


Abstract
DNA topoisomerases play essential roles in chromosome organization and replication. Most bacteria possess multiple topoisomerases which have specialized functions in the control of DNA supercoiling or in DNA catenation/decatenation during recombination and chromosome segregation. DNA topoisomerase I is required for the relaxation of negatively supercoiled DNA behind the transcribing RNA polymerase. Conflicting results have been reported on the essentiality of the topA gene encoding topoisomerase I in the model bacterium Bacillus subtilis. In this work, we have studied the requirement for topoisomerase I in B. subtilis. All stable topA mutants carried different chromosomal amplifications of the genomic region encompassing the parEC operon encoding topoisomerase IV. Using a fluorescent amplification reporter system we observed that each individual topA mutant had acquired such an amplification. Eventually, the amplifications were replaced by a point mutation in the parEC promoter region which resulted in a fivefold increase of parEC expression. In this strain both type I topoisomerases, encoded by topA and topB, were dispensable. Our results demonstrate that topoisomerase IV at increased expression is necessary and sufficient to take over the function of type 1A topoisomerases.
INTRODUCTION
The thorough control of DNA topology is an essential function in every living cell. In bacteria, DNA is usually present as circular covalently closed molecule that is compacted via the introduction of negative supercoils. Moreover, DNA replication and transcription involve the unwinding of the DNA double helix, and result in the introduction of positive supercoils ahead of the polymerizing complexes. In addition, transcription generates negative supercoils behind RNA polymerase (1). To ensure the faithful operation of genetic information, these alterations of the DNA topology state must be relaxed to put the chromosome to the status quo ante. DNA topology is not only important for packaging of the DNA, but also for gene expression, as DNA topology affects the accessibility of specific chromosomal regions for transcription (2).
All organisms possess DNA topoisomerases to either introduce or relax supercoils to maintain the topological state. In addition to the control of DNA supercoiling, the DNA topoisomerases are also involved in the resolution of DNA catenanes that are formed at the chromosomal terminus region during DNA replication and in the resolution of Holliday junctions during DNA recombination (3). To perform all these activities most bacteria, like the model organisms Escherichia coli and Bacillus subtilis, possess four topoisomerases that are members of two different families (see Table 1) (4). The type I topoisomerases are monomeric proteins that carry out an ATP-independent reaction that cleaves one DNA strand of the double helix, passes the second strand through, and finally reseals the DNA molecule (5). The paralogous type I topoisomerases are encoded by topA and topB and called topoisomerase (topo) I and III, respectively. Both proteins belong to the type 1A topoisomerases. The type II topoisomerases are ATP-dependent heterotetramers that cleave both strands of the DNA molecule, pass one region of the DNA through another, and rejoin the molecules by re-ligating both strands (5). The type II topoisomerases are encoded by gyrAB and parEC and are referred to as DNA gyrase and topo IV, respectively.
Table 1.
DNA topoisomerases in B. subtilis
| Name | Gene(s) | Essential | Activity |
|---|---|---|---|
| Type I topoisomerases | |||
| Topo I | topA | Yes | Relaxation of negatively supercoiled DNA behind RNA polymerase |
| Topo III | topB | No | Decatenation of DNA, resolution of Holliday junctions |
| Type II topoisomerases | |||
| DNA gyrase | gyrAB | Yes | Introduction of negative supercoils, relaxation of positive supercoils ahead of DNA and RNA polymerases |
| Topo IV | parEC | Yes | Decatenation/ segregation of replicating chromosomes |
DNA gyrase is required for introducing negative supercoils into the bacterial chromosomes, and thus essential for efficient DNA packaging. In addition, gyrase relaxes the positive supercoils that form ahead of the replication and transcription complexes (6). The relaxation of negatively supercoiled DNA behind the RNA polymerase is catalyzed by topo I. Topo III and topo IV are required for the resolution of Holliday junctions during recombination and for the segregation of replicating chromosomes, respectively (3). As expected from their critical functions in maintaining the proper DNA structure and allowing DNA replication, the two type II enzymes, gyrase and topo IV, are essential both in E. coli and in B. subtilis (7,8). Topo III, encoded by topB, is dispensable both in E. coli and B. subtilis reflecting the specialized function of this enzyme in DNA recombination. Some laboratory strains of B. subtilis (JH642 and its derivatives) even lack a chromosomal region of 18 kb that encompasses the topB gene (9). For topo I encoded by the topA gene, contradicting results have been reported: The gene is essential in E. coli, and two studies reported it to be essential in B. subtilis (7,10,11). In contrast, in a recent genome-wide essentiality study, the topA gene could be deleted; however only by replacing the gene with a kanamycin cassette whereas replacements with other resistance cassettes were not successful (8). Thus, no clear conclusion about the essentiality of topo I is possible.
Interestingly, the essentiality of the DNA topoisomerases is also reflected by their presence in bacteria. Most bacteria possess the four enzymes described above. However, Mycobacterium tuberculosis, Helicobacter pylori and Campylobacter jejuni only encode topoisomerase I and gyrase, and thus seem to lack decatenation activity. In these bacteria, decatenation at the end of replication can possibly be performed by gyrase (12,13). Moreover, a few obligate bacterial endosymbionts that have undergone extreme genome reduction and therefore are fully dependent on their insect host, such as Carsonella rudii, Hodkinia cicadicola and Trembleya princeps, have lost all their genes encoding DNA topoisomerases. Most likely, these bacteria acquire the enzymes from their hosts, as it is also the case in eukaryotic organelles (14). With genomes smaller than 160 kb, these bacteria belong to the organisms with the smallest genomes. Similarly, other host-adapted symbionts or pathogens have partially lost their topoisomerase complement. The endosymbionts Buchnera aphidicola, Wigglesworthia glossinidia and Sulcia muelleri have lost both type I topoisomerases, but do possess the essential enzymes gyrase and topo IV. In the bacteria of the genus Mycoplasma that are capable of living independently of host cells with the smallest known genomes, topo I, DNA gyrase and topo IV are present. These three enzymes and the encoding genes are also essential for Mycoplasma pneumoniae and Mycoplasma genitalium (15,16).
We are interested in the identification of the minimal set of genes that allows independent bacterial life. The identification of such a set of genes will be an important first step in our understanding of the functions of these genes and will undoubtedly facilitate the analysis of functional and physical interactions between the encoded proteins. Of course, there is not a single minimal gene set, but the solution to this problem depends on the lifestyle and cellular biology of the bacteria. So far, most advance in constructing minimal cells has been made with naturally genome-reduced bacteria of the genus Mycoplasma. With only 473 genes, the artificial organism Mycoplasma mycoides JCVI syn3.0 is the smallest independently viable bacterium. Among these 473 genes are the five genes encoding DNA gyrase (gyrA, gyrB), topo I (topA) and topo IV (parE, parC) (17). We approach genome reduction by a top-down approach using the model bacterium B. subtilis as the starting point. An analysis of a potential minimal genome of a B. subtilis-derived MiniBacillus revealed the requirement for the same set of topoisomerases (18,19). So far, we have been able to delete >36% of the genome, making B. subtilis the organism with the most advanced top-down genome reduction project (20).
Precise knowledge on essential genes and functions is the key to any successful minimal genome project. Essential genes by definition cannot be individually deleted in a wild type genetic background under standard growth conditions (18). In this study, we have addressed the issue of topo I essentiality in B. subtilis. While the topA gene could indeed be deleted, this was accompanied by the immediate acquisition of suppressor mutations that result in overexpression of the topo IV-encoding parEC operon. We demonstrate how these suppressor mutants eventually evolve from amplification of the parEC chromosomal region by acquiring a more stable point mutation in the parEC promoter. Our results suggest that topo IV, when overexpressed, can functionally replace topo I.
