

Abstract
PURPOSE:
To describe a distinct phenotypic outcome of outer retinal degeneration in a cohort of genetically confirmed patients with recessive Stargardt disease (STGD1).
DESIGN:
Retrospective case series.
METHODS:
Twelve patients, who were clinically diagnosed with STGD1 and exhibited a unique degenerative phenotype, were included in the study. Two disease-causing mutations were found in all patients by direct sequencing of the ABCA4 gene. Clinical characterization of patients were defined on fundus photographs, autofluorescence images (488-nm and 532-nm excitation), spectral-domain optical coherence tomography (SD-OCT), and full-field electroretinogram (ffERG) testing.
RESULTS:
Mean age at initial presentation was 67.8 years and reported age of symptomatic onset was 14.1 years (mean disease duration [ 53.8 years). Best-corrected visual acuity ranged from 20/400 to hand motion. All patients exhibited advanced degeneration across the posterior pole resulting in a reflectively pale, blonde fundus owing to unobstructed exposure of the underlying sclera. SD-OCT revealed complete loss of the outer retinal bands (external limiting membrane, ellipsoid zone, interdigitation zone, and retinal pigment epithelium) and choroidal layers. Scotopic and photopic waveforms on ffERG were nonrecordable or severely attenuated in 8 patients who were tested.
CONCLUSIONS:
Widespread scleral exposure is a clinical outcome in a subset of STGD1 following a long duration of disease progression (~50 years). The blonde fundus in such cases may exhibit phenotypic overlap and shared therapeutic implications with other aggressive chorioretinal dystrophies such as end-stage choroideremia, gyrate atrophy, or RPE65-Leber congenital amaurosis.
AUTOSOMAL RECESSIVE STARGARDT DISEASE (STGD1; MIM #248200) is the most common inherited retinal dystrophy, responsible for mostly adolescent-onset progressive central vision loss.1 The causal gene, the photoreceptor-specific ATP-binding cassette transporter, ABCA4, was identified in 19972; since then >1000 disease-associated variants have been reported.3 The disease phenotypes, resulting from biallelic mutations in ABCA4, vary extensively and sometimes exhibit phenotypic overlap with conditions caused by mutations in other genes. For example, bull’s-eye and occult maculopathy are well-described early clinical abnormalities detected in patients harboring the c.5882G>A (p.Gly1961Glu) mutation in ABCA4,4,5 as well as other conditions ranging from maculopathies caused by mutations in CRX6,7 and PROM1,8 central areolar choroidal dystrophy (RDS/PRPH2),8–10 RP1L1-occult macular dystrophy,11,12 and achromatopsia (CNGA3,13 CNGB3,14 GNAT2,15 PDE6C,16 PDE6H,17 ATF618) to drug-induced toxicities (chloroquine and hydroxychloroquine).19
Clinical precision decreases with disease progression as the manifestation of pathognomonic features, such as peripapillary sparing20 and the appearance of pisciform flecks,21,22 become indiscernible from gradual deterioration of retinal tissue. These features are subsequently replaced by the appearance of bone-spicule pigment deposition, vessel attenuation, optic disc pallor, and generalized attenuation of cone and rod function, which reflect characteristics of panretinal diseases such as retinitis pigmentosa.23,24 Comprehensively characterizing the expansive clinical presentation of a disease improves diagnostic accuracy in the clinic and provides invaluable scientific insight into disease etiology and natural history. Furthermore, detailed clinical characterization of STGD1 guides and facilitates the effective design of interventional trials, some of which are currently ongoing, including, gene therapy (NCT01345006) and the slowing of A2E formation by the oral ingestion of deuterated vitamin A (NCT02402660).
The current study describes a phenotypic outcome of advanced degeneration in the natural history of STGD1. Clinical documentation consists of multimodal retinal imaging in a study cohort with a mean disease duration of over 50 years.
METHODS
PATIENTS:
All study procedures were defined under protocol #AAAI9906 approved by the Institutional Review
Board at Columbia University Medical Center. The study adhered to tenets set out in the Declaration of Helsinki. A retrospective review of 300 patients with a clinical diagnosis and genetic confirmation (at least 2 disease-causing mutations in the ABCA4 gene) of STGD1 was conducted at the Department of Ophthalmology, Columbia University. Patients identified and selected for the study exhibited widespread chorioretinal degeneration of the posterior pole resulting in visibility of the underlying sclera. Patients with degeneration of the outer retina but not the choroid (ie, presence of continuously intact choroidal vessels) were not included in the characterization of this phenotype. Assessment of scleral visibility was made on digital color fundus photographs (50-degree field).
