

Abstract
The majority of patients with pancreatic ductal adenocarcinoma (PDA) develop metastatic disease after resection of their primary tumor. We found that livers from patients and mice with PDA harbor single, disseminated cancer cells (DCCs) lacking expression of cytokeratin-19 (CK19) and major histocompatibility complex class I (MHCI). We created a mouse model to determine how these DCCs develop. Intra-portal injection of immunogenic PDA cells into pre-immunized mice seeded livers only with single, non-replicating DCCs that were CK19- and MHCI-. The DCCs exhibited an endoplasmic reticulum (ER) stress response but, paradoxically lacked both inositol-requiring enzyme 1α activation and expression of the spliced form of transcription factor XBP1 (XBP1s). Inducible expression of XBP1s in DCCs, in combination with T cell-depletion, stimulated the outgrowth of macro-metastatic lesions that expressed CK19 and MHCI. Thus, unresolved ER stress enables DCCs to escape immunity and establish latent metastases.
Pancreatic ductal adenocarcinoma (PDA) is the fourth most common cause of death from cancer worldwide (1), and has a five year survival rate of 6% (2). The majority of patients are diagnosed after the disease has spread beyond the primary tumor site. Patients who show no clinical evidence of local invasion or distant metastasis are treated by surgery but approximately 75% of these patients develop metastatic disease within two years after resection of their primary tumors (3, 4), despite intra-operative examination of the liver confirming the absence of macro-metastatic lesions (5). These observations lead to the conclusion that latent metastases, detectable only microscopically, were present in these patients at the time of surgery and were responsible for the post-operative development of metastatic disease.
Latent metastases are thought to be lesions in which cancer cell proliferation is balanced by immune-mediated cancer cell death (6–8). A more recent hypothesis highlights the role of quiescent, single disseminated cancer cells (DCCs) (9–11). Single, non-replicating DCCs have been observed in several cancer types, most often in the bone marrow (12, 13), but whether quiescence is enforced by the microenvironment or is cancer cell-autonomous is not known (14). Immunity, both innate (15) and adaptive (7, 16, 17), also is likely to have a role in the selection and/or maintenance of latent DCCs. This has long been suspected based on the clinical observation that immunosuppressed recipients of allografts occasionally present with donor-derived cancer (18, 19). However, there is an unexplained paradox of immunity preventing the outgrowth of latent metastases while not eliminating latent metastases.
In the present study, we examine the nature of latent metastases in PDA by developing a mouse model that replicates the characteristics of hepatic DCCs that are found in human PDA and in spontaneously arising PDA in mice. We studied the metastatic process in the context of an ongoing adaptive immune response because of the occurrence of cancer cell-specific immunity in human and mouse PDA (20–23).
Results
Quiescent, single disseminated cancer cells in the livers of human and mice with PDA.
To determine whether hepatic DCCs occur in human PDA, we microscopically examined tissue sections from the primary tumors and livers of five patients with PDA who had no clinically detectable hepatic metastases. The clinicopathologic characteristics of the patients are shown in table S1. The tumors were genotyped as having p53 loss-of-heterozygosity, which permitted staining for mutant p53 accumulation as an identifier of cancer cells (24). p53+ cancer cells were present in both the primary tumors and livers of all five patients. The p53+ cancer cells resided in the livers as single cells that were consistently CK19-, Ki67-, and MHCI-, in contrast to the cancer cells in the primary tumors, which exhibited all three markers (Fig. 1A–C). We also examined the livers from mice bearing the autochthonous LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre; RosaYFP (KPCY) model of PDA (25–27), which recapitulates human PDA. In livers devoid of macro-metastases, we found both yellow fluorescent protein (YFP)+ micro-metastases and DCCs. While the micro-metastases always expressed CK19, Ki67 and MHCI, the single DCCs were mainly CK19- (32/40), Ki67- (22/22), and MHCI- (28/28) (Fig.1D-F). Therefore, the livers of patients and mice with PDA contain DCCs that share an unusual phenotype linking the loss of epithelial gene expression and quiescence with a potential for escape from T cell recognition.
Fig. 1. Single DCCs with a characteristic phenotype are present in the livers of humans and mice with PDA.
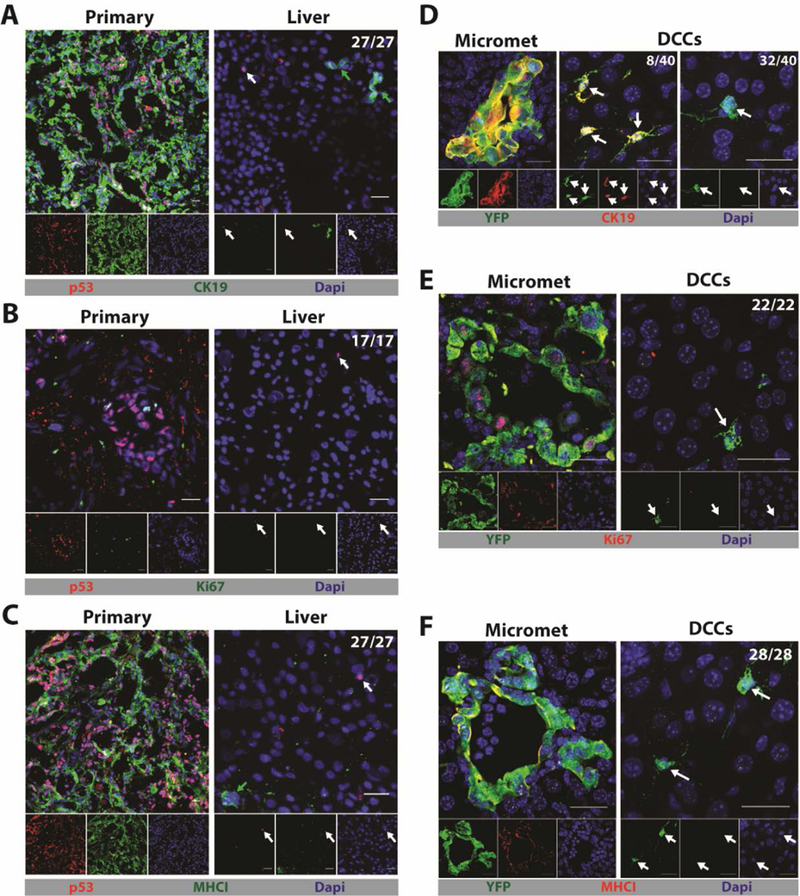
(A-C) Immunofluorescence (IF) of sections from the primary tumor and liver of a patient with PDA that have been stained with anti-p53 to reveal cancer cells (red) and (A) anti-CK19 (green), (B) anti-Ki67 (green) or (C) anti-MHCI (green). Photomicrographs are representative of five patients. (D-F) IF of sections from a liver of a KPCY mouse with spontaneous PDA and no hepatic macro-metastases. Sections were stained with anti-YFP to reveal cancer cells (green) and (D) anti-CK19 (red), (E) anti-Ki67 (red), and (F) anti-MHCI (red). Photomicrographs are representative of three mice. The ratios shown in the top right corners of the photomicrographs represent the frequency of the observed DCC phenotype relative to the total number of DCCs that were assessed. All frequencies are compiled in table S2. White arrows designate DCCs and, in the sections of human livers, green arrows designate normal, liver-resident CK19+ or MHCI+ cells. Scale bar = 25μm.
A mouse model of hepatic metastasis in the context of an adaptive immune response
The absence of expression of MHCI on DCCs and the presence of cancer-specific CD8+ T cells in the genetically engineered mice model of PDA (20) and possibly in patients with PDA (28) suggested that DCCs may be selected by an anti-cancer immune response during the metastatic process. Accordingly, we developed a mouse model of hepatic metastases that allowed us to assess the effect of a pre-existing immune response. The mM1 (mouse metastasis 1) cell line was derived from a spontaneous liver metastasis of a mouse bearing an autochthonous PDA, and was stably transfected with a transposon vector directing the expression of diphtheria toxin receptor (DTR), Herpes simplex thymidine kinase (HSV-TK), firefly luciferase, and mTagBFP2 to generate mM1DTLB cells. Syngeneic C57Bl/6 mice were injected subcutaneously with 106 mM1DTLB cells, tumors were grown for 14 days, and then eliminated by treatment with diphtheria toxin (DTx) and ganciclovir (GcV). These “pre-immunized” mice and naïve mice were challenged by intra-splenic injections of 106 mM1DTLB cells, followed by splenectomy, thereby seeding the liver via the portal vein, as in PDA (Fig. 2A).
Fig. 2. A mouse model for hepatic DCCs.
