


Abstract
Engineering allosteric transcriptional repressors containing an environmental sensing module (ESM) and a DNA recognition module (DRM) has the potential to unlock a combinatorial set of rationally designed biological responses. We demonstrated that constructing hybrid repressors by fusing distinct ESMs and DRMs provides a means to flexibly rewire genetic networks for complex signal processing. We have used coevolutionary traits among LacI homologs to develop a model for predicting compatibility between ESMs and DRMs. Our predictions accurately agree with the performance of 40 engineered repressors. We have harnessed this framework to develop a system of multiple toggle switches with a master OFF signal that produces a unique behavior: each engineered biological activity is switched to a stable ON state by different chemicals and returned to OFF in response to a common signal. One promising application of this design is to develop living diagnostics for monitoring multiple parameters in complex physiological environments and it represents one of many circuit topologies that can be explored with modular repressors designed with coevolutionary information.
INTRODUCTION
Engineering of biological behavior often requires reprogramming of biological and chemical cues that a biological system recognizes, and also the resulting response from these signals (1–4). Scientists have engineered various biological components that enable the use of multiple molecular signals to regulate the activity of a particular DNA-based promoter for driving gene expression, which provides a means to flexibly alter those pathways between signal detection and cellular response. These engineered components include the Tango system (5), chimeric antigen receptors (6), scaffold-based two-component systems (7), synNotch receptors (8), MESA receptors (9) and the dCas9-SynR system (10). These engineered parts rewire signal-response connections and generate new possibilities in practical applications. For instance, synNotch has been demonstrated as a powerful tool for engineering T-cells that participate in various cell-based therapies (11). In another example, chimeric antigen receptors have been utilized to reprogram the response of immune cells to eliminate specific cancer cells under clinical environments (6,12). These examples exhibit that modular parts for rewiring biological input-output connections are critical for developing cells with desirable functions and behavior.
In addition, allosterically regulated transcriptional repressors are good candidates for developing universal parts. Many of these repressors comprise an N-terminal DNA recognition module (DRM) that interacts with promoters and a C-terminal environmental sensing module (ESM) that senses molecular signals. They regulate transcription by binding to DNA-based promoters in response to small molecules that are permeable to most types of cells—their mechanism of action is simple and does not involve additional biological components from the host, rendering them functional for many biological systems. Indeed, some repressors, such as LacI and TetR, have been used to develop inducible expression systems in yeast (13), human cells (14), plants (15), animals (16), and many microbial organisms (17). Based on these previous studies, modular parts that developed from allosterically regulated transcriptional repressors are expected to serve as circuit parts in many cell types.
Several studies have demonstrated that transcriptional repressors are feasible for generating modular sensors with high efficiency (18–20). Swint-Kruse et al. have used an N-terminal region of the repressor, LacI, to replace homologous sequences from a number of repressors within the LacI family (18). They then randomized the amino acid fragments at the interface of the two regions in these chimeric proteins, and used a screening platform to identify mutants with desirable functions (19). We previously discovered a conserved boundary between DNA recognition modules and environmental sensing modules among some LacI family repressors (20), which led us to develop a module-swapping strategy to construct modular repressors: by fusing a DRM and an ESM from two different repressors, the resulting hybrid repressors possess the corresponding properties of DNA recognition and allosteric response from their respective native modules (Figure 1A). Based on this discovery, we previously used two DRMs and three ESMs to construct a set of six engineered repressors that enable flexible connections between small molecule sensing and promoter control. In doing so, we harnessed these engineered parts to establish a novel circuit topology that programs a cellular decision depending on three molecular signals in Escherichia coli (20). These studies show that transcriptional repressors are promising for creating robust, modular parts that facilitate cell engineering. Based upon this module-swapping strategy, we have extensively expanded the set of hybrid repressor in this current study. However, not all DRMs and ESMs are compatible to each other and the resulting hybrid repressors are poorly functional. This led us to use a computational approach based on coevolutionary information to model module compatibility. In this work, we aim to develop and use this computational model to predict the performance of hybrid repressors and subsequently guide the design of engineered hybrids.
Figure 1.

Development of a computational model to predict compatibility among DRMs and ESMs from LacI homologs. (A) Module swapping strategy. By combining DRMs and ESMs from different LacI family members, we can mix and match DNA recognition and allosteric response properties originated from different repressors (20). In this study, we used a computational strategy for the prediction of compatibility between DRMs and ESMs. (B–G). Computational strategy for the prediction of hybrid repressor performance. Direct Coupling Analysis was used to infer the parameters of the global joint probability distribution estimated for the LacI homologs. First, these parameters, eij and hi, were used to compute direct information values for residue pairs to discern the most important residue-residue pairs between the ESM and the DRM domains. Then, a compatibility score C(S) of a specific repressor sequence was calculated using eij, only for those residue position pairs with the highest DI values. C(S) provides a metric of functionality of the designed hybrid that is validated experimentally.
