

Abstract
Background:
Patients with cystic fibrosis (CF) experience variable lung disease phenotypes. The R117H mutation is often associated with preserved lung function. Our objective was to compare the rate of lung function decline in patients with the R117H mutation and patients homozygous for the F508del mutation.
Methods:
Rate of decline in percentage-of-predicted FEV1 (ppFEV1) was analyzed using the 2006-2010 US CF Foundation Patient Registry.
Results:
4-year rate of decline was slower in 156 R117H patients compared with 6251 F508del patients (−0.61 vs −2.03 ppFEV1/year, P<0.001). Rates of decline in children were slower in R117H vs F508del patients (6-12 year-olds: +0.73 vs −1.91 ppFEV1/year, P<0.001 and 13-17 year-olds: −1.55 vs −2.66 ppFEV1/year, P=0.046), whereas rates in adults were not significantly different (18-24 year-olds: −1.52 vs −2.12, P=0.26 and ≥25 year-olds: −1.17 vs −1.40, P=0.33).
Conclusions:
These findings are consistent with a delayed onset, but ultimately similar progression of lung disease in R117H and homozygous F508del patients.
Keywords: cystic fibrosis, R117H, F508del, lung function, lung function decline
GRAPHIC ABSTRACT
INTRODUCTION
Patients with cystic fibrosis (CF) experience a highly variable clinical disease. Some of this variation is explained by differences in function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein. This protein function is in turn determined largely by the specific mutations present in the CFTR gene. Since the 1970s, investigators have described certain patients as having “mild” CF lung disease.(1, 2) Patients with this less severe disease tend to be diagnosed at an older age, are more likely pancreatic sufficient, and are not homozygous for the F508del-CFTR mutation.(3) However, even patients with “mild” disease are noted to develop lung pathology similar to typical patients with CF and exhibit progressive, ultimately fatal lung disease in adulthood.(4)
One of the more common mutations associated with this less severe form of disease, the R117H-CFTR mutation, was identified in 1990.(5) With this mutation the CFTR protein is present at the cell surface, but exhibits both conductance and gating defects.(6) This results in reduced CFTR function, although the degree of reduction is further modified by a poly-thymidine (poly-T) sequence on intron 8.(7) Historically, patients with this mutation have been diagnosed later in life, often due to male infertility, and almost always demonstrate pancreatic sufficiency.(8) However, many of these patients develop progressive, life-shortening lung disease.
Although patients with less severe CF typically experience some degree of symptomatic lung disease, it is not clear if loss of lung function occurs gradually throughout life or is delayed in onset and progresses more rapidly in later life. We designed this study to compare lung function decline in patients with the R117H-CF mutation on at least one allele to that in patients with the common F508del-CFTR mutation on both alleles. In addition, we wanted to answer the question “In patients with CF and the R117H-CFTR mutation, is lung disease as manifested by airway obstruction, early in onset but gradual, or delayed in onset but rapid?”
METHODS
Rate of lung function decline over 4 years was analyzed using percentage of predicted forced expiratory volume in 1 second (ppFEV1)(9). Two retrospective cohorts of patients with CF and either ≥1 R117H-CFTR mutation or homozygous F508del-CFTR mutation were selected from the 2006 US CF Foundation Patient Registry (CFFPR). Data from 2006–2010 were used for the analysis since all CF centers were collecting encounter-based data by 2006 and we wanted to avoid any impact of patients enrolled in clinical trials of potential disease-modifying therapies (which started in 2011).
Since not all individuals with the R117H-CFTR mutation have clinical manifestations of CF, we excluded patients with presumed CFTR-related metabolic syndrome (CRMS).(10) This exclusion included individuals identified by newborn screening who had an R117H-CFTR mutation plus a sweat chloride level <60 mEq/mL or no sweat test result recorded. However, 2 patients meeting these criteria were included because they had been diagnosed due to the clinical presentation of failure to thrive prior to 1998 when R117H-CFTR mutation screening began.
Patients had to be ≥6 years old and have a baseline visit identified as the first recorded FEV1 measurement in 2006. The last visit within 12 months of this baseline visit was considered the last baseline year visit. In addition to the baseline FEV1, included patients had to have ≥3 values recorded with at least 1 during each of the second and fifth years of the study in order to assure all patients had 4 years of follow-up. All FEV1 values recorded after the last baseline year visit and during the 4-year time span (2007–2010) were used to calculate an individual patient’s annual ppFEV1 rate of decline. All calculations of ppFEV1 were based on Global Lung Initiative equations.(9) No post-lung transplant data were included.
