

Abstract
OBJECTIVE:
Understanding the role of endophenotypes is essential for process models of psychopathology. This study examined which candidate cognitive endophenotypes statistically mediate common variant genetic risk for ADHD.
METHOD:
A case-control design using community-recruited volunteer children 7–11 years old (n=656, n=435 ADHD) of which 514 were homogenous European ancestry for the primary models (n=337 ADHD, 177 non-ADHD). Children were assessed with a multi-informant, best-estimate diagnostic procedure and laboratory measures of working memory, response inhibition, executive functioning, arousal/attention, temporal information processing, and processing speed. Latent variables were created for the candidate cognitive measures and for parent and teacher-rated ADHD dimensions. Polygenic risk scores (PGS) were computed, using a discovery sample of 20,183 ADHD cases and 35,191 controls from the Psychiatric Genetics Consortium. Cognitive measures that survived multiple testing correction for association with the PGS were evaluated for mediation with ADHD using structural equation models.
RESULTS:
Results were essentially identical in the homogeneous European ancestry subgroup (n=514) and in the full sample (n=656). For the European population, the PGS was associated with ADHD diagnosis (Nagelkerke R2= .045; beta=.233, SE=.053, p=.000011) and multi-indicator dimensional ADHD latent variables by parent report (beta=.185, SE=.043) and teacher report (beta=.165, SE=.042). The PGS effect was statistically mediated by working memory (indirect effect, beta=.101, SE=.029, 95% CI=.05, .16, p=.00049, 43% of genetic effect accounted for) and arousal/alertness (indirect effect beta=.115, 95% CI=.04, .20, SE=.041, p=.005, 49% of genetic effect accounted for).
CONCLUSION:
This is the first clear demonstration from molecular genetic data that working memory and arousal regulation are promising cognitive endophenotypes for ADHD with regard to mediating genetic risk from common genetic variants.
INTRODUCTION
Attention Deficit Hyperactivity Disorder (ADHD) is associated with atypical cognitive functions. It remains unclear whether cognitive functions are directly related to the underlying pathophysiology of ADHD - an idea captured in the literature on endophenotypes, which are biological, cognitive, or other quantitative, reliable measures that fill gaps in the causal chain from genes to disorder. Endophenotypes are a crucial piece of a comprehensive strategy to map psychopathology and its causal processes.1–4 Cognitive endophenotypes, widely considered to be important to examine for multiple disorders, are expected to be non-specific yet informative clues to etiological process.3
Previous theory and data suggest several mediating cognitive functions for ADHD;5,6 we examined five. The first two are executive functions (EF) that have historically been the most reliably related to ADHD - working memory6 and response inhibition.5 These domains, and EF in general have been tightly linked to several frontal and parietal large-scale networks. These include the fronto-parietal network, cingulo-opercular network, and the dorsal and ventral attention systems.7–9 The third is attentional arousal (or vigilance), which is anchored in ascending noradrenergic neural systems7 and closely related to earlier ADHD-related proposals of arousal, activation, and gain efficiency, as well as to components of response-time variability.10 It is perhaps the most enduring cognitive mediator proposed for ADHD.11–13 The fourth domain, temporal information processing or “the mental clock,” allows for rapid time perception and reproduction, and is atypical in ADHD;14 it is related to cerebellar-cortical-basal-ganglia circuitry.15 The last domain is output speed, also of interest in ADHD.16 Longitudinal studies tentatively support a mediation model of development of aspects of EF and ADHD.17–20 We, therefore, test this claim in a path model environment using conventions accepted for this purpose21, and use the term “mediation” for simplicity, although EF and ADHD are measured concurrently herein. While individual cognitive measures suffer from weaker heritability than ADHD, latent variable measures of EF have stronger heritability than ADHD,22 so a latent variable approach was adopted. Because genetically-informed tests of the hypothesis that cognitive disruption mediates genetic influence on ADHD are few, this crucial conceptual proposal remains controversial.23,24
The genetic structure of ADHD is likely complex, including both common and rare genetic variants as well as epigenetic effects.25,26 Here we investigate molecular genetic effects of common DNA variants using a polygenic risk score (PGS),27 an approach that has proven fruitful in detecting the common variant signal for ADHD28–32 and other disorders. While candidate gene studies have previously looked at statistical mediation by cognitive measures on ADHD,33,34 only one used a polygenic score.35 They failed to detect mediation. We provide here a statistically more powerful test of the basic endophenotype model.
Our aims were: (a) update the evaluation of polygenic risk on ADHD using a larger discovery sample and more extensive phenotyping than in prior reports, and (b) evaluate, essentially for the first time, to what extent aggregate effects of common genetic variants support the claim to endophenotype status for five hypothesized cognitive candidates in ADHD: working memory, inhibition, vigilance, temporal processing, and output speed.
METHODS
Participants
Participants in the target sample were 656 unrelated children age 7–11 recruited from the community for a case-control study of ADHD. ADHD was deliberately oversampled to ensure adequate clinical range variation to detect genetic signal as recommended by others,35 and to enable us to examine ADHD heterogeneity later. To preserve the representativeness of the sample, we did not oversample for sex or other demographics. Thus, we expected groups to differ on sex ratio and possibly on socioeconomic standing, which are associated with ADHD.36
Recruitment and Diagnostic Assignment.
Human subjects and ethics approval was obtained from the local University Institutional Review Board. A parent/legal guardian provided written informed consent, and children provided written assent. After screening, a clinical evaluation was conducted using standardized, well-normed rating scales from parent and teacher, parent semi-structured clinical interview, child intellectual testing, and clinical observation. Best estimate research diagnoses and final eligibility were established by a team of two experienced clinicians (a child psychiatrist and a child psychologist) who independently arrived at the diagnosis. See the online supplement for further details.
Exclusion criteria.
Children were excluded for disallowed medications (see supplement), history of seizures or head injury, parent-teacher rating discrepancy making diagnosis uncertain, psychosis, mania, current major depressive episode, Tourette’s syndrome, autism and IQ<80.
Related individuals and Final Sample.
A resulting n=850 eligible children were scheduled for the cognitive battery (below); for the genetic analysis, related children were removed (see online supplement for details), resulting in the final sample of n=656 unrelated children, of which n=514 comprised the homogeneous European-ancestry sub-sample.
Data reduction: Diagnostic variables.