Computational methods have been useful in providing insights to study and engineer biomolecules based on biochemical and evolutionary principles (21–23). To guide the design of hybrid repressors from a large amount of possible combinations and to avoid blind experimental search for functional ones, we used a coevolutionary modeling approach for the first time to infer compatibility between DRMs and ESMs among LacI homologs. Coevolution among residues in a protein family plays an important role in stability and function of proteins. Diversifications of homologous proteins that bind to different effectors and promoters have an evolutionary pressure to preserve structural and functional constraints to maintain conformational dynamics and its allosteric regulation mechanism (24,25). A change in one residue site is coupled with the change in another site if these two sites are structurally or functionally associated (26). In addition to structural contacts (27), coevolving residues also participate in allosteric communication (28,29). Therefore, we proposed that coevolutionary cues between DRMs and ESMs contribute to repressor functions, which can be harnessed to design hybrid repressors with our module-swapping strategy. However, sequence covariation may arise from non-coevolving residues due to background signal or phylogenetic linkage (30,31). To find out essential coevolved residue positions between DRMs and ESMs in transcription repressors, we used direct coupling analysis (DCA) (27), which is able to separate the directly correlated residue pairs due to structural or functional constraints from other covariant amino acids. Coevolutionary analysis shows remarkable performance in the prediction of protein structures (32–34), conformational changes (35), and protein interactions (36,37) and its application has been further extended to other aspects recently, such as elucidating protein interaction networks from protein expression microarrays and gene-drug connectivity in pharmacogenomics (38,39), as well as RNA-protein interaction characterization (40). We have used a metric derived from DCA to infer protein binding specificity between histidine kinases and response regulators in two component system (TCS) (41,42) and found non-exclusive high binding preferences that yield crosstalk between proteins, SasA, CikA, RpaA, CikB, which are involved in circadian regulation in bacteria (43). Inspired by the fact that some hybrids of DRMs and ESMs exhibit comparable function to the native repressors (20), we devised a methodology to predict the compatibility of hybrid repressors with non-native DRMs and ESMs. The compatibility score used in this study evaluates the possibility of two modules from different origins to integrate and work as a functional protein that preserves properties of homodimerization, ligand and promoter binding, and allosteric transition.
Here, we applied our module swapping strategy to five ESMs and eight DRMs, generating a set of 40 repressors that were aimed for flexibly wiring five molecular signals to eight different promoters for controlling their transcriptional activities. Among these five native repressors and 35 engineered inducible systems, 22 of them generated >10-fold induction of protein expression, a dynamic range that is typically sufficient for regulating biological activities. Our compatibility model based on coevolution was able to predict with high accuracy (0.94 true positive predictive rate) for those hybrids that achieved >20-fold induction, suggesting useful applications of this model in guiding the construction of hybrids from other LacI family repressors. The ESMs and DRMs involved are highly orthogonal, in which no observable interfering activities were found among the five ESMs and their corresponding signaling molecules. Among the eight DRMs, crosstalk was only detected between two DRMs and their promoters. In our previous work, we used a unique property of these hybrid repressors to develop a passcode kill switch, where multiple environmental signal inputs are linked to a promoter for controlling a genetic output, in which this circuit can potentially be used for biocontainment (20). In this work, we introduced another novel circuit design by harnessing another unique property from these hybrid repressors: a single signal can be detected by multiple repressors that control different promoters. The resulting circuit is called Multiple Toggle Switches with a Master OFF Signal (hereinafter referred as MuTMOS), which programs engineered cells to detect two ON signals, in which each signal triggers the stable and continuous generation of a different biological activity. Whereas, the exposure to a master OFF signal switches off both biological activities, until ON signals are received again. While the master OFF signal strategy is broadly utilized in electrical and mechanical devices, this behavior may also provide significant benefits to biotechnological fields. As described in the Discussion section, we propose the use of the MuTMOS for developing living diagnostics that monitor an array of physiological parameters, which is expected to provide advantages unmatched by pioneering biological devices. Overall, we further demonstrated that the module swapping strategy can be robustly applied to a broad range of LacI homologs and can be rationally engineered to infer performance. We anticipate that these modular repressors will become enabling tools for constructing increasingly complex genetic circuits and for implementing unique circuit designs that could not be realized previously.
MATERIALS AND METHODS
Microbe strains
Characterization of modular repressor systems and toggle switches was performed in M9 minimal media with 0.2% glucose and 0.2% casamino acids. In all experiments, cells were grown at 37°C and 200 rpm, using appropriate antibiotics and ligands at the following concentrations: ampicillin (100 μg/ml), kanamycin (50 μg/ml), IPTG (1 mM), galactose (5 mM), cellobiose (10 mM), ribose (5 mM), fructose (5 mM) and ATc (50 μg/ml). Experiments were performed with E. coli MG1655 with ΔlacI, ΔgalR, ΔrbsR, and ΔpurR, and was created through P1 phage transduction of knockout strains from the Keio collection (44). For cloning and construction of genetic circuits, E. coli MG155 was used to grow in Luria-Bertani (LB) media.
Development of hybrid repressor systems
All plasmids were constructed using conventional molecular cloning protocols. Fragments of LacI, GalR, RbsR and PurR were cloned using E. coli MG1655 genome as a template and the template DNA fragments of other repressor genes were obtained by DNA synthesis. These templates and all primers were purchased from Eurofins Genomics. Construction of hybrid genes required overlap extension PCR to fuse DNA fragments from a DRM and an ESM. First, the two regions were cloned separately with PCR. For cloning the DRM, the reverse primer complementing the 3′ end contained a 5′ overhang of about 25 nucleotides, complementing the 5′ end of the ESM of the hybrid repressor. Similarly, the forward primer complementing the 5′ end of the ESM comprised a 5′ overhang that is identical to the last ∼25 nucleotides at the 3′ end of the DRM. Thus, in the two PCR products, the last ∼50 base pairs at the 3′ end of the DRM fragment matched the 5′ end of the ESM fragment. Then, these two DNA fragments were mixed to perform PCR again to generate a full-length hybrid gene. PCR products of hybrid repressor gene all contained a BamHI and a BsrGI restriction site at the 5′-end and the 3′-end, respectively, which were used for cloning these repressor genes into the plasmid, pTR, which contains a colE1 origin of replication, a kanamycin-resistance gene, and a PLtetO-1 promoter (20). Expression of the hybrid repressor gene was driven by the PLtetO-1, which expressed constitutively in our E. coli strains that did not contain tetR.
