

Abstract
The construction of biocatalytic cascades for the production of chemical precursors is fast becoming one of the most efficient approaches to multi-step synthesis in modern chemistry. However, despite the use of low solvent systems and renewably-resourced catalysts in reported examples, many cascades are still dependent on petrochemical starting materials, which as of yet cannot be accessed in a sustainable fashion. Herein we report the production of the versatile chemical building block cinnamyl alcohol from the primary metabolite and fermentation product L-phenylalanine. Through the combination of three biocatalyst classes (phenylalanine ammonia lyase, carboxylic acid reductase and alcohol dehydrogenase) the target compound could be reached in high purity, demonstrable at 100 mg scale achieving 53 % yield using ambient temperature and pressure in aqueous solution. This system represents a synthetic strategy in which all components present at time zero are biogenic and thus minimising damage to the environment. Further we extend this biocatalytic cascade by its inclusion in a L-phenylalanine overproducing strain of Escherichia coli. This metabolically engineered strain produces cinnamyl alcohol in mineral media using a glycerol and glucose as carbon source. This study demonstrates the potential to establish green routes to the synthesis of cinnamyl alcohol from a waste stream such as glycerol derived, for example, from lipase treated biodiesel.
In an effort to widen the remit of green chemistry in the production of materials, additives and pharmaceuticals, several avenues are being explored to reduce the long-term environmental impact often incurred in such processes. One strategy that has seen sustained attention is recent years is biocatalysis: the use of enzymes to complement and replace chemical routes to various target molecules.1, 2 The advantages of using enzymes as opposed to organic synthetic methods include mild reaction conditions, compatibility with aqueous media, sourcing of catalysts from renewable feedstocks and chemo-, regio- and enantioselectivity. These features often enable the efficient combination of multiple enzymes within a single reaction vessel under common conditions without unwanted cross-reactivity or the need for complex and wasteful purification of intermediate compounds.3–6 Despite this, a large number of biocatalytic routes make use of starting materials obtained from traditional sources, such as petroleum. As such, there is a drive to find methods employing biocatalysts for the conversion of sustainably-resourced substrates to products associated with multiple and / or industrially-important syntheses.
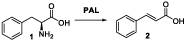
Cinnamyl alcohol is a simple versatile chemical implicated in the production of various compounds of applied and commercial interest. Simple esterification of this alcohol can afford a variety of cinnamyl esters, which find use in the flavour and fragrance industries,7, 8 as well as being used as precursors for the production of smart polymer materials.9 Examples of these include cinnamyl acetate, which is used to confer spicy and floral aromas, and cinnamyl methacrylate - a monomer which can be polymerised by both radical and photochemical means to create a dual crosslinked product. The amination of cinnamyl alcohol has also been reported, allowing the facile production of various cinnamyl amines.10–12 Notable examples of these include the clinically approved drugs flunarizine and naftifine (used for the treatment of fungal infections and peripheral vascular conditions, respectively). As well as one-step conversions yielding compounds of interest, cinnamyl alcohol is also reported as a starting material for the multistep synthesis of the drug dapoxetine and a widely used cancer treatment drug taxol (Figure 1).13, 14
Figure 1.

Examples of industrially-relevant chemicals which can be synthesized from cinnamyl alcohol. Cinnamyl amines including naftifine and flunarizine,10–12 cinnamyl esters including various fragrances7, 8 and photocrosslinking monomers, multi-step synthesis of compounds such as dapoxetine and taxol.13, 14
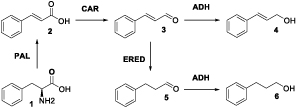
The current provision for bio-derived production of cinnamyl alcohol is poor, due to the low levels in which it occurs naturally as a metabolite. As such, synthesis of this compound usually involves the chemoselective reduction of the corresponding aldehyde using chemical reducing agents, finite and expensive metals or complex catalyst formulations, such as nanotubes or nanoparticles.15–18 Chemical reduction of allyl alcohols often requires fine control of reaction conditions, due to the possible non-selective reduction of both the hydroxyl and ene functionalities.19 Whilst there are examples of alcohol dehydrogenase (ADH) enzymes used to perform this same reaction in a more mild and chemoselective fashion,20, 21 access to cinnamaldehyde is not as easy as various primary metabolites, widely available as fermentation products. To this end, we envisaged the addition of well-documented carboxylic acid reductase (CAR)22, 23 and phenylalanine ammonia lyase (PAL) biocatalysts to allow production directly from the proteinogenic amino acid L-phenylalanine (L-Phe) (Figure 2).