We examined ADHD as a categorical diagnosis and as a latent dimension by parent report and by teacher report. For parents, the indicators were the relevant inattention and hyperactivity subscale scores on the ADHD-RS, K-SAD, CPRS-3, and SDQ; for the teachers, it was the ratings on the ADHD-RS, CPRS-3, and SDQ. These two measurement models fit well (parent: RMSEA=0.057, CFI=.996, TLI=.992; teacher: RMSEA=.000, CFI=1.00 and TLI=1.00).
Cognitive Measures
Children completed a second visit in which the cognitive measures were obtained after a medication washout of >= 7 half-lives. The battery was selected to capture multiple indicators of core constructs whenever possible. We administered the following tasks (a) Stop-Go task;37,38 (b) Identical Pairs Continuous Performance Task;39,40 (c) Spatial span forward and backward,41 (d) Digit span forward and backward from the WISC-IV, (e) N-back, including 0-back, 1-back, and 2-back conditions, (f) Delis, Kaplan, and Kramer(DKEF)42 version of the Stroop task (word, color, and color-word), (g) DKEF Trailmaking test (number, letter, and shifting), and (h) a motor time reproduction task at fast (500 ms) interval from which we derived clock precision (clock variation).43–45 See the online supplement for details of task procedures, data cleaning, quality control, and validity checks.
For the go-trials on the stop task, rather than use the difficult-to-interpret within-child standard deviation, we used a diffusion model decomposition to isolate drift rate10 as a more precise index of arousal/vigilance, and non-decision time as a measure of output speed. Other diffusion model components were ignored because they are not associated with ADHD.10 For the CPT, we computed two versions of the signal detection parameter d-prime (d’) as an index of arousal, one for difficult catch trials and one for easy standard trials (see online supplement). We used residual scores for Stroop and Trails conflict conditions, after removing their respective speed measures.
Missing Data.
15 children missed the task visit due to illness, no-show, or cancellation; they are included to improve the overall data matrix as recommended by methodologists.46 On each task, from 1–5% of data was removed for failing to pass our data quality checks. Missing data were handled using the full information maximum likelihood model in MPLUS 7.2.
Data reduction and latent cognitive constructs measured.
Table S2 (online) provides correlations among all the indicators. Table S-3 (online) summarizes how cognitive measures were conceptualized as indicating latent constructs, and their empirical correlations with the ADHD diagnosis and polygenic score. The latent variables were finalized while blind to the genetic data or PGS correlations, however. A confirmatory CFA model fit satisfactorily for both N=514 (X2 (107, N=656) = 297.50, CFI=.93, TLI=.91, RMSEA=.060) and N=656 (X2 (107, N= 656) = 372.37, CFI=.92, TLI=.90, RMSEA=.062) (a significant chi-square is not unexpected with this sample size, and remaining fit indices are satisfactory47–50). The final model is displayed in Figure 1. Correlations among the latent variables are in Table S-3 footnote.
Figure 1:
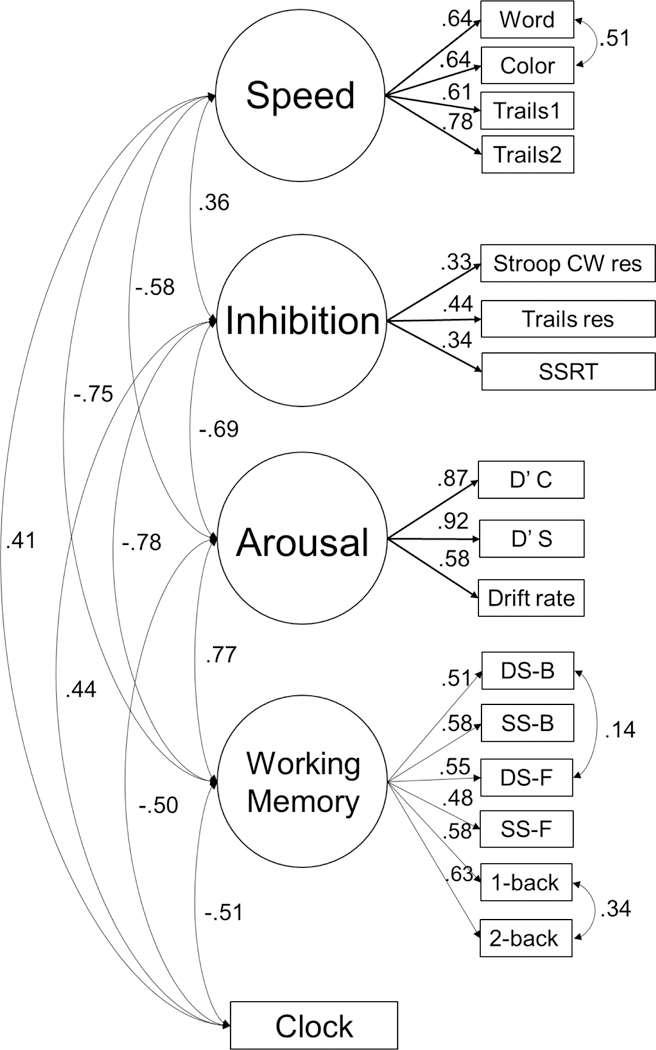
Confirmatory model with standardized coefficients. All estimates yield p<.01. Error terms not shown for readability. Fit for N=514: (X2(108)=297.50, CFI=.93, TLI=.91, RMSEA=.060). Fit for N=656: (X2(107)=365.74, CFI=.93, TLI=.90, RMSEA=.062) and the N=514 sample (X2(108)=297.50, CFI=.93, TLI=.91, RMSEA=.060). See Figure S4 for additional information about the measurement model with N=656. Word, Color are Stroop conditions. Trails 1=numbers, Trails 2=letters. Stroop CW res=Stroop color word interference trial with residual score regressed on Stroop color and word naming. SSRT=stop signal reaction time. D’C and D’S are d-prime scores on the continuous performance task for difficult (catch) and easy (stimulus) trials respectively. Drift rate is the diffusion model gain parameter. DS-B, DS-F=digit span back and forward. SS-B, SS-F=spatial span back, forward.
Genotyping and Polygenic score
Genotyping.
Salivary DNA samples were genotyped at the Stanley Center for Psychiatric Research (Broad Institute of MIT and Harvard, Cambridge, MA) using the PsychCHIP_v1–1 (N=603,132 SNPs), developed by Illumina, Inc (San Diego, CA) in collaboration with the Psychiatric Genomics Consortium (PGC). Processing and QC details are in the online Supplement.