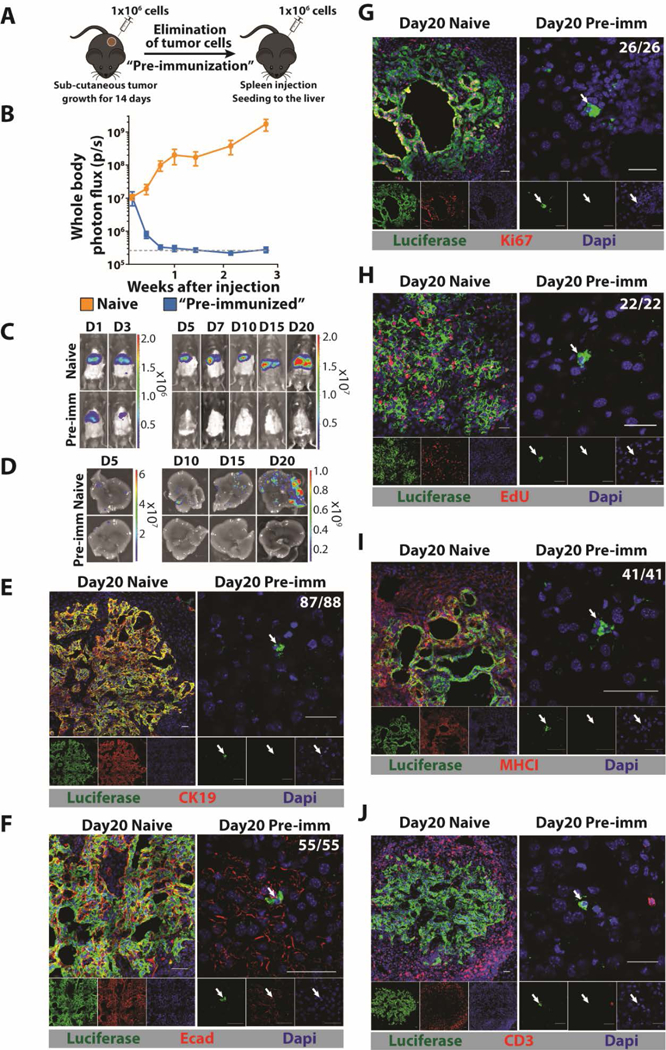
(A) Mice are pre-immunized by subcutaneous injection of 106 mM1DTLB PDA cells derived from a hepatic metastasis of a KPC mouse. After two weeks, tumors are eliminated by treating mice with DTx and GcV. For hepatic metastases, 106 mM1DTLB PDA cells are injected intra-splenically into naïve and pre-immunized mice, followed immediately by splenectomy. (B, C) Tumor growth was measured by whole body bioluminescence imaging. (D) Ex vivo photon flux of whole livers was measured at day 5, 10, 15 and 20 after cancer cell injection. Results are representative of three experiments with at least five mice per group. Dashed gray line represents the background luminescence in tumor-free mice. (E-J) IF of sections from a liver of a naive mouse (left panels) or a pre-immunized mouse (right panels) that have been stained with anti-luciferase (green) to identify cancer cells and (E) anti-CK19 (red), (F) anti-Ecad (red), (G) anti-Ki67 (red), (H) EdU (red), (I) anti-MHCI (red), and (J) CD3 (red). For EdU staining, mice were injected every 12 hours with EdU for three days. Photographs are representative of 20 mice from three independent experiments. The ratios shown in the top right corners of the photomicrographs represent the frequency of the observed DCC phenotype relative to the total number of DCCs that were assessed. All frequencies are compiled in table S2. White arrows designate DCCs. Scale bar = 25μm.
We found that in naïve mice, whole body bioluminescence increased from day 1 after injection, consistent with the growth of hepatic metastases. In pre-immunized mice, however, whole body bioluminescence decreased after day 1, and by day 7 luminescence was at background levels (Fig. 2B, C). In additional cohorts of naïve and pre-immunized mice, livers were removed at intervals after the intra-splenic injection of mM1DTLB cells, and assessed for bioluminescence. Both photon flux (Fig. 2B) and visually detectable metastases increased in the livers of naïve mice between days 5 and 20, while metastatic foci were barely detectable in pre-immunized mice at day 5, and were absent at later time points (Fig. 2D). A potential role for T cells in the elimination of mM1DTLB cancer cells was suggested by the finding that in the livers of pre-immunized mice, tumor cells were frequently surrounded by both CD8+ and CD8- CD3+ T cells by 24h (Fig. S2A). This possibility was confirmed by treating pre-immunized mice with depleting antibodies to CD4 and CD8, alone or together, or with isotype control antibody (Fig. S2B). Depleting either CD4+ or CD8+ T cells abrogated the ability of pre-immunization to suppress the development of macro-metastases (Fig. S2B), confirming this role of T cells.
Microscopic examination of livers from naïve and pre-immunized mice revealed the presence of macro-metastatic lesions in the former, but only single DCCs in the latter. The DCCs differed from cancer cells in the macro-metastases in the following ways: they did not express the epithelial markers CK19 and E-cadherin (Ecad), they did not express Ki67, they did not incorporate 5-ethynyl-2’-deoxyuridine (EdU), and they did not express MHCI (Fig. 2E-I). Importantly, the phenotype of the DCCs in this metastasis model is similar to that of DCCs in human PDA and the KPCY mouse. T cells in the vicinity of DCCs were infrequent, whereas they surrounded macro-metastatic lesions in naïve mice (Fig. 2J). The absence of CK19 and Ecad expression did not indicate an epithelial-mesenchymal transition (EMT) because DCCs did not express the EMT markers desmin, αSMA, Snail1, or Slug (Fig. S3).
When naïve and pre-immunized mice were challenged with mM1DTLB cells via the tail vein, lung macro-metastases developed only in the naïve mice, whereas CK19- DCCs were observed in the lungs of pre-immunized mice (Fig. S4). We also assessed the 1242 cell line derived from a primary PDA tumor of a KPC mouse (29), which had been modified with the same transposon vector, for its ability to generate DCCs. When naïve and pre-immunized mice were challenged by intra-splenic injection of 1242DTLB cells, hepatic macro-metastases developed in the naïve mice, whereas only CK19- DCCs were observed in the livers of pre-immunized mice (Fig. S5). To assess more stringently the proliferation of DCCs, we labelled the mM1DTLB cells with CFSE before intra-splenic injection into naïve or pre-immunized mice. Whereas all cancer cells in the macro-metastases of naïve mice had become CFSE-, DCCs in pre-immunized mice had retained CFSE (Fig. S6A). Mice that had received intra-splenic injections of mM1DTLB cells were also given EdU in the drinking water for 20 days. While cancer cells in macro-metastases in naïve mice had incorporated EdU, almost all DCCs in pre-immunized mice were EdU- (Fig. S6B) indicating the absence of proliferation. Therefore, the occurrence of quiescent, MHCI- DCCs in the absence of macro-metastases is a consequence of an ongoing cancer-specific immune response.
A latent capacity for outgrowth of DCCs is controlled by T cells.
We next examined whether a latent capacity of DCCs for outgrowth into macro-metastatic lesions might be revealed by T cell depletion. When depleting anti-CD4 and anti-CD8 antibodies were administered to pre-immunized mice three weeks after the establishment of DCCs, macro-metastases appeared in 10 of 15 mice. When T cells were depleted at nine weeks, macro-metastases appeared in 2 of 15 mice (Fig. 3). The metastases were composed of cancer cells that had re-expressed CK19 and MHCI, suggesting that DCCs revert to an epithelial phenotype to initiate the formation of macro-metastases. The lower frequency of macro-metastases in mice in which T cells were depleted at nine weeks suggests that DCCs with a capacity for reversion had decreased between three and nine weeks. Indeed we found that livers of pre-immunized mice have fewer DCCs at nine weeks than at three weeks (Fig. S7A). This loss of DCCs may reflect the killing by T cells of spontaneous revertants, as suggested by the occasional occurrence of a CK19+ DCC surrounded by T cells in the pre-immunized mice (Fig. S7B). The re-expression of MHCI by these growing metastases also suggests the means by which T cells control the outgrowth of spontaneously reverting DCCs. To demonstrate that T cells alone are both necessary and sufficient for controlling the growth of MHCI- DCCs, we depleted NK cells by administering anti-NK1.1 antibody at the time of mM1DTLB cell challenge. Pre-immunized mice lacking NK cells were indistinguishable, with respect to the occurrence of DCCs and absence of macro-metastases, from control antibody-treated mice (Fig. S8). This evidence for a dominant role of the T cell in controlling DCCs is supported by the finding that DCCs were never seen to be in contact with CD45+ (Fig. S9A), F4/80+ (Fig. S9B), CD19+ (Fig. S9C), CD31+ (Fig. S9D), αSMA+ (Fig. S9E) or Ly-6G+ (Fig. S9F) cells, the distribution of these cell types being similar to that in the normal liver (Fig. S9 G-N).