Figure 2.

A biocatalytic route to cinnamyl alcohol (4) from bio-derived L-phenylalanine (1) using a combination of phenylalanine ammonia lyase (PAL), carboxylic acid reductase (CAR) and alcohol dehydrogenase (AHD) enzymes. Deamination reaction does not require any additional cofactors however the following both reductions need NADPH and ATP for CAR enzyme activity and either NADPH or NADH for ADH.
Results and discussion
As an initial test of the feasibility of the three enzyme cascade proposed in Figure 2, separate biotransformations were set up to represent the first two steps. To this end the PAL from the cyanobacterium Anabaena variabilis was chosen due to its extensive use in biocatalytic literature.24–26 Previous studies report the use of this enzyme within lyophilised whole cells. As such a pET-16b plasmid containing the codon-optimised avpal gene was used to transform E. coli BL21(DE3) cells and subsequently produce the dry whole cell formulation as previously reported.26 Reactions with varying quantities of AvPAL incubted for 24 hr at 30°C with 10mM L-phenylalanine showed increasing conversions up to 90% as seen by reverse-phase HPLC (Table 1).
Table 1. Effect of catalyst loading on the conversion of L-phenylalanine (1) to trans-cinnamic acid (2) by PALa.
 | ||
|---|---|---|
| Catalyst Loading / mg mL-1 | % composition in aqueous phaseb | |
| 1 | 2 | |
| 0 | >99 | <1 |
| 1 | 44 | 56 |
| 2 | 36 | 74 |
| 3 | 18 | 82 |
| 4 | 13 | 87 |
| 5 | 10 | 90 |
1–5 mg of lyophilised E. coli cells producing AvPAL, 100 mM potassium phosphate buffer (pH 7.5), 10 mM L-Phe, final volume 1 ml, 30 °C, 250 rpm vertical shaking, 24 h.
determined via reverse-phase HPLC analysis on a non-chiral phase.
Next a CAR from Mycobacterium marinum (MCAR) was produced in its active form via co-transformation of the encoding gene from pET-16a vector with that for a phosphopantetheinyl transferase from Bacillus subtilis in pCDF-1b. This system was deemed suitable for cascade construction, due to its successful incorporation into a reported method for the multi-step enzymatic production of chiral piperidines.27 Initial studies of the whole lyophilised cells with trans-cinnamic 2 acid gave only 10 % conversion to the corresponding aldehyde. Indeed the major products formed gave a mass spectrum consistent with cinnamyl alcohol 4 and 3-phenylpropanol 6. It could be envisaged that these products were formed via the action of CAR and a host cell ene-reductase in either order, followed by host cell aldehyde reduction. It has been demonstrated that E. coli ADHs have a broad specificity28 and reduce cinnamaldehyde as defence mechanism.29 In addition various ene-reductases are known for their high affinity towards cinnamaldehyde.30 This situation was found to be remedied (in part) by the use of purified MCAR, with 84 % conversion of 2 seen, giving a product ratio of ∼20:1 (3:4) with steady state kinetic parameters exhibited in table 2. This was possibly due to the presence of co-purified E. coli ADH enzymes and / or use of a glucose dehydrogenase (GDH) with low activity for the aldehyde. Evidence of the action of endogenous ADH enzymes in both the whole cell systems prompted investigation of PAL-CAR combinations to ascertain whether cinnamyl alcohol could be produced without the addition of a third biocatalyst. The use of both AvPAL and MCAR in whole cell formulations was indeed found to yield both the unsaturated alcohol 4 and saturated by-product 6 at a ratio of ~1:2.4 within the extracted organic layer, with the rest predominantly staying as the acid 2. A more favourable ratio of ~4:1 (4:6) could be achieved through the use of purified MCAR in the presence of an additional GDH-based cofactor recycling system (Table 2). However, the conversion was found to be lower when using PAL enzyme in lyophilised and MCAR in purified enzyme form compared to both enzymes in lyophilised cells. The apparent reduction of ene-reductase activity within the system was expected due to the lower cell mass used for this biotransformation. Further investigation revealed the low impact of cinnamaldehyde reduction by the GDH recycling system (~1 % conversion of the substrate).