Baseline demographics and clinical characteristics were derived from the first baseline year visit (age, sex, year of diagnosis, genotype, sweat chloride, “first visit” ppFEV1), the last baseline year visit (height, weight, body mass index [BMI], microbiology) or from all recorded values during the baseline year (the highest value [“best”] ppFEV1, CF related diabetes [CFRD], liver disease, intravenous [IV] antibiotic-treated pulmonary exacerbations [PEx]). Lack of comprehensive poly-T data meant that no analysis of this sequence was conducted.
The ppFEV1 intercepts and slopes (rate of decline) were estimated for the R117H and F508del cohorts separately by age group and overall. These estimates reflect the linear component of lung function decline over the 4 years of the trend (ie. the average rate of decline). Estimation and significance testing of the difference between mutation cohorts were conducted using a repeated-measures model that accounted for the correlation of values within patient. The model included mutation, age group, time (in years) since the last FEV1 measurement in the baseline year, and all interactions. Overall estimates of intercept and slope were calculated by weighting each age group category according to the observed distribution pooled across mutation groups. In addition, intercept and slope were calculated separately for each age group from the same model. Final estimated intercepts and annual rates of decline with 95% confidence intervals were generated using the ESTIMATE statement within the MIXED procedure in SAS Version 9.4 (SAS Institute, Inc., Cary, NC).
A sensitivity analysis was performed using 2-year rates of lung function decline to include additional patients in the analysis that may have been excluded due to lack of complete 5-year data. For this analysis, a 2-year period was randomly chosen for each patient within each of the 4 age groups (6–12, 13–17, 18–24, and ≥25 years). Consequently, patients could contribute at most two 2-year periods. Patients were required to have ≥3 FEV1 values recorded during the 2 years (spanning a minimum of 6 months), and all values were included in the analysis. Separate age group-specific repeated-measures models that accounted for the correlation of values within patient were used to estimate intercept and slope. These models included mutation, the starting year of measurement, time (years) since the first FEV1 measurement in the 2-year period, and all interactions. Intercept and slope were calculated separately from each age group-specific model. Starting year was weighted equally.
RESULTS
The original dataset included 818 R117H-CFTR mutation patients and 13,632 homozygous F508del-CFTR mutation patients (Figure 1). After excluding 145 subjects with CRMS there were 673 R117H-CFTR mutation patients with CF. Of the patients ≥6 years old, 68% of the R117H patients were excluded due to missing data compared with only 38% of the F508del patients. Mortality or lung transplant accounted for exclusion in 5.7% of R117H patients and 13.3% of F508del patients. In total, 156 patients with the R117H-CFTR mutation and 6251 patients homozygous for the F508del-CFTR mutation met the selection criteria and were included in the 4-year analysis. At baseline, the R117H cohort was older, had a lower mean sweat chloride value, better nutritional status, and less CFRD or liver disease than the F508del cohort; however mean best ppFEV1 values from the baseline year were not significantly different between cohorts (Table 1). At the end of the baseline year, the R117H cohort had a lower frequency of Pseudomonas aeruginosa and Staphylococcus aureus infection than the F508del cohort (Table 1). Candida species was slightly more common and Aspergillus species was less common in the R117H cohort. The R117H cohort experienced fewer IV-treated PEx during the baseline year, resulting in fewer hospitalizations and fewer home IV-antibiotic treatments than did the F508del cohort (Table 1). However, for patients experiencing ≥1 IV-treated PEx, the event rates were similar between cohorts.
Figure 1.

STROBE diagram showing the derivation of the R117H-CFTR and homozygous F508del-CFTR mutation cohorts in the 4-year analysis.
CFTR, cystic fibrosis transmembrane conductance regulator; FEV1, forced expiratory volume in 1 second.
Table 1.