Plasmids for reporting hybrid repressor activities (pREPORT) were constructed from the plasmid pZA12, with a PLlacO-1 promoter driving the expression of a gfp gene, an ampicillin resistance gene, and a p15A origin of replication (45). To develop promoters that interact with each specific DRM, DNA fragments of these engineered promoters were generated with DNA synthesis and they were used to replace the PLlacO-1 in pREPORT via restriction sites, XhoI and EcoRI. Sequences of these synthetic promoters are present in Supplementary Table S1.
To assemble the inducible transcriptional system with each hybrid repressor, we co-transformed two plasmids into the E. coli strain, which are the pTR plasmid containing the repressor gene and the pREPORT plasmid containing the corresponding promoter-gfp gene construct. Resulting cells were grown in media with both kanamycin and ampicillin to maintain the two plasmids.
In vivo characterization of hybrid repressors
We picked single colonies containing the inducible expression system and inoculated them in M9 medium to grow overnight. The saturated cultures were diluted 100-fold with the same medium and they were grown in the same conditions. After OD600 reached 0.2–0.3, a volume of 200 μl of each culture was transferred to a well of a 96-well plate and a ligand was added to the culture. Cells in the plate were incubated at 37°C and 1000 rpm for 3 h before analyzing their cellular GFP fluorescence by using an ACEA NovoCyte 2030YB flow cytometer (ACEA Biosciences, Inc.). Flow cytometry data were gated by forward and side scatter to eliminate multi-cell aggregates, and the geometric means of GFP fluorescence distributions were calculated using the NovoExpress software (ACEA Biosciences, Inc.). At least 10 000 events were collected for each measurement. Similarly, to assess the activity of our engineered promoters (Supplementary Figure S1), this flow cytometry method was used to analyze cells containing only the promoter-gfp gene construct but without any hybrid repressors.
Construction of toggle switch and MuTMOS circuits
To develop plasmids that contain the toggle switches (TetR/RbsR, TetR/(RbsR-CelR), (MalR-RbsR)/LacI and (MalR-CelR)/LacI), the plasmid pKDL071 was used as the circuit backbone, which contains a TetR/LacI toggle switch, a colE1 origin of replication, and a kanamycin-resistance gene (46). For building the TetR/RbsR circuit, we removed the mcherry gene in pKDL071 that was regulated by LacI; lacI was replaced by rbsR via restriction sites, BsrGI and SacII; the promoters for driving tetR was replaced by PLrbsO via NcoI and SalI. Similarly, TetR/(RbsR-CelR) was constructed by using rbsR-celR and PLcelO instead of native rbsR and PLrbsO, respectively. For building the (MalR-RbsR)/LacI and (MalR-CelR)/LacI circuit, the gfp gene in pKDL071 regulated by tetR was removed; tetR was replaced with either malR-rbsR or malR-celR via NheI and SacI; and the promoter for driving lacI expression was replaced by PLmalO via BamHI and EagI. The two toggle switch circuits, (MalR-RbsR)/LacI and (MalR-CelR)/LacI, were cloned to a plasmid that contains a p15A origin of replication and an ampicillin-resistance gene via restriction sites, XhoI and AatII. As a result, cells containing TetR/RbsR or TetR/(RbsR-CelR) are resistant to kanamycin and those containing (MalR-RbsR)/LacI or (MalR-CelR)/LacI are resistant to ampicillin.
We optimized the performance of toggle switches by using different promoters and a range of ribosomal binding site (RBS) sequences to modulate the expression levels of repressor gene. RBS sequences were rationally designed by using the RBS calculator (https://salislab.net/software/) (47). For each version of the circuit, engineered cells were grown overnight in M9 cultural medium with the appropriate antibiotic as described above. The overnight cultures were diluted 200-fold in the same medium and grown on 96-well plates with the targeted ligand (IPTG, aTc, ribose or cellobiose) for 6 hours. Cells were then washed three time with the M9 cultural medium without the ligand, in which cells were spinned down and resuspended with fresh cultural medium. The washed cells were transferred to another 96-well plate to grow for another 6 hours. After that, cells were diluted 200-fold and were grown under the same conditions for 12 h. During this 24-h growth, we measured OD600 and GFP/mCherry fluorescence at time = 6, 12 and 24 h by using a Synergy H1 microplate reader (BioTek Instruments, Inc.). GFP fluorescence was measured with excitation at 488 nm and emission at 530 nm; and mCherry fluorescence with excitation at 561 nm and emission at 615 nm. To modulate gene expression in toggle switches, promoter and RBS fragments were generated by DNA synthesis. For toggle switches, TetR/RbsR and TetR/(RbsR-CelR), a range of RBS was used to control tetR expression and these RBS fragments were inserted into the circuit via restriction sites, SalI and NheI (Supplementary Figures S3 and S14). For optimizing the (MalR-RbsR)/LacI circuit, we first altered the RBS sequence of lacI via restriction sites, BamHI and BsrGI (Supplementary Figure S8). The version with the best performance was further engineered by modulating the expression of malR-rbsR with different promoters (via NcoI ans SalI) and RBS (via SalI and NheI) as shown in Supplementary Figure S9. For optimizing the (MalR-CelR)/LacI circuit, after malR-rbsR was replaced by the malR-celR gene, we incorporated different RBS, via SalI and NheI, to control malR-celR (Supplementary Figure S16).