Table 2. Conversion of 1, 2 and 3 by combinations of PAL, CAR and GDH biocatalysts.a.
 | ||||||
|---|---|---|---|---|---|---|
| Biocatalyst(s) | Substrate | % composition in extracted phaseb | ||||
| 2 | 3 | 4 | 5 | 6 | ||
| CARc | 2 | <1 | 11 | 33 | <1 | 56 |
| CARd | 2 | 17 | 79 | 4 | <1 | <1 |
| PAL/CARe | 1 | 18 | 1 | 57 | <1 | 24 |
| PAL/CARf | 1 | 45 | <1 | 44 | <1 | 11 |
| GDHg | 3 | - | 99 | 1 | <1 | <1 |
| Steady state kinetic parameters for conversion of 2 to 3 using MCAR.h | ||||||
| kcat (min-1) | KM (mM) | KI (mM) | kcat/KM (min-1 mM-1) |
|---|---|---|---|
| 184.54 ± 8.9 | 0.424 ± 0.059 | 24.2 ± 5.3 | 435.24 |
general reaction conditions containing PAL: 100 mM potassium phosphate buffer (pH 7.5), 5 mM substrate, 30 °C, 250 rpm vertical shaking. For any other reactions containing CAR enzymes additionally or GDH 10 mM MgCl2, 15 mM D-glucose, 10 mM ATP, 10 U GDH, 500 μM NADP+ were added.
determined via GC-MS on a non-chiral phase.
CAR biocatalyst used as a whole lyophilised cell formulation (1 mg mL-1)
CAR biocatalyst used as an isolated enzyme formulation (2 μM)
CAR and PAL biocatalyst used as a whole lyophilised cell formulation (1 and 3 mg mL-1 respectively)
CAR biocatalyst used as an isolated enzyme formulation and PAL as whole lyophilised cell formulation (2 μM and 3 mg mL-1)
GDH biocatalyst used in recycling reaction quantities. The conversions were calculated according to peak area on GC device. The conversions of PAL are not accounted in this table and supposed to be around ~70 % known from Table 1.
MCAR kinetic parameters were recorded by monitoring the rate of NADPH oxidation at 340 nm. Reaction conditions: 100 mM KH2PO4/K2HPO4, pH 7.5, 10 mM MgCl2, 1 mM ATP, 200 μM NADPH, 0.3 μM MCAR enzyme, 30 °C.
In order to ensure selective recovery of cinnamyl alcohol from the reaction mixture, the conversion of trans-cinnamic acid to the alcohol product was studied further with a combination of MCAR and a commercially-available ADH from Saccharomyces cerevisiae. It was reasoned that addition of this enzyme to biotransformations would act to remove cinnamaldehyde produced by CAR at a higher rate, pushing the overall conversion higher and minimising the effect of ene-reductase enzymes, where whole cells were used. The use of 0.25 mg mL-1 ScADH in conjunction with 1 mg mL-1 whole cells harbouring MCAR was found to give increasingly high consumption of cinnamate (>90% after 2 hours) to the desired alcohol, with a small amount of cinnamaldehyde also being detected after 8 and 24 hour time points. Increasing the MCAR whole cell biocatalyst component to 2.5 mg mL-1 was found to give almost complete conversion after 2 hours with only traces of cinnamate or cinnamaldehyde. Longer reaction times (4, 8 and 24 hours) resulted in accumulation first of cinnamaldehyde and then 3-phenylpropanol contaminants (Figure 3). These results indicate the importance of time course assays in the development an optimised cascade procedure and possibly highlight issues with the reversibility of ScADH in the mitigation of side product formation.
