubMed Central PMCID: PMCPMC3012984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.