Baseline Demographic and Clinical Characteristics by Mutation.
| Characteristic |
R117H (n=156) |
F508del (n=6251) |
P-Value* |
|---|---|---|---|
| Age, years, mean (SD) | 29.2 (17.2) | 17.0 (9.1) | <0.001 |
| Female, % | 51.9 | 47.3 | 0.26 |
| Year of CF diagnosis | |||
| median | 1998 | 1992 | <0.001 |
| min, max | 1958, 2006 | 1949, 2006 | |
| Sweat chloride | [n=126] | [n=5,729] | |
| mmol/L, mean (SD) | 77.8 (23.9) | 102.6 (16.3) | <0.001 |
| Height z-score, mean (SD) | 0.11 (1.03) | −0.51 (1.01) | <0.001 |
| Weight z-score, mean (SD) | 0.46 (1.14) | −0.41 (1.02) | <0.001 |
| BMI z-score, | [n=6,250] | ||
| mean (SD) | 0.41 (1.07) | −0.20 (0.96) | <0.001 |
| ppFEV1 baseline first visit, mean (SD) | 79.0 (22.2) | 77.3 (23.0) | 0.35 |
| ppFEV1 best baseline year value, mean (SD) | 83.9 (22.4) | 84.8 (22.3) | 0.61 |
| CF-related diabetes, % | 7.1 | 15.5 | 0.004 |
| Liver disease, % | 0.6 | 4.1 | 0.029 |
| Microbiology | [n=152] | [n=6,130] | |
| Staphylococcus aureus, % | 36.2 | 54.1 | <0.001 |
| Pseudomonas aeruginosa, % | 29.6 | 49.2 | <0.001 |
| Candida species, % | 7.2 | 3.3 | 0.007 |
| Stenotrophomonas maltophilia, % | 5.3 | 7.7 | 0.27 |
| Aspergillus species, % | 3.9 | 8.9 | 0.034 |
| Haemophilus influenzae, % | 3.3 | 6.1 | 0.15 |
| ≥1 IV treatment for PEx, % | 28.2 | 45.9 | <0.001 |
| ≥1 hospitalization, % | 21.2 | 40.8 | <0.001 |
| ≥1 home IV antibiotic treatment, % | 18.6 | 26.3 | 0.031 |
| IV treatment for PEx events/year, mean (SD) | 0.44 (0.84) | 0.87 (1.31) | <0.001 |
| Hospitalization events/year, mean (SD) | 0.32 (0.74) | 0.75 (1.22) | <0.001 |
| Home IV antibiotic events/year, mean (SD) | 0.25 (0.60) | 0.41 (0.84) | 0.020 |
| IV treatment in patients with ≥1 PEx, mean (SD) | 1.57 (0.87) | 1.90 (1.33) | 0.10 |
P-Value obtained from t-test or chi-square test.
CF, cystic fibrosis; BMI, body mass index; max, maximum; min, minimum; ppFEV1, percentage of predicted forced expiratory volume in 1 second; IV, intravenous; PEx, pulmonary exacerbation; SD, standard deviation.
R117H patients excluded from the analysis were similar to the included patients, although they had slightly lower sweat chloride levels and possibly milder disease as evidenced by significantly fewer being treated for a PEx during the baseline year (Supplemental Table 1). The CFTR mutation on the second allele for the excluded R117H patients was (86% Class I, II, or III; 7% Class IV or V; 7% unknown) and for the included patients was (92% Class I, II, or III; 3% Class IV or V; 6% unknown). On the other hand, F508del patients excluded from the final analysis were at a worse stage of disease than included patients in multiple areas: older age, minimally higher sweat chloride levels, worse nutritional results, much lower baseline ppFEV1, more CFRD and Pseudomonas aeruginosa culture positivity, but less Staphylococcus aureus, Haemophilus influenzae, and Aspergillus species culture positivity. Although fewer excluded patients had a PEx, they had an overall increased number of PEx and hospitalizations (Supplemental Table 1).
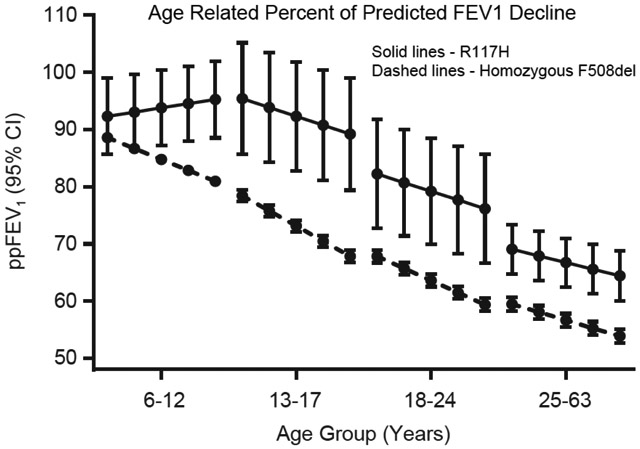
The recorded best baseline ppFEV1 was similar between cohorts, although after adjusting for age the estimated overall ppFEV1 intercept in the R117H cohort was significantly higher than in the homozygous F508del cohort (86.58 vs 76.54; P<0.001) (Table 2, Figure 2A). The estimated ppFEV1 slope was not as steep and therefore rate of decline was slower in the R117H cohort than in the F508del cohort (−0.61 vs -2.03 ppFEV1/year; P<0.001). However, estimated intercept and slope varied greatly by age group (Figure 2B). The rate of decline across age groups was consistently negative for the F508del cohort, ranging from −1.40 to −2.66 ppFEV1/year (Table 3). In contrast, the R117H cohort had an increasing ppFEV1 slope among 6–12 year olds, which became negative in the 13–17 year olds, although significantly less negative than in the F508del cohort (−1.55 vs −2.66 ppFEV1/year P=0.046). In the older 18–24 and ≥25 year old age groups the rates of decline were not statistically different between R117H and F508del cohorts (Table 3). It should be noted that for this analysis the number of R117H patients in each age group was small. Although a minimum of 3 FEV1 measurements was required, slope estimates were based on considerably more values in each cohort: R117H mean 13.5, median 12 (interquartile range of 9–17) and F508del mean 19.5, median 17 (interquartile range 13–23).