MATERIALS AND METHODS
Bacterial strains, growth conditions and phenotypic characterization
All B. subtilis strains used in this study are listed in Table 2. All strains are derived from the laboratory strain 168 (trpC2). B. subtilis was grown in Lysogeny Broth (LB medium) (21). LB plates were prepared by addition of 17 g Bacto agar/l (Difco) to LB and SP (sporulation medium), respectively (21,22). Quantitative studies of lacZ expression in B. subtilis were performed as described previously (22). One unit of β-galactosidase is defined as the amount of enzyme which produces 1 nmol of o-nitrophenol per min at 28°C.
Table 2.
B. subtilis strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| 168 | trpC2 | Laboratory collection |
| BKE04260 | trpC2 ΔtopB::ermC | 8 |
| BKK16120 | trpC2 ΔtopA::aphA3 | 8 |
| GP1948a | trpC2 ΔtopA::aphA3 (yneP-xynC)19 | This study |
| GP1952a | trpC2 ΔtopA::aphA3 (yneE-alsT)17 | This study |
| GP1958a | trpC2 ΔtopA::aphA3 (yneP-xynC)17 | This study |
| GP1963a | trpC2 ΔtopA::aphA3 (citB-alsT)16 | This study |
| GP1964a | trpC2 ΔtopA::aphA3 (yneP-xynC)28 ΔtopB::ermC | BKE04260 → GP1963 |
| GP1966 | trpC2 ΔynfC::(palf4-gfp ermC) | This study |
| GP1967 | trpC2 amyE::(pparE*-lacZ cat) b | This study |
| GP1974a | trpC2 ΔtopA::aphA3 (citB-alsT)15 | This study |
| GP1975a | trpC2 ΔtopA::aphA3 (citB-alsT)15 | This study |
| GP1979a | trpC2 ΔtopA::aphA3 (yneP … parEC … ΔynfC::(palf4-gfp ermC) … xynC)15 | This study |
| GP1983a | trpC2 ΔtopA::aphA3 (yneP … parEC … ΔynfC::(palf4-gfp ermC) … xynC)4codY (S129L) rocG (S61R) | This study |
| GP1985a,b | trpC2 ΔtopA::aphA3 ΔynfC::(palf4-gfp ermC) codY (S129L) rocG (S61R) gltA (V1181A) S672 (C5T) | This study |
| GP1896 | trpC2 ΔtopB::cat | 11 |
| GP1987a | trpC2 ΔtopA::aphA3 (yneP … parEC … ΔynfC::(palf4-gfp ermC) … xynC)4codY (S129L) rocG (S61R) | This study |
| GP2542 | trpC2 ΔrecA::spc | This study |
| GP2543 | trpC2 ΔtopA::aphA3 (…parEC … ΔynfC::(palf4-gfp ermC) …)n | This study |
| GP2544 | trpC2 ΔtopA::aphA3 (… parEC … ΔynfC::(palf4-gfp ermC) …)n | This study |
| GP2545 | trpC2 ΔtopA::aphA3 (… parEC … ΔynfC::(palf4-gfp ermC) …)n | This study |
| GP2546a | trpC2 ΔtopA::aphA3 (… parEC … ΔynfC::(palf4-gfp ermC) …)3 | This study |
| GP2547a | trpC2 ΔtopA::aphA3 ΔynfC::(palf4-gfp ermC) S672 (C5T) | This study |
| GP2548 | trpC2 ΔtopA::aphA3 ΔynfC::(palf4-gfp ermC) S672 (C5T) ΔtopB::cat | GP1896 → GP2547 |
| GP2549a | trpC2 ΔtopA::aphA3 ΔynfC::(palf4-gfp ermC) S672 (C5T) | This study |
| GP2550 | trpC2 amyE::(pparE-lacZ cat) | pGP3111 → 168 |
aThe genomic DNA of these strains was analyzed by whole genome sequencing.
bThe parE promoter region of this strain carries the C → T substitution in the -10 region of the promoter (see Figure 4).
DNA manipulation and genome sequencing
B. subtilis was transformed with plasmids, genomic DNA or PCR products according to the two-step protocol (21,22). Transformants were selected on SP or LB plates containing erythromycin (2 μg/ml) plus lincomycin (25 μg/ml), chloramphenicol (5 μg/ml), kanamycin (15 μg/ml), or spectinomycin (150 μg/ml). Phusion DNA polymerase (Thermo Fisher Scientific, Dreieich, Germany) was used as recommended by the manufacturer. DNA fragments were purified using the peqGOLD Cycle-Pure Kit (Peqlab, Erlangen, Germany). DNA sequences were determined by the dideoxy chain termination method (21). Chromosomal DNA from B. subtilis was isolated using the peqGOLD Bacterial DNA Kit (Peqlab, Erlangen, Germany). To identify the mutations in the suppressor mutant strains GP1948, GP1952, GP1958, GP1963, GP1964, GP1974, GP1975, GP1979, GP1983, GP1985, GP1987, GP2546, GP2547 and GP2549 (see Table 2), the genomic DNA was subjected to whole-genome sequencing. The reads were mapped on the reference genome of B. subtilis 168 (GenBank accession number: NC_000964) (23). Mapping of the reads was performed using the Geneious software package (Biomatters Ltd., New Zealand) (24). The resulting genome sequences were compared to that of our in-house wild type strain. Single nucleotide polymorphisms were considered as significant when the total coverage depth exceeded 25 reads with a variant frequency of ≥90%. All identified mutations were verified by PCR amplification and Sanger sequencing. Copy numbers of amplified genomic regions were determined by dividing the mean coverage of the amplified regions by the mean coverage of the remaining genome (25). The raw fastq reads of strains GP1948, GP1979 and GP1985 have been deposited in the NCBI-SRA database (https://www.ncbi.nlm.nih.gov/sra) and are accessible under the accession numbers SRR7967955, SRR7967956, and SRR7967957, respectively.
Construction of an amplification reporter system
To assay the amplification of the parEC genomic region, we replaced the adjacent ynfC gene by integrating a cassette composed of the gfp and ermC genes encoding the green fluorescent protein and erythromycin resistance determinant, respectively. In this construct, gfp expression is controlled by a weakly active promoter (pa) (26) and the ribosome binding site of the gapA gene. To fuse the gfp gene to the expression signals, we amplified the gfp gene using the oligonucleotides KG427 and KG429 (for oligonucleotide sequences, see Supplementary Table S1) and plasmid pBP43 (27) as the template. KG427 attaches the promoter and the ribosome binding site. The amplicon was inserted between the EcoRI and SalI sites of the expression vector pGP3101 resulting in plasmid pGP3102. pGP3101 in turn was obtained by cloning a DNA sequence specifying the immunodetectable FLAG-tag (obtained by PCR with primers ML05/ML06 and plasmid pGP1331 (28) as the template) into the HindIII site of the expression plasmid pBQ200 (29). To fuse the gfp gene with its expression signals to the ermC resistance gene and chromosomal regions up- and downstream of the anticipated insertion site (the ynfC gene), these four fragments were amplified by PCR (gfp with DR642/DR643 from pGP3102; ermC with mls-fwd(kan)/mls-rev(kan) from pDG647 (30); up- and downstream fragments with DR640/DR641 and DR644/DR645, respectively, from B. subtilis chromosomal DNA). The PCR products were purified, fused in another round of PCR using the oligonucleotides DR640 and DR645, and the 4.1 kb fusion PCR product was used to transform B. subtilis 168. The resulting strain was GP1966.
