

ABSTRACT
Translation factor a/eIF5A is highly conserved in Eukarya and Archaea. The eukaryal eIF5A protein is required for transit of ribosomes across consecutive proline codons, whereas the function of the archaeal orthologue remains unknown. Here, we provide a first hint for an involvement of Sulfolobus solfataricus (Sso) aIF5A in translation. CRISPR-mediated knock down of the aif5A gene resulted in strong growth retardation, underlining a pivotal function. Moreover, in vitro studies revealed that Sso aIF5A is endowed with endoribonucleolytic activity. Thus, aIF5A appears to be a moonlighting protein that might be involved in protein synthesis as well as in RNA metabolism.
KEYWORDS: Translation factor aIF5A, Archaea, RNase, Sulfolobus solfataricus
Introduction
The eukaryotic eIF5A and the bacterial orthologue EF-P were first identified as translation initiation factors [1]. However, subsequent studies revealed that they affect translational elongation by alleviating ribosome stalling at poly-proline stretches [2,3] as well as translational termination [3]. Both factors are post-translationally modified. The eukaryal protein is hypusinated [4], whereas the bacterial orthologue is β-lysinylated [5]. It was further shown that the archaeal protein is either hypusinated or deoxyhypusinated, and that some Archaea contain both versions of the protein [6,7]. The 3D structures of eIF5A and aIF5A are very similar, with a strong conservation of residues in the N-terminal moiety that contains the eIF5A hypusination site [8].
Eukaryal eIF5A was shown to possess RNA binding activity [9,10]. Studies of a temperature-sensitive eIF5A variant in yeast suggested that the protein plays a role in mRNA turnover, raising the possibility that it might be involved in degradation of a specific subset of mRNAs [11,12]. Moreover, Wagner et al. showed that eIF5A and aIF5A of the euryarchaeon Halobacterium sp. NRC-1, display RNase activity, which did not require post-translational modification of halobacterial aIF5A [13]. The RNase activity was dependent on specific charged aa residues and occurred preferentially between an adenine and a cytosine in single stranded regions of RNA. The halobacterial aIF5A was also shown to bind to RNAs in vitro. However, in contrast to the RNase activity, hypusination was required for formation of these RNA-protein complexes [13].
The mRNA decay pathways have been intensively studied in Bacteria and Eukarya [14]. However, the information on archaeal mRNA turnover is still rudimentary. Archaeal mRNAs are degraded with 5´ to 3´ as well as with 3΄ to 5΄ directionality. Members of the β-CASP ribonuclease family (aCPSF1, aCPSF2) often show a dual 5´ to 3´ exo- and endoribonucleolytic activity, and play a crucial role in RNA processing and decay in Archaea [14–17]. The archaeal exosome machinery exhibits a 3΄ to 5΄ exoribonucleolytic activity and can synthesize heteropolymeric RNA tails [18,19]. In rare cases, e.g. Halophiles and Methanococcales, which lack genes encoding the exosome, the 3΄ to 5΄ exoribonucleolytic activity is carried out by an RNase R bacterial-like enzyme [20]. In addition, several endoribonucleases, like RNase P, members of the RNase Z family [21,22], the splicing endonuclease End A [23] or the CRISPR Cas6 family [24] have been identified in Archaea.
Sso mRNAs are either polycistronic or monocistronic, and leaderless mRNAs lacking a Shine and Dalgarno (SD) motif are prevalent [25]. It has been shown that the trimeric translation initiation factor aIF2 can bind by means of its γ-subunit to the 5´-triphosphorylated end (5´-P3-end) of mRNAs [26,27]. Masking of the 5´-P3-end appears to counteract 5´ to 3´directional decay of RNA by Sso aCPSF2 [17,28], which preferentially degrades mono-phosphorylated RNA or RNA bearing a 5´-hydroxyl group [17].
Thus, aIF2 is a moonlighting protein required for translation initiation [27,29] as well as for the protection of mRNAs from exonucleolytic decay under certain conditions, e.g. under nutrient stress when translation comes to a halt [17,27]. Moreover, the translation recovery factor (Trf) was implicated in promoting resumption of translation upon outgrowth from stationary phase by triggering aIF2(γ) release from the 5´-P3-end of mRNAs [30].
Here, we provide new insights into the function of aIF5A in Sso. We show that the protein associates with ribosomal subunits in a Sso cell lysate programmed for protein synthesis, providing a first indication that aIF5A might participate in translation. In addition, we show that Sso aIF5A cleaves different RNA substrates, linking it as well to RNA metabolism.
Results
aIF5A associates with ribosomes
In contrast to its eukaryal and bacterial counterparts, so far no evidence has been provided for an involvement of aIF5A in translation. Therefore, we asked whether Sso aIF5A associates with the translational machinery. To assess the cellular distribution of aIF5A, ribosome profiling experiments were performed followed by immuno-localization of aIF5A. First, a cell lysate of Sso P2 was treated with formaldehyde and separated by 10–30% sucrose gradient centrifugation, and the individual fractions were probed for aIF5A with anti-aIF5A antibodies as well as with antibodies against aIF6, which served as a marker for 50S subunits [31]. As shown in Figure 1(a), under these conditions aIF5A partitioned with the ribosome free fraction. As Sso 70S monosomes and polysomes are known to dissociate during ultracentrifugation even after formaldehyde fixation [32,33], they were not detectable under these conditions. To achieve stabilization of 70S monosomes, the Sso cell lysate was programmed in vitro for protein synthesis with a bicistronic mRNA as described by Condo et al. [32], followed by cross-linking with formaldehyde. After separation by 10–30% sucrose gradient centrifugation aIF5A was found in the supernatant but also partitioned with 30S, 50S and 70S ribosomes (Figure 1(b)).
Figure 1.
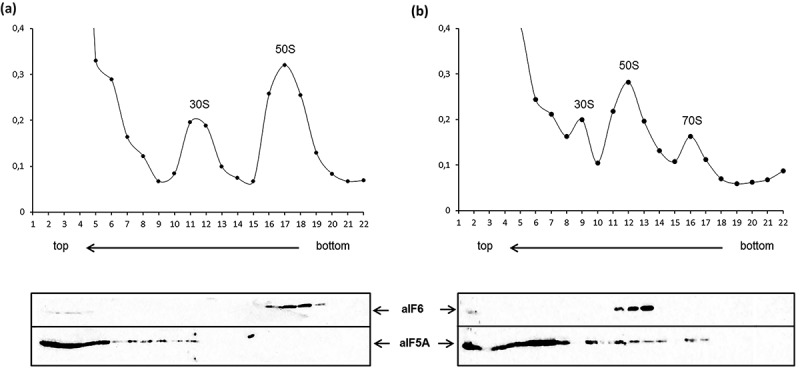
Cellular partitioning of aIF5A derived from Sso P2. (a) Top: Sedimentation profile showing 30S and 50S subunits derived from extracts of Sso P2 after fixation with formaldehyde. (b) Top: Sedimentation profile showing 30S and 50S subunits as well as 70S monosomes derived from extracts of Sso P2 programmed for translation with a bicistronic mRNA [32] after fixation with formaldehyde. (a, b) Bottom: Equal volumes of each protein fraction were subjected to western-blotting using anti-aIF6 or anti-aIF5A antibodies. The localization of aIF6, which is known to localize to 50S subunits [31], served as a control.