Fig. 3. T cells control outgrowth of latent DCCs.
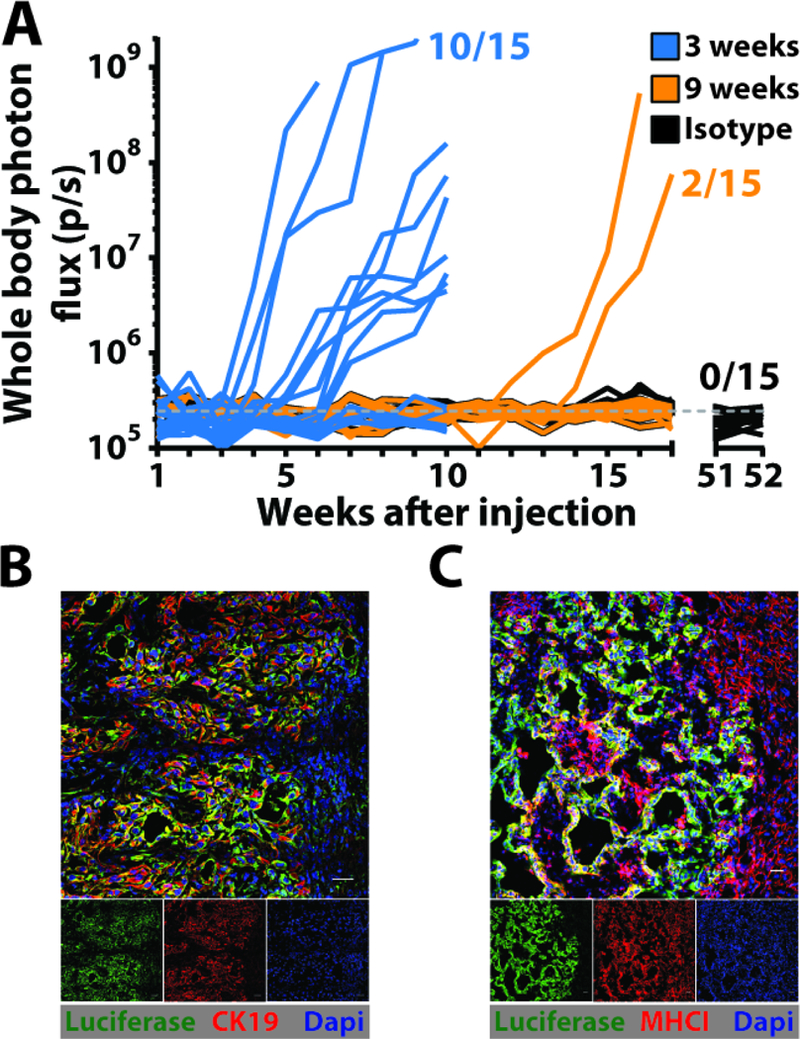
(A) The growth of hepatic metastases in pre-immunized mice that had been depleted of T cells by administration of antibodies to CD4 and CD8 beginning at three weeks or nine weeks after splenic injection of mM1DTLB PDA cells was assessed by bioluminescence imaging. One group of mice was also treated with isotype control antibody. (B and C) IF of sections containing macro-metastases from a liver of a pre-immunized mouse that had been depleted of T cells three weeks after splenic injection of mM1DTLB PDA cells. Anti-luciferase identifies cancer cells. Scale bar = 25μm.
A rare sub-population of PDA cells in vitro with the phenotype of DCCs
The absence of MHCI expression by DCCs raised the possibility that these cells were present in the injected PDA population and were negatively selected by T cells. Indeed, approximately 1% of the mM1DTLB cells in tissue culture were Ecad- and CK19-, and all Ecad- cells were MHCI- (Fig. 4A, B). The Ecad- cells resided in a non-proliferating sub-population of cells, as indicated by the resistance of these HSV-TK-expressing cells to GcV (Fig. 4C). A phenotypic plasticity of the mM1DTLB cells was shown by culturing FACS-purified Ecad+ and Ecad- cells for three days, and finding that they generated Ecad- and Ecad+ cells, respectively (Fig. 4D). The Ecad- MHCI- phenotype shared by PDA cells in vitro and the DCCs in vivo suggested that the former may be the precursors of the latter. We assessed this possibility by intra-splenically injecting 106 Ecad+ and 104 Ecad- cells, respectively, into naïve and pre-immunized mice. The growth of Ecad+ macro-metastases in the livers of the naïve mice receiving Ecad+ cells was similar to that of naïve mice receiving unsorted mM1DTLB cells (Fig. 4E). Microscopic examination of the livers of these mice, however, revealed no Ecad- DCCs (Fig. 4F). Injection of Ecad+ cells into pre-immunized mice resulted in a rapid decline in photon flux, with no specific signal by day 7 (Fig. 4E), and microscopic examination of these livers also demonstrated the absence of DCCs (Fig. 4F). Injection of Ecad- cells into naïve mice resulted in delayed development of hepatic macro-metastases (Fig. 4E), and microscopy revealed the presence of DCCs (Fig. 4F). Injection of Ecad- cells into pre-immunized mice led to DCCs but no macro-metastases confirming that an on-going T cell response controls DCC outgrowth (Fig. 4E, F). The macro-metastases found in naïve mice injected with Ecad- cells were CK19+ (Fig. 4G) and MHCI+ (Fig. 4H) indicating reversion to an epithelial phenotype. In summary, the origin of the DCC is the Ecad- cell, as DCCs were not present in naïve or pre-immunized mice following the injection of Ecad+ cells. Reversion from the quiescent, Ecad- state to the proliferating, Ecad+ phenotype is observed in naïve mice, but is masked by the ongoing T cell response in pre-immunized mice because reversion is associated with re-expression of MHCI. Therefore, the two states of mM1DTLB cells that are observed in vivo--the proliferating MHCI+/Ecad+/CK19+ macro-metastasis and the quiescent MHCI-/Ecad-/CK19- DCC--occur in vitro, reflecting a developmental plasticity that may be controlled by a cell-autonomous process.
Fig. 4. A subpopulation of PDA cells in vitro shares phenotypic features with DCCs.
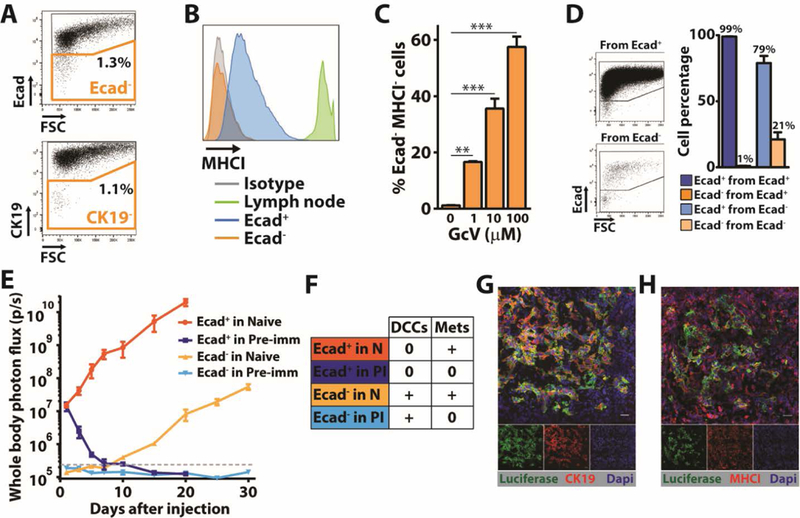
(A) Flow cytometry analysis of mM1DTLB PDA cells that have been stained with anti-CK19 or anti-Ecad. Results are representative of five independent experiments. (B) Flow cytometry measurement of anti-MHCI staining of Ecad+ and Ecad- mM1DTLB PDA cells, and of lymph node cells as a comparator. Results are representative of five independent experiments. (C) mM1DTLB PDA cells were treated in vitro for 48h with increasing doses of GcV to kill proliferating cells, and the proportion of viable cells that was Ecad-/MHCI- was measured by flow cytometry. Results are representative of two independent experiments. **=p<0.01, ***=p<0.001. (D) FACS analysis of purified Ecad+ and Ecad- mM1DTLB PDA cells, respectively, that have been cultured for three days. Dot plots (left panel) and histograms (right panel) are representative of three independent experiments. (E) Growth of hepatic metastases after intra-splenic injection of 106 Ecad+ or 104 Ecad- mM1DTLB PDA cells into naïve and pre-immunized mice was assessed by whole body bioluminescence imaging. n=5 mice per group. Dashed gray line represents the luminescence background in tumor-free mice. (F) Table summarizing the occurrence of DCCs and/or metastases in each group of mice thai had been injected with Ecad+ or Ecad- mM1DTLB cells. (G and H) IF of sections from a liver of a naïve mouse that had received an intra-splenic injection of Ecad- mM1DTLB cells. Anti-luciferase (green) identifies cancer cells. Photomicrographs are representative of five mice. Scale bar=25μm.