Table 2.
Estimated Percentage of Predicted FEV1 Intercept and Slope for R117H Patients (n=156) and F508del Patients (n=6,251)*
| Parameter | Estimate | Standard Error |
P-Value | Lower 95% CI |
Upper 95% CI |
|---|---|---|---|---|---|
| Intercept (ppFEV1) | |||||
| R117H | 86.58 | 2.03 | <0.001 | 82.60 | 90.55 |
| F508del | 76.54 | 0.26 | <0.001 | 76.04 | 77.05 |
| Difference | 10.04 | 2.04 | <0.001 | 6.03 | 14.04 |
| Slope (ppFEV1/year) | |||||
| R117H | −0.61 | 0.21 | 0.005 | −1.03 | −0.19 |
| F508del | −2.03 | 0.02 | <0.001 | −2.07 | −1.98 |
| Difference | 1.42 | 0.22 | <0.001 | 1.00 | 1.84 |
Repeated measures model adjusted for correlation within patient. Age group and mutation were fixed effects. All 2-way and 3-way interactions were included. Overall estimates of intercept and slope were calculated by weighting each age group category according to the observed distribution pooled across mutation groups.
CI, confidence interval; ppFEV1, percentage of predicted forced expiratory volume in 1 second.
Figure 2.

Estimated ppFEV1 intercept and slope by CFTR mutation (R117H or homozygous F508del) (Tables 2 and 3). Panel A: Overall estimates. R117H-CFTR mutation n=156, F508del-CFTR mutation n=6,251. Panel B: Estimates separated by age group. R117H-CFTR mutation ages 6–12 years, n=36; ages 13–17 years, n=17; ages 18–24 years, n=18; ages ≥25 years, n=85). F508del-CFTR mutation ages 6–12 years, n=2,398; ages 13–17 years, n=1,447; ages 18–24 years, n=1,278; ages ≥25 years, n=1,128.
Solid lines = R117H-CFTR mutation. Dashed lines = homozygous F508del-CFTR mutation. Shaded areas represent 95% Confidence Intervals.
ppFEV1, percentage of predicted forced expiratory volume in 1 second.(9)
Table 3.