Figure 3.

The effect of catalyst loading on composition of CAR-ADH cascade reactions over time. Reaction performed in 100 mM potassium phosphate buffer (pH 7.5) 5 mM trans-cinnamic acid, 10 mM MgCl2, MCAR dried cells, 30 mM D-glucose, 10 mM ATP, 10 U GDH, 0.05 mg/ml ADH, 500 µM NADP+, 500 µM NAD+ final volume 1 ml at 30 °C, 250 rpm.
Having demonstrated the possibility of high conversion with minimal side reactions, the system was extended to include AvPAL starting from L-phenylalanine. The effect of varying the AvPAL whole cell loading on the overall composition of the volatile reaction intermediates / products could be easily tested by extraction of these from any remaining phenylalanine in organic solvent. The full three enzyme reaction was performed both as a one-pot cascade and by implementation of the PAL and CAR-ADH portions under temporal separation (figure S1). Overall the partition reaction gave better results with higher percentage compositions of 4, no by-product 6 and only small amounts of 2 or 3 detected. The presence of the unwanted alcohol 6 and higher percentage of 2 in the cascade reaction were presumably due to the increased overall cell loading (with associated ene-reductase activity) and lower stability of CAR and/or ADH enzymes compared to AvPAL. In an attempt to increase flux through the second part of the partitioned reaction, loading of ScADH was varied between 0.025 to 0.25 mg mL-1 with either a single or double batch addition at t=0 min and t=300 min (Figure S2). These experiments revealed a siphoning effect resulting in increased purity of 6 for higher ADH concentrations, with batch addition also giving lower percentage compositions of 2 and 3. Additionally, the effect of altering ATP concentration was investigated, demonstrating that a molar ratio of 1:2 (substrate to ATP) was sufficient for conversion of the trans cinnamic acid to cinnamyl alcohol. Lower molar ratios of substrate to ATP result in insufficient conversion (Figure S3), which might be related to the substrate stability or to lower catalytic activity of enzymes.
Furthermore a full time course experiment was performed for the two biocatalytic systems using a combination of reverse phase HPLC (to monitor the initial PAL reaction) and GC (to follow product formation with the CAR-ADH cascade).The reaction was performed with 107 mg (in 65 ml) as starting material around 85% being converted to 2 after a 22 hour PAL reaction before complete double reduction (volume increased to 130 ml after PAL separation) after a further 5.5 hours (Figure 4). The reaction was performed in sequential manner separating PAL and CAR with ADH reactions. 6 hours after addition of the second and third biocatalysts, the product could be easily extracted and purified using flash chromatography from the remaining L-phenylalanine, with an isolated yield of 43.5 mg (molar yield 53 %). The cinnamyl alcohol was confirmed by 1H and 13C NMR as well as GC-MS chromatogram and ionisation spectrum. The high resolution MS however did not show any ionisation pattern (Figure S4).
Figure 4.

Composition profile for the conversion of 1 to 4 via addition of PAL (t=0)performed in 100 mM potassium phosphate buffer (pH 7.5), 3 mg/ml of AvPAL dried cells and 10 mM L-Phe (107 mg) at 30 °C, 250 rpm 65 ml final volume. (t=1320) reaction mixture was spun down and supernatant supplemented with 10 mM MgCl2, 1 mg mL-1 MCAR dried cells, 30 mM D-glucose, 10 mM ATP, 10 U GDH, 0.05 mg/ml ADH, 500 µM NADP+, 500 µM NAD+ final volume 130 ml at 30 °C, 250 rpm.