Unresolved ER stress characterizes PDA cells with the DCC phenotype
To identify the cell-autonomous “switch” regulating the developmental state of the metastases, we performed single-cell RNA sequencing (scRNA-seq) of in vitro sorted Ecad+ and Ecad- cells. The most upregulated pathway in Ecad- cells relative to Ecad+ cells is “Response to ER Stress” (Fig. 5A and S10C), and the most downregulated pathway is “Cell Division” (Fig. 5B and S10D). Network analysis of other upregulated pathways that distinguish the Ecad- and Ecad+ PDA populations, such as “Autophagy”, shows that they are linked to “Response to ER Stress”. Similarly, network analysis of other downregulated pathways demonstrates linkage to “Cell Division” (Fig. 5B). These two populations of the mM1DTLB PDA cells are also distinct by Principal Component Analysis (PCA), in which Ecad- cells appear to be more heterogeneous than Ecad+ cells (Fig. S10A). Indeed, analysis at the single-cell level identifies four sub-populations of Ecad- cells, in three of which the dominant upregulated pathway is related to “ER stress”, and the major downregulated pathway is “Cell Division” (Fig. S11). An EMT signature was not present among the 1639 genes that were differentially expressed between Ecad+ and Ecad- cells (Fig. S10B), confirming the immunofluorescence analysis of Ecad- cells (Fig. S6E). In addition, neither Ecad+ nor Ecad- cells express three of the four major NKG2D ligands, Ulbp1, H60b and H60c, and both strongly express the inhibitory ligand, Qa1, providing an explanation for the absence of a role for NK cells in controlling outgrowth of macro-metastases (Fig. S7C) (30). Genes that are involved in the processing of MHCI were not differentially expressed, which is consistent with reports that the ER stress response suppresses the expression of MHCI by a post-transcriptional mechanism (31, 32). Finally, support for the conclusion that the ER stress response alters the expression of MHCI and Ecad is the finding that treatment of mM1DTLB PDA cells with tunicamycin, an inducer of ER stress, increased the proportion of cells that were MHCI- and Ecad- (Fig. S12).
Fig. 5. The ER stress response in PDA cells that share phenotypic features of DCCs.
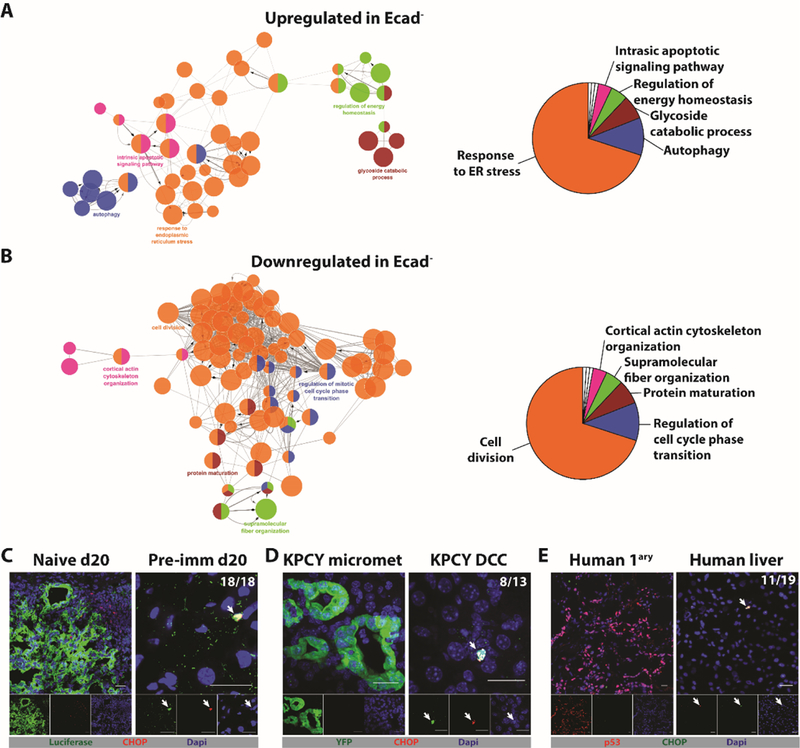
104 Ecad+ and 98 Ecad- mM1DTLB PDA cells were subjected to single-cell RNA-Seq. (A) Network analysis, following pathway enrichment analysis, shows ontology relationships between the pathways (left panel). Their relative representation is depicted as a pie chart (right panel) for (A) Upregulated pathways and (B) Downregulated pathways in the Ecad- cells relative to Ecad+ cells. Pathways are significant with an adjusted p<0.01 after Benjamini-Hochberg false discovery rate. (C) Liver sections from a naive mouse and a pre-immunized mouse were stained with anti-luciferase (green) to identify PDA cells and anti-CHOP (red) to identify cells exhibiting an ER stress response. (D) Liver sections from a KPCY mouse were stained with anti-YFP (green) to identify PDA cells and with anti-CHOP (red). (E) Sections from the primary tumor and liver of a patient with PDA were stained with anti-p53 to identify PDA cells (red) and with anti-CHOP (green). Photomicrographs are representative of five patients who had no detectable liver metastases. The ratios shown in the top right corners of the photomicrographs represent the frequency of the observed DCC phenotype relative to the total number of DCCs that were assessed. All frequencies are compiled in table S2. White arrows designate DCCs. Scale bar = 25μm.
The most differentially expressed gene was Ddit3/CHOP, the mRNA level of which was 18-fold higher in Ecad- cells than in Ecad+ cells. This gene encodes a transcription factor, C/EBP homologous protein (CHOP) that is part of the ER stress response. We examined the expression of CHOP protein by immunofluorescence in the Ecad- DCCs in human and mouse PDA. Anti-CHOP staining was demonstrated in hepatic DCCs in pre-immunized mice (Fig. 5C) and in KPCY mice (Fig. 5D), but not in PDA cells of macro-metastases of naïve mice or of micro-metastases of KPCY mice (Fig. 5E). Importantly, DCCs in the livers of 3/5 patients with PDA also stained with anti-CHOP antibody (Fig. 5E).
The higher expression level of CHOP in DCCs versus in growing PDA cells with an epithelial phenotype suggested that the response of DCCs to ER stress was not as effective as the response of growing CK19+/Ecad+/MHCI+ PDA cells. To restore protein homeostasis, cells undergoing ER stress activate the unfolded protein response (UPR) (33, 34)). CHOP is induced by the protein kinase RNA-like endoplasmic reticulum kinase (PERK) pathway of the UPR (33, 34), and, indeed, this pathway is activated in both Ecad+ and Ecad- cells, as shown by phosphorylation of PERK and EIF2α (Fig. S14A). In contrast, the Inositol-requiring kinase 1 (IRE1α) pathway is activated in Ecad+ cells but not in Ecad- cells, as shown by phosphorylation of IRE1α and splicing of the Xbox binding protein 1 (XBP1) transcription factor mRNA (Fig. S14A). Impaired activation of the IRE1α pathway in Ecad- cells was supported by scRNA-seq results showing decreased expression of XBP1 target genes relative to their expression levels in Ecad+ cells (Fig. S14B). The cancer cells in primary PDA tumors in patients and hepatic macro-metastases in mice also demonstrated phosphorylation of both EIF2α and IRE1α, but hepatic DCCs in patients and mice stained only with anti-pEIF2α (Fig. 6A-D). Thus, DCCs may not be able to resolve the ER stress response because they do not activate the IRE1α pathway, which is required to generate the spliced, active form of XBP1 (XBP1s) that regulate the transcription of multiple proteins that promote protein folding in the ER.
Fig. 6. Absence of IRE1α pathway activation in DCCs.