To generate the MuTMOS circuit that responds to ribose as the OFF signal (Figure 4), two plasmids, one containing the TetR/RbsR circuit and the other one containing the (MalR-RbsR)/LacI circuit, were co-transformed into the strain of E. coli. Since the plasmids of TetR/RbsR and (MalR-RbsR)/LacI contain resistance gene for kanamycin and ampicillin, respectively, the resulting cells were required to grow in the M9 cultural medium that contains both antibiotics. Similarly, to develop the other version of MutMOS system with cellobiose as the OFF signal (Supplementary Figure S11), plasmids containing TetR/(RbsR-CelR) and (MalR-CelR)/LacI were co-transformed into the cells.
Figure 4.

Construction and characterization of a MuTMOS system with ribose as the master OFF signal. (A) A schematic of the MutMOS circuit that uses MalR-RbsR and native RbsR for detecting ribose. Engineered cells exposed to IPTG and ATc are switched to mCherry-ON state and GFP-ON state, respectively. An exposure to ribose turns off the expression of both mCherry and GFP. (B) Stochastic simulation of concentration changes of 4 repressors after induction (ON) by IPTG plus ATc (in shade). Ribose induces a subsequent OFF signal (in shade). A.U. stands for arbitrary units. The simulation was conducted by using the Gillespie algorithm. The levels of RbsR and MalR-RbsR increase while the levels of TetR and LacI decrease with the induction of IPTG plus ATc for the first 100,000 steps, and then maintained the equilibrium state even after the removal of ligands (upper row). The addition of ribose after 500,000 steps switched the toggle switches back to the state with high levels of LacI and TetR (lower row). (C) Experimental characterization of the MuTMOS system with ribose as the OFF signal. As shown on the first row, cells at the OFF state were exposed to IPTG only to switch to a stable mCherry-ON only state (left column); exposing cells only at the mCherry-ON state to ribose switched them back to a stable OFF state (right column). Similarly, results shown on the second row illustrates that ATc switched cells to a stable GFP-ON state (left), and ribose switched cells back to the OFF state (right). When cells were exposed to both ATc and IPTG (third row), cells became both mCherry-ON and GFP-ON (left), whereas ribose deactivated the expression of both mCherry and GFP to return cells back to a stable OFF state (right). Representative flow cytometry data are illustrated on the fourth row for the switching process between the state with both mCherry-ON and GFP-ON and the OFF state. All data points represent the mean ± S.D. of three biological replicates.
Characterization of the MuTMOS system
Cells with the MuTMOS system were grown overnight in M9 medium with kanamycin and ampicillin. To prepare cells at different GFP and mCherry states for assessing MuTMOS behavior, the saturated cultures were diluted 200-fold into 96-well plates containing M9 medium with the appropriate ligand(s) and cultured for 6 hours. After that, they were washed three times with the same medium without ligands. These cells were then diluted 200-fold with medium that did not contain any ligands and were cultured on 96-well plates for additional 12 h. At this point, cells were at different states of GFP and mCherry and this time point was considered as time = 0 h for MuTMOS characterization. Cells were diluted 200-fold again with the presence of the appropriate ligands. At time = 6 h, cells were washed three times to remove ligand(s). At time = 12 h, to prevent cells from getting into a dormant state at stationary phase, they were passaged once by diluting 200 folds and were cultured until time = 24 h. During the growth, cells were collected at time = 0, 2, 4, 6, 12 and 24 h for flow cytometry analysis as described above (Figure 4 and Supplementary Figure S11). The final version of each individual toggle switch was also characterized by using this flow cytometry method (Supplementary Figures S4, S10, S15 and S17).
Multiple sequence alignment (MSA) of LacI homologs
For the development of the coevolutionary model, we first collected an extensive set of LacI homologs and aligned them to define homologous positions among these proteins. For this purpose, we used the hmmbuild function from HMMER 3.1b2 to build a hidden Markov model (HMM) profile with the LacI sequence (UniProt ID: P03023) as the seed sequence. The HMM profile was then used to search against Uniprot database with the command hmmsearch. Significant hits with E-value ≤10 were reported as one alignment in Stockholm format, which was then converted to FASTA format by using esl-reformat, in which ∼70 000 LacI homologs were identified and were used for building our coevolutionary model as described below.