In order to convert the L-phenylalanine to cinnamyl alcohol in an economical and environmentally friendly way, the enzymatic reactions were transferred from an in vitro setup into an in vivo system. The substrate, L-phenylalanine was produced by E. coli NST strain31 using glycerol / glucose mixture as carbon source (1 g L-1).32 Additionally, E. coli NST was transformed with the vector pZZ-Eva2 (Fig. S6) enabling the new strain to produce cinnamyl alcohol in TB media. Production was measured by the extraction of cinnamyl alcohol from the culture medium using ethyl acetate, further derivatization and final quantification performed by GC, following 24h, 48h and 72h of cell growth (Figure 5a). A maximum of 300 mg cinnamyl alcohol per litre of culture was produced after 24h of incubation, with an estimated 30 % conversion of L-phenylalanine 32 to cinnamyl alcohol. Cinnamyl aldehyde production was constant at 75mg L-1 over the entire 72h, while the production of trans-cinnamic acid increased over the course of the experiment to a maximum of 65 mg L-1. Alternatively we tested the production in mineral M9 media, supplemented with the same glycerol / glucose mix and with either glycerol or glucose as carbon source (Figure 5b The production of cinnamyl alcohol in M9 media was the highest with glucose as carbon source (80 mg L-1), followed by production with the glycerol/glucose mix (40mg L-1), and glycerol as the sole carbon source yielded (20mg L-1). Regardless of the relatively low cinnamyl alcohol titres, the major advantage of using mineral M9 media is that no identifiable side product formation was observed in any of the biological replicas over the whole course of the experiment.
Figure 5.

In-vivo production of cinnamyl alcohol in E. coli NST using the plasmid pZZ-Eva2, measured by GC after 24h, 48h AND 72h. (A) Production in TB media, and (B) production of cinnamyl alcohol comparing different carbon sources in M9 media. Error bars represent standard deviation (SD) of three biological replicates.
Here we demonstrate cinnamyl alcohol production via a three step biocatalytic cascade and by metabolic engineering in E. coli NST. The production of cinnamyl alcohol using the biocatalytic cascade might be advantageous as a result of the high loading capacity of enzymes, which is consistent with their use with higher concentrations of substrate / product. However, such an approach is potentially disadvantageous regarding the additional cost and reduced simplicity in an industrial setting (e.g. attributed to biocatalyst preparation and cofactor supplementation). Nevertheless recent improvements in the field of biocatalysis,34 particularly regarding coimmobilization35 of enzymes and cofactors and / or using coimmobilization to enable reuse of different reaction components,36 might present a future green and economically viable solution.
We have also demonstrated that metabolic engineering in E. coli NST can be applied to produce cinnamyl alcohol from analytical grade glucose and / or glycerol. Crude glycerol33 is a by-product of biodiesel production and is therefore a potential attractive carbon source for E. coli37 in metabolic engineering programmes that aim to establish green routes to the production of valuable chemicals. Most crude glycerol is derived from alkali treatment of biodiesel. Crude glycerol however contains different contaminants that might interfere with fermentation. However, lipase treatment of biodiesel38 yields a multitude of different plant derived oils and has reached industrial production levels.39 Glycerol as a by-product of enzymatic (lipase) biodiesel production, is easily removable,40 contains reduced levels of contaminants,41 and should therefore be directly suitable as a green carbon source for the production of cinnamyl alcohol.
Conclusions
The chemical building block cinnamyl alcohol has many uses in the literature as a precursor to various fragrance compounds, smart materials and commercially-available pharmaceuticals. Through use of a three enzyme cascade, we have demonstrated the simple production of this compound in good yield and high purity using biocatalytic functional group interconversion. The optimised method has several attractive features including a bio-derived starting material (L-phenylalnine), renewably resourced catalysts (phenylalanine ammonia lyase, carboxylic acid reductase and alcohol dehydrogenase enzymes), ambient, low energy reaction conditions and facile, inexpensive extraction of the final product. This method opens up routes to the conversion of biomass to useful products in an industrial setting as well as synthetic biology approaches to create designer organisms for the direct fermentative production of cinnamyl alcohol from primary metabolic processes.