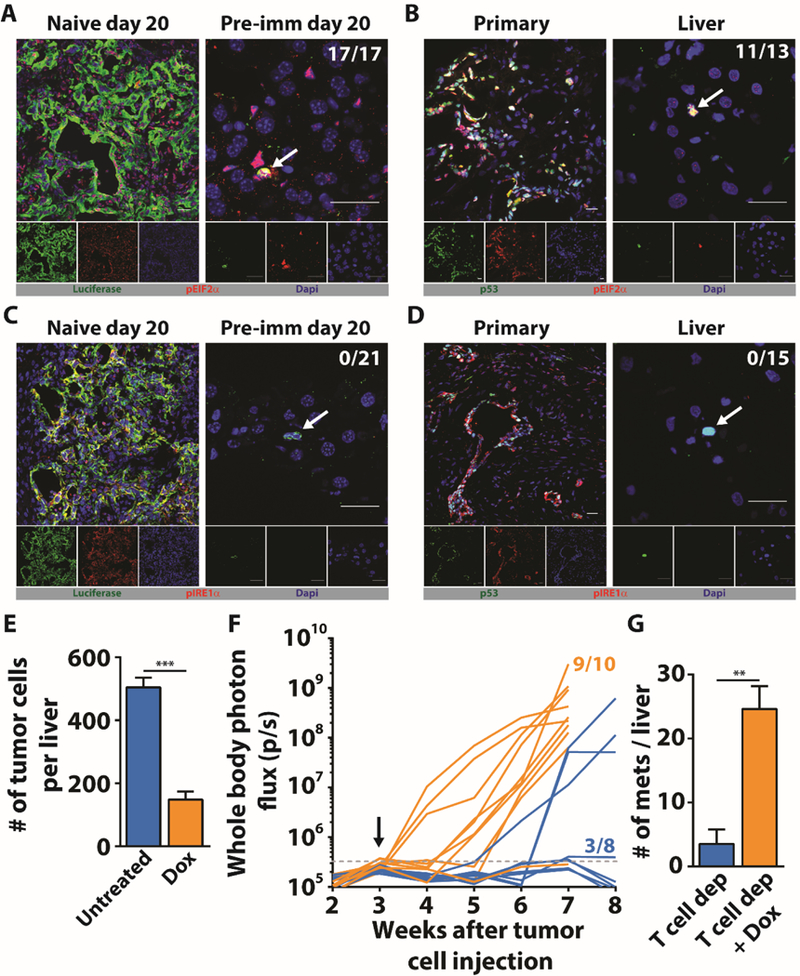
(A) Liver sections from naive and pre-immunized mice were stained with anti-luciferase (green) to identify PDA cells and anti-pEIF2α (red) to assess activation of the PERK pathway. (B) Sections from the primary tumor and liver of a patient with PDA were stained with anti-p53 to identify PDA cells (green) and anti-pEIF2α (red). (C) Liver sections from naive and pre-immunized mice were stained with anti-luciferase (green) to identify PDA cells and anti-pIRE1α (red). (D) Sections from the primary tumor and liver of a patient with PDA were stained with anti-p53 to identify PDA cells (green) and anti-pIRE1α (red). Photomicrographs are representative of five patients who had no detectable liver metastases. The ratios shown in the top right corners of the photomicrographs represent the frequency of the observed DCC phenotype relative to the total number of DCCs that were assessed. All frequencies are compiled in table S2. White arrows designate DCCs. Scale bar = 25μm. (E) Pre-immunized mice (n=5) that had received splenic injections of mM1TetXBP1s PDA cells were treated with Dox starting on the day of injection. The number of hepatic DCCs was determined three weeks later and compared to that of mice not receiving Dox (n=5). (F) Pre-immunized mice that had received splenic injections of mM1TetXBP1s PDA cells were treated (n=10) or not treated (n=8) with Dox beginning three weeks later. T cells were depleted by administering anti-CD4 and anti-CD8 antibodies. Growth of hepatic metastases was assessed by whole body bioluminescence imaging (G) The mean number of bioluminescent metastases formed with or without Dox treatment was determined in resected livers. ** = p<0.01, *** = p<0.001.
To determine a whether this unresolved ER stress of DCCs contributes to their phenotype, we treated mM1DTLB PDA cells with the chemical chaperone, 4-phenylbutyrate (4-PBA), which binds to solvent-exposed hydrophobic segments of unfolded or improperly folded proteins, thereby “protecting” them from aggregation and relieving ER stress (35). 4-PBA treatment decreased the proportion of cells lacking expression of Ecad (Fig. S15A), and increased MHCI expression in both Ecad+ and Ecad- cells (Fig. S15B). We also assessed the effect of 4-PBA on the proliferative capability of mM1DTLB PDA cells by first eliminating proliferating mM1DTLB PDA cells by treatment with GcV, and then pulsing the residual quiescent cells with EdU overnight in the presence or absence of 4-PBA. Relieving ER stress with 4-PBA increased the proportion of cells incorporating EdU by 10-fold (Fig. S15C). We extended the analysis to DCCs by administering 4-PBA to pre-immunized mice, beginning on the day of the mM1DTLB cell injection and continuing for three weeks. This treatment decreased the number of hepatic DCCs by 4-fold relative to the number in control mice (Fig. S15D), consistent with the possibility that promoting the conversion of DCCs to replicating, MHCI+ cells leads to their T cell-mediated immune elimination. To confirm this interpretation, we began 4-PBA treatment of pre-immunized mice three weeks after the intra-splenic injection of mM1DTLB cells and depleted the mice of T cells. Within two weeks of 4-PBA treatment, all mice had developed MHCI+ macro-metastases (Fig. S15E, F), and the number of macro-metastatic lesions in the livers of 4-PBA-treated, T cell-depleted mice was 6-fold higher than the number in the livers of mice subjected only to depletion of T cells (Fig. S15E).
We also assessed the role specifically of the IRE1α pathway in generating PDA cells with the DCC phenotype. In a loss-of-function experiment, mM1DTLB cells were treated with the IRE1α inhibitor, Kira6 (36), which converted the mM1DTLB cells from an Ecad+, MHCI+ state to an Ecad-, MHCI- phenotype in a dose responsive manner (Fig. S14C). We then performed gain-of-function experiments with the mM1TetXBP1s cell line expressing a doxycycline (Dox) inducible form of XBP1s. This allowed us to test the hypothesis that circumventing the defect in IRE1α activation by expression of the spliced form of XBP1 would resolve ER stress in a more specific and physiological manner than was achieved with 4-PBA (Fig. S13). Overnight treatment of mM1TetXBP1s cells with Dox induced the expression of XBP1s and almost eliminated the Ecad-, MHCI- sub-population (Fig. S14D). To examine the role of the IRE1α pathway in DCCs in vivo, pre-immunized mice were continuously treated with Dox beginning on the day that mM1TetXBP1s cells were injected intra-splenically. After three weeks, the frequency of hepatic DCCs was significantly decreased relative to their frequency in control mice not treated with Dox (Fig. 6E). The possibility that this decrease in XBP1s-expressing hepatic DCCs indicated killing by immune T cells was confirmed by repeating the experiment with the additional intervention of T cell-depletion. Within three weeks of Dox-induced XBP1s expression in the DCCs, 9 of 10 T cell-deficient mice had developed hepatic macro-metastases whereas only 1 of 8 of the T cell-depleted mice without Dox treatment developed these lesions (Fig. 6F). Furthermore, the number of macro-metastatic lesions in the livers of Dox-treated, T cell-depleted mice was 7-fold higher than in the livers of mice subjected only to depletion of T cells (Fig. 6G). These results lead us to conclude that unresolved ER stress has a non-redundant, cell-autonomous role in the maintenance of quiescent, immune-resistant DCCs.
Discussion
This clinical observation that PDA metastases develop in the majority of patients following the surgical removal of their primary tumors, despite no evidence of metastases at the time of surgery, indicates that these patients had harbored latent metastatic lesions. The nature of these latent metastases was suggested by our finding of single DCCs in the livers of patients and KPCY mice with PDA that have a distinctive phenotype of absent CK19, Ecad, and MHCI. The absence of two typical markers of epithelial ductal adenocarcinoma cells, without the occurrence of characteristic markers of EMT, indicates that these DCCs are distinct from the PDA cells that comprise growing macro-metastases (37). Moreover, the absence of MHCI implies an unusual relationship of DCCs to the adaptive immune system (38), as previously suggested studies of in breast, stomach and colon carcinoma (12, 39). These descriptive findings provided the rationale for developing a mouse model that replicates DCCs with this distinctive phenotype and allows mechanistic studies defining both the cell-autonomous response responsible for the phenotype and the role of the immune system.