PGS computation.
The polygenic risks score (PGS) was constructed using the 2016–2017 PGC meta-analysis51 as the discovery data set (20,183 ADHD cases and 35,191 controls), genotyped with the same chip. The PGS was calculated in the target sample by multiplying the number of risk alleles (0, 1, or 2) by the log (odds ratio) of that SNP in the discovery data set and averaging over all SNPs (details in online supplement; odds ratios by decile are in Figure S1 online). The PGS created from the full PGC meta-analysis was tested in a homogeneous European-ancestry sub-sample (n=514) of our dataset. As a further check on population ancestry artifact, we computed a PGS using only the European population (19,099 ADHD cases and 34,194 controls) of the PGC data and tested it in our European test-sample. Alternative ways of computing the PGS at different thresholds (Figure S2, online) did not alter results.
Data analysis
Analyses relied on binomial logistic regression to examine effects on ADHD diagnosis, and structural equation modeling (SEM) to examine mediation effects of cognitive measures. SEMs with categorical outcomes were analyzed using the robust weighted least squares estimator while those with dimensional outcomes used the maximum likelihood estimator. For the logistic regression model, we report the Nagelkerke pseudo-R2 change. For the SEM models, standard indices of model fit are reported (CFI, TLI, and RMSEA).52 Statistical mediation was tested using the model indirect command in MPLUS 7.2 which uses the delta method for calculating the statistical significance of indirect effects. Bootstrapping methods (1000 bootstrap samples) were used to calculate 95% confidence intervals for these indirect effects.
Correction for multiple testing.
With five correlated outcome neuropsychological latent variables, a Bonferroni correction is underpowered53 whereas an FDR test yields excessive type I error.54 Blakesley et al54 recommended the Hochberg correction55 in this case, which was therefore employed here. Mediation tests were planned only if simple effects with the polygenic score had corrected P<.050. We report uncorrected mediation tests to avoid correcting the same effect twice. Because the ADHD parent and teacher scores are versions of the same construct (and thus not new hypotheses), correcting for them would be too conservative.
Population Stratification.
To ensure that results were not due to population stratification (on one hand) and to ensure that results were not attributable to low power (on the other hand), we carried out the analysis three times. The primary model used the full discovery sample to compute the PGS but was restricted to the European-only test sample (n=514). Due to the potential loss of power in that sample, analyses were repeated using the full sample (n=656). Finally, we computed a modified PGS from only the European subset of the PGC.
Covariates.
We covaried sex and age in all results reported. In the analyses that considered the full sample (n=656), the first 10 genetic principal components were also controlled (from both the cognitive and ADHD measures), in order to control for effects of population stratification. In the European-only test sample, no principal components were correlated with outcomes so they were not covaried. We did not covary IQ as it is inappropriate to do so because reduced IQ may be part of the ADHD syndrome and may be a consequence of reduced executive functioning. SES was not covaried for similar reasons.56 Medication was washed out for ≥7 half-lives prior to cognitive testing (Table S1 provides details); prescription status was not covaried to avoid inappropriately removing ADHD severity (prescription status correlation with ADHD severity within the ADHD group, r=.79, p<.001).
RESULTS
Overview and sample description
Table 1 provides clinical and demographic description of the sample. As expected, boys are over-represented in the ADHD group. ADHD presentations by DSM-5: combined (72.1%), inattentive (25.8%), hyperactive (2.1%).
Table 1:
Sample descriptive data
| European-only(N=514) | Complete Sample(N=656) | |||
|---|---|---|---|---|
| Control | ADHD(all) | Control | ADHD(all) | |
| N | 177 | 337 | 221 | 435 |
| % male* | 54% | 72% | 52% | 71% |
| % non-Hispanic white | 100% | 100% | 83% | 80% |
| Age at intake | 9.4(1.5) | 9.5(1.5) | 9.4(1.5) | 9.5(1.5) |
| Age when EF measured | 9.6(1.5) | 9.7(1.5) | 9.6(1.5) | 9.7(1.5) |
| Income (thousands $)** | 96.8(33.2) | 90.0(34.1) | 94.5(20) | 85.3(24) |
| Estimated full scale IQ* | 115(12.8) | 109.7(13.5) | 114.9(13) | 108.5(14) |
| Reading* | 113.9(11.1) | 106.4(14) | 113.5(11) | 106.4(13) |
| ADHD-RS Parent H* | 45.2(7.5) | 67.9(14.4) | 45.5(7.4) | 67.7(14.5) |
| ADHD RS Parent I* | 44.5(7.1) | 72.3(12.3) | 44.6(7.0) | 72.2(12.2) |
| Ever any ADHD med* | 0% | 44% | 0% | 41% |
| Current stimulant* | 0% | 39% | 0% | 36% |
Footnote to Table 1:
ADHD differs from controls, p<.05.
In the complete sample (N=656) only, ADHD < controls, p<.05. C=ADHD combined subtype (presentation); I=ADHD primarily inattentive presentation; H=ADHD primarily hyperactive-impulsive presentation. ADHD-RS are T-scores based on national norms; ADHD medications are listed in Table S2 (online). PGS=polygenic score as explained in the text.
Primary Analyses
First analysis: ADHD and polygenic score.
The first aim was to validate, using data from a large discovery sample,51 the association of cumulative genetic signal with ADHD in this sample. We begin with our primary analysis using the European-only subsample (n=514): For ADHD diagnosis, the association with the PGS was robust, beta=.233, SE=.053, OR=17.57, P=. 000011, Nagelkerke pseudo R2 = .045. (Incremental R2 values for the other models are in online Table S-4.) For the parent and teacher ADHD latent dimensional variables, effects were similar though very slightly attenuated (Table 2, top rows). Results were similar for the two symptom dimensions looked at separately (not shown). The PGS was not related to sex in the overall sample (p=.95) or within the ADHD (boys m=.405, girls m=.414, p=.57). Sensitivity analyses repeated with all available children (N=656) and our Euro-only subsample (n=514) was then re-tested with the polygenic score derived from an Euro-only subsample of the discovery data; all yielded very similar results, lending confidence to these observations (top 3 rows of Table 2, and Table S-4 online).