CLINICAL EXAMINATION AND CHARACTERIZATION:
Each patient underwent a complete ophthalmic examination by a retinal physician (S.H.T.), which included a slit-lamp and dilated funduscopy examination, best-corrected visual acuity (BCVA; Snellen), color fundus photography, fundus autofluorescence (AF, 488 nm, 532 nm, and 787 nm), spectral-domain optical coherence tomography (SD-OCT) scanning and full-field electroretinogram (ffERG) testing. Imaging across all modalities was conducted following pupil dilation (>7 mm) with tropicamide (1%) and phenylephrine hydrochloride (2.5%). Fundus autofluorescence (488 nm) images and 9 mm horizontal foveal SD-OCT scans were acquired with the Spectralis HRA+OCT (Heidelberg Engineering, Heidelberg, Germany). Ultra-widefield autofluorescence images were acquired with an Optos 200 Tx (Optos PLC, Dunfermline, United Kingdom). The ffERGs were recorded with silver-impregnated fiber electrodes (DTL; Diagnosys LLC, Littleton, Massachusetts, USA) on the Espion Visual Electrophysiology System (Diagnosys LLC) in accordance with International Society for Clinical Electrophysiology of Vision (ISCEV) standards.25
MOLECULAR CHARACTERIZATION:
Screening of the ABCA4 gene was performed by next-generation sequencing (NGS) as previously described.26,27 All detected possibly disease-associated variants were confirmed by Sanger sequencing and analyzed with the Alamut software (http://www.interactive-biosoftware.com). Segregation of the new variants with the disease was analyzed in families if family members were available. Functional annotation of variants was determined using computational software including Annovar using pathogenicity scores of MCAP, REVEL, Eigen, CADD, DANN, and SPIDEX.26–32 As a general guideline, pathogenic consequences are predicted for variants with scores over 0.025 for MCAP, 0.5 for REVEL, 0.5 for Eigen, 20 for CADD, 0.97 for DANN, and more than −2 or less than 2 for SPIDEX (psi z-score).28–34 The allele frequencies of all variants were compared with The Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/gene/ENSG00000198691; accessed January 2018).
RESULTS
ALL PATIENTS (N = 12) INCLUDED IN THE STUDY PRESENTED with a long history of retinal degeneration. Mean cohort age at presentation was 67.8 years (range, 48–85 years). The reported age of symptomatic onset varied from 5 to 29 years of age (mean = 14.1 years), giving the study cohort an average disease duration of 53.8 years. Visual acuities were not correctable outside 20/400 to hand motion (HM) in all patients. Table 1 further summarizes demographic, clinical, and genetic characteristics. Funduscopic examinations in each patient were remarkable for features consistent with advanced chorioretinal degeneration including optic disc pallor, attenuation of the retinal vasculature, and dark pigment migration in the macula and periphery. Wide-spread loss of retinal pigment epithelium (RPE) and choroidal vessels was observed in the posterior pole of all patients, resulting in exposure of underlying scleral tissue (Figure 1). The fundus at this stage was highly reflective, exhibiting a blonde hue and an irregularly tessellated appearance in certain regions. These profound areas of degeneration extended into the far periphery, in some cases, beyond which residual retinal tissue and large choroidal vessels became visible. Widespread nummular and bone-spicule (nonparavascular) pigment deposition was noted in all patients (Figure 1, blue arrowheads).
TABLE 1.
Demographic, Clinical, and Genetic Characteristics of Patients in the Scleral Exposure Stage of Stargardt Disease
| BCVA |
ffERG |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Age (y) | AO (y) | DD (y) | Sex | Race | OD | OS | Scotopic | Max | Flicker | Photopic | Follow-up Duration (y) |
| 1 | 85 | 20 | 65 | M | White | CF | CF | NR | NR | NR | NR | 10 |
| 2 | 68 | 29 | 39 | M | White | 20/400 | 20/400 | ↓↓ | ↓↓ | ↓↓ | ↓↓ | 10 |
| 3 | 74 | 14 | 60 | M | White | 20/400 | 20/400 | 1 | ||||
| 4 | 55 | 9 | 46 | F | White | 20/400 | 20/400 | NR | ↓↓ | NR | NR | 10 |
| 5 | 65 | 8 | 57 | F | White | CF | CF | NR | NR | NR | NR | 6 |
| 6 | 48 | 5 | 43 | F | White | 20/400 | 20/400 | NR | NR | NR | NR | 6 |
| 7 | 75 | 25 | 50 | M | White | CF | CF | ↓↓ | ↓↓ | ↓↓ | ↓↓ | 4 |
| 8 | 62 | 14 | 48 | M | White | CF | 20/400 | NR | NR | NR | NR | N/A |
| 9 | 72 | 17 | 55 | M | White | HM | HM | NR | NR | NR | NR | 2 |
| 10 | 68 | 18 | 50 | F | White | 20/400 | 20/400 | 2 | ||||
| 11a | 70 | 5 | 65 | F | White | CF | 20/400 | 5 | ||||
| 12a | 72 | 5 | 67 | M | White | CF | CF | 5 | ||||
y = years; AO = age of onset; CF = counting fingers; DD = disease duration; ffERG = full-field electroretinogram; HM = hand motion; N/A = not available; NR = nonrecordable; ↓↓ = severely attenuated.
Patients 11 and 12 are siblings.
FIGURE 1.
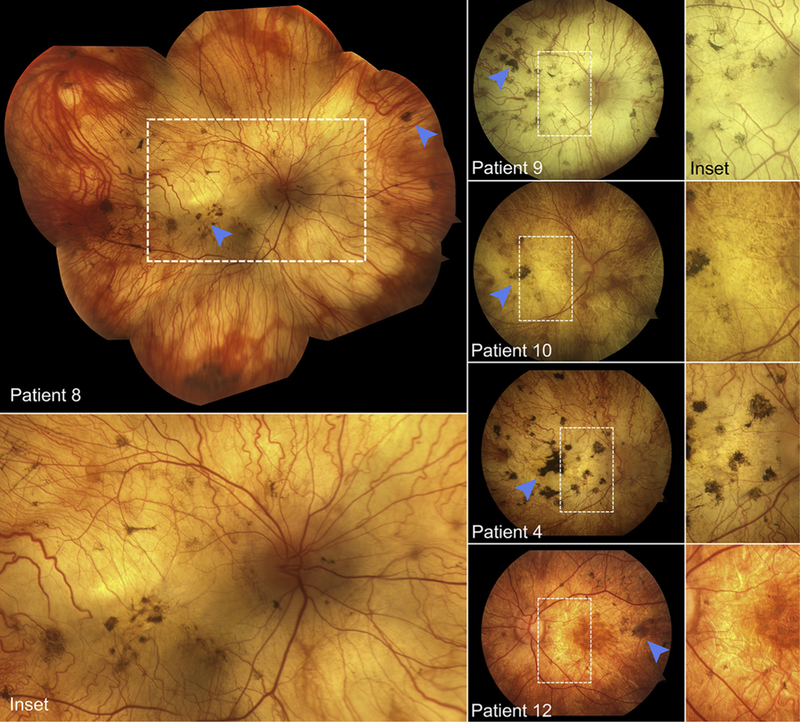
Fundus montage and photographs of Stargardt disease patients exhibited advanced retinal degenerative features such as optic disc pallor, retinal vasculature attenuation, and pigment deposition (blue arrowheads). The fundus of each patient, following a prolonged duration of disease progression, is characterized by a reflectively pale, blonde appearance owing to exposure of the underlying sclera.