Construction of parE-lacZ fusions
Plasmid pAC5 (31) was used to construct a translational fusion of the parE promoter region with the lacZ gene. For the construction of plasmid pGP3111 containing a parE-lacZ fusion, the region upstream of parE was amplified using the oligonucleotides PF25/PF26. The PCR product was digested with EcoRI and BamHI, and cloned into pAC5 linearized with the same enzymes. Plasmid pGP3112 for the mutant upstream region was constructed accordingly using chromosomal DNA of GP1985 as the template. The fusion constructs were integrated into the amyE site of the B. subtilis chromosome by transformation of B. subtilis 168 with linearized plasmids. The resulting strains were GP2550 and GP1967 for the wild type and mutant promoter, respectively.
Construction of a recA mutant
Deletion of the recA gene was achieved by transformation with PCR products constructed using oligonucleotides (see Supplementary Table S1) to amplify DNA fragments flanking the target genes and intervening antibiotic resistance cassettes as described previously (30,32).
Overexpression of E. coli topB
To express the E. coli topB gene in B. subtilis, we amplified the gene using the oligonucleotides PF79 and PF80, and cloned it between the XbaI and BamHI sites of the expression vector pHT01 (33). B. subtilis GP1966 was transformed with the plasmid, and recombinant cells were selected for chloramphenicol resistance. Expression of topB was induced by the addition of IPTG (1 mM) to the culture.
Staining and microscopy
For microscopy, cells were grown at 37°C in liquid LB medium overnight. Cells were harvested and re-suspended in PBS buffer (pH 7.4). DAPI and Nile Red were added to a final concentration of 20 μg/μl and staining occurred for 15 min in the dark. The cells were washed three times in PBS buffer (pH 7.4). Images were acquired using an Axioskop 40 FL fluorescence microscope, equipped with digital camera AxioCam MRm and AxioVision Rel (version 4.8) software for image processing (Carl Zeiss, Göttingen, Germany) and Neofluar series objective at ×100 primary magnification. We used the Filter set 49 (G 365, FT 395, BP 445/50; Carl Zeiss) for DAPI detection and Filter set 43 (BP 545/25, FT 570, BP 605/70) for Nile Red detection. Overlays of stained images were created with ImageJ 1.44p (National Institutes of Health, Bethesda, USA).
Primer extension
Total RNA was isolated from B. subtilis strains 168 (wild-type) and GP1985 grown in complex TY medium by the hot-phenol method (34) and treated with RNase-free DNase I (NEB) for 1 h at 37°C. 10 μg total RNA were hybridized to 5′-labeled-oligonucleotide SB3204 by incubation at 65°C for 5 min followed by quick cooling on ice. Reverse transcription was performed with Superscript IV RT (Invitrogen) in three subsequent 20 min steps at 50°C, 55°C and 65°C. Products were heat-denatured with formamide loading dye at 95°C for 5 min and separated on a 6% denaturing PAA gel along a Sanger sequencing reaction of plasmid pGP3111 with oligonucleotide SB3204 (Supplementary Table S1).
RESULTS
Inactivation of the topA gene results in the amplification of the parEC chromosomal region
Contradictory results have been reported about the essentiality of the topA gene encoding topo I in B. subtilis. While topA was found to be essential in two studies (10,11), the gene could be deleted in other genome-wide analyses (8,35). However, Koo et al. (8), were able to replace the topA gene by a kanamycin resistance gene, but not by other resistance cassettes, highlighting the importance of the gene. In an attempt to verify the possibility of topA deletion, the ΔtopA::aphA3 cassette from the mutant strain BKK16120 was amplified by PCR and used to transform our wild type strain B. subtilis 168. This transformation yielded two distinct types of colonies: the smaller colonies lysed rapidly whereas the larger colonies were stable. To test whether the deletion of the topA gene had occurred without further mutations, the chromosomal DNAs of two of the large transformants were subjected to whole genome sequencing. As expected, the topA gene was deleted and replaced by the aphA3 resistance gene in the two transformants. For both strains, the genome sequence revealed the amplification of a genomic region (see Figure 1A). In case of strain GP1948, the 12 kb yneP-xynC chromosomal region was amplified, whereas the 18 kb sirA-alsT region was amplified in the second strain, GP1952. The amplified regions of both strains overlap. The yneQ and yneR genes encoding proteins of unknown function, the essential plsY gene encoding the acylphosphate:glycerol-phosphate acyltransferase required for phospholipid biosynthesis, the yneT gene encoding a predicted coenzyme A-binding protein, the parEC operon encoding the two subunits of topo IV, and the ynfC gene of unknown function form the common core of the amplified region. Strikingly, the deletion of the topA gene generates a selective pressure to maintain the amplification of a topoisomerase-encoding operon. In contrast, the paralogous topB gene, encoding the second type I topoisomerase (topo III), was not affected by any mutation. In addition, we found several point mutations. Both strains carried a point mutation in the mreB gene, and an insertion of an A in the terminator region of the yfhJ gene. Mutations in mreB are frequently encountered in suppressor screens (11), and the mutation in the yfhJ terminator is not expected to have a phenotypic effect. Therefore, these mutations can be regarded as genetic hitchhikers. Moreover, strain GP1952 had a mutation in the panC gene resulting in an amino acid substitution (V128G) in the pantothenate synthase. The relevance of this mutation is unknown.
Figure 1.

Gene amplifications in ΔtopA suppressor strains. (A) Scheme of the parEC region of the B. subtilis wild type chromosome and of the amplifications in the suppressor mutants. (B) Scheme of the parEC region of the amplification reporter strain GP1966. The gfp gene replaces the ynfC gene. The suppressor strain GP1979 was isolated after deletion of topA in GP1966. The blue curves indicate the genome coverage of the corresponding genomic regions. Copy numbers are indicated on the right. Pictures of genome coverage were created using the Geneious Software version 10.0 (Biomatters Ltd., New Zealand).
The suppressor strains grew well on LB plates, however, the colonies lysed after prolonged incubation. To observe the further evolution of one of the topA mutants, we passaged strain GP1948 several times on LB plates. This resulted in the appearance of papilla-like colonies. One of them (GP1958) was selected, and subjected to further rounds of passaging on LB plates. This again gave rise to papillae that survived longer periods of incubation on plates (GP1963). The latter strain was subsequently passaged several times in liquid medium, and single colonies were isolated after eight passages. Two of these colonies (GP1974 and GP1975) were chosen for further analysis. The genomes of the four suppressor strains were sequenced. Interestingly, all these strains carried the amplification around the parEC operon, however, both the boundaries and the copy numbers of the amplified regions differed from strain to strain indicating ongoing dynamic evolution (see Figure 1A). In the first evolved strain (GP1958), the amplified region was identical to that of the parent strain. The next round of evolution exemplified by GP1963 resulted in a shift of the amplified region to the 12 kb citB-alsT region with about 16 copies of the amplified region per genome. Further cultivation had no effect on the boundaries and copy number of the amplified region (see Figure 1A). This suggests that this state of the amplification provided sufficient genome stability for the bacteria.
Topoisomerase I is essential for B. subtilis
The results described above suggest that the deletion of the topA gene results in selection for amplification of the parEC genomic region. To get further evidence for this conclusion, we decided to construct a screening system that allows the rapid detection of the parEC amplification. For this purpose, we inserted a cassette encoding the green fluorescent protein and an erythromycin resistance determinant (gfp-ermC) under the control of a weak constitutive promoter immediately downstream of the parEC operon (see Materials and Methods for the details of strain construction). The resulting reporter strain, GP1966, exhibited a faint fluorescence (see Figure 2). We then deleted the topA gene, and tested fluorescence of the emerging colonies. Upon topA deletion from GP1966, all resulting colonies immediately showed an intense fluorescence suggestive of an amplification of the parEC chromosomal region. Indeed, whole genome sequencing of one of the resulting mutants (GP1979) confirmed the amplification of parEC chromosomal region (see Figures 1B and 2). In this strain, the 12 kb yneP-xynC region was amplified 15-fold. The observation that no single stable transformant with the deletion of topA showed a faint fluorescence, i.e. had the parEC gfp region in the single copy state, indicates that the topA mutant is not viable unless this genomic region has been amplified. Thus, our results support the conclusion that the topA gene is essential and suggest that topo IV encoded by parEC might be able to substitute for topo I.