Supplementary Material
Electronic Supplementry Infornmation (ESI) available: complete experimental section, molecular biology protocols, analytical methods, product characterisation. See DOI: 10.1039/x0xx00000x
Acknowledgements
This work was funded by the Biotechnology and Biological Sciences Research Council (BBSRC) and Glaxo-SmithKline (GSK) under the Strategic Longer and Larger (sLoLa) grant initiative ref. BB/K00199X/1. NSS is an Engineering and Physical Sciences Research Council (EPSRC) Established Career Fellow. NJT acknowledges the ERC for an Advanced Grant.
Notes and References
- 1.Regil R, Sandoval G. Biomolecules. 2013;3:812–847. doi: 10.3390/biom3040812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 3.France SP, Hepworth LJ, Turner NJ, Flitsch SL. ACS Catalysis. 2016 acscatal.6b02979-acscatal.02976b02979. [Google Scholar]
- 4.Schoffelen S, Van Hest JCM. Current Opinion in Structural Biology. 2013;23:613–621. doi: 10.1016/j.sbi.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Ricca E, Brucher B, Schrittwieser JH. Advanced Synthesis and Catalysis. 2011;353:2239–2262. [Google Scholar]
- 6.Lopez-Gallego F, Schmidt-Dannert C. Current Opinion in Chemical Biology. 2010;14:174–183. doi: 10.1016/j.cbpa.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 7.Belsito D, Bickers D, Bruze M, Calow P, Greim H, Hanifin JM, Rogers AE, Saurat JH, Sipes IG, Tagami H. Food and Chemical Toxicology. 2007;45 doi: 10.1016/j.fct.2007.09.067. [DOI] [PubMed] [Google Scholar]
- 8.Reuss G, Disteldorf W, Gamer AO, Hilt A. Ulmann's Encyclopedia of iIndustrial Chemistry. 2012;15:735–768. [Google Scholar]
- 9.Lerari D. Macromolecular Research. 2015;18:1008–1014. [Google Scholar]
- 10.Emayavaramban B, Roy M, Sundararaju B. Chemistry (Weinheim an der Bergstrasse, Germany) 2016;22:3952–3955. doi: 10.1002/chem.201505214. [DOI] [PubMed] [Google Scholar]
- 11.Kanno H, Taylor RJK. Tetrahedron Letters. 2002;43:7337–7340. [Google Scholar]
- 12.Ohshima T, Miyamoto Y, Ipposhi J, Nakahara Y, Utsunomiya M, Mashima K. Journal of the American Chemical Society. 2009;131:14317–14328. doi: 10.1021/ja9046075. [DOI] [PubMed] [Google Scholar]
- 13.Bonini C, Righi G. J Chem Soc, Chem Commun. 1994:2767–2768. [Google Scholar]
- 14.Venkatesan K, Srinivasan KV. ARKIVOC. 2008;2008:302–310. [Google Scholar]
- 15.Castillejos E, Debouttière PJ, Roiban L, Solhy A, Martinez V, Kihn Y, Ersen O, Philippot K, Chaudret B, Serp P. Angewandte Chemie - International Edition. 2009;48:2529–2533. doi: 10.1002/anie.200805273. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Chen X, Wang M, Xu Y. Applied Catalysis A: General. 2002;225:117–130. [Google Scholar]
- 17.Ma H, Wang L, Chen L, Dong C, Yu W, Huang T, Qian Y. Catalysis Communications. 2007;8:452–456. [Google Scholar]
- 18.Mitsudome T, Mikami Y, Funai H, Mizugaki T, Jitsukawa K, Kaneda K. Angewandte Chemie - International Edition. 2008;47:138–141. doi: 10.1002/anie.200703161. [DOI] [PubMed] [Google Scholar]
- 19.Gallezot P, Richard D. Selective Hydrogenation of α,β-Unsaturated Aldehydes. 1998 [Google Scholar]
- 20.Chamouleau F, Hagedron C, May O, Groger H. Flavour and Fragrance Journal. 2007;22:169–172. [Google Scholar]