Areas of degeneration seen on fundus examination corresponded to homogenously hypoautofluorescent lesions with well-delineated, scalloped edges, continuous across a large area (Figure 2, Top) or in a diffuse pattern of numerous, round coalescing foci (Figure 2, Bottom) on AF (488-nm and 532-nm) imaging. Nonatrophic areas adjacent to degeneration were heterogeneously atrophic, exhibiting a punctate appearance of alternating hyper-and hypoautofluorescent flecks reaching postequatorial regions. Macular SD-OCT scans showed extensive retinal thinning and a visible loss of the characteristic foveal contour in all cases except Patient 6 (Figure 3). A residual presence of the retinal nerve fiber layer (RNFL), ganglion layer, and plexiform layer was noted in all patients. A closer examination was required to detect the intermittent presence of, albeit considerably thinned, remnants of the outer nuclear layer (ONL). The most notable finding on SD-OCT was the complete absence of all hyperreflective outer retinal bands that are attributable to photoreceptor inner/outer segments, ellipsoid zone (EZ) and interdigitation zone (IZ), and RPE, as well as the underlying choroid, resulting in increased signal transmission into the sclera throughout the length of the B-scan (Figure 3, red arrows). Longitudinal assessment was possible in all except Patient 8, through follow-up visits ranging between 1 and 10 years after initial presentation. Further lesion growth and the appearance of peripheral degeneration were noted at subsequent visits; however, the pale regions of visible sclera remained effectively unchanged over time (ie, no increase or loss of pigment deposition) (Figure 4). ffERG testing revealed virtually nonrecordable cone and rod responses in 6 out of 8 patients tested, while severely attenuated cone and rod responses were measured in Patient 2 and Patient 7 (Table 1).
FIGURE 2.
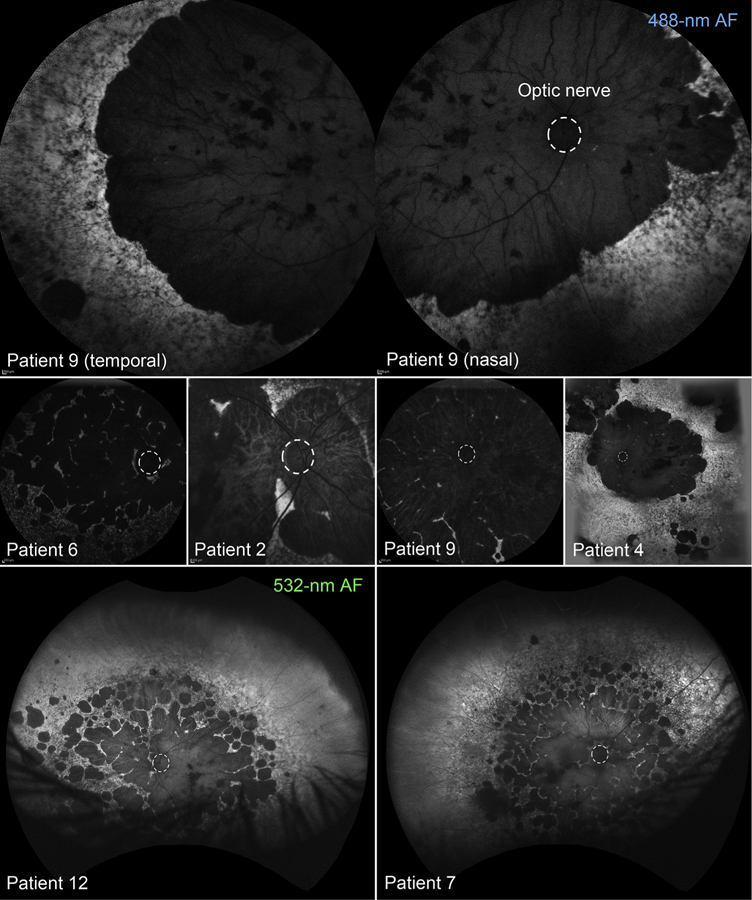
Ultra-widefield fundus autofluorescence (AF) imaging (488-nm and 532-nm) of patients in the scleral exposure stage of Stargardt disease. Regions of atrophy are hypoautofluorescent with well-defined edges, covering the posterior pole as either (Top) a continuous region or (Bottom, left and right) multiple, coalescing foci. An extensive heterogeneity in AF attributable to confluent accumulations of flecks is visible in the mid to far periphery. The optic disc in each fundus image is outlined (dotted white line).
FIGURE 3.