Figure 2.

Visualization of the parEC copy number using the amplification reporter system. Colony images (upper panel) and GFP intensities (lower panel) of the different strains are shown. The indexed numbers in the genotypes indicate the copy number of the parEC chromosomal region. Please note that the single copy parEC operon present in the suppressor strain GP1985 carries a promoter-up mutation.
The amplification of chromosomal regions depends on the functional recombination protein RecA. It has been shown previously that the lack of the RecA protein results in a drastic reduction of the amplification frequency in response to a selective pressure (25). In turn, the absence of RecA facilitates the detection of rare point mutations that may also suppress the original mutation. To test this idea for the suppression of the topA deletion, we intended to construct a recA topA double mutant to isolate new suppressor mutants. However, all attempts to construct such a strain failed, supporting the idea that topA suppression can only occur by amplification of the parEC region.
Interestingly, in E. coli loss of topA can also be suppressed by overexpression of topB (36). However, we failed to detect any mutants with increased topB expression. Therefore, we attempted to test suppression of the B. subtilis topA mutant by overexpression of E. coli topB. For this purpose, we have overexpressed plasmid-borne E. coli topo III (using plasmid pGP3121) in B. subtilis, and then attempted deletion of topA (see Figure 3). In this case, we observed that initially only a small part of the mutants had the parEC amplification (as judged by the GFP signal). In contrast, all topA mutants carrying the empty vector pHT01 had acquired the parEC amplification in the parallel control experiment. However, upon cultivation in liquid medium and re-isolation, many colonies exhibited fluorescence suggesting that they had acquired the suppressing amplification of the parEC region. This indicates that E. coli topB can indeed partially compensate for the loss of topA in B. subtilis. However, the eventual acquisition of the parEC amplification suggests that the suppression by topB is not sufficient and that the cells still experience a significant selective pressure to overexpress parEC.
Figure 3.

Suppression of a topA mutant by overexpression of E. coli topB. Strain GP1966 carrying the amplification reporter (ΔynfC::gfp) was transformed with a plasmid expressing the E. coli topB gene (A) or with the empty vector pHT01 (B). In these plasmid-bearing strains, the topA gene was deleted. The consequences for the amplification of the parEC chromosomal region were investigated using the gfp reporter gene inserted next to the parEC locus. Upper panel: initial transformants were photographed. Lower panel: Colonies exhibiting weak fluorescence were re-isolated, passaged in liquid LB medium, and re-analyzed for fluorescence.
Evolution of a strain carrying multiple parEC copies results in de-amplification of the parEC chromosomal region and the acquisition of point mutations
As shown above, it was impossible to obtain a topA mutant without concomitant amplification of the parEC locus suggesting that overexpression of topo IV compensates for the loss of topo I. However, strains with amplifications are genetically less stable, and we reasoned that our suppressor strains might become more stable with reduced copy number of the parEC locus. If this would be the case, one would expect a decrease in the brightness of fluorescence of the suppressor strain GP1979 upon long-term cultivation. To test this hypothesis, GP1979 (with 15 copies of parEC) was passaged ten times in LB medium. After each passage, a dilution series of the culture was plated on LB medium, and the colonies were screened for fluorescence intensity. After ten passages, we observed colonies with strongly reduced intensity of fluorescence, and one colony was as faint as the original strain GP1966 that contains only a single copy of the parEC region (see Figure 2). The strain with the strongest loss of fluorescence intensity (GP1985) as well as two of the strains (GP1983 and GP1987) with reduced intensity were analyzed by genome sequencing. Strikingly, only one copy of the parEC region was present in strain GP1985 whereas the other two strains still harbored four copies of this genomic region. This finding confirms our hypothesis that the strain with the amplification undergoes evolution to reduce the copy number of the amplified region and thus to stabilize the genome. The reduction of the parEC chromosomal region copy number in the suppressor strains was accompanied by the appearance of novel point mutations. In GP1985, mutations resulting in amino acid substitutions in CodY (Ser129Leu), GltA (Val1181Ala) and RocG (Ser61Arg) were detected. In addition, this strain had a mutation in the upstream region of the parEC operon. This operon is preceded by a 315 nucleotides long leader RNA (RNA feature S672 according to 37), and the mutation changed the C at position 5 of the RNA to a T (see Figure 4A, note that primer extension experiments revealed that the mutation affected the promoter upstream of this RNA feature, see below). The mutations affecting CodY and RocG were also found in the strains with intermediate levels of amplification (GP1983 and GP1987). Interestingly, these strains were ambiguous concerning the sequence of the parEC upstream region. In these strains, we detected both the wild type sequence as well as the C5T variant of the S672 RNA feature. It is likely that the multiple copies of the parEC chromosomal region in these strains encode these different variants of the S672 RNA feature resulting in a mixed sequence at this position. Taken together, our results suggest that we have observed distinct steps in the evolution pathway of the topA suppressor mutants: The amplification of the parEC region is followed by the reduction in copy number (de-amplification) and the concomitant acquisition of point mutations that allow compensation of the topA deletion. In the second step, only one copy of the amplified region is retained, and the selected point mutations allow growth of the bacteria in the absence of topo I.
Figure 4.

Genetic organization of the parE promoter region. (A) Structure of the parE promoter region. Sequence of the promoter region showing the -35 and -10 regions; as well as the transcription start site. (B) Reverse-transcriptase mapping of the transcriptional start point of the parE gene using the primer SB3204. Sequencing reactions were performed using the same oligonucleotide as primer.
In the strain with the finalized de-amplification of the parEC region (GP1985), we found point mutations upstream of the parEC coding sequence as well as in genes encoding three presumably unrelated proteins (CodY, GltA, RocG). The mutation in the parEC leader mRNA might result in higher expression of this operon, similar to the amplification of the region. This hypothesis suggests that the point mutation might be sufficient for the suppression. To test this idea, we used chromosomal DNA of the suppressor mutant GP1985 to transform the wild type strain B. subtilis 168. Transformants were selected for both kanamycin and erythromycin resistance, i.e. for the simultaneous transfer of both the topA deletion and the amplification reporter cassette downstream of the parEC operon. All transformants exhibited faint fluorescence excluding amplification of the parEC region. In 13 of these transformants, the chromosomal regions carrying point mutations in GP1985 (codY, gltA, rocG, S672) were sequenced. The C5T mutation upstream of parEC was present in all sequenced transformants, whereas gltA and rocG were wild type (one exception of transfer of the rocG mutation from GP1985). The codY alleles of the sequenced transformants exhibited variability: In most cases, the mutation from GP1985 was transferred with the resistance markers, and only in 25% of the transformants we observed the wild type allele of codY. The frequent transfer of the codY mutation is likely caused by the genetic linkage between topA (replaced by the aphA3 resistance gene) and codY rather than by a selective pressure. One of the strains with only the point mutation upstream of parEC was selected for further experiments and designated GP2547. This strain was subjected to whole genome sequencing. As expected, we found a single copy of the parEC region and the C5T point mutation upstream of parEC. Moreover, a SNP was present in the yhcF gene. Since this mutation did not result in an amino acid exchange, it is unlikely to be related to the suppressor phenotype.