- 21.Zucca P, Littarru M, Rescigno A, Sanjust E. Bioscience, biotechnology, and biochemistry. 2009;73:1224–1226. doi: 10.1271/bbb.90025. [DOI] [PubMed] [Google Scholar]
- 22.Akhtar MK, Turner NJ, Jones PR. Proceedings of the National Academy of Sciences. 2013;110:87–92. doi: 10.1073/pnas.1216516110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gahloth D, Dunstan MS, Quaglia D, Klumbys E, Lockhart-Cairns MP, Hill AM, Derrington SR, Scrutton NS, Turner NJ, Leys D. Nature chemical biology. 2017;13:975–981. doi: 10.1038/nchembio.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moffitt MC, Louie GV, Bowman ME, Pence J, Joseph P, Moore BS. Biochemistry. 2008;46:1004–1012. doi: 10.1021/bi061774g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parmeggiani F, Lovelock SL, Weise NJ, Ahmed ST, Turner NJ. Angewandte Chemie (International ed. in English) 2015;54:4608–4611. doi: 10.1002/anie.201410670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weise NJ, Ahmed ST, Parmeggiani F, Siirola E, Pushpanath A, Schell U, Turner NJ. Catalysis Science & Technology. 2016;6:4086–4089. [Google Scholar]
- 27.France SP, Hussain S, Hill AM, Hepworth LJ, Howard RM, Mulholland KR, Flitsch SL, Turner NJ. ACS Catalysis. 2016;6:3753–3759. [Google Scholar]
- 28.Atsumi S, Wu T-y, Eckl E-m, Hawkins SD, Buelter T, Liao JC. 2010:651–657. doi: 10.1007/s00253-009-2085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visvalingam J, Hernandez-doria JD, Holley A. 2013;79:942–950. doi: 10.1128/AEM.02767-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toogood HS, Mansell D, Gardiner JM, Scrutton NS. 1 edn. Elsevier Science; 2012. pp. 216–260. [Google Scholar]
- 31.Tribe DE. Google Patents. 1987
- 32.Yakandawala N, Romeo T, Friesen AD, Madhyastha S. Applied microbiology and biotechnology. 2008;78:283–291. doi: 10.1007/s00253-007-1307-z. [DOI] [PubMed] [Google Scholar]
- 33.Konstantinović SS, Danilović BR, Ćirić JT, Ilić SB, Savić DS, Veljković VB. Chemical Industry and Chemical Engineering Quarterly. 2016:19–19. [Google Scholar]
- 34.Bilal M, Iqbal HM, Shuqi G, Hu H, Wang W, Zhang X. International Journal of Biological Macromolecules. 2017 doi: 10.1016/j.ijbiomac.2017.01.133. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt-Dannert C, Lopez-Gallego F. Microbial biotechnology. 2016;9:601–609. doi: 10.1111/1751-7915.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peirce S, Virgen-Ortíz JJ, Tacias-Pascacio VG, Rueda N, Bartolome-Cabrero R, Fernandez-Lopez L, Russo ME, Marzocchella A, Fernandez-Lafuente R. RSC Advances. 2016;6:61707–61715. [Google Scholar]
- 37.Srinophakun P, Reakasame S, Khamduang M, Packdibamrung K, Thanapi A. Chiang Mai J Science. 2012;39:59–68. [Google Scholar]
- 38.Hama S, Noda H, Kondo A. Current Opinion in Biotechnology. 2018;50:57–64. doi: 10.1016/j.copbio.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Price J, Nordblad M, Martel HH, Chrabas B, Wang H, Nielsen PM, Woodley JM. Biotechnology and bioengineering. 2016;113:1719–1728. doi: 10.1002/bit.25936. [DOI] [PubMed] [Google Scholar]
- 40.Vicente G, Coteron A, Martinez M, Aracil J. Industrial crops and products. 1998;8:29–35. [Google Scholar]
- 41.Garcia E, Laca M, Pérez E, Garrido A, Peinado J. Energy & fuels. 2008;22:4274–4280. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.