Age Group Specific Estimated Percentage of Predicted FEV1 Intercept and Slope for R117H Patients and F508del Patients.*
| Parameter | Estimate | Standard Error |
P-value | Lower 95% CI |
Upper 95% CI |
|---|---|---|---|---|---|
| 6-12 year olds (R117H n=36, F508del n=2,398) | |||||
| Intercept (ppFEV1) | |||||
| R117H | 92.32 | 3.41 | <0.001 | 85.65 | 99.00 |
| F508del | 88.59 | 0.42 | <0.001 | 87.77 | 89.40 |
| Difference | 3.74 | 3.43 | 0.28 | −2.99 | 10.46 |
| Slope (ppFEV1/year) | |||||
| R117H | 0.73 | 0.34 | 0.032 | 0.06 | 1.39 |
| F508del | −1.91 | 0.04 | <0.001 | −1.98 | −1.84 |
| Difference | 2.64 | 0.34 | <0.001 | 1.97 | 3.31 |
| 13-17 year olds (R117H n=17, F508del n=1,447) | |||||
| Intercept (ppFEV1) | |||||
| R117H | 95.40 | 4.99 | <0.001 | 85.62 | 105.18 |
| F508del | 78.43 | 0.53 | <0.001 | 77.38 | 79.47 |
| Difference | 16.97 | 5.02 | <0.001 | 7.13 | 26.82 |
| Slope (ppFEV1/year) | |||||
| R117H | −1.55 | 0.55 | 0.005 | −2.63 | −0.47 |
| F508del | −2.66 | 0.04 | <0.001 | −2.74 | −2.57 |
| Difference | 1.11 | 0.55 | 0.046 | 0.02 | 2.19 |
| 18-24 year olds (R117H n=18, F508del n=1,278) | |||||
| Intercept (ppFEV1) | |||||
| R117H | 82.23 | 4.84 | <0.001 | 72.73 | 91.72 |
| F508del | 67.81 | 0.57 | <0.001 | 66.70 | 68.93 |
| Difference | 14.41 | 4.88 | 0.003 | 4.85 | 23.97 |
| Slope (ppFEV1/year) | |||||
| R117H | −1.52 | 0.53 | 0.004 | −2.56 | −0.49 |
| F508del | −2.12 | 0.05 | <0.001 | −2.22 | −2.02 |
| Difference | 0.60 | 0.53 | 0.26 | −0.45 | 1.64 |
| ≥25 year olds (R117H n=85, F508del n=1,128) | |||||
| Intercept (ppFEV1) | |||||
| R117H | 69.05 | 2.22 | <0.001 | 64.69 | 73.41 |
| F508del | 59.42 | 0.61 | <0.001 | 58.23 | 60.62 |
| Difference | 9.63 | 2.31 | <0.001 | 5.11 | 14.15 |
| Slope (ppFEV1/year) | |||||
| R117H | −1.17 | 0.23 | <0.001 | −1.62 | −0.71 |
| F508del | −1.40 | 0.06 | <0.001 | −1.51 | −1.29 |
| Difference | 0.23 | 0.24 | 0.33 | −0.24 | 0.70 |
Repeated measures model adjusted for correlation within patients. Age group and mutation were fixed effects. All 2-way and 3-way interactions were included.
CI, confidence interval; ppFEV1, percentage of predicted forced expiratory volume in 1 second.
The 2-year sensitivity analysis included more patients (R117H n=329; F508del n=9,961) and showed similar trends in ppFEV1 rate of decline by age group (Supplemental Table 2). Again the youngest R117H patients had a positive slope and the slope in older patients was not significantly different from F508del patients.
DISCUSSION
These analyses demonstrate that progressive lung disease occurs in patients with CF and either the R117H-CFTR mutation or homozygous F508del-CFTR mutation. Overall the ppFEV1 rate of decline in the R117H cohort was significantly less steep than that in the F508del cohort. However, lung function decline varied greatly by age in both cohorts. Comparing this age-related pattern of decline between cohorts suggests that the deterioration of lung function in patients with CF and the R117H-CFTR mutation may only be delayed, and not necessarily lessened once chronic lung disease becomes established, compared with that in patients homozygous for the F508del-CFTR mutation.
Many factors influence lung function decline in patients with CF. In general, patients with higher lung function experience a more rapid rate of decline.(11, 12) Average baseline recorded lung function was similar between these 2 cohorts, indicating they were at a similar stage of disease, although the R117H patients were significantly older than the F508del patients, suggesting they had a less severe disease. Additional risk factors for lung function decline (e.g. CF-related diabetes, Pseudomonas aeruginosa infection, and PEx) were present in both the R117H and F508del cohorts, although fewer patients in the R117H cohort had these known risk factors for lung function decline. Again, the difference in age between the cohorts is important since CF-related diabetes and Pseudomonas aeruginosa infection are both generally more common in older patients with CF.(13) Finally, one of the strongest predictors of mortality and a major risk factor for lung function decline is the frequency of PEx.(14) Patients with the R117H-CFTR mutation were nearly 40% less likely to experience a PEx, although if they did have an exacerbation in the prior year, the annual event rate was similar to that of the F508del patients. This suggests that once clinical lung disease is established in R117H patients, they progress in much the same way as F508del patients.