CRISPR-mediated aif5A knockdown results in growth retardation
With the rationale that aIF5A might be of importance for translation, we next asked whether depletion of aIF5A would affect cell viability as seen before in yeast [34]. All our attempts to delete the aif5A gene by established genetic methods failed. Therefore, we employed CRISPR-mediated (Clustered Regularly Interspaced Short Palindromic Repeats) post-transcriptional silencing, using a minimal CRISPR (miniCR) locus supplied on a viral shuttle vector, and a native CRISPR type III ribonuclease (Figure 2(a)) [35]. Two different silencing miniCR were designed, miniCR-aIF5A-I and miniCR-aIF5A-II, each expressing a single crRNA species (CRISPR-RNA, transcribed from spacer) targeting the aif5A mRNA at position PS-I or PS-II (Figure 2(a)), respectively. MiniCR- aIF5A-I (targeting at PSI) transformed cultures were not further processed, as all screened cultures had lost the aif5A-targeting spacer already at early growth stages (Figure S1). Loss or mutation of targeting spacers is generally observed when essential genes are strongly silenced [I. Zink, unpublished], again indicating a certain threshold of aIF5A protein necessary for cell survival. Furthermore, a control construct miniCR-MoE, carrying the same miniCR locus but lacking a host-targeting spacer, was constructed (Figure 2(a)). When compared with the control construct, expression of miniCR-aIF5A-II resulted in ∼40% reduction of aif5A mRNA (Figure 2(b)) and aIF5A protein (Figure 2(c)) in three biological replicates of Sso P1 compared to the MoE control cultures. As shown in Figure 2(d), the Sso P1 aif5A knock-down strain displayed a severe growth phenotype. The lag phase was prolonged and at the end point the cells did not grow to the high cell density as observed with the control strain (Figure 2(d)). Hence, this finding might point to a retardation of translation in the aif5A knock-down strain.
Figure 2.
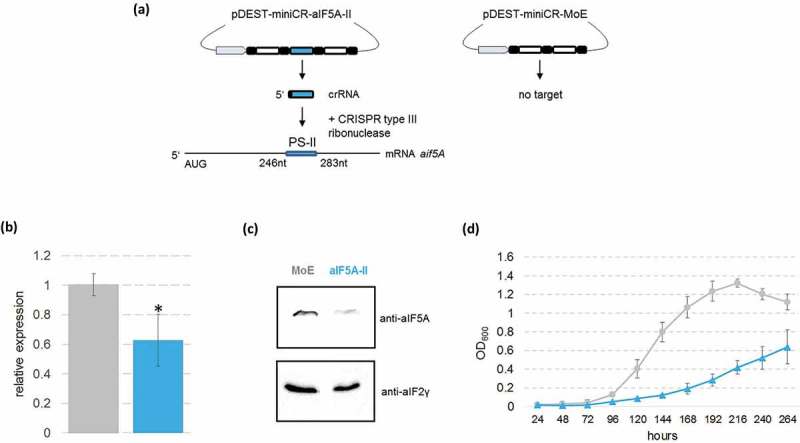
CRISPR-mediated post-transcriptional silencing of the aif5A gene in Sso P1-18. (a) Schematic representation of the silencing procedure with the miniCR- aif5A-II silencing vector. aif5A spacer II is transcribed to a crRNA, which incorporates into a CRISPR type III complex and binds to protospacer II region (PS II) within the aif5A mRNA. The protospacer spanning sequence (nt +246 −283) with regard to the A (+1) of the start codon of the aIF5A gene is indicated. The MoE – control vector is composed of the miniCRISPR backbone array without a host targeting spacer. Grey: promoter, black: CRISPR-repeats, white: non-targeting spacers according to Zebec et al. [35]; Blue: aif5A spacer II. (b) RT-qPCR performed with total RNA extracts of pDEST-miniCR- aif5A-II and MoE – transfected cultures harvested at an OD600 = 0.2. aif5A mRNA was quantified with the qaIF5A -FW/RV primer pair relatively to the housekeeping reference gene SSOP1_3283 amplified with q3194-FW/RV primers. The asterisk indicates significant difference to MoE (tow-tailed t-test, P < 0.01). Error bars: SD (n ≥ 3). (c) pDEST-miniCR- aIF5A-II and MoE – transfected cultures (obtained from three different plaques) were harvested at an OD60 0 = 0.2, and the aIF5A protein levels were determined by western-blot analysis. One representative western-blot is shown for MoE and aIF5A-II, respectively. aIF2(γ) protein served as a loading control. (d) Growth curve of cultures transfected with pDEST-miniCR- aIF5A-II (blue) and pDEST-miniCR-MoE (grey). The first measurement was taken 24 hours after inoculation of plaques into liquid medium. Error bars: SD (n = 3, i.e. three different single plaques picked per transfection).
Sso aIF5A displays RNase activity
More than 95% of aIF5A was detected in the ribosome free fraction when the ‘programmed lysate’ was subjected to fractionation (Figure 1(b)). Thus, it was conceivable that aIF5A fulfils additional function(s). As Halobacterium sp. NRC-1 aIF5A was shown to display ribonucleolytic activity [13], we next asked whether Sso aIF5A is also endowed with RNase activity. To detect RNase activities in Sso P2 a zymogram assay [36] was performed. In vitro transcribed 2508sh RNA was incorporated into a gel and a Sso P2 lysate was separated. Degraded RNA, i.e. the presence of RNase activity, was visible as white bands (Figure 3(a)) on dark background as digested RNA diffuses out of the gel [36]. In addition to a ~ 55 kDa band, which was previously identified as the aCPSF2 5´ to 3´ exoribonuclease [17,28], a band was visible at a position, which corresponded to 15 kDa, approximating to the size of aIF5A (Figure 3(a)).
Figure 3.
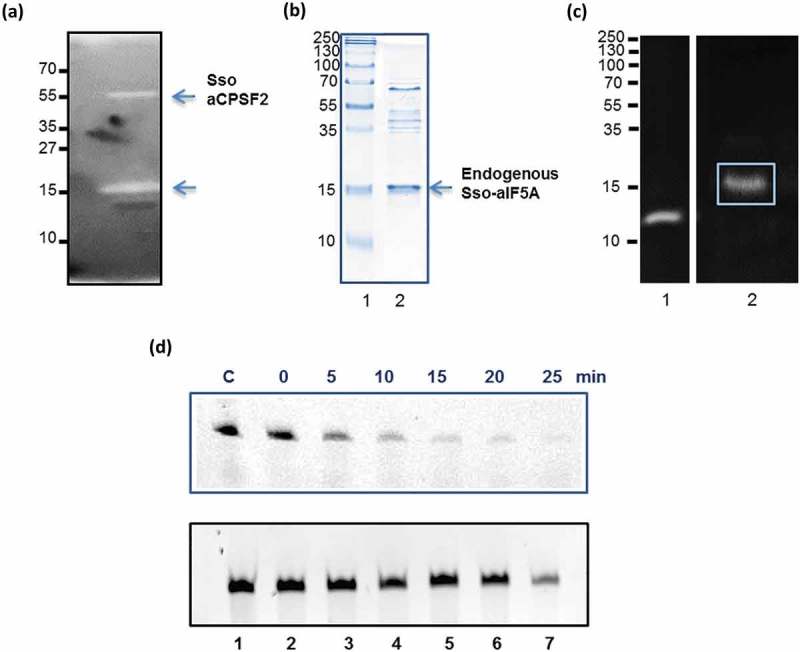
Ribonucleolytic activity of native aIF5A and recombinant N-His-aIF5A derived from Sso P2. (a) Zymogram assay for the identification of RNase activities in Sso P2. 20 µg of total protein of the Sso P2 S100 extract were separated on a 15% SDS polyacrylamide gel, which was polymerized together with Sso 2508sh RNA. The gel was stained with ethidium bromide. Bands displaying ribonucleolytic activity appear as white bands. Molecular weight standards are indicated at the left. (b) Lane 1, molecular weight standards. Lane 2, 15% SDS-PAGE with an enriched aIF5A preparation derived from Sso P2. The position of aIF5A is indicated by an arrow. The gel was stained with Coomassie brilliant blue. (c) Lane 1, the zymogram assay was performed with RNase A from bovine pancreas (positive control) and Sso aIF5A purified from Sso P2 (Lane 2) as described under (a). (d) Upper panel: Degradation of full length Sso 2508sh RNA with recombinant N-His-aIF5A purified from Sso PH1-16(pMJ05-aIF5A-C-His). Lane 1, incubation of 2508sh RNA for 25 min at 65°C in the absence of aIF5A. Lanes 2–7, time course of degradation of full length Sso 2508sh RNA with recombinant N-His-aIF5A. Lower panel: Lane 1, incubation of Sso 2508sh RNA for 25 min at 65°C. Lanes 2–7, time-dependent incubation of 2508sh RNA with the eluate fraction derived from the mock control (Sso PH1-16(pMJ05-ptf55α)).