Identification of important coevolving DRM-ESM residue pairs
The amino acid distribution along the LacI homologous sequences was modeled as below:
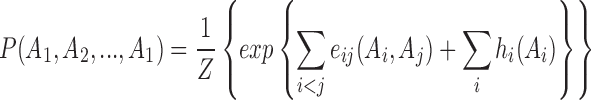 |
(1) |
where, n = 360 (the total number of amino acid residues in LacI), 1 ≤ i, j ≤ 360. Z is a normalization factor. The detailed inference of the probability distribution model and the parameters can be found in supplementary methods and elsewhere (27). The final expression of pairwise couplings, eij(Ai, Aj), which provide information on the coevolutionary strength between residue position i and j in the case where the amino acids in these two positions are Ai and Aj, respectively, is calculated from the cross-correlation matrix C:
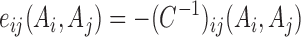 |
(2) |
Then a direct information (DI) metric was computed by using the derived probability with estimated parameters to estimate the coevolution between i th and j th residues:
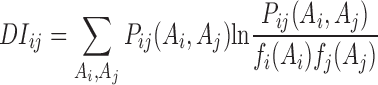 |
(3) |
High DI values indicate a strong dependency or functional relevance between these two residue positions among the LacI homologs.
Coevolutionary based compatibility model
Among the DRM-ESM residue pairs (47 DRM residues × 313 ESM residues = 14,711 pairs), 1500 residue-residue pairs with the highest DI values were included for compatibility model development. Only the top 1500 amino acid pairs were selected since considering extra residues induces noise to the predictive performance of the metric. The above-derived coupling parameter, eij, was further used to evaluate the compatibility between DRMs and ESMs originated from different LacI family members. The eij of each residue pair is a 21 × 21 submatrix with coupling values of any combination of amino acid pairs for residue i and j (20 amino acids plus a gap from sequence alignment). For a given sequence, such as a hybrid protein consisting of DRM from one repressor and ESM from another repressor, we introduced a quantity named compatibility score, C(S), to estimate the compatibility of the two modules by summing the coupling strength values (eij(Ai, Aj)) of those 1500 pairs with the highest DI values. The C(S) is defined as:
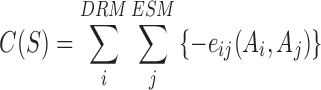 |
(4) |
with the constraint of DIij ≥ DI(1500th), 0 < i ≤ 47 and 47 < j ≤ 360.
A highly negative C(S) indicates strong compatibility between the DRM and ESM, and high potential of developing a functional hybrid repressor from these two modules.
Toggle switch models and stochastic simulation
To computationally evaluate the performance of MuTMOS system, we built a series of ODE function models for the transcription, translation, and molecular degradation processes for the toggle switch systems. Gillespie algorithm (48) then was implemented in the MATLAB to solve the equations and simulate the concentration changes of repressors used in MuTMOS system by using parameters estimated from the literature (49) and experiments. Equations and detailed process can be found in Supplementary Methods section.
Statistical evaluation of predictive model
We used statistical methods to compare coevolutionary model predictions and experimental results. The ROC curve of the model was generated by testing various thresholds for C(S). At each C(S) threshold level, the positive prediction rate and false positive prediction rate are computed. ROC curve evaluates the sensitivity and specificity of the prediction model. The sensitivity refers to true positive rate, which represents the ratio of number of true positive prediction to the number of repressors with fold increase ≥20. Specificity equals to 1 – false positive rate, which is the ratio of number of false positive prediction to the number of repressors with fold increase <20. For repressors containing the same DRM or ESM, we ranked them based on their C(S) scores and then evaluated the positive prediction rate for the repressors at each rank (ranking 1 to 5 for repressors with the same DRM and ranking 1 to 8 for repressors with the same ESM).
RESULTS
Computational strategy for the prediction of hybrid repressor performance
We developed a coevolutionary modeling approach (Figure 1B–F) to predict the compatibility between DRMs and ESMs among members of the LacI family. This model is expected to filter out the non-functional and low functional hybrid repressors prior to experiments in the future. We hypothesized that key connectivity between the DRM and the ESM in each LacI homolog is necessary to execute the repressor function and thereby, amino acid residues involved in these inter-module communication should be coevolving—residual changes in one module should be coupled with changes in its coupling partner in the other module. Thus, by capturing the coevolutionary pattern among members of the LacI family, we expect to reveal residue pairs key to the repressor function and to use them to predict compatibility between DRMs and ESMs originated from different homologs.
We first identified >70 000 homologs of Escherichia coli LacI (UniProt ID P03023) from the UniProt Database (50) using domain definitions from hidden Markov models developed for the Pfam database (51) and organized in multiple sequence alignment (Figure 1B) (52). Based upon our previous studies, we defined sequences that are homologous to LacI residues 1 to 47 as DRMs and LacI residues 48 to the C-terminus as ESMs; a DRM contains a helix-turn-helix domain and an ESM contains a hinge helix and a regulatory domain (20). We then estimated the global joint probability distribution of amino acids at each position of the aligned LacI homologs (Figure 1C; Equation (1)), and used direct coupling analysis (DCA) to determine the parameters of the distribution (pairwise couplings, eij, and local fields, hi). The pairwise couplings, eij, quantify the coupling strength between any two residue positions for all possible combinations of amino acids (Figure 1D) and hi is a proxy of amino acid propensities at position i (27). To identify residue pairs with high potential in contributing to protein function, we computed a quantity, direct information (DI), from the global joint probability distribution (Figure 1E; Equation (3)) to determine the residue pairs that are strongly coupled among LacI homologs. High DI values indicate strong dependencies between two residues, where the variation of amino acids are coupled during evolutionary history, suggesting their associations are important for protein structure and function and the compromise on the coupling strength of those co-evolved residues may have large impact. Since our goal is to understand interactions between DRMs and ESMs, we used DI values only for inter-domain pairs (DRM-ESM) excluding intradomain DI values. The amino acid changes on those residue pairs with high DI values, due to rewiring of a DRM and an ESM, may change the coupling strength for important pairs, which in turn affects protein functionality. Therefore, the top DI pairs were selected and their eij values for all amino acid pairings were used to develop a compatibility prediction model (Figure 1F; Equation (4)), which assigns compatibility score, C(S), to estimate compatibility between a DRM and an ESM among LacI homologs. In this framework, DRMs and ESMs with highly negative scores C(S) are predicted as highly compatible for constructing functional hybrid repressors. At last, we validated this predictive model with experimental results from hybrid repressors (Figure 1G).