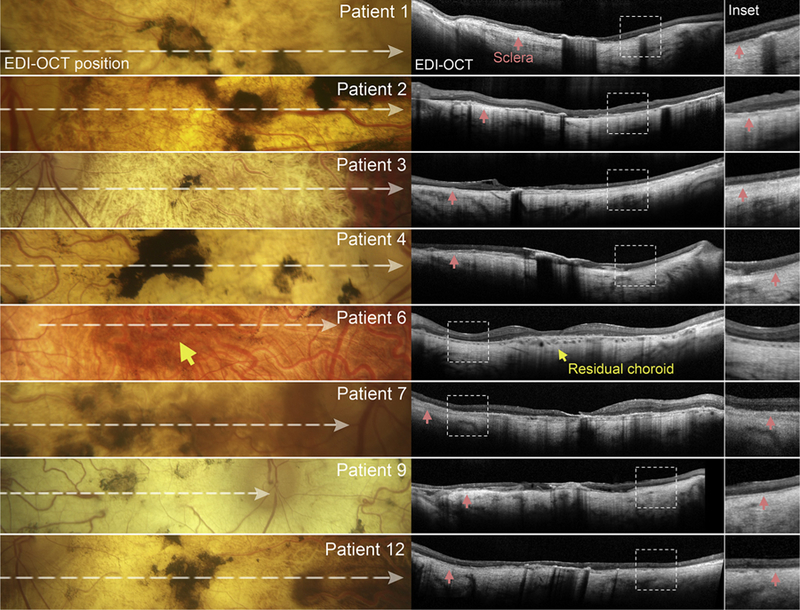
Spectral-domain optical coherence tomography (SD-OCT, center column) with corresponding fundus regions (left column) of patients in the scleral exposure stage of Stargardt disease. Horizontal SD-OCT scans through the fovea reveal a complete loss of the reflective outer retinal bands, external limiting membrane, ellipsoid zone, interdigitation zone, and retinal pigment epithelium, as well as choroidal layers. Hypertransmission of the OCT signal is visible throughout the scan, revealing the underlying sclera (red arrowhead) in maginfied SD-OCT insets (right column). A central region of residual choroid (yellow arrow) was observed in Patient 6.
FIGURE 4.
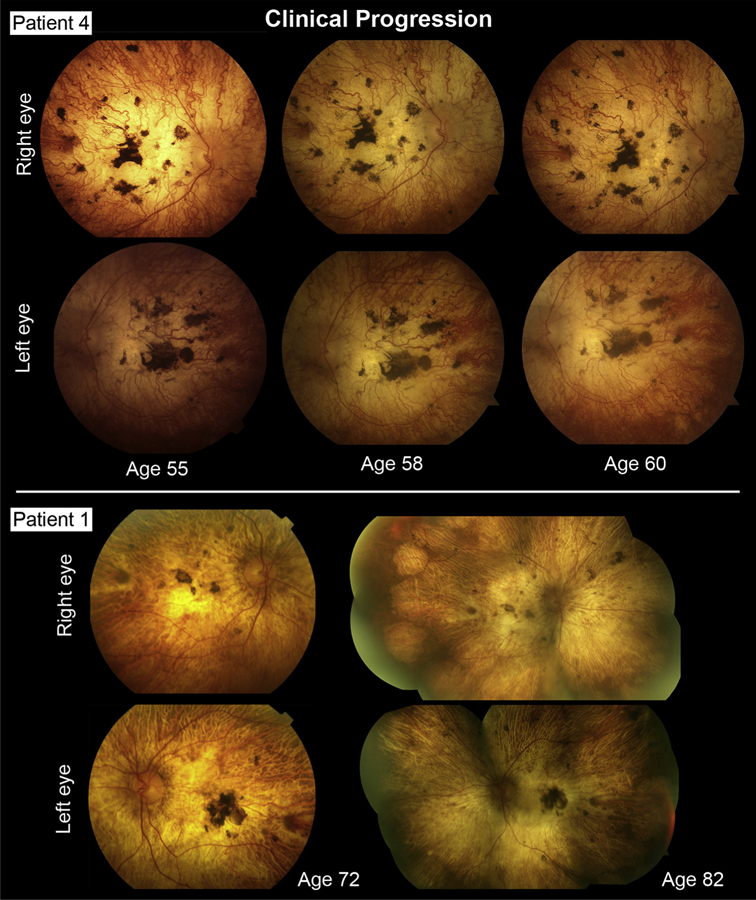
Longitudinal documentation of Stargardt disease patients exhibiting widespread, scleral-deep chorioretinal degeneration. No detectable changes in pigment deposition or further vascular deterioration were noted after 5–10 years in Patient 4 (Top rows) and Patient 1 (Bottom rows), respectively, indicating the complete absence of chorioretinal tissue.
Complete sequencing of the ABCA4 gene identified at least 2 (expected) disease-causing variants in all patients (Table 2). Variants found in the cohort included 17 missense variants, of which 4 were complex alleles, including c.[2588G>C; 5603A>T] (p.[Gly863Ala; Asn1868Ile]),35 c.[1253T>C; 5603A>T] (p.[Phe418Ser; Asn1868Ile]), and c.[4594G>A; 5603A>T] (p.[Asp1532Asn; Asn1868Ile]) twice. All missense variants were predicted to be ‘‘deleterious’’ by SIFT (score = 0) and MutationTaster (p = 1) and pathogenic by M-CAP (scores = 0.391–0.791), REVEL (scores = 0.77–0.98), and CADD (scores = 27–42). Other variants included a stop-gain, c.6088C>T (p.Arg2030*) variant in Patient 3 and 8 noncoding variants: 5 variants in canonical splice site sequences (c.4773+3A>G, c.2160+1G>C, c.768G>T, c.3050+5G>A, and c.5714+5A>G) and 3 deepintronicvariants(c.302+68C>T, c.4539+2028C>T and c.4539+2001G>A) that likely have a negative effect on exon splicing.36 The sibling pair Patient 11 and Patient 12 harbored a deep intronic variant, c.4539+2028C>T, and a deletion/insertion, c.6148–698_c.6670del/insTGTGCACCTCCCTAG, described in a previous report.37 Patient 5 harbors another deep intronic variant, c.4539+2001G>A.