Hypusinated aIF5A was then enriched from Sso P2 [6] (Figure 3(b)). To test further whether the observed RNase activity (Figure 3(a)) originated from Sso aIF5A the zymogram assay described above was exploited again. A unique white band was observed at the position that corresponded to a molecular weight of ~ 15 kDa (Figure 3(c), lane 2). The corresponding band was excised from the gel and analyzed by mass spectrometry. The majority of the detected peptides corresponded to aIF5A (Table S1, Figure S2). Nevertheless, the mass spectrometry analysis also identified several other proteins (Table S1, Figure S2).
Therefore, we also purified a hypusinated C-terminally His-tagged variant of aIF5A [6] from Sso strain PH1-16(pMJ05-aIF5A-C-His). As shown in Figure 3(d) (upper panel) the 2508sh RNA was degraded in the presence of the aIF5A-C-His variant, whereas no degradation was visible (Figure 3(d), lower panel) when the RNA was incubated with a concentrated eluate obtained after mock purification using a lysate of the control strain Sso PH1-16(pMJ05-ptf55α), suggesting that aIF5A is indeed endowed with RNase activity.
Next, we asked whether unmodified Sso N-His-aIF5A [6] purified to near homogeneity from E. coli (Figure 4(a)) is able to degrade 2508sh RNA. As shown in Figure 4(b), the degradation of the RNA substrate occurred at 65°C either in presence (Panel I) or absence of Mg++ (Panel II). In contrast, degradation of 2508sh RNA did not occur at 37°C (Figure 4(b), Panel III) suggesting that the aIF5A ribonucleolytic activity is only exerted under thermophilic conditions. To further verify that the ribonucleolytic activity of N-His-aIF5A preparation purified from E. coli is intrinsic to N-His-aIF5A, 2508sh RNA was incubated with a His-tagged version of recombinant Sso deoxyhypusine synthase (DHS), which was purified from E. coli using the same procedure as for N-His-aIF5A [6]. As shown in Figure 4(b), panel IV, no ribonucleolytic activity was observed, revealing that the ribonucleolytic activity of N-His-aIF5A derived from E. coli is not attributable to contaminant ribonucleases.
Figure 4.
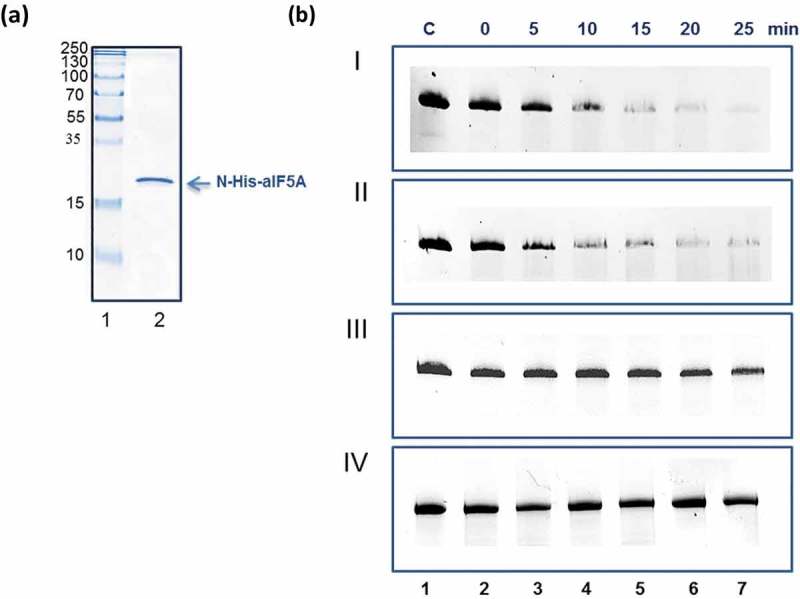
Ribonucleolytic activity of recombinant N-His-aIF5A purified from E. coli. (a) Lane 1, molecular weight marker. Lane 2, Lane 2, 15% SDS-PAGE with purified N-His-aIF5A from E. coli. The position of N-His-aIF5A is indicated by an arrow. The gel was stained with Coomassie brilliant blue. (b) Degradation of full length Sso 2508sh RNA with recombinant N-His-aIF5A purified from E. coli. Lane 1, incubation of 2508sh RNA for 25 min at 65°C, in the absence of proteins. Panel I, lanes 2–7, time course of degradation of full length Sso 2508sh RNA with recombinant N-His-aIF5A at 65°C in the presence of Mg++. Panel II, lanes 2–7, time course of degradation of full length Sso 2508sh RNA with recombinant N-His-aIF5A at 65°C in the absence of Mg++. (Panel III), time-dependent incubation of 2508sh RNA with N-His-aIF5A at 37°C. Panel IV, lanes 2–7, time-dependent incubation of 2508sh RNA with Sso aDHS-C-His, purified from E. coli (negative control).
Sso aIF5A displays endoribonucleolytic activity
To further study the ribonucleolytic activity of aIF5A, recombinant N-His-aIF5A produced in E. coli was incubated with a 41-nt-long RNA (5´-PPP-GGA*-″-3´ RNA), containing a single radioactively labelled adenine at the 5´-end. The RNA was designed to reveal a 5´ to 3´ exonuclease activity as shown before for the aCPSF2 RNase [17]. The degradation pattern of the RNA by N-His-aIF5A differed from that observed with aCPSF2 [17] in that no single A nt, indicative for 5´ to 3´ directional decay, was visible (Figure 5(a)). In contrast, a stable decay product occurred. Similarly, when Sso N-His-aIF5A produced in E. coli was incubated with 5´-end labelled 2508sh RNA, the increased accumulation of a specific degradation product was observed with time (Figure 5(b)). The stable decay product obtained after aIF5A cleavage of 5′-PPP-GGA*-″-3′ RNA appeared also in the presence of aIF2(γ) (Figure S3), which binds to the 5´-P3 end of mRNAs and was shown to prevent 5´ to 3´ directional cleavage by aCSPF2 [28]. Taken together, these experiments suggested that aIF5A is either endowed with an endoribonucleolytic or with 3´ to 5´ exoribonucleolytic activity, the latter of which is impeded at several positions, leading to the occurrence of several decay intermediates (Figure 5(b)).
Figure 5.

Endoribonucleolytic activity of Sso aIF5A. (a) Lane 1, [α-32P] ATP was loaded on the gel. Lane 2, incubation of 5′-PPP-GGA*-″-3′ RNA for 22 min at 65°C. Lanes 3–14, time course of degradation of 5′-PPP-GGA*-″-3′ RNA at 65°C in the presence of N-His-aIF5A purified from E. coli. The blue arrows indicate the full-length RNA and the single A nucleotide. The red arrow indicates a stable degradation product. (b) Lane 1, RNA size marker. Lane 2, incubation of 5´end-labelled 2508sh RNA for 22 min at 65°C. Lanes 3–14, time course of degradation of 5´end-labelled 2508sh RNA at 65°C in the presence of N-His-aIF5A purified from E. coli. The arrow indicates the accumulation of a stable degradation product. Only the relevant part of the autoradiogram is shown.