Construction and characterization of a LacI family hybrid repressor library
In parallel to developing the coevolutionary model, we constructed and characterized a set of hybrid repressors that were generated with LacI homologs from diverse organisms. We used five ESMs selected from a list of collated LacI family proteins with known allosteric response properties. These 5 ESMs are originated from LacI, GalR, CelR, RbsR, and ScrR, which respond to ligands, Isopropyl β-d-1-thiogalactopyranoside (IPTG), galactose, cellobiose, ribose, and fructose, respectively. Each of the five ESMs was combined with each of the eight selected DRMs from LacI family members with well-studied DNA binding properties, including CelR, GalR, LacI, MalR, RbsR, ScrR, XltR, and PurR. A systematic combination of the five ESMs with the eight DRMs leads to the construction of a library of 35 hybrid repressors and five native repressors (Figure 2A). For genes that are not originated from E. coli, they were codon optimized for expression in this bacterial species and constructed using DNA synthesis; sources and sequences of the 40 repressors are illustrated in Supplementary Table S1 and primers used to generate hybrid repressor genes are listed in Supplementary Table S2.
Figure 2.

Characterization of LacI family hybrid repressors via experimental design and coevolutionary models. (A) Experimental characterization of hybrid repressors. We assessed the ability of hybrid repressors in gene expression regulation and allosteric response, by using a transcriptional reporter assay in a strain of E. coli with GFP as a reporter of transcription activities. A number of 40 repressors (including 35 hybrids) were characterized with this assay. Results shown in each column and each row are from repressors with the same DRM and ESM, respectively. Repressors with the same ESM were exposed to the same signaling molecule as indicated in the bracket on the left of each row. Repressors with the same DRM were used to control GFP expression driven by the same promoter (shown for each column). Data points in bar graphs represent GFP fluorescence in cells that were uninduced (grey) and induced (red). The blue number above each plot represents the corresponding fold-change of GFP induction. All data points represent mean ± S.D. of three biological replicates. (B) Heatmap of compatibility scores, C(S), predicted from coevolutionary information for 40 hybrid repressors. The hybrid repressors with the same ESM are presented in rows, while the repressors with the same DRM are shown in columns. The more negative score is shown in darker color, indicating more favorable compatibility. (C) Agreement between experimental observations and coevolutionary compatibility predictions. Orange and red cells indicate successful prediction of negative or positive for hybrid functionality (induction threshold at 20), respectively. White cells indicate disagreement between computational predictions and experimental results.
We then developed synthetic promoters that interact specifically with the hybrid repressors. A strong constitutive promoter, PL of phage lambda (45), was used for activating transcription and the operator sequence was placed upstream of the −35 region and between the −10 and −35 regions. Binding of the corresponding DRM to the operators is expected to repress gene expression driven by the engineered promoter. With this strategy, eight promoters were developed for the eight different DRMs in our studies (Supplementary Table S1). To characterize promoter activities, each engineered promoter was used to drive the expression of a gfp gene in E. coli cells in the absence of a corresponding repressor. Cells containing any one of the eight promoters possessed high GFP fluorescence, indicating that all these engineered promoters are transcriptionally active (Supplementary Figure S1). We then used these promoter-gfp constructs to characterize all 40 repressors in terms of their allosteric response and transcription regulatory properties (Figure 1G). Each repressor was constitutively expressed in E. coli cells harboring a GFP transcriptional reporter driven by an engineered promoter; the promoter contained operators that were recognized by the DRM of the hybrid repressor. Binding interactions between the hybrid repressor and the engineered promoter lead to the repression of GPF expression. Cells containing the synthetic expression system were exposed to a signaling molecule according to the ESM of the hybrid repressor (i.e. repressors containing an ESM originated from LacI, GalR, CelR, ScrR or RbsR were exposed to IPTG, galactose, cellobiose, fructose, or ribose, respectively). GFP fluorescence of ligand-exposed and unexposed cells from each strain was measured by flow cytometry. The fold induction was reported as the ratio between fluorescence of exposed cells to unexposed cells (Figure 2A).