We hypothesized that metastases in patients with PDA may occur in the context of a cancer-specific adaptive immune response (20–23). The absence of MHCI expression at the surface of DCCs in human and mouse PDA, and the absent or relatively infrequent occurrence of hepatic macro-metastases raised the possibility that immunity prevents the outgrowth of macro-metastases while ignoring MHCI- DCCs. This prediction was based also on the observation that DCCs in human and mouse PDA, in contrast to cancer cells in macro-metastases, were quiescent. The finding that pre-existing immunity prevented the occurrence of hepatic macro-metastases while permitting the seeding of non-replicating, MHCI- hepatic DCCs verified this prediction, and provided a potential explanation of how quiescent metastases can persist in the presence of an adaptive immune response that is capable of suppressing the growth of macro-metastases.
This selective effect of adaptive immunity on macro-metastases required that the cell-autonomous mechanism that is responsible for the phenotype of DCCs invariably links the expression of MHCI to a capacity for cellular replication. This association was not only demonstrated in additional cell lines from primary and metastatic PDA tumors from KPC mice, but also was supported by the finding that ER stress, which inhibits MHCI expression (31, 32) and cell division (40) were the major up-regulated and down-regulated transcriptional signatures that distinguish Ecad- cells from Ecad+ PDA cells. Thus, MHCI expression is “off” in quiescent cells and “on” in replicating cells. In addition, if the markedly increased expression in Ecad- PDA cells of CHOP, a transcription factor that is induced by the PERK pathway of the UPR (33, 34), is taken as an indicator of the ER stress response, then the finding that in human and mouse PDA, hepatic DCCs are MHCI-, Ki67- and CHOP+ links quiescence and immune concealment to an ongoing, unresolved ER stress in vivo. Our results confirm the previous suggestion that sustained activation of the UPR allows persistence of DCCs in the bone marrow of breast cancer patients (41), and may be related to the recent observation that knock-down of IRE1α inhibits tumor growth (42). We showed that resolution of ER stress by a chemical chaperone or, more definitively, by the induced expression of XBP1s cause reversion of DCCs to an epithelial, proliferating, MHCI+ phenotype. Thus the DCC phenotype was caused by unresolved ER stress secondary to a block in IRE1α activation. Our study does not address how PDA cells avoid activating IRE1α, but other studies suggest that under high ER stress, IRE1α acquires endonucleolytic activity against RNA targets in addition to the mRNA encoding XBP1 in a reaction termed, regulated IRE1-dependent decay of mRNA (RIDD) (43). These additional endonucleolytic events may be associated with apoptosis, so that the ability of the PDA cell to adopt the DCC phenotype may not only protect it from death caused by immunity, but also death by the RIDD reaction.
A latent capacity of hepatic DCCs for reversion to growing cancer cells was revealed by the outgrowth of macro-metastases following the depletion of T cells. This finding also implied that T cells eliminated those DCCs spontaneously reverting to a replicating, MHCI+, epithelial phenotype. T cell killing of reverting DCCs must be efficient and occur before an immune suppressive microenvironment is established since no macro-metastases were observed during a 12 month period of observation of T cell-replete mice with hepatic DCCs. The causal link between reversal of ER stress in the DCC to a replicating, MHCI+, epithelial phenotype was established by the use of the chemical chaperone, 4-PBA, and by the induced expression of XBP1s in DCCs. Relief of ER stress by each intervention enhanced the expression of Ecad and MHCI and the proliferation by DCCs in vitro, and, more importantly, caused the outgrowth of macro-metastases from DCCs in T cell-depleted mice. This observation, coupled with the capacity of 4-PBA and XBP1s to decrease hepatic DCCs in T cell-replete mice, supports the essential role of unresolved ER stress response in maintaining the DCC phenotype. Other factors, such as CXCR2 (C-X-C chemokine receptor type 2)-expressing neutrophils, may also have a role in this response (44).
The implications for therapy to prevent the occurrence of metastatic disease in patients following the surgical removal of their primary PDAs may be two-fold. First, outgrowth of latent DCCs in the mouse model requires suppression of T cell immunity. Elevations of plasma cortisol following pancreatectomy in patients with PDA (45) are in the range that was found to be immune suppressive in cachectic mice with PDA (46). This stimulation of the hypothalamic-pituitary-adrenal axis may also occur with the caloric deprivation that commonly occurs in patients after this surgical procedure (47). Therefore, post-operative parenteral hyper-alimentation may be an effective means to decrease the occurrence of metastatic disease in patients following surgical removal of their primary PDAs. Second, and more speculative, the administration of a chemical chaperone, like 4-PBA, pre-operatively when tumor immunity is intact might purge organs of latent DCCs, thereby decreasing the likelihood of post-operative metastatic disease.
Material and Methods
Animals
Male C57Bl/6 mice 10 to 12 weeks of age purchased from The Jackson Laboratory were used. All procedures were approved by the Cold Spring Harbor Laboratory Institutional Animal Care and Use Committee (IACUC) and were conducted in accordance with the NIH “Guide for the Care and Use of Laboratory Animals”.
Cell culture
Cell lines used in that study were a gift of Dr. David Tuveson (Cold Spring Harbor Laboratory). The metastatic cell line mM1 and the primary cells line 1242 were derived from a KPC mice liver metastasis and a primary tumor, respectively. Cells were cultured in DMEM medium (#10–013-CV, Cellgro) supplemented with 10% fetal calf serum (FCS) (#1500–500, Seradigm), 100 units/mL penicillin, and 100 μg/mL streptomycin.
Human samples
Postmortem tissues were obtained following the Iacobuzio-Donahue laboratory rapid autopsy program previously described in detail (48). Patients were selected based on two criteria: p53 loss-of-heterozygosity, which allowed the staining for accumulation of mutant p53 protein, and absence of clinically detectable liver metastases.
Cell culture reagents
Ganciclovir (GcV, Sigma-Aldrich #G2536), was used at concentration ranging from 1–100mM. Tunicamycin (#11445, Cayman chemicals) was used at 5μg/ml. Sodium 4-Phenylbutyrate (4-PBA, #11323, Cayman chemicals) was used at 5mM. 5-Ethynyl-2’-deoxyuridine (EdU, #sc-284628, Santa Cruz biotechnology) was used at 10mM.
EdU labelling
For short term EdU pulse, mice were injected every 12 hours with 1.4 mg of EdU over a 3-day period. For long term experiments, EdU was given in the drinking water at 0.82 mg/ml + 2.5% sucrose. Revelation of EdU containing cells was performed with a kit (Click-iT™ Plus EdU Imaging Kit, Molecular probes, #C10640) following the manufacturer protocol.
CFSE labelling
Cells were harvested from the cell culture and CFSE (CellTrace™ CFSE Cell Proliferation Kit, Molecular probes, #C34554) was used according to the manufacturer protocol.
Immune cells depletion
200μg of anti-CD4 (clone GK1.5, BioXcell) and/or anti-CD8 (clone 53.6–7, BioXcell) or anti-NK1.1 (clone PK136, BioXcell) were injected intraperitoneally 2 times a week.
Antibodies for immunofluorescence and flow cytometry
| Antibody | Clone or Cat. # | Provider | Dilution |
|---|---|---|---|
| GFP | D5.1 | Cell signaling technology | 1/100 |
| CK19 (mouse and human) | EP1580Y | Abcam | 1/300–1/1000 |
| Ki67 (mouse) | #NB500–170 | Novus | 1/100 |
| Ki67 (human) | #ab15580 | Abcam | 1/800 |
| MHCI | M¼2 | Biolegend | 1/100 |
| CD3 | 17A2 | Biolegend | 1/100 |
| CD8 | 53.6–7 | Biolegend | 1/50 |
| E-cadherin | Decma-1 | Biolegend | 1/200 |
| Firefly Luciferase | #200–101-C44 | Rockland | 1/200 |
| CHOP | 9C4 | Novus | 1/100 |
| Desmin | #ab15200 | Abcam | 1/200 |
| aSMA | 1A4 | Abcam | 1/200 |
| Snail1 | C15D3 | Cell signaling technology | 1/50 |
| Slug | C1967 | Cell signaling technology | 1/50 |
| NKp46 | 29A1.4 | Biolegend | 1/100 |
| CD45 | 30-F11 | Novus | 1/100 |
| F4/80 | BM8 | Biolegend | 1/100 |
| CD19 | 1D3 | Biolegend | 1/100 |
| CD31 | 390 | Molecular probes | 1/50 |
| p53 (human) | DO1 | Invitrogen | 1/100 |
| MHCI (human) | HCA2 | Acris | 1/100 |
| Donkey anti-Goat | #A-11055 | Life technologies | 1/500 |
| Donkey anti-Rabbit | #A-31573 | Life technologies | 1/500 |
| Donkey anti-Rat | #ab175475 | Abcam | 1/500 |
| Goat anti-mouse | #A21424 or A21235 | Invitrogen | 1/600 |
| pIRE1α (mouse and human) | #NB100–2323 | Novus biologicals | 1/100 |
| pEIF2α (mouse and human) | #ab84980 | Abcam | 1/100 |
| XBP1s | #83418 | Cell signaling technology | 1/1000 |
| pPERK | #MA5–15033 | ThermoFisher | 1/100 |
Fluorochrome conjugation
For some applications (Ki67 staining in Fig. 1 and CHOP staining in Fig. 5), the anti-p53 antibody was conjugated using Zenon labeling reagent 555 (Life Sciences, # Z25005) following the manufacturer’s protocol.