TABLE 2.
Summary and Pathogenicity Analysis of ABCA4 (NM_000350.2) Variants in the Study Cohort
| Patient | cDNA Variant | Protein Variant | Type | Coding Effect | M-CAP | REVEL | Eigen | CADD13 | DANN |
|---|---|---|---|---|---|---|---|---|---|
| 1 | c.3322C>T | p.(R1108C) | Substitution | Missense | 0.797 | 0.89 | 0.81 | 35 | 1.00 |
| c.4139C>T | p.(P1380L) | Substitution | Missense | 0.391 | 0.87 | 0.70 | 28 | 1.00 | |
| 2 | c.4139C>T | p.(P1380L) | Substitution | Missense | 0.391 | 0.87 | 0.70 | 28 | 1.00 |
| c.5714+5G>A | p.(?) | Substitution | ? | - | - | - | - | - | |
| 3 | c.2588G>Ca | p.(G863A) | Substitution | Missense | - | 0.80 | 0.58 | 27 | 1.00 |
| c.5603A>Ta | p.(N1868I) | Substitution | Missense | - | 0.40 | 0.03 | 26 | 0.92 | |
| c.6088C>T | p.(R2030*) | Substitution | Nonsense | - | - | 0.54 | 42 | 1.00 | |
| 4 | c.3322C>T | p.(R1108C) | Substitution | Missense | 0.797 | 0.89 | 0.81 | 35 | 1.00 |
| c.1253T>C | p.(F418S) | Substitution | Missense | 0.582 | 0.93 | 0.81 | 29 | 1.00 | |
| 5 | c.161G>A | p.(C54Y) | Substitution | Missense | - | 0.98 | 0.87 | 29 | 1.00 |
| c.2160+1G>C | p.(?) | Substitution | ? | - | - | - | - | - | |
| 6 | c.768G>T | p.(?) | Substitution | ? | - | - | - | - | - |
| c.4539+2001G>A | p.(?) | Substitution | ? | - | - | - | - | - | |
| 7 | c.3050+5G>A | p.(?) | Substitution | ? | - | - | - | - | - |
| c.4594G>Aa | p.(D1532N) | Substitution | Missense | 0.722 | 0.77 | 0.80 | 28 | 1.00 | |
| c.5603A>Ta | p.(N1868I) | Substitution | Missense | - | 0.40 | 0.03 | 26 | 0.92 | |
| 8 | c.3056C>T | p.(T1019M) | Substitution | Missense | 0.611 | 0.96 | 1.10 | 33 | 1.00 |
| c.3056C>T | p.(T1019M) | Substitution | Missense | 0.611 | 0.96 | 1.10 | 33 | 1.00 | |
| 9 | c.161G>A | p.(C54Y) | Substitution | Missense | - | 0.98 | 0.87 | 29 | 1.00 |
| c.4773+3A>G | p.(?) | Substitution | ? | - | - | - | - | - | |
| 10 | c.4139C>T | p.(P1380L) | Substitution | Missense | 0.391 | 0.87 | 0.70 | 28 | 1.00 |
| c.4594G>Aa | p.(D1532N) | Substitution | Missense | 0.722 | 0.77 | 0.80 | 28 | 1.00 | |
| c.5603A>Ta | p.(N1868I) | Substitution | Missense | - | 0.40 | 0.03 | 26 | 0.92 | |
| 11 | c.302+68C>Ta | p.(?) | Substitution | ? | - | - | - | - | - |
| c.4539+2028C>Ta | p.(?) | Substitution | ? | - | - | - | - | - | |
| c.6148–698_ c.6670del - 4770 bp del | p.(?) | Deletion | ? | - | - | - | - | - | |
| 12 | c.302+68C>Ta | p.(?) | Substitution | ? | - | - | - | - | - |
| c.4539+2028C>Ta | p.(?) | Substitution | ? | - | - | - | - | - | |
| c.6148–698_ c.6670del - 4770 bp del | p.(?) | Deletion | ? | - | - | - | - | - |
Variants in cis; predicted pathogenicity: M-CAP (>0.025); REVEL (>0.5), Eigen (>0.5), CADD13 (>20), DANN (>0.97).
DISCUSSION
THE CURRENT STUDY DESCRIBES AN END-STAGE PHENOTYPE in a genetically confirmed cohort (n = 12) of STGD1 patients with disease duration of >50 years. All patients presented with advanced disease characteristics such as optic disc pallor, retinal vasculature attenuation, and wide-spread pigment deposition. Poor visual acuity (20/400 to HM) in the cohort was attributable to widespread degeneration across the posterior pole with near-complete loss of the outer retinal layers, as evidenced by the visible absence of the hyperreflective RPE, IZ, EZ, and ELM bands on SD-OCT, and choroidal vasculature across the posterior pole. Consequently, the fundus in these patients exhibited a reflectively pale, blonde hue resulting from an unobstructed view of the underlying sclera. The absence of observable changes within these regions (eg, further pigment accumulation) over many years after initial examination reflects the complete loss of chorioretinal tissue and is indicative of the end of a longstanding degenerative process in the retina.