To test whether aIF5A displays 3´ to 5´ exoribonucleolytic activity, Sso N-His-aIF5A produced in E. coli was incubated with 2508sh RNA that was labelled at the 3´-end with pCp. The degradation pattern did not reveal a single C nt (not shown), but the presence of several degradation products (Figure S4), indicating an endoribonucleolytic rather than a 3´ to 5´ exonuclease activity. Moreover, in line with an endoribonucleolytic activity, aIF5A showed no preference for 5ʹ-hydroxyl- over RNA substrates with a 5´-P3 end (Figure S5) as observed for aCSPF2 [17].
A subset of RNAs co-purify with Sso aIF5A are cleaved by the protein
The observations that (i) eukaryal eIF5A interacts with RNA [9,10] and that (ii) archaeal aIF5A displays ribonucleolytic activity (Figure 3–5) [13], prompted us to identify RNAs bound to Sso aIF5A in vivo. To address this, aIF5A-C-His, was isolated from Sso PH1-16(pMJ05-aIF5A-C-His) using Ni-NTA magnetic beads and the identity of the co-purifying RNAs was revealed by deep-sequencing. Unspecific binding to the affinity matrix was controlled by a mock purification using a lysate of the control strain Sso PH1-16(pMJ05-ptf55α). The amount of RNA eluted in the presence of aIF5A-C-His was ten-fold higher with respect to the RNA isolated from the mock purification (not shown). The Sso PH1-16 genome has not been completely sequenced. Therefore, the reads were mapped against the genome of the close relative Sso 98/2. By setting the threshold at 100 reads and at an eight-fold difference in the TPM values (Materials and Methods), only a small set of RNAs was found to associate with aIF5A-C-His (Table S2). Among them only few RNAs encode known functions (Table S2). Then, we asked whether recombinant N-His-aIF5A would degrade some of the identified RNA substrates. Three Sso P2 mRNAs (Sso 2184/0910/0118), homologs of the Sso 98/2 mRNAs (SSOL_RS00015, SSOL_RS09370, SSOL_RS05430), which presented the highest number of reads in the aIF5A-C-His eluate (Table S2), were transcribed in vitro and used as substrates for N-His-aIF5A produced in E. coli. As shown in Figure S6, all three RNAs were degraded by the protein.
Discussion
This study provide for the first time an indication that Sso aIF5A is a moonlighting protein that associates with the translational machinery and act as an endoribonuclease. The factor aIF5A partitioned with the soluble fraction in translationally silent Sso extracts (Figure 1(a)), whereas a minor fraction was bound to ribosomal particles in lysates programmed for translation (Figure 1(b)). The Sso aif5A knockdown strain showed a slow-growth phenotype (Figure 2(d)). Whether this phenotype results from a role of aIF5A in translation or whether this defect is associated with the RNase activity requires further experimentation (see below). It is also formally possible that ribosome bound aIF5A exerts RNase activity, and that it serves in a quality control pathway to cope with translational errors.
The function of eukaryal IF5A and bacterial EF-P has been linked with the translation of proteins containing consecutive poly-proline stretches [2,3]. Nevertheless, a genome-wide analysis [37] has shown that these proteins are not common in both, Bacteria and Archaea (2.0–2.5%). The genome of the crenarchaeon Sulfolobus solfataricus P2 encodes 2,977 proteins [38] and only 2.2% are proline-rich proteins. Thus, further studies are warranted to test whether bacterial EF-P and archaeal aIF5A have an additional and/or another function in translation.
The hypusinated aIF5A, purified from Sso [6] as well as the recombinant unmodified His-tagged aIF5A purified from E. coli were able to cleave different RNA substrates with a 5´-P3 end as well as a substrate with a 5´hydroxyl end (Figure S5). In addition, endonucleolytic cleavage occurred, when the 5´end was blocked in the presence of aIF2(γ) (Figure S3). This 5´-end independence can be reconciled with the apparent endonucleolytic activity of the protein (Figure 5). It was shown that specific amino acid exchanges in the protein sequence affect the ribonucleolytic activity of Halobacterium sp. NRC-1 aIF5A protein [13]. In particular, the ribonucleolytic activity of aIF5A was reduced when amino acids at position 9 (N-terminal domain), amino acids at positions 72/73 (hinge region), or amino acids at position 117 or 122/123 (C-terminal domain), were exchanged (Figure S7). The alignment of aIF5A from Halobacterium sp. NRC-1 and from Sso P2 revealed that the three residues in the C-terminal domain are conserved (Figure S7). Hence, they may likewise be required for the RNAse activity of Sso aIF5A. Nonetheless, bioinformatics analyzes did not reveal typical signatures for ribonucleases (not shown). Thus, it remains to be shown whether aIF5A is endowed with a true endonucleolytic activity or whether this activity is rather unspecific. However, the limited number of RNA species bound to aIF5A seem to argue against non-specific binding. The ribonucleolytic activity of Halobacterium aIF5A seems to be restricted to single-stranded CA motifs [13]. We were unable to delineate a common cleavage motif for the 41 nt RNA and the 2508sh RNA substrates (Figure 5). Similarly, bioinformatic analyzes of the aIF5A co-purifying RNAs (Table S2) did not reveal a consensus motif with regard to substrate specificity. However, this does not exclude the possibility of structure recognition as known for archaeal splicing endonucleases [14]. The lack of defined cleavage sequences is reminiscent to the un-specificity described for the small ~ 6.5 kDa Sso7d ribonuclease, which was shown to cleave a RNA substrate internally, and even in double stranded regions [39]. However, from the cleavage patterns and the predicted secondary structures of the substrates used in this study it is more likely that cleavage occurs in single stranded regions or bulges (not shown).
What role in RNA metabolism might be assigned to the endoribonucleolytic activity of aIF5A? Several rRNA and tRNA processing enzymes as well as splicing endonucleases with endoribonuleolytic activity have been identified in Archaea, orthologues of which are present in Sulfolobales [14]. Given that the respective substrates were not predominant among the RNAs bound to aIF5A (Table S2), it appears rather unlikely that aIF5A is involved in processing of stable RNAs or dedicated to specific tasks, which would also argue against the idea that the growth defect observed with the aif5A knock-down strain results from the lack of formation of an essential RNA molecule.
In addition to the above mentioned processing enzymes, there are only a few examples for putative and confirmed riboendonucleolytic activities in Archaea and Sso. Among them are the putative orthologues of aPelota and aCPSF1, the latter of which might be endowed with exo- and endoribonuclease activity [14]. The only confirmed endoribonuclease with a broad substrate specificity is Sso7d [39]. Although much has to be learned with regard to mRNA decay pathways in Archaea, it is conceivable that endonucleolytic cleavage by aIF5A, and perhaps by other ribonucleases such as aPelota or Sso7d provides substrates suitable for 5´ to 3´directional decay by aCPSF1 [14] and/or aCPSF2 [17,28] as both, aCPSF1 [40] and aCPSF2 [17], require 5´-monophosphorylated RNA substrates to exert 5´ to 3´ exonuclease activity. In contrast to Bacteria, which harbour a dedicated phosphohydrolase [41], this activity was not found in Sso [17]. Moreover, it was shown that translation initiation factor aIF2(γ) can bind to the 5´-P3-end of mRNAs [27], which in turn counteracted the 5´ to 3´exonuclease activity of Sso aCPSF2. Thus, endonuleolytic cleavage of mRNAs containing a 5´-P3-end or a blocked 5´ end could pave the way for subsequent 5´ to 3´directional decay by aCPSF1 and/or CPSF2.
Materials and methods
Oligonucleotides and strains
Oligonucleotides and strains used in this work are listed in Table S3.