Table 2:
Polygenic Score Association with ADHD Construct (as Diagnosis and as Parent and Teacher rated Latent Variable Dimension) and with Five Endophenotypes as Latent Variables
| Full PGC Discovery Sample | Euro-Only Discovery Sample | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Euro-Only test sample (N=514) |
P-value | Adjusted P-Value |
Full test sample (n=656) |
P-value | Adjusted P-Value |
Euro-Only test sample (N=514) |
P-Value | Adjusted P-Value |
|
| B(SE) | P | PAdj | B(SE) | P | PAdj | B(SE) | P | PAdj | |
| ADHD | |||||||||
| ADHD Diagnosis | .233(.053) | 1.10E-05 | -- | .236(.050) | 2.36E-06 | -- | .207(.052) | 6.87E-05 | -- |
| Teacher Latent Variable | .165(.042) | 8.55E-05 | -- | .176(.040) | 1.08E-05 | -- | .158(.042) | 1.69E-04 | -- |
| Parent Latent Variable | .185(.043) | 1.69E-05 | -- | .196(.041) | 1.75E-06 | -- | .177(.043) | 3.85E-05 | -- |
| ENDOPHENOTYPES | |||||||||
| Working Memory* | .227(.040) | 1.39E-08 | 6.95E-08 | .190(.044) | 1.57E-05 | 7.85E-05 | .199(.047) | 2.03E-05 | 1.02E-04 |
| Vigilance/Arousal* | .130(.049) | 0.0079 | 0.0316 | .116(.046) | 0.0116 | 0.0464 | .123(.048) | 0.0104 | 0.0416 |
| Slow output speed | .093(.043) | 0.0305 | 0.0915 | .096(.041) | 0.0192 | 0.0576 | .089(.042) | 0.0341 | 0.1023 |
| Mental clock | .087(.043) | 0.0430 | 0.0806 | .068(.041) | 0.0921 | 0.1842 | .087(.043) | 0.0430 | 0.0860 |
| Response Inhibition | .079(.074) | Ns | ns | .037(.076) | ns | ns | .136(.075) | 0.0697 | 0.0697 |
Notes to Table 2: Values are standardized regression coefficients (SE), adjusted age and sex (and for the full test sample, the first 10 genetic principal components).
indicates reliable effect in all models after correction.
Second analysis: Cognitive latent variables and polygenic score.
Consistent with a massive literature, all latent cognitive variables were robustly related to ADHD diagnosis and latent dimensional ADHD variables. Results for the polygenic score varied sharply, however. As shown in the lower five rows of Table 2, the PGS was not related to the inhibition latent variable. “Mental clock” and slow response speed, while marginally related to the PGS at least in some models, did not survive correction in any models. Working memory and vigilance-arousal had reliable and strong associations with the PGS in all models (Table 2) and so were carried forward to mediation tests.
Third analysis: Path modeling of mediation effects.
Statistical mediation was tested for working memory and arousal. Results are shown in Table 3 and were essentially identical (although slightly attenuated) in the sensitivity analyses, lending confidence that results were not attributable to population stratification or to power. Overall, reliable partial mediation was seen for the working memory and vigilance latent variables regardless of how ADHD was defined and accounted for 34% to 50% of the polygenic effect on ADHD across models. Figure 2 displays these two models in relation to ADHD diagnosis.
Table 3:
Mediation Results, Three Models of polygenic score and Three ADHD Definitions
| Working Memory | Vigilance/Arousal | |||||
|---|---|---|---|---|---|---|
| Full PGC Discovery Sample | Euro-Only Discovery |
Full PGC Discovery Sample | Euro-Only Discovery |
|||
| Euro-Only Test Sample (n=514) |
Full Test Sample (n=656) |
Euro-Only Test Sample (n=514) |
Euro-Only Test Sample (n=514) |
Full Test Sample (n=656) |
Euro-Only Test Sample (n=514) |
|
| ADHD Diagnosis | ||||||
| Main effect to ADHD | .450(.064) | .512(.054) | .450(.064) | .643(.060) | .605(.055) | .643(.060) |
| Total PGS to ADHD effect | .233(.054) | .236(.050) | .208(.055) | .233(.054) | .236(.050) | .207(.055) |
| Total indirect effect | .101(.029) | .104(.027) | .099(.027) | .115(.041) | .095(.036) | .106(.044) |
| Indirect effect p-value | 0.00049 | 0.0001 | 0.00024 | 0.005 | 0.0083 | 0.0159 |
| Indirect % PGS explained | 43.30% | 44.10% | 47.60% | 49.40% | 40.30% | 51.20% |
| Model Fit (RMSEA) | 0.026 | 0.000 | 0.024 | 0.047 | 0.000 | 0.043 |
| Teacher Latent Variable | ||||||
| Main effect to ADHD | .404(.059) | .446(.050) | .404(.059) | .449(.053) | .532(.056) | .449(.053) |
| Total PGS to ADHD effect | .161(.044) | .174(.043) | .157(.043) | .160(.043) | .170(.042) | .156(.043) |
| Total indirect effect | .075(.023) | .080(.023) | .073(.022) | .071(.027) | .063(.025) | .068(.027) |
| Indirect effect p-value | 0.00011 | 0.0005 | 0.00091 | 0.0077 | 0.0118 | 0.0118 |
| Indirect % PGS explained | 46.60% | 47.10% | 46.50% | 44.40% | 37.10% | 43.60% |
| Model Fit (RMSEA) | 0.037 | 0.04 | 0.037 | 0.058 | 0.049 | 0.058 |
| Parent Latent Variable | ||||||
| Main effect to ADHD | .401(.051) | .419(.043) | .401(.051) | .495(.048) | .481(.042) | .495(.048) |
| Total PGS to ADHD effect | .184(.043) | .194(.041) | .177(.044) | .185(.043) | .196(.044) | .177(.044) |
| Total indirect effect | .093(.027) | .091(.024) | .090(.025) | .080(.030) | .068(.027) | .077(.031) |
| Indirect effect p-value | 0.0011 | 0.00015 | 0.00032 | 0.0077 | 0.0118 | 0.0129 |
| Indirect % PGS explained | 50.50% | 46.40% | 50.80% | 43.20% | 34.70% | 43.50% |
| Model Fit (RMSEA) | 0.067 | 0.074 | 0.099 | 0.125 | 0.088 | 0.126 |
Footnote to Table 3: PGS=polygenic score. Values are standardized regression coefficients (SE), after adjustment for age and sex (and for the full test sample, the first 10 genetic principal components). Mediation effect = total indirect effect in the SEM model. All P-values are 2-tailed.