The finding of sclera-deep degeneration broadens the list of differential diagnoses to conditions that are characterized by aggressive chorioretinal deterioration and rod-cone dystrophies such as choroideremia (CHM, MIM #303100),38,39 gyrate atrophy of the choroid and retina (OAT, MIM #258870),40 Leber congenital amaurosis (RPE65, MIM #613794),41 and clinically advanced cases of C2orf71-related retinopathy.42 Ocular history and ffERG testing would most effectively differentiate STGD1 from these other listed conditions, as patients affected with the latter generally report progressive visual field constriction and nyctalopia (rod-cone attenuation on ffERG) as opposed to early central vision loss and a cone-rod dysfunction on ffERG. Anatomically, the current study cohort bears the most clinical resemblance to patients with end-stage choroideremia, who exhibit the similar pale fundus appearance from exposed scleral reflectance. The precise pathophysiology of choroideremia remains largely unknown despite promising advances in adeno-associated viral (AAV) gene therapy (NCT01461213).43,44 The protein product of CHM, Rab escort protein-1 (REP1), has been localized exclusively to rods,45 although numerous studies have presented histopathologic and adaptive optics–scanning light ophthalmoscopy data supporting RPE as the primary site of degeneration.46–52 The outcome of partial or complete deterioration of the outer retina and underlying choroid owing to severely incapacitated RPE is therefore a conceivable eventuality given the physiological interdependence of adjacent layers in this part of the retina.53–55 Likewise, such a pathway can be recapitulated by ABCA4 dysfunction in which the formation of bisretinoid fluorophores in phagocytized photoreceptor outer segments perpetuates the rapid demise of lipofuscin-laden RPE.6,56–59
Consistent with other rod-cone degenerative conditions, patients with choroideremia experience early, progressive night blindness but retain visual acuity until relatively late in the disease.49 A recent study of 56 consecutive patients at Oxford Eye Hospital found the median survival (Kaplan-Meier analysis) of 20/20 acuity, bilaterally, to be 39 years,60 whereas the patients in the present STGD1 cohort all experienced a comparatively earlier age of central visual acuity loss (mean 14.1 years). This difference in ocular history can thus serve as a diagnostic aid in situations of phenotypic overlap between the 2 conditions. A further point of distinction between STGD1 and choroideremia is the frequency at which this phenotypic outcome presents to the clinic. While scleral visibility within areas of degeneration is routinely observed in choroideremia,38,39 only 12 out of a database of 300 (~4%) STGD1 patients presented with this finding. It is unclear whether specific genotypes are an underlying factor behind this clinical outcome. Most ABCA4 variants in these patients are either deleterious or very severe. Each patient would be appropriately diagnosed as cone-rod dystrophy with a group 3 ffERG classification61; however, their overall disease trajectory is not as severe as in patients with the rapid-onset chorioretinopathy sub-phenotype who are exclusively biallelic null cases (ie, completely lack functional ABCA4 protein) and report symptomatic onset within the first decade of life. The most significant underlying factor could be the long duration of disease progression (>50 years) in these patients, who comprise a STGD1 demographic (>60 years of age) that is poorly characterized in the current literature.
The outcome of complete chorioretinal degeneration has serious implications for therapeutic approaches to end-stage STGD1. The pathophysiology of ABCA4 dysfunction will require replacement of both photoreceptors and RPE to restore function in the retina; however, the absence of a native choroid, as described in this study, presents an added obstacle involving tissue revascularization. Macular translocation of RPE-choroid grafts has been demonstrated in patients with age-related macular degeneration, with considerable success.62–67 Such an approach, coupled with gene therapy, if the tissue source is autologous, may be a feasible strategy for STGD1 if the neurosensory retina can also be incorporated. Further applications of gene therapy include imparting photosensitivity to secondary neurons in the inner retina using optogenetic therapy.68 Preliminary efficacy of delivering channelrhopsin-2 (ChR2), a light-gated cation channel isolated from the green alga Chlamydomonas reinhardtii, to patients with advanced retinitis pigmentosa is currently being evaluated (NCT02556736). Visual restoration can also be artificially achieved by the implantation of a bionic prosthesis, although as with the current potential of optogenetic therapy, the promise of high-resolution vision remains a work in progress. Nevertheless, long-term safety and improved visual function with the Argus II Retinal Prosthesis System was reported in a group of 30 subjects, which included a patient with choroideremia (NCT00407602).69
In summary, an end-stage sub-phenotype of genetically confirmed STGD1 characterized by complete loss of the outer retina and choroid, resulting in widespread scleral exposure, is associated with long disease duration (>50 years) in older patients. This clinical stage exhibits significant phenotypic overlap with aggressive chorioretinal dystrophies such as choroideremia, but can be distinguished, in addition to genetic screening, by an ocular history of central vision loss and a cone-rod pattern of functional attenuation on ffERG.
Acknowledgments
FUNDING/SUPPORT: THIS WORK WAS SUPPORTED, IN PART, BY GRANTS FROM THE NATIONAL EYE INSTITUTE/NIH EY021163, EY019861, and EY019007 (Core Support for Vision Research); and unrestricted funds from Research to Prevent Blindness (New York, New York, USA) to the Department of Ophthalmology, Columbia University. Financial Disclosures: The following authors have no financial disclosures: Winston Lee, Jana Zernant, Takayuki Nagasaki, Stephen H. Tsang, and Rando Allikmets. All authors attest that they meet the current ICMJE criteria for authorship.
Contributor Information
WINSTON LEE, Departments of Ophthalmology, Columbia University, New York, New York, USA..
JANA ZERNANT, Departments of Ophthalmology, Columbia University, New York, New York, USA..