Ribosome profiling and immuno-localization
Sso P2 was grown at 75°C in Brock’s medium [42] supplemented with 0.2% NZ amine and 0.2% sucrose. The cells were harvested at an OD600 of 0.8 and 1,5 g (wet weight) of cells were disrupted by grinding in the presence of alumina [33]. The cell lysate was resuspended in extraction buffer (20 mM Tris/HCl pH 7.4, 40 mM NH4Cl, 10 mM Mg(CH3COO)2, 1 mM DTT, 2.5 μg/mL DNase I) and centrifuged to remove cellular debris. After two centrifugation steps at 26000 g for 30 min at 4°C, the supernatant (S30 extract) was collected. The S30 extract (500 μg total proteins), was pre-heated at 75°C for 10 min and then incubated in the presence of 2% formaldehyde for 1h on ice (control). The same sample was programmed for protein synthesis with 4 μg of the in vitro transcribed bicistronic mRNA encoding the Sso ORFs 104 and 143 [32], in a final volume of 50 μL containing 10 mM KCl, 20 mM Triethanolamine-HCl (TEA) pH 7.0, 18 mM MgCl2, 3 mM ATP, 1 mM GTP, 1,5 μg of bulk Sso tRNA and an amino acid mixture (0.1 mM each). The sample was incubated for 30 min at 75°C and then on ice for 1h with 2% formaldehyde. The samples were layered on a 10%-30% linear sucrose density gradient and centrifuged at 100000 g for 17 h at 4°C. 500 μL fractions were collected by continuously monitoring the absorbance at 260 nm, and then TCA precipitated. The proteins were separated on 12% SDS-polyacrylamide gels and analyzed by western-blotting for the presence of aIF5A and aIF6 (internal control) using specific antibodies as described below.
Western-blot analyzes
The rabbit polyclonal antibodies directed against Sso aIF5A, Sso aIF6 and aIF2(γ) have been described [6,27,31]. The proteins were separated on 12.5% SDS polyacrylamide gels and electro-transferred at 15V for 20 min onto a 0.2 μm nitrocellulose membrane (GE Healthcare) using transfer buffer (25 mM Tris base, 192 mM glycine, 0.1% (w/v) SDS, 20% (v/v) methanol). The blot was incubated with the respective antibodies. Anti-rabbit IgG coupled to horseradish peroxidase (Cell Signaling Technology) was used as a secondary antibody and the blot was developed using a chemiluminescent reagent (SuperSignal West Pico PLUS, Thermo Scientific). The signal was detected using the BioRad ChemiDoc™ MP Imaging system.
CRSPR-mediated aif5A knockdown
Two targeting aif5A-crRNAs (to be transcribed from the miniCR spacers aIF5A-I and -II, resp., see below) were designed with complementarity to 37 bp regions (SSOP1_1003, genome coordinates 849296–849332 and 849382–849418, resp.; GenBank: LT549890.1) within the aif5A gene. Protospacers were selected by the presence of a flanking 3´ PAS (protospacer adjacent sequence) motif [43]. aIF5A-silencing miniCRISPR- arrays were constructed analogously to the protocol described by Zebec et al. [44] using primers aIF5A_PS1-FW and aIF5A_PS1-RV assembling spacer aIF5A-I (5ʹ TCAACTTGCTGATCTACTGGAGCCATTAGTGTCTTTT 3ʹ), as well as aIF5A_PS2-FW and aIF5A_PS2-RV assembling spacer aIF5A-II (5ʹ TCTCATAACTTTCTAAATCCATTACTTGTATTTTATT 3ʹ), respectively (Table S3). A non-targeting miniCRISPR array ‘MoE’ was constructed using primers MoE-FW, MoE-RV assembling the miniCRISPR backbone array only by fusing backbone spacers D1 and D5 [44] (Table S3). MiniCRISPR arrays were finally inserted into the SSV1-virus based pDEST-MJ vector [35]. For the aif5A silencing experiments, Sso P1-18, an uracil – auxotrophic derivate of Sso P1 [45], was cultivated at 75°C in basic Brock T/S/U medium [44]. Sso P1-18 cells were transfected with the pDEST-miniCR vectors by electroporation [46]. The electroporation solutions were used in inverse plaque assays (on uracil – free Y/S gellan gum plates) after recovery [44]. Three plaques per transfectant were isolated from the plate and transferred to fresh Brock T/S medium (referred to as plaque cultures). Growth of control and of the silenced plaque cultures was monitored. 5 mL aliquots were harvested at an OD600 of 0.2 for RNA, DNA preparation and analysis of aIF5A protein levels. RNA and DNA preparations were performed as described in Zebec et al. [35]. RNA was further purified from DNA (TURBO DNA-free kit ThermoFisher, Scientific), reverse transcribed (ProtoScript II Reverse transcriptase, NEB) and 5 ng of cDNA were used for quantitative PCR (qPCR). All qPCRs were performed using GoTaq® qPCR Master Mix (Promega) in an Eppendorf Mastercycler epgradient S relplex2 (Eppendorf). Three biological replicates and three technical replicates of aif5A-silenced and MoE control cultures were measured, respectively. Transcripts of aif5A and of the SSOP1_3283 housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase) were quantified from the same cDNA preparations using primer pairs qaIF5A-FW/-RV and q3194-FW/-RV, respectively (Table S3). Relative expression of the aif5A gene was assessed by calculating the ΔCt of the two primer pairs and normalizing the values to the MoE control cultures [47]. qPCR efficiency was between 100% and 94% in all runs. MiniCR integrity was verified by culture PCR on 2 µL of fresh culture using 406-FW and 406-RV primers (Table S3), which bind to the pDEST vector backbone, ~100 bp up- and downstream of the miniCR array. Total proteins in 0.2 OD600 units of pDEST-miniCR- aIF5A-II and MoE – transfected cultures were subjected to SDS-PAGE followed by western-blot analysis with anti-aIF5A and anti-aIF2(γ) specific antibodies, as described above.
RNA preparation
A segment of Sso 2508 mRNA (nt 1–253 with regard to the A (+1) of the start codon; 2508sh RNA), encoding the leaderless acetyl-CoA-acetyltransferase gene (SSO2508), was prepared using Sso P2 chromosomal DNA together with the oligonucleotides 2508sh-FW/-RV (Table S3) to generate a PCR template for in vitro transcription with the AmpliScribe™ T7-Flash™ Transcription Kit (Epicentre). From the A (+1) of the start codon, Sso 2184/0910/0118 mRNAs were also in vitro transcribed as described above, using primer pairs 2184-FW/-RV, 0910-FW/-RV and 0118-FW/-RV (Table S3). The RNAs were gel purified and an aliquot of Sso 2508sh mRNA was treated with FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific) as specified by the manufacturer. 10 pmol of dephosphorylated 2508sh RNA were radiolabeled at the 5´-end with T4 Polynucleotide Kinase (Thermo Scientific) in the presence of 15 pmol of [α-32P]ATP (3000 Ci/mmol, Hartmann Analytic GmbH). Similarly, 10 pmol of gel purified Sso 2508sh mRNA were radioactively labelled at the 3′ end with [α-32P]pCp (3000 Ci/mmol, Hartmann Analytic GmbH) and T4 RNA ligase (Thermo Scientific). Radiolabeled 2508sh RNAs were purified with a ProbeQuant™ G-50 Micro Column (GE Healthcare) to remove unincorporated radiolabeled nucleotides, and then phenol/chloroform extracted. Two different 41-nt-long synthetic RNAs, harboring a single radiolabeled A at the 5´-triphosphorylated end (5′-PPP-GGA*-″-3′ RNA) or a 5′-hydroxyl (5′-OH) end (5′-OH-GGA*-″-3′ RNA) (Microsynth Austria GmbH) were generated as previously described [17].