Although the overall rate of decline was quite different between the R117H and homozygous F508del cohorts, most striking was the ppFEV1 slope when analyzed by age group. Marked differences were present in the youngest patients. During childhood, patients with CF and the R117H-CFTR mutation experienced almost no decline in lung function. However during adulthood, there was no evidence to suggest that the R117H patients lost function at a rate different from adults homozygous for the F508del-CFTR mutation. Because these adults with the R117H-CFTR mutation began with higher lung function, their parallel drop with the F508del patients suggests that, although they remain with higher overall lung function in adulthood, they experience a significant rate of decline. Thus when the clinician is faced with an adult patient who has the R117H-CFTR mutation, close monitoring is essential. Complacency with patients thought to have “mild” mutations may lead to missing early, potentially controllable lung disease, and risks the patient experiencing rapid lung function decline.
No attempt was made to match patients in these analyses given that these 2 mutation groups are known to have different clinical manifestations of CF disease. Additionally, no adjustments were made for sex or other factors when comparing these groups. We felt general population descriptions were useful to understand the clinical course of disease in these 2 mutation groups, recognizing that one cannot use the analysis to infer that any differences are independent effects of the R117H genotype in the individual patient. The estimated baseline ppFEV1 was significantly higher in the R117H cohort in all age groups. However in adult patients, the estimated slope of lung function decline was not different between R117H and homozygous F508del patients. Older adults with typical CF lung disease are known to have a slower rate of lung function decline compared to younger patients, partly due to earlier death in patients with more rapid decline.(12) Indeed, in both of the F508del and R117H adult age groups, lung function decline was slightly less than in the adolescents. The fact that patient death or lung transplant excluded over twice as many homozygous F508del patients compared to the R117H patients potentially biases the results by excluding these patients with more severe lung disease.
An additional selection bias that might explain the appearance of delayed lung function decline in the R117H patients is the fact that the adult R117H patients were frequently diagnosed due to clinical symptoms, while younger patients were often detected by newborn screening. Newborn screening for CF, including the R117H-CFTR mutation, is a recent development and the younger patients in this study probably include cases that will not develop clinical symptoms until a much later age, if at all. Thus the younger R117H patients probably represent a different distribution of disease severity from the older patients. However, since the drop in lung function appears to begin during adolescence and early adulthood, and since this study does not quantify risk factors for lung function decline, it is probably important that all children with the R117H-CFTR mutation be closely monitored.
This analysis does not address why this rapid loss of lung function appears to be delayed into adulthood for patients with the R117H-CFTR mutation. In addition to the potential for selection bias noted previously, differences in pathophysiology may be contributing. Patients with CF and the R117H-CFTR mutation have limited CFTR protein function, as opposed to almost no function in patients homozygous for the F508del-CFTR mutation. One theoretical possibility for rapid lung function decline would be a trigger event or sequence overwhelming this limited CFTR function. Following the trigger event, the patient might then experience lung function deterioration similar to typical patients with CF. A second theoretical possibility is that lung disease has been present throughout childhood but is not detected by spirometry lung function measurements until adulthood. In some ways this is similar to what occurs in patients with CF who have spirometry results in the normal range, yet still have neutrophilic airway inflammation.(15) Often this inflammation begins in early childhood and more sensitive lung function measurement techniques may be necessary to detect changes.(16) However, if the R117H patient has airway inflammation present throughout childhood, there would still be a need to explain the rapid decline in later life. Further study of younger patients with CF and the R117H-CFTR mutation is needed to better understand this delayed decline in ppFEV1.
As with all epidemiologic studies, this analysis is limited by the available data. In the primary analysis there was a large discrepancy in the number of excluded patients with the R117H-CFTR mutation compared to the F508del-CFTR mutation. R117H patients had less frequent lung function testing, suggesting that they were not clinically followed as closely as the F508del patients. For this reason, a sensitivity analysis requiring only 2 years of data was conducted. This analysis more than doubled the number of included R117H and F508del patients yet showed similar patterns in the rate of lung function decline. These results suggest that the exclusion of milder R117H patients (lower sweat chloride and fewer PEx) and more severe F508del patients (lower lung function, more PEx, higher mortality) did not change the conclusion that the slope of FEV1 decline was different in children, but similar in adult patients with either an R117H-CFTR mutation or homozygous for the F508del-CFTR mutation.