TAKAYUKI NAGASAKI, Departments of Ophthalmology, Columbia University, New York, New York, USA..
STEPHEN H. TSANG, Departments of Ophthalmology, Columbia University, New York, New York, USA.
RANDO ALLIKMETS, Departments of Pathology & Cell Biology, Columbia University, New York, New York, USA..
REFERENCES
- 1.Stargardt K Über familiäre, progressive Degeneration in der. Maculagegend des Auges. Graefes Arch Clin Exp Ophthalmol 1909;71:534–549. [Google Scholar]
- 2.Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997; 15(3):236–246. [DOI] [PubMed] [Google Scholar]
- 3.Cornelis SS, Bax NM, Zernant J, et al. In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum Mutat 2017;38(4):400–408. [DOI] [PubMed] [Google Scholar]
- 4.Noupuu K, Lee W, Zernant J, Tsang SH, Allikmets R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Invest Ophthalmol Vis Sci 2014; 55(11):7217–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cella W, Greenstein VC, Zernant-Rajang J, et al. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp Eye Res 2009;89(1):16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto S, Jaiswal M, Charng WL, et al. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 2014;159(1):200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paunescu K, Preising MN, Janke B, Wissinger B, Lorenz B. Genotype-phenotype correlation in a German family with a novel complex CRX mutation extending the open reading frame. Ophthalmology 2007;114(7):1348–1357.e1. [DOI] [PubMed] [Google Scholar]
- 8.Michaelides M, Gaillard MC, Escher P, et al. The PROM1 mutation p.R373C causes an autosomal dominant bull’s eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest Ophthalmol Vis Sci 2010;51(9):4771–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renner AB, Fiebig BS, Weber BH, et al. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am J Ophthalmol 2009; 147(3):518–530 e1. [DOI] [PubMed] [Google Scholar]
- 10.Hoyng CB, Heutink P, Testers L, Pinckers A, Deutman AF, Oostra BA. Autosomal dominant central areolar choroidal dystrophy caused by a mutation in codon 142 in the peripherin/RDS gene. Am J Ophthalmol 1996;121(6):623–629. [DOI] [PubMed] [Google Scholar]
- 11.Fujinami K, Kameya S, Kikuchi S, et al. Novel RP1L1 variants and genotype-photoreceptor microstructural phenotype associations in cohort of Japanese patients with occult macular dystrophy. Invest Ophthalmol Vis Sci 2016;57(11): 4837–4846. [DOI] [PubMed] [Google Scholar]
- 12.Davidson AE, Sergouniotis PI, Mackay DS, et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum Mutat 2013;34(3):506–514. [DOI] [PubMed] [Google Scholar]
- 13.Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet 1998;19(3):257–259. [DOI] [PubMed] [Google Scholar]
- 14.Kohl S, Baumann B, Broghammer M, et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 2000;9(14):2107–2116. [DOI] [PubMed] [Google Scholar]
- 15.Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet 2002; 71(2):422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiadens AA, den Hollander AI, Roosing S, et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet 2009;85(2):240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohl S, Coppieters F, Meire F, et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet 2012;91(3):527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohl S, Zobor D, Chiang WC, et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat Genet 2015;47(7): 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen E, Brown DM, Benz MS, et al. Spectral domain optical coherence tomography as an effective screening test for hydroxychloroquine retinopathy (the ‘‘flying saucer’’ sign). Clin Ophthalmol 2010;4:1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci 2005;46(12):4739–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sparrow JR, Marsiglia M, Allikmets R, et al. Flecks in recessive Stargardt disease: short-wavelength autofluorescence, near-infrared autofluorescence, and optical coherence tomography. Invest Ophthalmol Vis Sci 2015;56(8):5029–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cukras CA, Wong WT, Caruso R, Cunningham D, Zein W, Sieving PA. Centrifugal expansion of fundus autofluorescence patterns in Stargardt disease over time. Arch Ophthalmol 2012;130(2):171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet 1998;7(3):355–362. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998;18(1):11–12. [DOI] [PubMed] [Google Scholar]
- 25.McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015;130(1):1–12. [DOI] [PubMed] [Google Scholar]
- 26.Zernant J, Schubert C, Im KM, et al. Analysis of the ABCA4 gene by next-generation sequencing. Invest Ophthalmol Vis Sci 2011;52(11):8479–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zernant J, Xie YA, Ayuso C, et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum Mol Genet 2014; 23(25):6797–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015;347(6218):1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016;48(12): 1581–1586. [DOI] [PubMed] [Google Scholar]
- 32.Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet 2016;48(2): 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015;31(5):761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99(4):877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zernant J, Lee W, Collison FT, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet 2017;54(6):404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sangermano R, Khan M, Cornelis SS, et al. ABCA4 midigenes reveal the full splice spectrum of all reported non-canonical splice site variants in Stargardt disease. Genome Res 2018;28(1):100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee W, Xie Y, Zernant J, et al. Complex inheritance of ABCA4 disease: four mutations in a family with multiple macular phenotypes. Hum Genet 2016;135(1):9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aleman TS, Han G, Serrano LW, et al. Natural history of the central structural abnormalities in choroideremia: a prospective cross-sectional study. Ophthalmology 2017;124(3): 359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCulloch C Choroideremia: a clinical and pathologic review. Trans Am Ophthalmol Soc 1969;67:142–195. [PMC free article] [PubMed] [Google Scholar]