Purification of aIF5A from Sso P2
Sso P2 cell extract was prepared using 20 g of cells [48]. The purification of native aIF5A was carried out as described [7] with some modifications. S100 extract in buffer A (20 mM Tris-HCl pH 7.5, 10 mM Mg(CH3COO)2, 40 mM NH4Cl, 6 mM β-mercaptoethanol) was subjected to affinity chromatography on Cibacron blue sepharose 6-fast flow (GE Healthcare, 50 mL bed volume, 25 × 2.5 cm, 20 mL/h), equilibrated in buffer B (20 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 150 mM KCl). The proteins were eluted in buffer B containing 6 mM spermine. 10 mL fractions were collected and 10 µL of each fraction were analyzed for the presence of aIF5A using dot-blots (Bio-Rad) and anti-aIF5A antibodies. aIF5A positive fractions were concentrated using a 3 kDa Amicon® Ultra centrifugal filter device. The subsequent chromatographic steps were performed using an FPLC AKTA purification system (GE Pharmacia). The concentrated protein pool was subjected to gel filtration chromatography on a superose 12 column (GE Healthcare, 25 mL bed volume, 30 × 1.0 cm, 0.5 mL/min) equilibrated in buffer B. 0.5 mL fractions were eluted with the same buffer and 2 µL of each fraction were analyzed for the presence of aIF5A. aIF5A positive fractions were concentrated using 3 kDa Amicon® Ultra centrifugal filter device and the buffer was exchanged to buffer B (pH 9.0). The last chromatographic step was performed on a Mono Q (1 mL bed volume, 0.5 mL/min) equilibrated in buffer B (pH 9.0). The proteins were eluted using a liner gradient of KCl (50 mM – 1 M). 0.5 mL fractions were collected and 2 µL of each fraction were analyzed for the presence of aIF5A. aIF5A-positive fractions were concentrated using a 3 kDa Amicon® ultra centrifugal filter device, dialyzed against aIF5A storage buffer (50 mM Tris-HCl pH 7.5, 150 mM KCl, 5% glycerol), and analyzed by SDS-PAGE. The protein concentration was determined using the Bradford test (Sigma-Aldrich).
Zymogram assay
In vitro transcribed Sso 2508sh RNA was incubated in DEPC water at 50°C for 5 min and incorporated into the gel matrix (15% SDS polyacrylamide gel) to a final concentration of 0.15 mg/mL. 20 μg total protein of a Sso P2 S100 extract were loaded onto the gel and separated. In addition, 3 μg of Sso aIF5A and 1 ng of bovine pancreatic RNase A (Merck Millipore; positive control) were also assayed. The gel was incubated overnight in 25% isopropanol on a shaking platform at room temperature. The following renaturing steps were carried out at room temperature as previously described [17]: 2 × 15 min, 6 M guanidine chloride (GnCl) in incubation buffer (IB; 5 mM HEPES, pH 7.0, 10 mM KCl, 10 mM Mg(CH3COO)2) and 0.1 mM DTT; 15 min, 3 M GnCl in IB and 0.1 mM DTT; 15 min, 1.5 M GnCl in IB and 0.1 mM DTT; 15 min, 0.75 M GnCl in IB and 0.1 mM DTT; 15 min, 0.375 M GnCl in IB and 0.1 mM DTT; 15 min, 0.1 M GnCl in IB and 0.1 mM DTT; 15 min, IB and 0.1 mM DTT. The last incubation step was performed for 90 min at 65°C in IB and 1 mM DTT. To assay the ribonucleolytic activity of bovine pancreatic RNase A, the incubation step was carried out for 90 min at 37°C in IB in the presence of 1 mM DTT. The gel was stained with ethidium bromide and the signal was detected using the BioRad ChemiDoc™ MP Imaging system.
RNA cleavage assays
The RNA degradation assays were performed with either Sso aIF5A, recombinant Sso N-His-aIF5A produced in Sso PH1-16(pMJ05-aIF5A-C-His) [6] or recombinant Sso N-His-aIF5A produced in E. coli [6]. Prior to the assay, the proteins were pre-heated for 10 min at 65°C and the in vitro transcribed RNA substrates were denatured for 5 min at 85°C. Sso aIF5A (10 µM) and the recombinant His-tagged aIF5A proteins (10 µM) were incubated in a total reaction volume of 5 µL together with 2508sh RNA (2 µM) in incubation buffer (10 mM HEPES pH 8.0, 100 mM KCl, 5 mM MgCl2, 5 mM β-mercaptoethanol, 5% glycerol). RNA degradation was allowed to proceed between 0 and 25 min at 65°C. As a control 2508sh RNA was incubated for 25 min at 65°C in the absence of Sso aIF5A. The reactions were terminated by addition of 5 µL of 95% formamide/20 mM EDTA. The samples were loaded on a 8% PAA-8M urea gel and the gel was stained with ethidium bromide. The assay with recombinant N-His-aIF5A produced in E. coli was also performed in the absence of Mg++ and in the presence of the concentrated eluate obtained from an E. coli lysate in which the recombinant Sso N-His-aIF5A protein was not produced (mock control). The assay was also performed in the presence of the concentrated eluate obtained from an Sso PH1-16(pMJ05) lysate in which the recombinant Sso N-His-aIF5A protein was not produced (mock control).
The RNase activity of recombinant N-His-aIF5A produced in E. coli (6 µM) towards 2508sh RNA (10 nM 5´-end labeled RNA and 1.5 µM cold RNA) was allowed to proceed between 0 and 22 min at 65°C as described above. As a control 5´-end labeled 2508sh RNA was incubated for 22 min at 65°C in the absence of recombinant N-His-aIF5A. Likewise, 2508sh RNA (10 nM 3´-end labeled RNA and 1.5 µM cold RNA) was incubated with recombinant N-His-aIF5A (6 µM) produced in E. coli between 0 and 18 min at 65°C, using the experimental conditions described above. As a control 3´-end labeled 2508sh RNA was incubated for 18 min at 65°C in the absence of recombinant N-His-aIF5A. The reactions were terminated by addition of 5 µL of 95% formamide/20 mM EDTA. The samples were loaded on a 20% PAA-8M urea gel, which was analyzed with a Typhoon FLA 9500 PhosphorImager (GE Healthcare).
The RNA cleavage assay with recombinant N-His-aIF5A produced in E. coli (75 µM) was also performed with 5′-PPP-GGA*-″-3′ RNA (10 nM radiolabeled RNA and 1.5 µM cold RNA), pre-incubated for 10 min at 65°C with the γ-subunit of the trimeric factor aIF2 (3 µM), produced in E.coli as previously described [29]. The degradation assays with 5′-PPP-GGA*-″-3′ RNA were performed between 0 and 22 min at 65°C in incubation buffer and in a total reaction volume of 5 µL. 5′-PPP-GGA*-″-3′ RNA was also incubated for 22 min at 65°C in the absence of recombinant N-His-aIF5A produced in E.coli. The reactions were terminated by addition of 5 µL of 95% formamide/20 mM EDTA. The samples were loaded on a 20% PAA-8M urea gel. The gel was dried and the radioactive signals were detected with a Typhoon FLA 9500 PhosphorImager (GE Healthcare). In addition, 5′-PPP-GGA*-″-3′ RNA (2 µM) or 5′-OH-GGA*-″-3′ RNA (2 µM) was incubated in a total reaction volume of 5 µL together with recombinant N-His-aIF5A produced in E. coli (10 µM). The degradation assays with 41-nt-long synthetic RNAs were performed between 0 and 22 min at 65°C in incubation buffer. As a control 5′-PPP-GGA*-″-3′ RNA or 5′-OH-GGA*-″-3′ RNA was incubated for 22 min at 65°C in the absence of recombinant N-His-aIF5A.The reactions were terminated by addition of 5 µL of 95% formamide/20 mM EDTA. The samples were loaded on a 20% PAA-8M urea gel and the gel was stained with ethidium bromide.