To evaluate whether these hybrid repressors are capable to serve as parts for genetic circuit construction, we compared their performance with that of the native LacI repressor, since LacI has been used robustly in synthetic biology. Among these 40 inducible systems (Figure 2A), 9 of them generated a fold induction <3, which are not efficient for controlling gene expression. These inducible systems involve repressors GalR-LacI, XltR-LacI, MalR-GalR, GalR-CelR, CelR-ScrR, LacI-ScrR, RbsR-ScrR, XltR-ScrR and LacI-RbsR (hybrid repressors are named based on the DRM and ESM that they contain and in the form of ‘DRM-ESM’). Most of these systems provided high basal GFP levels in unexposed cells relative to the LacI system, which suggests that they do not form strong binding to the engineered promoter. The only exception is MalR-GalR, in which the engineered cells generated a low basal GFP florescence level but the level did not significantly increase upon induction. This implies that the ligand galactose does not interact efficiently with MalR-GalR to trigger an allosteric response. Another 9 hybrid repressor/engineered operator pairs generated 3 to 10 folds increase in GFP florescence upon induction, which shows that these systems possess expected biological activities but the dynamic range of genetic regulation is relatively narrow. Among these 9 repressors, MalR-LacI, RbsR-LacI, RbsR-GalR, PurR-GalR and CelR-RbsR provided low basal GFP levels that are comparable to that from native LacI; and native GalR, XltR-GalR, GalR-ScrR and GalR-RbsR generated high basal expression levels. The rest 22 repressors interacted with their corresponding ligands to produce a fold induction above 10, including CelR-LacI, native LacI, ScrR-LacI, PurR-LacI, CelR-GalR, LacI-GalR, ScrR-GalR, native CelR, LacI-CelR, MalR-CelR, RbsR-CelR, ScrR-CelR, XltR-CelR, PurR-CelR, MalR-ScrR, native ScrR, PurR-ScrR, MalR-RbsR, native RbsR, ScrR-RbsR, XltR-RbsR and PurR-RbsR. These 22 inducible systems are great candidates as genetic circuit parts because each of them generated low basal expression and a significant range of induction.
To test the interoperability between these inducible systems, the specificity of promoter-DRM interaction was evaluated by using the same GFP transcriptional reporter assay. Among the set of 40 repressors, we picked one out of the five repressors containing the same DRM, in which a total of eight repressors were selected. We then assessed interactions between the eight DRM-specific promoters and these eight repressors (Supplementary Figure S2). Each repressor was expressed in 8 different strains of cells, which contained a gfp gene driven by a different promoter. GFP fluorescence in resulting cells was measured at induced and uninduced states to assess repressor-promoter interactions. These eight DRMs are highly orthogonal and the only cross-interactions were observed in GalR DRM/PLmalO and MalR DRM/PLgalO; it is noteworthy that the GalR and MalR are originated from two different organisms but their operator sequences are only different by 2 base pairs (Supplementary Table S1). Similarly, we also evaluated the ligand-ESM interaction specificity, in which five inducible systems that involve the 5 different ESMs were selected (Supplementary Figure S2). These inducible systems responded specifically to their expected ligands and did not respond to the signals for the other ESMs. We sampled a subset of the 40 repressors and the results demonstrate that this assay described here can be used robustly for characterizing interoperability of these inducible expression systems. Generally, these results support that the DRMs and ESMs used in our repressor library are capable to function in the same cells with negligible interference.
The coevolutionary model accurately predicts hybrids with broad genetic inducibility
To validate our coevolutionary model, we compared the model prediction with experimental results from the 40 repressors (Figure 2A). Compatibility scores C(S) computed for these repressors are presented in Figure 2B and Supplementary Table S3. As expected, highly favorable C(S) values were assigned to all native repressors, which agrees with the fact that these native proteins are functional. For hybrid repressors, coevolutionary model predictions are also largely accurate when compared to experimental results. We attempted to predict the performance of these repressors by using the dynamic range of GFP expression as our evaluation parameter. As shown in Figures 2C and 3A, our model is able to predict the performance of 35 of the 40 repressors using 20-fold induction as a threshold. The performance of this model was evaluated with the receiver operating characteristic (ROC) analysis (Figure 3B), in which the area under the curve was 0.88 and the corresponding compatibility score C(S) was −69 at the optimal operating point. Using these thresholds, the true positive prediction rate of our model was 0.94, while the false positive rate was 0.17, revealing high sensitivity and specificity of this compatibility model.
Figure 3.