To get independent confirmation for the reduction of the parEC copy number after the initial amplification, we passaged three further independently isolated suppressor mutants (GP2543, GP5244, GP2545) that carried the parEC amplification, until the intensity of the fluorescence caused by the concomitant gfp amplification was reduced to the levels observed for the original strain GP1966. For each of the three suppressor mutants, we tested several of the evolved strains for the presence of the parEC point mutation. Indeed, each independently isolated suppressor mutant gave rise to evolved strains that had acquired the same point mutation upstream of parEC as described above. Moreover, two of the evolved strains (one with the promoter mutation and one lacking it) were subjected to genome sequencing. In strain GP2549, we found the parEC point mutation accompanied by a point mutation in rpoC affecting the β’ subunit of RNA polymerase (Thr891Ile). In the second strain, three copies of the parEC region (yneP … xynC, see Figure 1) were present, and the parEC upstream region was identical to that of the wild type. This indicates that a triplication of the parEC genomic region is sufficient for the suppression of the loss of the topA gene.
Taken together, these results demonstrate that the original suppressor mutants reduce the copy number of the amplified region, and replace the amplification by a point mutation upstream of the parEC operon. This mutation is sufficient for the suppression of the loss of topo I in B. subtilis. This conclusion is supported by the observation of similar DNA supercoiling in the wild type strain 168 and in the suppressed topA mutant GP1985 (see Supplementary Figure S1).
The observed evolution of the strains upon topA deletion suggests that the physiological properties of the bacteria in the distinct states of evolution were different. To test this idea, we compared growth of the wild type strain B. subtilis 168, the wild type strain carrying the amplification reporter system GP1966, as well as of the topA mutant with the amplification of parEC or the point mutation upstream of the parEC operon (GP1979 and GP2547, respectively) (see Figure 5). The growth of the wild type strain and the wild type strain carrying the reporter construct was indistinguishable indicating that the loss of the ynfC gene and the introduction of the gfp-ermC reporter cassette did not compromise the physiology of the bacteria. In contrast, deletion of topA accompanied by the amplification of the parEC genomic region resulted in a severe delay of growth. Moreover, we observed a biphasic growth of the bacteria indicating a re-organisation of metabolism during growth. As expected for evolved bacteria, the strain with a single parEC operon copy but the point mutation in the upstream region (GP2547), grew significantly faster compared to the strain with the amplification, but slightly slower than the isogenic wild type strain (GP1966). However, this strain also exhibited diauxic growth indicative of a re-adaptation of the bacteria during cultivation (see Discussion). The growth properties of GP2547 are in good agreement with the observation that no further suppressor mutations could be isolated indicating that this strain has found the best possible response to the loss of topo I.
Figure 5.
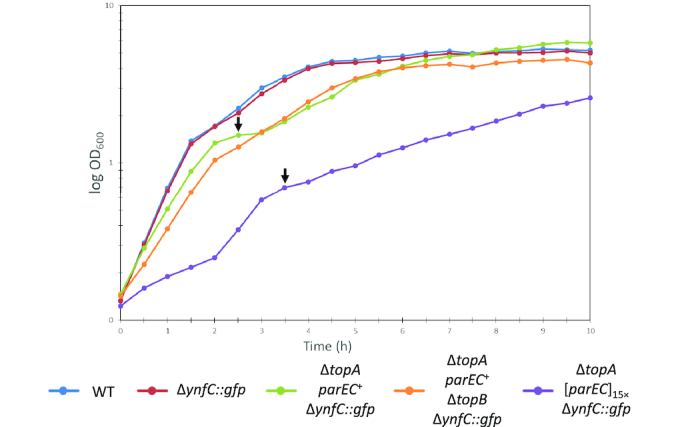
ΔtopA suppressor strains exhibit reduced growth. Strains were grown in liquid LB medium for 10 h at 37°C and 240 rpm. The OD600 was measured every 30 min. Curves represent the average of three independent replicates. Arrows indicate a second lag phase indicating diauxic growth of the mutants. Genotypes: 168: Wild type. GP1966: ΔynfC::Palf4-gfp-ermC. GP2547: ΔynfC::Palf4-gfp-ermC ΔtopA::aphA3 [parEC]1× C→T mutation in -10 region of parEC promoter. GP1979: trpC2 ΔynfC::Palf4-gfp-ermC ΔtopA::aphA3 [parEC]15×. GP2548: ΔynfC::Palf4-gfp-ermC ΔtopA::aphA3 ΔtopB::cat [parEC]1× C→T mutation in –10 region of parEC promoter.
The mutation in the parEC upstream region affects expression of the parEC operon by increasing promoter strength
The results presented above suggest that the parEC amplification and the point mutation in the upstream region might result in increased parEC expression which then causes suppression of the topA mutation. To test this hypothesis, we compared the promoter activities of the wild type and mutant parEC upstream regions using fusions to a promoterless lacZ reporter gene encoding β-galactosidase. The reporter strains GP2550 and GP1967 carrying the wild type and mutant promoter fusions, respectively, were cultivated in LB medium and the β-galactosidase activities were determined. The wild type promoter gave rise to 85 ± 13 units mg−1 of protein. In contrast, the mutant promoter region exhibited a fivefold increased activity (498 ± 26 units mg−1 of protein). These results suggest that the expression of parEC is indeed increased in the mutant.
The increased promoter activity of the mutant upstream region present in the suppressor strain may result from the generation of a novel promoter or from increased activity of the original promoter. To distinguish between these possibilities, we determined the transcription start points in the wild type and the suppressor strain. Primer extension experiments with total RNA isolated from wild type strain 168 and the mutant strain GP1985 revealed that transcription started in both strains at the same position, a single A residue (see Figure 4B). Moreover, the signal intensity was strongly increased in the mutant strain indicating that the mutation results in a stronger promoter activity. Indeed, the mutation has altered the –10 box of the Sigma A-type promoter from TACAAT to the perfect consensus –10 box TATAAT resulting in increased strength of the transcription initiation signal.
Overexpression of topo IV allows growth of B. subtilis in the absence of both type I topoisomerases
The B. subtilis genome encodes two paralogous type I DNA topoisomerases, the essential topo I (TopA) and the non-essential topo III (TopB). Our results indicate that B. subtilis is viable in the absence of the essential enzyme, provided that topo IV is overexpressed. However, it is not known whether B. subtilis is viable in the absence of both type I topoisomerases. To construct a ΔtopA ΔtopB double knockout mutant, we used chromosomal DNA of a topA mutant to transform the topB mutant BKE04260. No transformants were obtained in this way. In contrast, transformation of the topA mutant GP1963 which exhibited amplification of the parEC region with chromosomal DNA of the topB mutant was possible and yielded the ΔtopA ΔtopB double mutant GP1964 (see Discussion). Similarly, the double mutant lacking both type I topoisomerases could be generated based on the suppressor strain carrying the mutation upstream of parEC resulting in strain GP2548. These results suggest that overexpression of topo IV encoded by parEC is sufficient to allow growth of B. subtilis without any type I topoisomerase.
In E. coli, deletion of both topA and topB results in severe growth and chromosome segregation defects (38,39). To test whether this is also the case in B. subtilis, we determined the growth behaviour of the suppressed topA topB double mutant GP2548 in liquid medium and on plates. As shown in Figure 5, growth of the double mutant was very similar to the otherwise isogenic suppressed topA mutant GP2547. However, as the topA single mutant, the double mutant exhibited long filamentous cells. DAPI staining indicated that in some of these filamentous cells the chromosomes were segregated as in the wild type, whereas some cells had a segregation defect (see Figure 6). This segregation defect was not observed in the suppressed topA mutant (GP2547), and is therefore a result of the simultaneous loss of both type 1A topoisomerases. This observation suggests that loss of topo III in addition to deletion of the topA gene also results in additional problems for the cell (DNA segregation), and this problem can not be solved by overexpression of topo IV.