Liquid chromatography – mass spectrometry (LC-MSMS)
Mass Spectrometry analyzes were performed by the MFPL Mass Spectrometry Facility using the VBCF instrument pool (https://www.mfpl.ac.at/research/scientific-facilities/mass-spectrometry.html). The proteins were in-gel digested with trypsin according to the protocol of Mair et al. [49]. The peptide solution was desalted on custom-made C18 StageTips [50,51]. The tryptic digests were separated by reversed-phase liquid chromatography (LC) on an Ultimate 3000 RSLC nano-flow chromatography system (Thermo Fisher Scientific), using a pre-column for sample loading (PepMapAcclaim C18, 2 cm × 0.1 mm, 5 μm) and a C18 analytical column (PepMapAcclaim C18, 50 cm × 0.75 mm, 2 μm, Dionex-Thermo-Fisher Scientific). The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in 80% acetonitrile). The proportion of solvent B was linearly increased from 2% to 35% wíthin 60 min at a flow rate of 230 nL/min. Eluted peptides were separated on a Q Exactive HFX Orbitrap mass spectrometer, equipped with a Proxeon nanospray source (Thermo Fisher Scientific) and operated in a data-dependent mode. Scans were obtained in a mass range of 375–1500 m/z with lock mass on, at a resolution of 60000 at 200 m/z and an AGC target value of 3E6. The 10 most intense ions were selected with an isolation width of 1.6 Da, fragmented in the HCD cell at 27% collision energy, and the spectra were recorded at a target value of 1E5 and a resolution of 30000. Peptides with a charge of +1 were excluded from fragmentation, the peptide match and exclude isotope features were enabled, and selected precursors were dynamically excluded from repeated sampling for 15 s. The raw data were processed with MaxQuant software package (version 1.6.0.16, www.maxquant.org) [51] by searching against the sequence of aIF5A in the background of the Sulfolobus solfataricus uniprot (www.uniprot.org) and sequences of common contaminants, with tryptic specificity allowing 2 missed cleavages. The protein list was further filtered for a minimum of 2 unique and razor peptides.
Deep sequencing analysis
4L of Sso PH1-16(pMJ05-aIF5A-C-His) [6] and 4L Sso PH1-16(pMJ05-ptf55α) [6], were grown at 75°C in Brock’s medium [42], supplemented with 0.2% NZ amine and 0.2% sucrose. At an OD600 of 0.8, the cells were harvested and lysed by sonication in 40 mL lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 15 mM imidazole, 10 mM β-mercaptoethanol, 1 mM PMSF), and the lysates were centrifuged at 25000 g for 30 min at 4°C. The clear lysates were incubated overnight at 4°C with 300 µl of pre-equilibrated Ni–NTA agarose resin (Qiagen). The lysates were transferred to Poly-Prep chromatography columns (Bio-Rad, Hercules, CA, USA) and washed with 30 mL of washing buffer (50 mM Tris–HCl pH 7.4, 300 mM NaCl, 40 mM imidazole in DEPC-water). The proteins were eluted with 1 mL elution buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 250 mM imidazole in DEPC-water). The eluates were dialyzed (50 mM Tris–HCl pH 7.4, 150 mM KCl, 5% glycerol in DEPC-water), concentrated using 3 K Amicon® Ultra-0.5 centrifugal filter devices, and analyzed by SDS-PAGE. The RNA present in the eluates was extracted with phenol/chloroform, precipitated and resuspended in 50 μL of DEPC-water. The samples were treated with DNase I (RNase-free, Roche) and complete degradation of chromosomal DNA was assessed by PCR using the 16S rRNA-specific oligonucleotides (16S-FW/-RV, Table S3). The quality and the quantity of the RNA samples was determined with an Agilent 2100 Bioanalyzer (Agilent Technologies) and the RNA 6000 Pico Kit (Agilent Technologies), respectively. cDNAs libraries were constructed using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech) and a total of 50 bp single end sequence reads were generated by the next generation sequencing facility at the Vienna Biocenter Core Facilities GmbH (VBCF) (https://www.vbcf.ac.at/facilities/next-generation-sequencing/) using the Illumina HiSeq 2000 platform.
Bioinformatic analyzes
Adaptor sequences were removed with cutadapt [52] from the raw reads and mapping of the samples against the Sso 98/2 reference genome (NC_017274.1) was performed with segemehl (http://www.bioinf.uni-leipzig.de/Software/segemehl/). Reads for coding regions and for non-coding regions were counted using BEDtools [53] and processed for automatic visualization with Vienna NGS toolbox [54]. The reads were normalized to obtain the TPM (Transcripts per million) values, which facilitate measures of RNA abundance within and between samples [55]. For a comparison of transcripts levels, bound to either Sso aIF5A-C-His or co-eluted in the mock control, a threshold of eight-fold difference in the TPM values (TPM Sso-aIF5A-C-His/TPM mock) was used and all RNAs with more than 100 reads in the enriched eluate of Sso aIF5A-C-His were selected. The raw sequencing data were deposited in the NCBI sequence read archive (SRA) as a study under the accession number PRJNA505653.
Funding Statement
Funds provided by Austrian Science Fund (FWF). This work was supported by Grant 22888 from the Austrian Science Fund to UB and by funds (RSA2017) from the Polytechnic University of Marche to ALT.
Acknowledgments
This work was supported by Grant 22888 from the Austrian Science Fund to UB and by funds (RSA2017) from the Polytechnic University of Marche to ALT. We thank Dr´s Paola Londei, Dario Benelli, Roberto Spurio and Attilio Fabbretti for their support with the ribosome profiling experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Gutierrez E, Shin BS, Dever TE.. The hypusine-containing translation factor eIF5A. Crit Rev Biochem Mol Biol. 2014;49:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ude S, Lassak J, Starosta AL, et al. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science. 2013;339:82–85. [DOI] [PubMed] [Google Scholar]
- [3].Schuller AP, Wu -C-C-C, Dever TE, et al. eIF5A functions globally in translation elongation and termination. Mol Cell. 2017;66:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shiba T, Mizote H, Kaneko T, et al. Hypusine, a new amino acid occurring in bovine brain. Isolation and structural determination. Biochim Biophys Acta. 1971;244:523–531. [DOI] [PubMed] [Google Scholar]
- [5].Yanagisawa T, Sumida T, Ishii R, et al. A paralog of lysyl-tRNA synthetase aminoacylates a conserved lysine residue in translation elongation factor P. Nat Struct Mol Biol. 2010;17:1136–1143. [DOI] [PubMed] [Google Scholar]
- [6].Bassani F, Romagnoli A, Cacciamani T, et al. Modification of translation factor aIF5A from Sulfolobus solfataricus. Extremophiles. 2018;22:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bartig D, Lemkemeier K, Frank J, et al. The archaebacterial hypusine-containing protein: structural features suggest common ancestry with eukaryotic translation initiation factor 5A. Eur J Biochem. 1992;204:751–758. [DOI] [PubMed] [Google Scholar]
- [8].Yao M, Ohsawa A, Kikukawa S, et al. Crystal structure of hyperthermophilic archaeal initiation factor 5A: a homologue of eukaryotic initiation factor 5A (eIF-5A). J Biochem. 2003;133:75–81. [DOI] [PubMed] [Google Scholar]
- [9].Xu A, Jao DL, Chen KY. Identification of mRNA that binds to eukaryotic initiation factor 5A by affinity co-purification and differential display. J Biochem. 2004;384:585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Xu A, Chen KY. Hypusine is required for a sequence-specific interaction of eukaryotic initiation factor 5A with post systematic evolution of ligands by exponential enrichment RNA. J Biol Chem. 2001;276:2555–2561. [DOI] [PubMed] [Google Scholar]
- [11].Schrader R, Young C, Kozian D, et al. Temperature-sensitive eIF5A mutant accumulates transcripts targeted to the nonsense-mediated decay pathway. J Biol Chem. 2006;281:35336–35346. [DOI] [PubMed] [Google Scholar]