Figure 2.
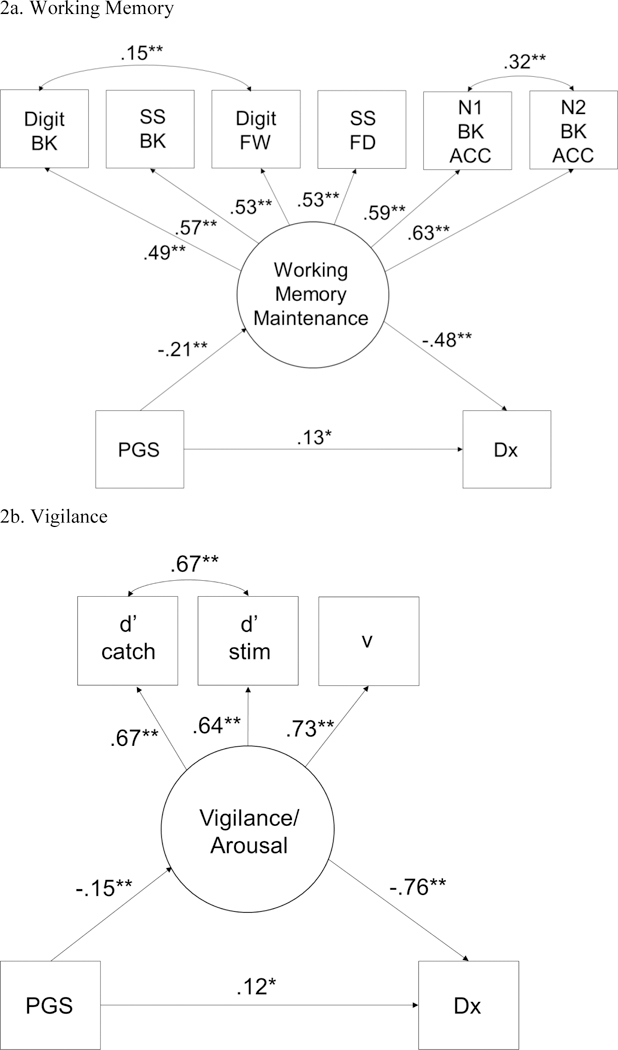
PGS=polygenic score; DX=ADHD diagnosis versus controls. BK=back, FW=forward; SS=spatial span, Digit=Digit Span; N1 BK ACC=1-back accuracy across all trials. 2A shows that working memory maintenance latent variable mediates the association between the PGS and ADHD. 2B shows the effect for Arousal/Vigilance. For the working memory model, χ2 (27, N=514) = 36.55, p=.10, CFI=.99, TLI=.98, RMSEA=.026. Indirect effect β=.101 (SE=.029), 95% CI=.05,.16. For the vigilance model, χ2 (7, N=514) = 14.96, p=.04, CFI=.99, TLI=.97, RMSEA=.047. Indirect effect β=.12(SE=.041), 95% CI=.04,.20. Based on the modification indices errors were correlated for the two d-prime scores in these models.
DISCUSSION
These results show for the first time that putative cognitive endophenotypes statistically mediate genetic risk for ADHD and thus may serve as useful components of models for genetic effects in ADHD. They are here supported as endophenotypes. Put another way, part of the polygenic risk from common SNP variants on ADHD is statistically accounted for by alterations in working memory and vigilance. These two domains are empirically and conceptually related, however, and it remains to be seen whether shared dependence on executive attention or other cognitive capacities further simplifies this picture.57 In contrast, response inhibition showed less promise; mediation effects for output speed and time reproduction were less pronounced and did not survive statistical correction at this sample size. A strong neuroimaging literature implicates several fronto-parietal and subcortical structures in working memory, and a consistent literature highlights the role of ascending noradrenergic systems in attentional vigilance. These neural systems appear to be prime targets for future work relating genetic effects to neuroimaging.
Other potential intermediate phenotypes were not examined here and may be equally or more effected by polygenic risk (e.g., reward discounting, delay aversion, set shifting). The negative results for response inhibition appear inconsistent with twin data suggesting shared genetic influences on ADHD and response inhibition58 and may suggest either alternative genetic mechanisms than those studied here, or that our measures of inhibition were sub-optimal for genetic studies. Notably, the inhibition effect approached significance using a restricted discovery sample and test sample, this construct may be more vulnerable to population effects. The working memory latent variable captured a mix of short-term store and maintenance, but not complex cognition or planning. However, these indicators may share association with the executive attention aspect of working memory,57,59 explaining the excellent fit of the WM latent variable.
Reaction time variability, of considerable interest in ADHD genetics,33,60,61 was decomposed into constituent functions in our diffusion decomposition to improve it’s biological applicability.10,62 We emphasized the drift rate parameter because it has shown the largest ADHD effects in prior studies. Consistent with theory, it loaded on an arousal/vigilance factor with similar measures from signal detection theory (d’) and mediated genetic effects on ADHD. Arousal mechanisms contributing to RT variability may serve as an endophenotype.10 The arousal results conform well to prior twin and molecular studies suggesting shared genetic influences on ADHD and reaction time variability.33,63
The present study is among the first of its kind, so there is little precedent for comparison. Kamradt34 reported in a different sample using a candidate gene approach that response inhibition mediated the effect of polymorphisms at DRD4 and DAT1. Those mechanisms may be distinct from the SNP effects summed here. Benca et al35 failed to find reliable EF mediation of PGS effects on ADHD but had a smaller discovery and target sample. Additionally, their model emphasized response inhibition; ours included a broader set of domains. Like them, we prioritized a latent variable approach. Latent variables reflect only variance correlated across tasks and so are free from measurement error due to unreliability. They are also theorized to remove the inherent task impurity in laboratory tasks; twin data suggest they provide the strongest genetic signal versus individual task variables.58
The magnitude of relationships between phenotype measures and PGS in our independent test sample is stronger than reported in prior studies using independent samples. Three advantages accrued to the present study over prior reports. First, ours is the first report of PGS, ADHD and cognition to capitalize on the latest and largest ADHD meta-analysis from the Psychiatric Genomics Consortium.51 Second, we used a case-control design, not a population sample; this enabled us to enrich our sample for ADHD cases and ensure a stronger variation of ADHD psychopathology than otherwise. Third, ADHD phenotyping here included a best-estimate diagnostic procedure, teacher ratings, and semi-structured clinical interviews that were not available in some prior studies.