Figure 6.

Phenotypic analysis of B. subtilis topoisomerase mutants. The wild type strain B. subtilis 168, the topA mutants GP1979 and GP2547, the topB mutant GP1896, and the topA topB double mutant GP2548 were analyzed for colony morphology (left and second left panel). Moreover, the cells were analyzed by phase contrast microscopy, for the nucleoid (DAPI stain), and for the cell membrane (Nile Red stain). The right panel shows an overlay of the DAPI and Nile Red stains.
DISCUSSION
DNA topoisomerases are the key players in the global control of chromosome conformation. Given their important functions in chromosome compaction and segregation during DNA replication, it is not surprising that these enzymes are essential for bacterial life. In the current study, we have investigated the essentiality of topo I encoded by the topA gene in the Gram-positive model organism B. subtilis. Our results demonstrate that topo I is essential unless topo IV can compensate for the loss of the topA gene.
In principle, all bacterial DNA topoisomerases of both types have both the relaxation and the catenation/ decatenation activity. However, each of the enzymes has different kinetic properties that finally result in the prevailing activities for controlling DNA supercoiling (topo I and gyrase) and DNA catenation/decatenation (topo III and topo IV) (40–43, see Table 1). Topo I, topo III and topo IV are all able to relax hyper-negative supercoils behind RNA polymerase, and therefore both topo III and topo IV are candidates for the suppression of the loss of topo I. However, in our experiments we consistently observed that overexpression of topo IV may overcome the lack of topo I, whereas overexpression of topo III was never observed. A particular feature that distinguishes the two type I topoisomerases is their C-terminal domain. While topo I contains a zinc ribbon domain at the C-terminus, this domain is not present in topo III. In E. coli, the zinc ribbon domain interacts with the β’ subunit of RNA polymerase thus bringing the relaxing activity of topo I to the site of transcription where its activity is needed (44). Interestingly, an interaction between topo I and RNA polymerase has also been observed in B. subtilis (45). Moreover, the C-terminal domain of topo I is also required for the recognition of hyper-negatively supercoiled DNA, the preferred catalytic substrate of topo I (46). The ability of multi-copy E. coli topo III to compensate for the loss of topo I both in E. coli and B. subtilis (36, see Figure 3) which was not observed with the B. subtilis protein suggests important differences between the topo III enzymes of the two bacteria.
As for B. subtilis, contradicting results have been obtained about the essentiality of topo I in E. coli. While a deletion of the topA gene was not possible in a genome-wide inactivation study (7), other studies reported that topA mutants were viable if the deletion of topA was accompanied by the simultaneous acquisition of suppressor mutations (47,48). Finally, one study suggested that E. coli topA mutants might be viable without any suppressing mutations (49). However, the genomes of the latter mutants have not been sequenced indicating that the presence of compensatory mutations cannot be excluded. In E. coli, gyrA or gyrB mutations resulting in reduced DNA gyrase activity and thus reduced negative DNA supercoiling support growth of the topA mutant (48,50). We did never observe mutations affecting DNA gyrase in our suppressor screens. Among all DNA topoisomerase genes, the gyrAB operon is most strongly expressed in B. subtilis (37) suggesting that mutations that reduce gyrase activity may not be tolerated in this organism. In addition, overexpression of either topo III encoded by topB or of topo IV encoded by the parE and parC genes allowed growth of the E. coli topA mutant (36,51,52). Similarly, overexpression of topo IV compensated for loss of topo I in Shigella flexneri (53). Based on our results using the amplification screen, we conclude that in B. subtilis the topA mutant is only viable if it acquires compensatory mutations, and that these compensatory mutations without any exception result in overexpression of the parEC operon encoding topo IV. Thus, the mutation landscape of possible topA suppressors is much narrower in B. subtilis than it is in E. coli. The reason for this difference is so far unknown.
Most bacteria contain type I and type II topoisomerases. While the type I enzyme TopA as well as both type II enzymes are individually essential in B. subtilis, the type I protein TopB (topo III) is not (8). It was therefore of particular interest whether the deletion of topA would make topB essential. Importantly, this was not the case if the effect of the topA deletion was suppressed by overexpression of ParEC. Similar to the E. coli topA topB double mutant (38), the B. subtilis strain lacking both type 1A topoisomerases exhibited filamentous morphology and had DNA segregation defects. As described earlier, this segregation defect may result from a redundant function of topo I and topo III during constitutive stable DNA replication to allow chromosome segregation (38).
In our work, we have observed either amplification of the parEC genes or a promoter-up mutation that results in about fivefold increased expression of the parEC operon. Moreover, DNA supercoiling was similar in the wild type and in the topA deletion mutant with increased parEC expression (see Supplementary Figure S1). Taken together, these data demonstrate that overexpression of topo IV is sufficient to compensate at least partially for the loss of topo I. This conclusion is in agreement with the results of in vivo studies in E. coli that demonstrated a five-fold reduced DNA relaxation activity of topo IV as compared to topo I (54).
One interesting result of this work is the observed diauxic growth of the suppressor mutants in complex medium (Figure 5). This observation suggests that the bacteria rapidly exhaust a particular nutrient from the medium, and that they experience difficulties in swiftly adapting their metabolism. In this context, it is interesting to note that we have observed large scale alterations in central metabolism in B. subtilis as a result of changes in DNA topology (11). Strikingly, glutamate biosynthesis was more efficient as a result of increased topo I activity which in turn may suggest that the expression of the gltAB operon encoding glutamate synthase is reduced upon loss of topo I. Interestingly, we found point mutations in two genes involved in glutamate metabolism in the originally isolated suppressor strain that had lost the amplification of the parEC region. In this strain, GP1985, the GltA and RocG proteins involved in glutamate synthesis and degradation, respectively, carried mutations. While these mutations were not essential for the suppression they may contribute to improved growth. Indeed, previous work has shown that B. subtilis tightly controls glutamate homeostasis, by the regulation of gene expression, but also by the rapid acquisition of suppressor mutations (25,55,56). Interestingly, DNA topology also affects the synthesis of glutamine, an amino acid directly derived from glutamate in E. coli (57), suggesting a more general link between DNA topology and the synthesis of central amino acids.
In this work, we have established a screening system to detect chromosomal amplifications. Using this system, we were able to demonstrate that each individual stable topA mutant had acquired the amplification of the parEC region. Moreover, this system was very helpful in the analysis of the further evolution of the strains: after several passages we observed the reduction of the parEC copy number accompanied by the acquisition of a parEC promoter up mutation in some of the copies. In the final step, only one copy of the parEC region was present, and overexpression of parEC was now achieved by the stronger promoter. Based on the fact that about 10% of all cells in a bacterial population carry duplicated chromosomal segments (58), it is not surprising that strains with distinct amplifications of the parEC chromosomal region did readily appear. Similar results have been obtained with a B. subtilis strain lacking the transcriptional activator for the glutamate biosynthetic genes. In such strains, the gltAB operon encoding glutamate synthase can be amplified up to 20-fold to compensate for the lack of transcription activation (25). Using our reporter system, the origin of mutations can be studied at the single cell level to the molecular details.