- [12].Zuk D, Jacobson A. A single amino acid substitution in yeast eIF-5A results in mRNA stabilization. Embo J. 1998;17:2914–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wagner S, Klug G. An archaeal protein with homology to the eukaryotic translation initiation factor 5A shows ribonucleolytic activity. J Biol Chem. 2007;282:13966–13976. [DOI] [PubMed] [Google Scholar]
- [14].Clouet-d’Orval B, Batista M, Bouvier M, et al. Insights into RNA-processing pathways and associated RNA-degrading enzymes in Archaea. FEMS Microbiol Rev. 2018;42:579–613. [DOI] [PubMed] [Google Scholar]
- [15].Evguenieva-Hackenberg E, Bläsi U. Attack from both ends: mRNA degradation in the crenarchaeon Sulfolobus solfataricus. Biochem Soc Trans. 2013;41:379–383. [DOI] [PubMed] [Google Scholar]
- [16].Dominski Z, Carpousis AJ, Clouet-d’Orval B. Emergence of the β-CASP ribonucleases: highly conserved and ubiquitous metallo-enzymes involved in messenger RNA maturation and degradation. Biochim Biophys Acta. 2013;1829:532–551. [DOI] [PubMed] [Google Scholar]
- [17].Hasenöhrl D, Konrat R, Bläsi U. Identification of an RNase J ortholog in Sulfolobus solfataricus: implications for 5′-to-3′ directional decay and 5′-end protection of mRNA in Crenarchaeota. Rna. 2011;17:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Portnoy V, Evguenieva-Hackenberg E, Klein F, et al. RNA polyadenylation in Archaea: not observed in Haloferax while the exosome polynucleotidylates RNA in Sulfolobus. EMBO Rep. 2005;6:1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Evguenieva-Hackenberg E, Walter P, Hochleitner E, et al. An exosome-like complex in Sulfolobus solfataricus. EMBO Rep. 2003;4:889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Portnoy V, Schuster G. RNA polyadenylation and degradation in different Archaea; roles of the exosome and RNase R. Nucleic Acids Res. 2006;34:5923–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jarrous N, Gopalan V. Archaeal/eukaryal RNase P: subunits, functions and RNA diversification. Nucleic Acids Res. 2010;38:7885–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Späth B, Canino G, Marchfelder A. tRNase Z: the end is not in sight. Cell Mol Life Sci. 2007;64:2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li H, Abelson J. Crystal structure of a dimeric archaeal splicing endonuclease. J Mol Biol. 2000;302:639–648. [DOI] [PubMed] [Google Scholar]
- [24].Wang R, Preamplume G, Terns MP, et al. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011;19:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wurtzel O, Sapra R, Chen F, et al. A single-base resolution map of an archaeal transcriptome. Genome Res. 2010;20:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Arkhipova V, Stolboushkina E, Kravchenko O, et al. Binding of the 5′-Triphosphate end of mRNA to the γ-subunit of translation initiation factor 2 of the crenarchaeon Sulfolobus solfataricus. J Mol Biol. 2015;427:3086–3095. [DOI] [PubMed] [Google Scholar]
- [27].Hasenöhrl D, Lombo T, Kaberdin V, et al. Translation initiation factor a/eIF2 (-γ) counteracts 5ʹ to 3ʹ mRNA decay in the archaeon Sulfolobus solfataricus. Proc Natl Acad Sci USA. 2008;105:2146–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Märtens B, Amman F, Manoharadas S, et al. Alterations of the transcriptome of Sulfolobus acidocaldarius by exoribonuclease aCPSF2. PLoS One. 2013;8:e76569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pedulla N, Palermo R, Hasenöhrl D, et al. The archaeal eIF2 homologue: functional properties of an ancient translation initiation factor. Nucleic Acids Res. 2005;33:1804–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Märtens B, Manoharadas S, Hasenöhrl D, et al. Back to translation: removal of aIF2 from the 5′-end of mRNAs by translation recovery factor in the crenarchaeon Sulfolobus solfataricus. Nucleic Acids Res. 2014;42:2505–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Benelli D, Marzi S, Mancone C, et al. Function and ribosomal localization of aIF6, a translational regulator shared by archaea and eukarya. Nucleic Acids Res. 2009;37:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Condo I, Ciammaruconi A, Benelli D, et al. Cis-acting signals controlling translational initiation in the thermophilic archaeon Sulfolobus solfataricus. Mol Microbiol. 1999;34:377–384. [DOI] [PubMed] [Google Scholar]
- [33].Londei P, Altamura S, Cammarano P, et al. Differential features of ribosomes and of poly(U)-programmed cell-free systems derived from sulphur-dependent archaebacterial species. Eur J Biochem. 1986;157:455–462. [DOI] [PubMed] [Google Scholar]
- [34].Kang HA, Hershey JW. Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. J Biol Chem. 1994;269:3934–3940. [PubMed] [Google Scholar]
- [35].Zebec Z, Manica A, Zhang J, et al. CRISPR-mediated targeted mRNA degradation in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2014;42:5280–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Scadden ADJ, Naaby-Hansen S. Analysis of ribonucleases following gel electrophoresis. Methods Enzymol. 2001;341:126–141. [DOI] [PubMed] [Google Scholar]
- [37].Mandal A, Mandal S, Park MH. Genome-wide analyses and functional classification of proline repeat-rich proteins: potential role of eIF5A in eukaryotic evolution. PLoS One. 2014;9:e111800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].She Q, Singh RK, Confalonieri F, et al. The complete genome of the crenarchaeon Sulfolobus solfataricus P2. Proc Natl Acad Sci USA. 2001;98:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shehi E, Serina S, Fumagalli G, et al. The Sso7d DNA-binding protein from Sulfolobus solfataricus has ribonuclease activity. FEBS Lett. 2001;497:131–136. [DOI] [PubMed] [Google Scholar]
- [40].Levy S, Portnoy V, Admon J, et al. Distinct activities of several RNase J proteins in methanogenic archaea. RNA Biol. 2011;8:1073–1083. [DOI] [PubMed] [Google Scholar]
- [41].Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008;451:355–358. [DOI] [PubMed] [Google Scholar]
- [42].Brock TD, Brock KM, Belly RT, et al. Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch Mikrobiol. 1972;84:54–68. [DOI] [PubMed] [Google Scholar]
- [43].Manica A, Zebec Z, Steinkellner J, et al. Unexpectedly broad target recognition of the CRISPR-mediated virus defence system in the archaeon Sulfolobus solfataricus. Nucleic Acids Res. 2013;41:10509–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zebec Z, Zink IA, Kerou M, et al. Efficient CRISPR-mediated post-transcriptional gene silencing in a hyperthermophilic archaeon using multiplexed crRNA expression. G3 (Bethesda). 2016;6:3161–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Martusewitsch E, Sensen CW, Schleper C. High spontaneous mutation rate in the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by transposable elements. J Bacteriol. 2000;182:2574–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Manica A, Zebec Z, Teichmann D, et al. In vivo activity of CRISPR-mediated virus defence in a hyperthermophilic archaeon. Mol Microbiol. 2011;80:481–491. [DOI] [PubMed] [Google Scholar]
- [47].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Benelli D, Londei P. In vitro studies of archaeal translational initiation. Methods Enzymol. 2007;430:79–109. [DOI] [PubMed] [Google Scholar]
- [49].Mair A. SnRK1-triggered switch of bZIP63 dimerization mediates the low-energy response in plants. Elife. 2015;11:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Rappsilber J. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. [DOI] [PubMed] [Google Scholar]
- [51].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. [DOI] [PubMed] [Google Scholar]
- [52].Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10. [Google Scholar]
- [53].Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wolfinger MT, Fallmann J, Eggenhofer F, et al. ViennaNGS: a toolbox for building efficient next- generation sequencing analysis pipelines. F1000Res. 2015;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.