- 40.Simell O, Takki K. Raised plasma-ornithine and gyrate atrophy of the choroid and retina. Lancet 1973;1(7811): 1031–1033. [DOI] [PubMed] [Google Scholar]
- 41.Lorenz B, Gyurus P, Preising M, et al. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci 2000;41(9):2735–2742. [PubMed] [Google Scholar]
- 42.Gerth-Kahlert C, Tiwari A, Hanson JVM, et al. C2orf71 mutations as a frequent cause of autosomal-recessive retinitis pigmentosa: clinical analysis and presentation of 8 novel mutations. Invest Ophthalmol Vis Sci 2017;58(10):3840–3850. [DOI] [PubMed] [Google Scholar]
- 43.Edwards TL, Jolly JK, Groppe M, et al. Visual acuity after retinal gene therapy for choroideremia. N Engl J Med 2016; 374(20):1996–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase ½ clinical trial. Lancet 2014;383(9923):1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Syed N, Smith JE, John SK, Seabra MC, Aguirre GD, Milam AH. Evaluation of retinal photoreceptors and pigment epithelium in a female carrier of choroideremia. Ophthalmology 2001;108(4):711–720. [DOI] [PubMed] [Google Scholar]
- 46.Bonilha VL, Trzupek KM, Li Y, et al. Choroideremia: analysis of the retina from a female symptomatic carrier. Ophthalmic Genet 2008;29(3):99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flannery JG, Bird AC, Farber DB, Weleber RG, Bok D. A histopathologic study of a choroideremia carrier. Invest Ophthalmol Vis Sci 1990;31(2):229–236. [PubMed] [Google Scholar]
- 48.Lazow MA, Hood DC, Ramachandran R, et al. Transition zones between healthy and diseased retina in choroideremia (CHM) and Stargardt disease (STGD) as compared to retinitis pigmentosa (RP). Invest Ophthalmol Vis Sci 2011; 52(13):9581–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacDonald IM, Russell L, Chan CC. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol 2009;54(3):401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan JI, Han G, Klinman E, et al. High-resolution adaptive optics retinal imaging of cellular structure in choroideremia. Invest Ophthalmol Vis Sci 2014;55(10):6381–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mura M, Sereda C, Jablonski MM, MacDonald IM, Iannaccone A. Clinical and functional findings in choroideremia due to complete deletion of the CHM gene. Arch Ophthalmol 2007;125(8):1107–1113. [DOI] [PubMed] [Google Scholar]
- 52.Rodrigues MM, Ballintine EJ, Wiggert BN, Lee L, Fletcher RT, Chader GJ. Choroideremia: a clinical, electron microscopic, and biochemical report. Ophthalmology 1984; 91(7):873–883. [PubMed] [Google Scholar]
- 53.Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A 2009;106(44):18751–18756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redmond TM, Yu S, Lee E, et al. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet 1998;20(4):344–351. [DOI] [PubMed] [Google Scholar]
- 55.Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol 1969;42(2):392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cideciyan AV, Aleman TS, Swider M, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet 2004;13(5):525–534. [DOI] [PubMed] [Google Scholar]
- 57.Kim SR, Jang YP, Jockusch S, Fishkin NE, Turro NJ, Sparrow JR. The all-trans-retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc Natl Acad Sci U S A 2007; 104(49):19273–19278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999;98(1):13–23. [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto K, Yoon KD, Ueda K, Hashimoto M, Sparrow JR. A novel bisretinoid of retina is an adduct on glycerophos-phoethanolamine. Invest Ophthalmol Vis Sci 2011;52(12): 9084–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jolly JK, Xue K, Edwards TL, Groppe M, MacLaren RE. Characterizing the natural history of visual function in choroideremia using microperimetry and multimodal retinal imaging. Invest Ophthalmol Vis Sci 2017;58(12):5575–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol 2001;119(3):359–369. [DOI] [PubMed] [Google Scholar]
- 62.Joussen AM, Heussen FM, Joeres S, et al. Autologous translocation of the choroid and retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol 2006; 142(1):17–30. [DOI] [PubMed] [Google Scholar]
- 63.Maaijwee K, Van Den Biesen PR, Missotten T, Van Meurs JC. Angiographic evidence for revascularization of an RPE-choroid graft in patients with age-related macular degeneration. Retina 2008;28(3):498–503. [DOI] [PubMed] [Google Scholar]
- 64.MacLaren RE, Bird AC, Sathia PJ, Aylward GW. Long-term results of submacular surgery combined with macular translocation of the retinal pigment epithelium in neovascular age-related macular degeneration. Ophthalmology 2005; 112(12):2081–2087. [DOI] [PubMed] [Google Scholar]
- 65.Peyman GA, Blinder KJ, Paris CL, Alturki W, Nelson NC Jr, Desai U. A technique for retinal pigment epithelium trans-plantation for age-related macular degeneration secondary to extensive subfoveal scarring. Ophthalmic Surg 1991;22(2): 102–108. [PubMed] [Google Scholar]
- 66.Stanga PE, Kychenthal A, Fitzke FW, et al. Retinal pigment epithelium translocation after choroidal neovascular membrane removal in age-related macular degeneration. Ophthalmology 2002;109(8):1492–1498. [DOI] [PubMed] [Google Scholar]
- 67.van Meurs JC, Van Den Biesen PR. Autologous retinal pigment epithelium and choroid translocation in patients with exudative age-related macular degeneration: short-term follow-up. Am J Ophthalmol 2003;136(4):688–695. [DOI] [PubMed] [Google Scholar]
- 68.Busskamp V, Picaud S, Sahel JA, Roska B. Optogenetic therapy for retinitis pigmentosa. Gene Ther 2012;19(2):169–175. [DOI] [PubMed] [Google Scholar]
- 69.Ho AC, Humayun MS, Dorn JD, et al. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology 2015;122(8):1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]