There are additional limitations to interpreting the data since this analysis is focused on patients with CF and the R117H-CFTR mutation. First, limited information was available concerning the polythymidine sequence in these patients. In the few patients with this information, 5T and 7T variants were present in all age groups; however, we were unable to evaluate these groups separately due to the small numbers. Second, patients with the R117H-CFTR mutation often fulfilled the diagnostic criteria for CRMS instead of CF. Recently, Ren and colleagues(17) studied the CFFPR to better understand this diagnosis and found potential misclassification of these 2 diagnoses. We chose to use a strict definition of CF, requiring newborn screen detected patients to have a documented abnormal sweat chloride value in order for patients to be included in the analysis.(18) There is a possibility that patients with lower sweat chloride values may actually have clinical CF disease instead of CRMS. Third, not all subjects in the CFFPR had their genotype recorded. In 2010, 90.7% of patients in the CFFPR had genotype information and 1.4% had the R117H-CFTR mutation.(19) Fourth, the R117H-CFTR mutation is fairly common, and it is clear that not all individuals who have CF and the R117H-CFTR mutation have been diagnosed. Experience from newborn screening programs for CF has identified a much larger population of individuals with this mutation than historically have been diagnosed following clinical presentation. Since the adult R117H cohort in our analysis was diagnosed based on symptoms or family history and not neonatal screening, many individuals with less severe lung disease may exist, but have not yet been diagnosed. Additionally, the high prevalence of the R117H-CFTR mutation and lack of clinical disease in patients has led some newborn screening programs to exclude this mutation.(20) Finally, we used only one allele to define the R117H population, yet required the F508del patients to be homozygous for their mutation. Most of the R117H-CFTR mutation patients had an F508del-CFTR mutation on the second allele, although 4 of the 156 patients were paired with another “mild” Class IV or V mutation.(21)
Several conclusions can be drawn from these results. First, CRMS is fairly common and needs to be differentiated from CF in individuals with the R117H-CFTR mutation. Recently published guidelines recommend a complete evaluation of infants detected by newborn screening before giving them the diagnosis of CF.(18) Second, children with the R117H-CFTR mutation generally have stable lung function. Since we were unable to study if these patients were being treated with various therapies, it is not possible to say if therapies developed for typical CF are also of value in young R117H patients. Third, certain risk factors for lung function decline are less common in R117H patients and during adolescence and adulthood lung function is higher than that in homozygous F508del patients. However this does not mean that clinical monitoring should be lessened since, particularly for adults with the R117H-CFTR mutation, a rapid loss of lung function may occur. Finally, although there are differences between the ppFEV1 rates of decline in the R117H and F508del cohorts in the younger age groups, there is no evidence to suggest they are different in adult patients. These findings are consistent with a delayed onset but ultimately similar lung disease progression in older patients with the R117H-CFTR mutation. Importantly, although this pattern occurs in R117H patients, further study is needed to see if a similar pattern exists in patients with CF and other “mild” CFTR gene mutations.
Supplementary Material
HIGHLIGHTS.
We questioned how the rate of lung function decline compares between patients with cystic fibrosis who have “mild” lung disease (related to the R117H CF gene mutation) and those with “typical” lung disease (homozygous for the F508del CF gene mutation)?
We discovered that patients with cystic fibrosis and the R117H CF gene mutation experience a delayed, but ultimately similar decline in lung function.
This suggestion of a delayed progression of lung disease provides an opportunity for better understanding the pathophysiology of cystic fibrosis related lung disease and clinically may lead to more effective intervention and monitoring of these patients.
ACKNOWLEDGEMENTS
The authors thank the US Cystic Fibrosis Foundation and the Cystic Fibrosis Foundation Patient Registry team for providing the data used in these analyses. Study advice and coordination were provided by Barry Lubarsky, PhD, and medical writing and editorial coordination were provided by Dhrupad Patel, PharmD and Gauri Dixit, PhD. BL was previously, and DP and GD are currently, employed by Vertex Pharmaceuticals Incorporated and may own stock or stock options in the company.
Footnotes
DISCLOSURES
This study was funded by Vertex Pharmaceuticals Incorporated. JSW, EFM, MWK, and RBM have received consultancy fees and their institutions have received financial support from Vertex Pharmaceuticals Incorporated for participation in clinical trials. NM-H, GSS, CHG, and WJM are members of the US CFF Patient Registry committee and their institutions have received financial support from Vertex Pharmaceuticals Incorporated for participation in clinical trials. CHG was also part of a research group that received a clinical research grant from Vertex Pharmaceuticals Incorporated. JSW is a former employee of, but currently does not consult for or own stock or stock options in Vertex Pharmaceuticals Incorporated. DJP and SJM are employees of ICON Clinical Research, which was paid by Vertex Pharmaceuticals Incorporated to provide analytical services for this study. No author declares any competing interests with this manuscript.