Taken together, our work demonstrates that the topA gene is essential unless the loss of topo I is compensated by the overexpression of topo IV. Increased expression of topo IV is even sufficient to replace both type 1A topoisomerases, TopA and TopB. Interestingly, very similar results have very recently been reported for E. coli. In these bacteria, increased levels of topo IV that can relax excess negative supercoiling and prevent R-loop formation can substitute both type 1A topoisomerases (59). Based on the results presented here, a set of three topoisomerases including topo I, DNA gyrase, and topo IV should be included in any minimal genome to obtain a strain that is able to grow rapidly on a complex medium.
DATA AVAILABILITY
The raw fastq reads of strains gp1948, gp1979 and gp1985 have been deposited in the NCBI-SRA database (https://www.ncbi.nlm.nih.gov/sra) and are accessible under the accession numbers srr7967955, srr7967956 and srr7967957, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Fabian M. Commichau and Christina Herzberg for advice and helpful discussion.
Author contributions: Design of the study: D.R.R. and J.S.; experimental work: D.R.R., P.F., P.J.M., A.P. and B.M.K.; sequence analysis: D.R.R., A.P. and R.D.; wrote the paper: D.R.R., B.M.K., C.A.G., P.F., S.B. and J.S.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
EU Horizons 2020 program [Rafts4Biotech, 720776 to J.S.]; NIH [R35 GM118061 to C.A.G.]. Funding for open access charge: European Commission.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002; 3:430–440. [DOI] [PubMed] [Google Scholar]
- 2. Steck T.R., Franco R.J., Drlica K.. Topoisomerase mutations affect the relative abundance of many Escherichia coli proteins. Mol. Microbiol. 1993; 10:473–481. [DOI] [PubMed] [Google Scholar]
- 3. Chen S.H., Chan N.L., Hsieh T.. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013; 82:139–170. [DOI] [PubMed] [Google Scholar]
- 4. Forterre P., Gribaldo S., Gadelle D., Serre M. C.. Origin and evolution of DNA topoisomerases. Biochimie. 2007; 89:427–446. [DOI] [PubMed] [Google Scholar]
- 5. Ghilarov D. A., Shkundina I. S.. DNA topoisomerases and their functions in a cell. Mol. Biol. 2011; 46:47–57. [PubMed] [Google Scholar]
- 6. Nöllmann M., Crisona N.J., Arimondo P.B.. Thirty years of Escherichia coli DNA gyrase: from in vivo function to single-molecule mechanism. Biochimie. 2007; 89:490–499. [DOI] [PubMed] [Google Scholar]
- 7. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006; 2:doi:10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koo B.M., Kritikos G., Farelli J.D., Todor H., Tong K., Kimsey H., Wapinski I., Galardini M., Cabal A., Peters J.M. et al.. Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst. 2017; 4:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Srivatsan A., Han Y., Peng J., Tehranchi A.K., Gibbs R., Wang J.D., Chen R.. High-precision, whole genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet. 2008; 4:e1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi K., Ehrlich S.D., Albertini A., Amati G., Andersen K.K., Arnaud M., Asai K., Ashikaga S., Aymerich S., Bessières P. et al.. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reuß D.R., Rath H., Thürmer A., Benda M., Daniel R., Völker U., Mäder U., Commichau F.M., Stülke J.. Changes of DNA topology affect the global transcription landscape and allow rapid growth of a Bacillus subtilis mutant lacking carbon catabolite repression. Metab. Eng. 2018; 45:171–179. [DOI] [PubMed] [Google Scholar]
- 12. Aubry A., Fisher L.M., Jarlier V., Cambau E.. First functional characterization of a singly expressed bacterial type II topoisomerase: the enzyme from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 2006; 348:158–165. [DOI] [PubMed] [Google Scholar]
- 13. Debowski A.W., Carnoy C., Verbrugghe P., Nilsson H.O., Gauntlett J.C., Fulurija A., Camilleri T., Berg D.E., Marshall B.J., Benghezal M.. Xer recombinase and genome integrity in Helicobacter pylori, a pathogen without topoisomerase IV. PLoS One. 2012; 7:e33310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wall M.K., Mitchenall L.A., Maxwell A.. Arabidopsis thaliana DNA gyrase is targeted to chloroplasts and mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Glass J.I., Assad-Garcia N., Alperovich N., Yooseph S., Lewis M.R., Maruf M., Hutchison C.A. 3rd, Smith H.O., Venter J.C.. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lluch-Senar M., Delgado J., Chen W.H., Lloréns-Rico V., O’Reilly F.J., Wodke J.A.H., Unal E.B., Yus E., Martínez S., Ferrar T. et al.. Defining a minimal cell: essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 2015; 11:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hutchison C.A. 3rd, Chuang R.Y., Noskov V.N., Assad-Garcia N., Deerinck T.J., Ellisman M.H., Gill J., Kannan K., Karas B.J., Ma L. et al.. Design and synthesis of a minimal bacterial genome. Science. 2016; 351:aad6253. [DOI] [PubMed] [Google Scholar]
- 18. Commichau F.M., Pietack N., Stülke J.. Essential genes in Bacillus subtilis: a re-evaluation after ten years. Mol. BioSyst. 2013; 9:1068–1075. [DOI] [PubMed] [Google Scholar]
- 19. Reuß D.R., Commichau F.M., Gundlach J., Zhu B, Stülke J.. The blueprint of a minimal cell: MiniBacillus. Microbiol. Mol. Biol. Rev. 2016; 80:955–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reuß D.R., Altenbuchner J., Mäder U., Rath H., Ischebeck T., Sappa P.K., Thürmer A., Guérin C., Nicolas P., Steil L. et al.. Large-scale reduction of the Bacillus subtilis genome: consequences for the transcriptional network, resource allocation, and metabolism. Genome Res. 2017; 27:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sambrook J., Fritsch E.F., Maniatis T.. Molecular Cloning: A Laboratory Manual. 1989; 2nd ednNY: Cold Spring Harbor Laboratory, Cold Spring Harbor. [Google Scholar]
- 22. Kunst F., Rapoport G.. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J. Bacteriol. 1995; 177:2403–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barbe V., Cruveiller S., Kunst F., Lenoble P., Meurice G., Sekowska A., Vallenet D., Wang T., Moszer I., Médigue C. et al.. From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology. 2009; 155:1758–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C. et al.. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012; 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dormeyer M., Lübke A.L., Müller P., Lentes S., Reuß D.R., Thürmer A., Stülke J., Daniel R., Brantl S., Commichau F.M.. Hierarchical mutational events compensate for glutamate auxotrophy of a Bacillus subtilis gltC mutant. Environ. Microbiol. Rep. 2017; 9:279–289. [DOI] [PubMed] [Google Scholar]
- 26. Gundlach J., Herzberg C., Kaever V., Gunka K., Hoffmann T., Weiß M., Gibhardt J., Thürmer A., Hertel D., Daniel R. et al.. Control of potassium homeostasis is an essential function of the second messenger cyclic di-AMP in Bacillus subtilis. Sci. Signal. 2017; 10:eaal3011. [DOI] [PubMed] [Google Scholar]
- 27. Cascante-Estepa N., Gunka K., Stülke J.. Localization of components of the RNA-degrading machine in Bacillus subtilis. Front. Microbiol. 2016; 7:1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lehnik-Habrink M., Pförtner H., Rempeters L., Pietack N., Herzberg C., Stülke J.. The RNA degradosome in Bacillus subtilis: identification of CshA as the major helicase in the multiprotein complex. Mol. Microbiol. 2010; 77:958–971. [DOI] [PubMed] [Google Scholar]