It is unclear to what extent population stratification may bias polygenic risk score analyses. To address the possibility of bias due to mixed ancestry populations, we tested our models in multiple ways, with our primary model restricted to a homogeneous Caucasian-European ancestry test sample. However, to ensure those results were not attributable to reduced power in that restricted sample, we repeated analyses with the full sample while covarying the genetic principal components as a partial (but potentially inadequate) control of stratification (see online supplement for more discussion of this point). Finally, because we do not know whether a heterogeneous discovery sample may bias results, a further sensitivity analysis was conducted with a modified polygenic score using only a European-ancestry discovery sample. Results were nearly identical in all models, so population stratification cannot account for these results.
While effects were relatively consistent across formulations of ADHD here, ADHD is likely to be heterogeneous with regard to mediating cognitive mechanisms64,65 as well as with regard to comorbidity, severity, clinical course, temporal variation, environmental exposures, and probably genetic variation. We did not see sex differences in the PGS in the total sample or within the ADHD group, perhaps consistent with twin evidence of shared specific genetic influences in boys and girls.66 However, potential sex differences in mechanisms associated with polygenic risk remains a key area for future work in view of sex differences in ADHD incidence. Likewise, ADHD phenotypic characterization continues to be refined.67 While we used a case-control design to match the design of the PGC discovery sample and compare to prior literature, the PGS association on a putative ADHD trait in the general population cannot be inferred. In this design, it is also possible that differential distribution of EF scores or of the PGS in the ADHD and control groups could lead to false negative results for some measures (such as response inhibition). A check on this possibility suggested this was not the case, as histograms appear approximately normal across groups (See Figure S3 and S6 online) and interactions of ADHD group with PGS in predicting working memory or arousal were not significant (all p>.25). Finally, although prior longitudinal studies without genetic measures have supported mediation as noted earlier, and our path modeling is in line with accepted conventions in the field,21 in cross-sectional data the reverse model (that ADHD causes changes in EF) is mathematically equivalent.
Overall, the present study provides the first compelling molecular genetic evidence that cumulative common genetic risk loading for ADHD is partially accounted for by breakdowns in working memory maintenance and in vigilance. These measures, therefore, are promising intermediate phenotypes for ADHD.
Supplementary Material
Acknowledgments:
The authors thank Dr. Benjamin Neal, Ph.D., of Harvard Medical School (HMS), the Broad Institute, and the Analytic and Translational Genetics Unit at Massachusetts General Hospital (MGH) for consultation on the statistical analysis.
Funding: This project was supported by R01-MH099064 and R01-MH5104.
Footnotes
Disclosures:
Dr. Nigg has received royalties from Guilford Press for What Causes ADHD (2007) and Getting Ahead of ADHD (2017).
Dr. Faraone has received grant or research support from the K.G. Jebsen Centre for Research on Neuropsychiatric Disorders, the University of Bergen, Bergen, Norway, the European Union’s Seventh Framework Programme for research, technological development, and demonstration, the European Union’s Horizon 2020 research and innovation programme, and the National Institute of Mental Health. He has received income, potential income, travel expenses, continuing education support, research support from, and/or has served on the advisory boards of/as a consultant to Lundbeck, Rhodes, Arbor, KenPharm, Ironshore, Neurovance, Impact, Takeda, Shire, Akili Interactive Labs, CogCubed, Alcobra, VAYA Pharma, Sunovion, Genomind, and NeuroLifeSciences. In previous years, he has received income or research support from Shire, Neurovance, Alcobra, Otsuka, McNeil, Janssen, Novartis, Pfizer, and Eli Lilly and Co. He has served as editor of the American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. His institution (SUNY) has US patent US20130217707 A1 for the use of sodium-hydrogen exchange inhibitors in the treatment of ADHD. He has received royalties from books published by Guilford Press (Straight Talk about Your Child’s Mental Health), Oxford University Press (Schizophrenia: The Facts), and Elsevier (ADHD: Non-Pharmacologic Interventions). He has held stock in CogCubed and Ironshore. He is the principal investigator of http://adhdinadults.com/.
Dr. Mooney reports no biomedical financial interests or potential conflicts of interest.
Dr. Fair reports no biomedical financial interests or potential conflicts of interest.
Dr. Wilmot reports no biomedical financial interests or potential conflicts of interest
Dr. Gustafsson reports no biomedical financial interests or potential conflicts of interest.
Dr. Karalunas reports no biomedical financial interests or potential conflicts of interest.
Dr. Ryabinin reports no biomedical financial interests or potential conflicts of interest.
Dr. McWeeney reports no biomedical financial interests or potential conflicts of interest.
Contributor Information
Joel T. Nigg, Oregon Health and Science University, Portland..
Hanna C. Gustafsson, Oregon Health and Science University, Portland..
Sarah L. Karalunas, Oregon Health and Science University, Portland..
Peter Ryabinin, Oregon Health and Science University, Portland..
Shannon McWeeney, Oregon Health and Science University, Portland..
Stephen V. Faraone, State University of New York Upstate Medical University, Syracuse..
Michael Mooney, Oregon Health and Science University, Portland..
Damien A. Fair, Oregon Health and Science University, Portland..
Beth Wilmot, Oregon Health and Science University, Portland..