Chemical chaperone treatment
4-PBA was given to mice in drinking water at 1g/kg/day.
Doxycycline treatment
Dox was given to mice in drinking water at 2mg/ml + 5% sucrose
Plasmid preparation and transfection
For the generation of mM1DTLB PDA cells: Sequences of the genes of diphtheria toxin receptor (D), HSV-TK (T), firefly luciferase (L) and mTagBFP2 (B) were generated by gene synthesis (Invitrogen GeneArt) with appropriate restriction sites. For HSV-TK and firefly luciferase, sequences were digested and migrated with agarose gel electrophoresis. Appropriate size bands were purified with gel extraction kit (Qiagen, #28706) and cloned into the piggyback transposon vector (PB-EF1-MCS-IRES-Neo cDNA Cloning and Expression Vector, System Bioscience #PB533A-2). For mTagBFP2, the sequence was amplified by PCR and cloned into the linearized piggyback vector already containing HSV-TK and firefly luciferase with Gibson assembly master mix (New England Biolabs #E2611L). Plasmids were purified using Zippy plasmid miniprep kit (Zymo research, #D4037). Cells were transfected with the resulting vector and the plasmid of the super piggyback transposase (System Bioscience, #PB210PA-1) with Lipofectamine 3000 (Thermofisher, #L3000015). Cells were selected for stable transfectant with G418 (Sigma-Aldrich #A1720) and sorted for the highest expression of mTagBFP2(Fig. S1A). The functionality of Luciferase, DTR and HSV-TK was verified in vitro (Fig. S1B and C).
For the generation of mM1TetXBP1s PDA cells: The sequence of spliced XBP1 (XBP1s) and mCherry linked by a 2A peptide sequence was generated by gene synthesis (Genscript) with appropriate restriction sites. The sequence was digested and migrated with agarose gel electrophoresis. Appropriate size bands were purified with gel extraction kit (Qiagen, #28706) and cloned into an inducible TetON plasmid (gift of Dr. Scott Lyons). Plasmids were purified using Zippy plasmid miniprep kit (Zymo research, #D4037). mM1DTLB PDA cells were transfected with the resulting vector with Lipofectamine 3000 (Thermofisher, #L3000015). Cells were selected for stable transfectant with puromycin (Sigma-Aldrich #P8833) and sorted first for no mCherry expression without doxycycline (Dox) to remove cells with potentially leaking expression and then for the highest expression of mCherry upon Dox treatment to select inducible cells (Fig. S13A). Tight inducible over-expression of XBP1s was confirmed in vitro by western blot analysis (Fig. S13B).
Generation of pre-immunized mice
Mice were anesthetized by isoflurane and dorsal fur was removed with a clipper. A sub-cutaneous injection of 106 PDA cells was performed. Tumors were grown for 2 weeks. In order to eliminate the tumors, diphtheria toxin (DTx, List Biologicals, #150) was injected intraperitoneally on two consecutive days at a dose of 25ng/g. GcV was injected intraperitoneally on two consecutive days at a dose of 0.3mg/g. In approximately 20% of the cases, one round of DTx and GcV injection was not sufficient to eliminate the tumors, and a second round of injection was performed one week later. On rare instances two rounds of injections were insufficient, and these animals were not used. The absence of remaining cancer cells was confirmed by bioluminescence (Fig. S1D) and mice were used for experiments at least three weeks after the last injection of DTx and GcV.
Pancreatic cancer metastasis model
For the liver metastasis model, animals were anesthetized by isoflurane, a 1 cm incision was made in the left subcostal region and the spleen was exposed. A suspension of 106 pancreatic cancer cells in 50 μl of PBS was injected into the body of the spleen. Immediately following injection, mice were splenectomized to prevent growing of extra-hepatic tumors. The peritoneum was closed with a 5–0 absorbable surgical suture (Vicryl, Ethicon) and the skin with wound clips (Roboz surgical instruments). For the lung metastasis model 106 cells in 100 μl were injected through the tail vein.
Bioluminescence imaging
Mice were anesthetized by isoflurane and ventral fur was removed with a clipper. 150 μl of a 30mg/ml D-Luciferin K+ salt (Perkin-Elmer, #122799) solution was injected intraperitoneally. Mice were imaged with an IVIS Spectrum in Vivo Imaging System (Perkin-Elmer) 14 min after the injection.
Immunofluorescence
Organs were harvested and fixed with PLP buffer (1% PFA, 80mM L-lysin, 10mM NaIO4); through portal vein perfusion in the case of livers. Organs were embedded in Tissue-Tek OCT compound (Sakura, #4583) and section of 10 μm were cut on a Leica cryostat. Between each of the following steps, sections were washed three times for five min with PBS. Tissue sections were post-fixed for 15min at room temperature with PLP buffer and permeabilized for 15min at room temperature with 0.1% Triton X100 in PBS for mouse tissues or 30min at room temperature with 0.2% Triton X100 in PBS for human tissues. Sections were surrounded with hydrophobic barrier pen (ImmEdge, Vector labs, #H-4000) and unspecific antibody labelling was blocked for 1hr at room temperature with 10% donkey serum (Jackson immunoresearch, #017–000-121) in PBS for mouse tissues or 10% goat serum (Thermo Fisher, #16210–064) in PBS for human tissues. Primary antibodies were incubated overnight at 4 °C in the dark. Secondary antibodies were incubated for 2hr and room temperature in the dark. Nuclei were counterstain with dapi (Molecular probes, #R37606) for 10 min at room temperature in the dark and sections were mounted with ProLong diamond antifade mountant (Molecular probes, #P36965) for mouse tissues or ProLong gold antifade mountant (Molecular probes, #P36934). Images were acquired using a spinning disk confocal microscope (Perkin-Elmer UltraVIEW VoX, High speed spinning disk Yokogawa® CSU-X1) with 20X or 60X objectives. Images were analyzed with ImageJ. CFSE (CellTrace™ CFSE Cell Proliferation Kit, Molecular probes, #C34554) and EdU (Click-iT™ Plus EdU Imaging Kit, Molecular probes, #C10640) staining were performed following the manufacturer protocol.
Flow cytometry analysis and sorting
Cells were harvested with TrypLE (Gibco, #12605036), distributed in 96-well round-bottom plate and centrifuged at 2000 rpm for 1 min at 4 °C. Supernatant was removed and the cells incubated in ice cold FACS Buffer (1% FBS and 0.02% Sodim Azide in PBS) with Fc Block (Biolegend clone 93) at 4 °C for 15 min. After a wash in FACS Buffer, the cells were incubated with primary and secondary antibodies for 15 min at 4 °C in the dark. The cells were then washed twice in FACS Buffer and resuspended in 450 ml PBS before analysis using a LSR Fortessa (BD Biosciences) operated by a FACSDIVA (BD Biosciences) software. Data analysis was performed on FlowJo 10 (FlowJo, LLC). For sorting, cells were submitted to the same procedure as for flow cytometry analysis and processed using BD FACSAria (BD Biosciences).
Disseminated cancer cell count
Sections were taken every 100μm through the entire thickness of the liver. The thickness of the liver was measured at the same time. DCCs were identified by luciferase expression and counted in every section. The number of DCCs per whole liver was then calculated.
scRNAseq
scRNA libraries were generated from viable Ecad+ and Ecad- tumor cells obtained by flow cytometry sorting. PDA cells were adjusted to 200 cells/μL and applied to the C1 system for single-cell capture with a 10–17μm IFC (Fluidigm). In the C1, whole-transcriptome amplification was performed with the SMARTer kit (Clontech), and the product was converted to Illumina sequencing libraries using Nextera XT (Illumina). RNA-seq was performed on a NextSeq instrument (Illumina) single read 75. Quality control was performed using FastQC and cells with low read numbers were eliminated. Reads were aligned against reference mouse genome (EnsMart72) and transcript expression values were determined after transcript normalization (transcript per million; TPM) with AltAnalyze. Cells with aberrant expression for Gapdh, Actb and Hprt were eliminated leaving 104 Ecad+ and 98 Ecad- cells for further analysis. Transcripts were considered significantly expressed if TPM ≥ 1. Differential expression analysis was performed considering fold-change between Ecad+ and Ecad- cells ≥ 2 fold as a cut-off. Pathway enrichment analysis and network maps were performed with Cytoscape v3.5.1 and the ClueGo v2.3.3 plugin. Pathways with p<0.01 after Benjamini-Hochberg procedure for false discovery rate were considered significant.