- 29. Martin-Verstraete I., Débarbouillé M., Klier A., Rapoport G.. Interactions of wild-type and truncated LevR of Bacillus subtilis with the upstream activating sequence of the levanase operon. J. Mol. Biol. 1994; 241:178–192. [DOI] [PubMed] [Google Scholar]
- 30. Guérout-Fleury A.M., Shazand K., Frandsen N., Stragier P.. Antibiotic-resistance cassettes for Bacillus subtilis. Gene. 1995; 167:335–336. [DOI] [PubMed] [Google Scholar]
- 31. Martin-Verstraete I., Débarbouillé M., Klier A., Rapoport G.. Mutagenesis of the Bacillus subtilis “-12, -24” promoter of the levanase operon and evidence for the existence of an upstream activating sequence. J. Mol. Biol. 1992; 226:85–99. [DOI] [PubMed] [Google Scholar]
- 32. Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996; 12:259–265. [DOI] [PubMed] [Google Scholar]
- 33. Nguyen H.D., Phan T.T., Schumann W.. Expression vectors for the rapid purification of recombinant proteins in Bacillus subtilis. Curr. Microbiol. 2007; 55:89–93. [DOI] [PubMed] [Google Scholar]
- 34. Licht A., Preis S., Brantl S.. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in Bacillus subtilis. Mol. Microbiol. 2005; 58:189–206. [DOI] [PubMed] [Google Scholar]
- 35. Thomaides H.B., Davison E.J., Burston L., Johnson H., Brown Hunt A.C., Errington J., Czaplewski L.. Essential bacterial functions encoded by gene pairs. J. Bacteriol. 2007; 189:591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Broccoli S., Phoenix P., Drolet M.. Isolation of the topB gene encoding topoisomerase III as a multicopy suppressor of topA null mutations in Escherichia coli. Mol. Microbiol. 2000; 35:58–68. [DOI] [PubMed] [Google Scholar]
- 37. Nicolas P., Mäder U., Dervyn E., Rochat T., Leduc A., Pigeonneau N., Bidnenko E., Marchadier E., Hoebeke M., Aymerich S. et al.. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012; 335:1103–1106. [DOI] [PubMed] [Google Scholar]
- 38. Martel M., Balleydier A., Sauriol A., Drolet M.. Constitutive stable DNA replication in Escherichia coli cells lacking type 1A topoisomerase activity. DNA Repair (Amst). 2015; 35:37–47. [DOI] [PubMed] [Google Scholar]
- 39. Zhu Q., Pongpech P., DiGate R.J.. Type I topoisomerase activity is required for proper chromosomal segregation in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:9766–9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Terekhova K., Gunn, Marko K.H., Mondragón J.F., Bacterial A.. topoisomerase I and topoisomerase III relax supercoiled DNA via distinct pathways. Nucleic Acids Res. 2012; 40:10432–10440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seol Y., Hardin A.H., Strub M.P., Charvin G., Neuman K.C.. Comparison of DNA decatenation by Escherichia coli topoisomerase IV and topoisomerase III: implications for non-equilibrium topology simplification. Nucleic Acids Res. 2013; 41:4640–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zechiedrich E.L., Khodursky A.B.. Cozzarelli N.R. Topoisomerase IV, not gyrase, decatenates products of site-specific recombination in Escherichia coli. Genes Dev. 1997; 11:2580–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ullsperger C., Cozzarelli N.R.. Contrasting enzymatic activities of topoisomerase IV and DNA gyrase from Escherichia coli. J. Biol. Chem. 1996; 271:31549–31555. [DOI] [PubMed] [Google Scholar]
- 44. Cheng B., Zhu C.X., Ji C., Ahumada A., Tse-Dinh Y.C.. Direct interaction between Escherichia coli RNA polymerase and the zinc ribbon domains of DNA topoisomerase I. J. Biol. Chem. 2003; 278:30705–30710. [DOI] [PubMed] [Google Scholar]
- 45. Delumeau O., Lecointe F., Muntel J., Guillot A., Guédon E., Monnet V., Hecker M., Becher D., Polard P., Noirot P.. The dynamic protein partnership of RNA polymerase in Bacillus subtilis. Proteomics. 2011; 11:2992–3001. [DOI] [PubMed] [Google Scholar]
- 46. Tan K., Zhou Q., Cheng B., Zhang Z., Joachimiak A., Tse-Dinh Y.C.. Structural basis for suppression of negative supercoiling by E. coli topoisomerase I. Nucleic Acids Res. 2015; 43:11031–11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stockum A., Lloyd R.G., Rudolph C.J.. On the viability of Escherichia coli cells lacking DNA topoisomerase I. BMC Microbiol. 2012; 12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DiNardo S., Voelkel K.A., Sternglanz R., Reynolds A.E., Wright A.. Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell. 1982; 31:43–51. [DOI] [PubMed] [Google Scholar]
- 49. Stupina V.A., Wang J.C.. Viability of Escherichia coli topA mutants lacking DNA topoisomerase I. J. Biol. Chem. 2005; 280:355–360. [DOI] [PubMed] [Google Scholar]
- 50. Heddle J.G., Lu T., Zhao X., Drlica K., Maxwell A.. gyrB-225, a mutation of DNA gyrase that compensates for topoisomerase I deficiency: investigation of its low activity and quinolone hypersensitivity. J. Mol. Biol. 2001; 309:1219–1231. [DOI] [PubMed] [Google Scholar]
- 51. Dorman C.J., Lynch A.S., Ni Bhriain N., Higgins C.F.. DNA supercoiling in Escherichia coli: topA mutations can be suppressed by DNA amplifications involving the tolC locus. Mol. Microbiol. 1989; 3:531–540. [DOI] [PubMed] [Google Scholar]
- 52. Kato J., Nishimura Y., Imamura R., Niki H., Hiraga S., Suzuki H.. New topoisomerase essential for chromosome segregation in E. coli. Cell. 1990; 63:393–404. [DOI] [PubMed] [Google Scholar]
- 53. McNairn E., Ni Bhriain N., Dorman C.J.. Overexpression of the Shigella flexneri genes coding for DNA topoisomerase IV compensates for loss of DNA topoisomerase I: effect on virulence gene expression. Mol. Microbiol. 1995; 15:507–517. [DOI] [PubMed] [Google Scholar]
- 54. Zechiedrich E.L., Khodursky A.B., Bachellier S., Schneider R., Chen D., Lilley D.M.J., Cozzarelli N.R.. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000; 275:8103–8113. [DOI] [PubMed] [Google Scholar]
- 55. Commichau F.M., Gunka K., Landmann J.J., Stülke J.. Glutamate metabolism in Bacillus subtilis: gene expression and enzyme activities evolved to avoid futile cycles and to allow rapid responses to perturbations of the system. J. Bacteriol. 2008; 190:3557–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gunka K., Commichau F.M.. Control of glutamate homeostasis in Bacillus subtilis: a complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol. Microbiol. 2012; 85:213–224. [DOI] [PubMed] [Google Scholar]
- 57. Hayashi M., Tabata K.. Metabolic engineering for L-glutamine overproduction by using DNA gyrase mutations in Escherichia coli. Appl. Environ. Microbiol. 2013; 79:3033–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Anderson R.P., Roth J.R.. Tandem genetic duplications in phage and bacteria. Annu. Rev. Microbiol. 1977; 31:473–505. [DOI] [PubMed] [Google Scholar]
- 59. Brochu J., Vlachos-Breton É., Sutherland S., Martel M., Drolet M.. Topoisomerases I and III inhibit R-loop formation to prevent unregulated replication in the chromosomal Ter region of Escherichia coli. PLoS Genet. 2018; 14:e1007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw fastq reads of strains gp1948, gp1979 and gp1985 have been deposited in the NCBI-SRA database (https://www.ncbi.nlm.nih.gov/sra) and are accessible under the accession numbers srr7967955, srr7967956 and srr7967957, respectively.