REFERENCES
- 1.Gottesman II, Shields J. Genetic theorizing and schizophrenia. The British Journal of Psychiatry. 1973;122(566):15–30. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160(4):636–645. [DOI] [PubMed] [Google Scholar]
- 3.Miller GA, Rockstroh B. Endophenotypes in psychopathology research: where do we stand? Annu Rev Clin Psychol. 2013;9:177–213. [DOI] [PubMed] [Google Scholar]
- 4.Kendler KS, Neale MC. Endophenotype: a conceptual analysis. Mol Psychiatry. 2010;15(8):789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barkley RA. Behavioral inhibition, sustained attention, and executive functions: constructing a unifying theory of ADHD. Psychol Bull. 1997;121(1):65–94. [DOI] [PubMed] [Google Scholar]
- 6.Castellanos FX, Tannock R. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci. 2002;3(8):617–628. [DOI] [PubMed] [Google Scholar]
- 7.Petersen SE, Posner MI. The attention system of the human brain: 20 years after. Annu Rev Neurosci. 2012;35:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyake A, Friedman NP, Emerson MJ, Witzki AH, Howerter A, Wager TD. The unity and diversity of executive functions and their contributions to complex “Frontal Lobe” tasks: a latent variable analysis. Cogn Psychol. 2000;41(1):49–100. [DOI] [PubMed] [Google Scholar]
- 9.Diamond A Executive functions. Annu Rev Psychol. 2013;64:135–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karalunas SL, Geurts HM, Konrad K, Bender S, Nigg JT. Annual research review: Reaction time variability in ADHD and autism spectrum disorders: measurement and mechanisms of a proposed trans-diagnostic phenotype. J Child Psychol Psychiatry. 2014;55(6):685–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang-Pollock CL, Karalunas SL, Tam H, Moore AN. Evaluating vigilance deficits in ADHD: a meta-analysis of CPT performance. J Abnorm Psychol. 2012;121(2):360–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sergeant J The cognitive-energetic model: an empirical approach to attention-deficit hyperactivity disorder. Neurosci Biobehav Rev. 2000;24(1):7–12. [DOI] [PubMed] [Google Scholar]
- 13.Zentall SS, Meyer MJ. Self-regulation of stimulation for ADD-H children during reading and vigilance task performance. J Abnorm Child Psychol. 1987;15(4):519–536. [DOI] [PubMed] [Google Scholar]
- 14.Hwang-Gu SL, Gau SS. Interval timing deficits assessed by time reproduction dual tasks as cognitive endophenotypes for attention-deficit/hyperactivity disorder. PLoS One. 2015;10(5):e0127157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mauk MD, Buonomano DV. The neural basis of temporal processing. Annu Rev Neurosci. 2004;27:307–340. [DOI] [PubMed] [Google Scholar]
- 16.Kuntsi J, Wood AC, Rijsdijk F, et al. Separation of cognitive impairments in attention-deficit/hyperactivity disorder into 2 familial factors. Arch Gen Psychiatry. 2010;67(11):1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabinovitz BBONS, Rajendran K, Halperin JM. Temperament, executive control, and attention-deficit/hyperactivity disorder across early development. J Abnorm Psychol. 2016;125(2):196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajendran KRD, O’Neill S, Marks DJ, Nomura Y, Halperin JM. Neuropsychological functioning and severity of ADHD in early childhood: a four-year cross-lagged study. J Abnorm Psychol. 2013;122:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brocki KCEL, Thorell LB, Bohlin G. Interrelations between executive function and symptoms of hyperactivity/impulsivity and inattention in preschoolers: a two year longitudinal study. J Abnorm Child Psychol. 2010;38(2):163–171. [DOI] [PubMed] [Google Scholar]
- 20.Tillman CBK, Sørensen L, Lundervold AJ. A longitudinal examination of the developmental executive function hierarchy in children with externalizing behavior problems. J Atten Disord. 2015;19:163–171. [DOI] [PubMed] [Google Scholar]
- 21.Kenny DA. Correlation and Causality. New York: John Wiley & Sons; 1979. [Google Scholar]
- 22.Friedman NP, Miyake A, Young SE, DeFries JC, Corley RP, Hewitt JK. Individual differences in executive functions are almost entirely genetic in origin. J Exp Psychol Gen. 2008;137(2):201–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyle AE, Faraone SV, Seidman LJ, et al. Are endophenotypes based on measures of executive functions useful for molecular genetic studies of ADHD? J Child Psychol Psychiatry. 2005;46(7):774–803. [DOI] [PubMed] [Google Scholar]
- 24.Thissen AJ, Rommelse NN, Hoekstra PJ, et al. Attention deficit hyperactivity disorder (ADHD) and executive functioning in affected and unaffected adolescents and their parents: challenging the endophenotype construct. Psychol Med. 2014;44(4):881–892. [DOI] [PubMed] [Google Scholar]
- 25.Martin J, O’Donovan MC, Thapar A, Langley K, Williams N. The relative contribution of common and rare genetic variants to ADHD. Transl Psychiatry. 2015;5:e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesch KP. Maturing insights into the genetic architecture of neurodevelopmental disorders - from common and rare variant interplay to precision psychiatry. J Child Psychol Psychiatry. 2016;57(6):659–661. [DOI] [PubMed] [Google Scholar]
- 27.Wray NR, Lee SH, Mehta D, Vinkhuyzen AA, Dudbridge F, Middeldorp CM. Research review: polygenic methods and their application to psychiatric traits. Journal of Child Psychology and Psychiatry. 2014;55(10):1068–1087. [DOI] [PubMed] [Google Scholar]
- 28.Hamshere ML, Langley K, Martin J, et al. High loading of polygenic risk for ADHD in children with comorbid aggression. Am J Psychiatry. 2013;170(8):909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin J, Hamshere ML, Stergiakouli E, O’Donovan MC, Thapar A. Genetic risk for attention-deficit/hyperactivity disorder contributes to neurodevelopmental traits in the general population. Biol Psychiatry. 2014;76(8):664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riglin L, Collishaw S, Thapar AK, et al. Association of Genetic Risk Variants With Attention-Deficit/Hyperactivity Disorder Trajectories in the General Population. JAMA Psychiatry. 2016;73(12):1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stergiakouli E, Martin J, Hamshere ML, et al. Shared genetic influences between attention-deficit/hyperactivity disorder (ADHD) traits in children and clinical ADHD. J Am Acad Child Adolesc Psychiatry. 2015;54(4):322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carey CE KA, Conley ED, Hariri AR, Bogdan R. Reward-related ventral striatum activity links polygenic risk for attention-deficit/hyperactivity disorder to problematic alcohol use in young adulthood. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging. 2017;2(2):180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinto R, Asherson P, Ilott N, Cheung CH, Kuntsi J. Testing for the mediating role of endophenotypes using molecular genetic data in a twin study of ADHD traits. Am J Med Genet B Neuropsychiatr Genet. 2016;171(7):982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamradt JM, Nigg JT, Friderici KH, Nikolas MA. Neuropsychological performance measures as intermediate phenotypes for attention-deficit/hyperactivity disorder: A multiple mediation analysis. Dev Psychopathol. 2016:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benca CE, Derringer JL, Corley RP, et al. Predicting Cognitive Executive Functioning with Polygenic Risk Scores for Psychiatric Disorders. Behav Genet. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faraone SV, Asherson P, Banaschewski T, et al. Attention-deficit/hyperactivity disorder. Nat Rev Dis Primers. 2015;1:15020. [DOI] [PubMed] [Google Scholar]