REFERENCES
- 1.Shwachman H Gastrointestinal manifestations of cystic fibrosis. Pediatr Clin North Am. 1975;22(4):787–805. [DOI] [PubMed] [Google Scholar]
- 2.al-Jader LN, Meredith AL, Ryley HC, Cheadle JP, Maguire S, Owen G, et al. Severity of chest disease in cystic fibrosis patients in relation to their genotypes. J Med Genet. 1992;29(12):883–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon-Bouy B, Mornet E, Taillandier A, Serre JL, Boue J, Boue A. The delta F508 mutation in mild adult forms of cystic fibrosis (CF). Clin Genet. 1991;39(4):304–5. [DOI] [PubMed] [Google Scholar]
- 4.McKone EF, Emerson SS, Edwards KL, Aitken ML. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet. 2003;361(9370):1671–6. [DOI] [PubMed] [Google Scholar]
- 5.Dean M, White MB, Amos J, Gerrard B, Stewart C, Khaw KT, et al. Multiple mutations in highly conserved residues are found in mildly affected cystic fibrosis patients. Cell. 1990;61(5):863–70. [DOI] [PubMed] [Google Scholar]
- 6.Yu YC, Sohma Y, Hwang TC. On the mechanism of gating defects caused by the R117H mutation in cystic fibrosis transmembrane conductance regulator. J Physiol. 2016;594(12):3227–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kiesewetter S, Macek M Jr., Davis C, Curristin SM, Chu CS, Graham C, et al. A mutation in CFTR produces different phenotypes depending on chromosomal background. Nat Genet. 1993;5(3):274–8. [DOI] [PubMed] [Google Scholar]
- 8.Kristidis P, Bozon D, Corey M, Markiewicz D, Rommens J, Tsui LC, et al. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet. 1992;50(6):1178–84. [PMC free article] [PubMed] [Google Scholar]
- 9.Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40(6):1324–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren CL, Desai H, Platt M, Dixon M. Clinical outcomes in infants with cystic fibrosis transmembrane conductance regulator (CFTR) related metabolic syndrome. Pediatr Pulmonol. 2011;46(11):1079–84. [DOI] [PubMed] [Google Scholar]
- 11.Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr. 2007;151(2):134–9, 9 e1. [DOI] [PubMed] [Google Scholar]
- 12.Konstan MW, Wagener JS, Vandevanter DR, Pasta DJ, Yegin A, Rasouliyan L, et al. Risk factors for rate of decline in FEV1 in adults with cystic fibrosis. J Cyst Fibros. 2012;11(5):405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cystic Fibrosis Foundation Patient Registry 2005 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation, 2006. [Google Scholar]
- 14.Liou TG, Adler FR, Fitzsimmons SC, Cahill BC, Hibbs JR, Marshall BC. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol. 2001;153(4):345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care Med. 1994;150(2):448–54. [DOI] [PubMed] [Google Scholar]
- 16.Ramsey KA, Foong RE, Grdosic J, Harper A, Skoric B, Clem C, et al. Multiple Breath Washout Outcomes Are Sensitive to Inflammation and Infection in Children with Cystic Fibrosis. Ann Am Thorac Soc. 2017. [DOI] [PubMed] [Google Scholar]
- 17.Ren CL, Fink AK, Petren K, Borowitz DS, McColley SA, Sanders DB, et al. Outcomes of infants with indeterminate diagnosis detected by cystic fibrosis newborn screening. Pediatrics. 2015;135(6):e1386–92. [DOI] [PubMed] [Google Scholar]
- 18.Farrell PM, White TB, Howenstine MS, Munck A, Parad RB, Rosenfeld M, et al. Diagnosis of Cystic Fibrosis in Screened Populations. J Pediatr. 2017;181S:S33–S44 e2. [DOI] [PubMed] [Google Scholar]
- 19.Cystic Fibrosis Foundation Patient Registry 2009 Annual Data Report. Bethesda, MD: Cystic Fibrosis Foundation, 2010. [Google Scholar]
- 20.Thauvin-Robinet C, Munck A, Huet F, Genin E, Bellis G, Gautier E, et al. The very low penetrance of cystic fibrosis for the R117H mutation: a reappraisal for genetic counselling and newborn screening. J Med Genet. 2009;46(11):752–8. [DOI] [PubMed] [Google Scholar]
- 21.Sosnay PR, Salinas DB, White TB, Ren CL, Farrell PM, Raraigh KS, et al. Applying Cystic Fibrosis Transmembrane Conductance Regulator Genetics and CFTR2 Data to Facilitate Diagnoses. J Pediatr. 2017;181S:S27–S32 e1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.