Statistical analysis
Two column comparisons were performed with an unpaired t test. Comparison of three or more columns was analyzed using a one-way ANOVA followed by Tukey’s procedure. A p-value less than 0.05 was considered significant, ***p<0.001, **p<0.01 and *p<0.05. All statistical analyses were performed using GraphPad Prism software version 6.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. Tuveson for providing mM1 and 1242 cells as well as the KPCY livers tissues. We thank Rajya Kappagantula for her help in processing patients’ information.
Funding: This work was supported by a Distinguished Scholar Award to D.T.F from the Lustgarten Foundation, an award from the Cedar Hill Foundation, and 5P30CA45508–29, NIH-NCI. A.P. was supported by the PHilippe Foundation.
Footnotes
Competing interests: D.T. F. is a Co-Founder of Myosotis LLC (a company developing cancer immunotherapies) and is on the Scientific Advisory Boards of iTEOS Therapeutics (a company developing immuno-oncology drugs), IFM Therapeutics, LLC (a company developing therapies targeting the innate immune system), and Kymab (a company developing therapeutic antibodies).
Data availability: scRNA sequencing data were deposited on GEO with the accession number GSE108811.
References
- 1.Zhang Q et al. , Pancreatic Cancer Epidemiology, Detection, and Management. Gastroenterol Res Pract 2016, 8962321 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hariharan D, Saied A, Kocher HM, Analysis of mortality rates for pancreatic cancer across the world. HPB (Oxford) 10, 58 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groot VP et al. , Patterns, Timing, and Predictors of Recurrence Following Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Ann Surg, (March 23, 2017). [DOI] [PubMed] [Google Scholar]
- 4.Van den Broeck A et al. , Patterns of recurrence after curative resection of pancreatic ductal adenocarcinoma. Eur J Surg Oncol 35, 600 (June, 2009). [DOI] [PubMed] [Google Scholar]
- 5.Kim R et al. , PET/CT Fusion Scan Prevents Futile Laparotomy in Early Stage Pancreatic Cancer. Clin Nucl Med 40, e501 (November, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koebel CM et al. , Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903 (December 06, 2007). [DOI] [PubMed] [Google Scholar]
- 7.Eyles J et al. , Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J Clin Invest 120, 2030 (June, 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller M et al. , EblacZ tumor dormancy in bone marrow and lymph nodes: active control of proliferating tumor cells by CD8+ immune T cells. Cancer Res 58, 5439 (December 01, 1998). [PubMed] [Google Scholar]
- 9.Kang Y, Pantel K, Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573 (May 13, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein CA, Selection and adaptation during metastatic cancer progression. Nature 501, 365 (September 19, 2013). [DOI] [PubMed] [Google Scholar]
- 11.Sosa MS, Bragado P, Aguirre-Ghiso JA, Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14, 611 (September, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pantel K et al. , Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res 51, 4712 (September 01, 1991). [PubMed] [Google Scholar]
- 13.Mohme M, Riethdorf S, Pantel K, Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol 14, 155 (March, 2017). [DOI] [PubMed] [Google Scholar]
- 14.Massague J, Obenauf AC, Metastatic colonization by circulating tumour cells. Nature 529, 298 (January 21, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malladi S et al. , Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 165, 45 (March 24, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller-Hermelink N et al. , TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 13, 507 (June, 2008). [DOI] [PubMed] [Google Scholar]
- 17.Eyob H et al. , Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov 3, 751 (July, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strauss DC, Thomas JM, Transmission of donor melanoma by organ transplantation. Lancet Oncol 11, 790 (August, 2010). [DOI] [PubMed] [Google Scholar]
- 19.Martin DC, Rubini M, Rosen VJ, Cadaveric Renal Homotransplantation with Inadvertent Transplantation of Carcinoma. JAMA 192, 752 (May 31, 1965). [DOI] [PubMed] [Google Scholar]
- 20.Feig C et al. , Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212 (December 10, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poschke I et al. , Identification of a tumor-reactive T-cell repertoire in the immune infiltrate of patients with resectable pancreatic ductal adenocarcinoma. Oncoimmunology 5, e1240859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Connor AA et al. , Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol 3, 774 (June 01, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balli D, Rech AJ, Stanger BZ, Vonderheide RH, Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin Cancer Res 23, 3129 (June 15, 2017). [DOI] [PubMed] [Google Scholar]
- 24.Maitra A et al. , Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol 16, 902 (September, 2003). [DOI] [PubMed] [Google Scholar]
- 25.Hingorani SR et al. , Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469 (May, 2005). [DOI] [PubMed] [Google Scholar]
- 26.Rhim AD et al. , Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735 (June 16, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aiello NM et al. , Metastatic progression is associated with dynamic changes in the local microenvironment. Nat Commun 7, 12819 (September 15, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR, T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res 5, 978 (November, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boj SF et al. , Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324 (January 15, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fang L et al. , MICA/B expression is inhibited by unfolded protein response and associated with poor prognosis in human hepatocellular carcinoma. J Exp Clin Cancer Res 33, 76 (September 18, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granados DP et al. , ER stress affects processing of MHC class I-associated peptides. BMC Immunol 10, 10 (February 16, 2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Almeida SF, Fleming JV, Azevedo JE, Carmo-Fonseca M, de Sousa M, Stimulation of an unfolded protein response impairs MHC class I expression. J Immunol 178, 3612 (March 15, 2007). [DOI] [PubMed] [Google Scholar]
- 33.Clarke HJ, Chambers JE, Liniker E, Marciniak SJ, Endoplasmic reticulum stress in malignancy. Cancer Cell 25, 563 (May 12, 2014). [DOI] [PubMed] [Google Scholar]
- 34.Vanacker H et al. , Emerging Role of the Unfolded Protein Response in Tumor Immunosurveillance. Trends Cancer 3, 491 (July, 2017). [DOI] [PubMed] [Google Scholar]
- 35.Cuadrado-Tejedor M, Garcia-Osta A, Ricobaraza A, Oyarzabal J, Franco R, Defining the mechanism of action of 4-phenylbutyrate to develop a small-molecule-based therapy for Alzheimer’s disease. Curr Med Chem 18, 5545 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Ghosh R et al. , Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534 (July 31, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aiello NM et al. , Upholding a role for EMT in pancreatic cancer metastasis. Nature 547, E7 (July 05, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T, The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 39, 44 (April, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pantel K et al. , Differential expression of proliferation-associated molecules in individual micrometastatic carcinoma cells. J Natl Cancer Inst 85, 1419 (September 1, 1993). [DOI] [PubMed] [Google Scholar]
- 40.Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA, Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv Exp Med Biol 734, 73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartkowiak K et al. , Disseminated Tumor Cells Persist in the Bone Marrow of Breast Cancer Patients through Sustained Activation of the Unfolded Protein Response. Cancer Res 75, 5367 (December 15, 2015). [DOI] [PubMed] [Google Scholar]
- 42.Li XX et al. , Knockdown of IRE1alpha inhibits colonic tumorigenesis through decreasing beta-catenin and IRE1alpha targeting suppresses colon cancer cells. Oncogene 36, 6738 (November 30, 2017). [DOI] [PubMed] [Google Scholar]
- 43.Hollien J, Weissman JS, Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104 (July 7, 2006). [DOI] [PubMed] [Google Scholar]
- 44.Steele CW et al. , CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 29, 832 (June 13, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naito Y et al. , Responses of plasma adrenocorticotropic hormone, cortisol, and cytokines during and after upper abdominal surgery. Anesthesiology 77, 426 (September, 1992). [DOI] [PubMed] [Google Scholar]
- 46.Flint TR et al. , Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab 24, 672 (November 08, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keim V, Klar E, Poll M, Schoenberg MH, Postoperative care following pancreatic surgery: surveillance and treatment. Dtsch Arztebl Int 106, 789 (November, 2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Embuscado EE et al. , Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther 4, 548 (May, 2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.