- 37.Nigg JT. The ADHD response-inhibition deficit as measured by the stop task: replication with DSM-IV combined type, extension, and qualification. J Abnorm Child Psychol. 1999;27(5):393–402. [DOI] [PubMed] [Google Scholar]
- 38.Schachar R, Tannock R, Marriott M, Logan G. Deficient inhibitory control in attention deficit hyperactivity disorder. J Abnorm Child Psychol. 1995;23(4):411–437. [DOI] [PubMed] [Google Scholar]
- 39.Cornblatt BA, Malhotra AK. Impaired attention as an endophenotype for molecular genetic studies of schizophrenia. Am J Med Genet. 2001;105(1):11–15. [PubMed] [Google Scholar]
- 40.Curko Kera EA, Marks DJ, Berwid OG, Santra A, Halperin JM. Self-report and objective measures of ADHD-related behaviors in parents of preschool children at risk for ADHD. CNS Spectr. 2004;9(9):639–647. [DOI] [PubMed] [Google Scholar]
- 41.De Luca CR, Wood SJ, Anderson V, et al. Normative data from the CANTAB. I: development of executive function over the lifespan. J Clin Exp Neuropsychol. 2003;25(2):242–254. [DOI] [PubMed] [Google Scholar]
- 42.Delis DC, Kaplan E, Kramer JH. Delis-Kaplan Executive Function System (D-KEFS). San Antonio, TX: The Psychological Corporation; 2001. [Google Scholar]
- 43.Wing AM. Voluntary timing and brain function: an information processing approach. Brain Cogn. 2002;48(1):7–30. [DOI] [PubMed] [Google Scholar]
- 44.Wing AM, Kristofferson A. The timing of interresponse intervals. Percept Psychophys. 1973;13(3):455–460. [Google Scholar]
- 45.Wing AM, Kristofferson AB. Response delays and the timing of discrete motor responses. Percept Psychophys. 1973;14(1):5–12. [Google Scholar]
- 46.Schafer JL, Graham JW . Missing data: our view of the state of the art. Psychol Methods. 2002;7(2):147–177. [PubMed] [Google Scholar]
- 47.Tucker LR, Lewis C. A reliability coefficient for maximum likelihood factor analysis. Psychometrika. 1973;38(1):1–10. [Google Scholar]
- 48.Hoyle RH. Structural equation modeling: Concepts, issues, and applications. Sage; 1995. [Google Scholar]
- 49.Browne MW, Cudeck R, Bollen KA, Long JS. Alternative ways of assessing model fit. Sage focus editions. 1993;154:136–136. [Google Scholar]
- 50.Bentler PM, Bonett DG. Significance tests and goodness of fit in the analysis of covariance structures. Psychol Bull. 1980;88(3):588. [Google Scholar]
- 51.Demontis D, Walters RK, Martin J, et al. Discovery of the first genome-wide significant risk loci for ADHD. bioRxiv. 2017;DOI: 10.1101/145581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Browne MW, Cudeck R. Alternative ways of assessing model fit‥ In: Testing structural equation models. Vol 154 Newbury Park, CA: Sage; 1993:136–162. [Google Scholar]
- 53.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811–818. [DOI] [PubMed] [Google Scholar]
- 54.Blakesley RE, Mazumdar S, Dew MA, et al. Comparisons of methods for multiple hypothesis testing in neuropsychological research. Neuropsychology. 2009;23(2):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hochberg Y A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75(4):800–802. [Google Scholar]
- 56.Loe IM, Feldman HM. Academic and educational outcomes of children with ADHD. J Pediatr Psychol. 2007;32(6):643–654. [DOI] [PubMed] [Google Scholar]
- 57.Shipstead Z, Harrison TL, Engle RW. Working memory capacity and the scope and control of attention. Attention, Perception, & Psychophysics. 2015;77(6):1863–1880. [DOI] [PubMed] [Google Scholar]
- 58.Friedman NP, Miyake A, Young SE, Defries JC, Corley RP, Hewitt JK. Individual differences in executive functions are almost entirely genetic in origin. J Exp Psychol Gen. 2008;137(2):201–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Unsworth N, Engle RW. On the division of short-term and working memory: an examination of simple and complex span and their relation to higher order abilities. Psychol Bull. 2007;133(6):1038. [DOI] [PubMed] [Google Scholar]
- 60.Kuntsi J, Frazier-Wood AC, Banaschewski T, et al. Genetic analysis of reaction time variability: room for improvement? Psychol Med. 2013;43(6):1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin HY, Hwang-Gu SL, Gau SS. Intra-individual reaction time variability based on ex-Gaussian distribution as a potential endophenotype for attention-deficit/hyperactivity disorder. Acta Psychiatr Scand. 2015;132(1):39–50. [DOI] [PubMed] [Google Scholar]
- 62.Karalunas SL, Huang-Pollock CL, Nigg JT. Decomposing attention-deficit/hyperactivity disorder (ADHD)-related effects in response speed and variability. Neuropsychology. 2012;26(6):684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McLoughlin GPJ, Rijsdijk F, Makeig S. Genetic overlap between evoked frontocentral theta-band phase variability, reaction time variability, and attention-deficit/hyperactivity disorder symptoms in a twin study. Biol Psychiatry. 2014;75(3):238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nigg JT. Neuropsychologic theory and findings in attention-deficit/hyperactivity disorder: the state of the field and salient challenges for the coming decade. Biol Psychiatry. 2005;57(11):1424–1435. [DOI] [PubMed] [Google Scholar]
- 65.Fair DA, Bathula D, Nikolas MA, Nigg JT. Distinct neuropsychological subgroups in typically developing youth inform heterogeneity in children with ADHD. Proc Natl Acad Sci U S A. 2012;109(17):6769–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rhee SH, Waldman ID, Hay DA, Levy F. Sex differences in genetic and environmental influences on DSM-III-R attention-deficit/hyperactivity disorder. J Abnorm Psychol. 1999;108(1):24–41. [DOI] [PubMed] [Google Scholar]
- 67.Martel MM, von Eye A, Nigg JT. Revisiting the latent structure of ADHD: is there a ‘g’ factor? J Child Psychol Psychiatry. 2010;51(8):905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.