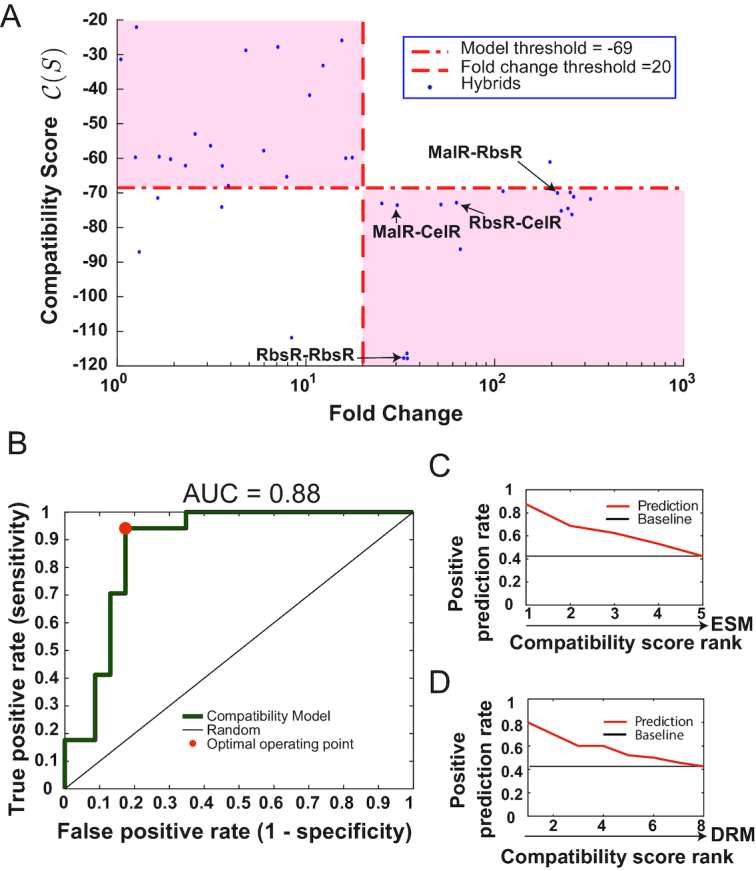
Global inference performance metrics of the hybrid compatibility score C(S) on predicting functional hybrid repressors. (A) Collection of hybrids with their corresponding experimental fold change measurements (x-axis) and C(S) scores (y-axis). The dashed line depicts the threshold for the experimental fold change, classifying the hybrids into two classes: inducible hybrids with fold increase ≥20 and non-inducible hybrids with fold change <20. The dash-dotted line denotes the threshold for compatibility score, classifying the hybrids into two classes: predicted to be compatible for hybrids with score ≤–69 and non-compatible hybrids with C(S) > –69. The shaded areas delimited by C(S) and fold response thresholds are regions where the model was able to correctly classify the response of the hybrid. The repressors used for building double toggle system described are indicated with arrows. (B) The ROC curve of the compatibility score model with fold change threshold at 20. The area under the curve is 0.88. The optimal operating point is highlighted by a dot in the curve. The black line indicates the performance of a random predictor. (C) Positive prediction rate of a C(S)-ranked set of hybrid repressors sharing a common DRM. There are 8 sets that contain five hybrids with different ESMs for each set. The positive prediction rate (y-axis) was calculated as the proportion, out of these eight repressors, with fold change ≥20 for each compatibility score rank category (x-axis). The best-ranked hybrid has a high probability to be functional. (D) Positive prediction rate of a C(S)-ranked set of hybrid repressors sharing a common ESM. In this case, there are five groups of hybrids with the same ESM and for each set there are eight different DRMs. The positive prediction rate is computed as in (C) for these hybrids. The best ranked scores indicate with high probability that the hybrid will be functional.
Next, we evaluated whether our model is able to predict the relative performance of repressors containing the same DRM or ESM. Repressors with the same DRM were grouped and for each of the eight DRM groups, the repressors were ranked based on their C(S) value. For repressors with the most favorable C(S) score in their group, 88% of them (seven out of eight repressors with eight DRMs) produced >20-fold induction in gene expression; and for the top two-ranked repressors in each group, the positive prediction rate was 0.69 (11 repressors out of 16 repressors containing 8 different DRMs) (Figure 3C). Similarly, ESM groups were formed for C(S) ranking and the positive prediction rate of top-ranked and top two-ranked repressors were 0.8 and 0.7, respectively (Figure 3D). These results support that C(S) scores, either absolute or relative, are indicative of functional performance and can serve as a reference for designing hybrid repressors by selecting DRMs and ESMs to achieve high compatibility.
A system of multiple toggle switches with a master OFF signal
With our hybrid repressor library, new connections can be generated between signaling molecules and a variety of promoters for regulating gene expression. As a result, a ligand is able to control the activities of multiple promoters and also, a promoter can be regulated by a range of chemical signals. This flexibility in wiring genetic pathways opens up new possibilities in genetic circuit topology, in which we have harnessed these properties to develop a system of Multiple Toggle Switches with a Master OFF Signal (MuTMOS; Figure 4A). In this system, each of these bistable toggle switches responds to a different signal to switch to the ON state and all of them respond to the same signal to switch to the OFF state.
A genetic toggle switch generates two genetic states that can be switched back and forth by using two molecular signals and the circuit can maintain its respective genetic state after removal of the signals (53). We have built a bistable genetic toggle switch with two transcriptional repressors, TetR and RbsR, which reciprocally repress each other (Supplementary Figures S3 and S4); additionally, TetR represses the expression of a gfp gene. With this design, induction with anhydrotetracycline (ATc) releases TetR from the tet promoter, leading to high expression of RbsR and GFP (GFP-ON state), whereas induction with ribose relieves repression from RbsR and generates high expression of TetR only, which represses the RbsR and GFP expression (GFP-OFF state).
Before constructing the TetR/RbsR toggle switch, we first devised a computational simulation to predict the dynamics and performance of the circuit and assessed if these components are capable of generating bistable states by responding to two ligands. We simulated changes in TetR and RbsR production during cellular response by using an ordinary differential equation (ODE) model (see Methods section and Supplementary Methods section) to deterministically interpret pseudo-reactions for the production and degradation of molecules in the MuTMOS system (49). Our mathematical model used compound Hill equations, in which one decreasing hill equation describes repressor-promoter interactions using parameters, free repressor concentration (R), repressor-DNA binding affinity (θ), and binding cooperativity (n). The second Hill function describes ligand-repressor interaction that affect free repressor concentrations, which includes the ligand concentration (l) and its kinetics, θ and n (Equation (8) in Supplementary Methods). The processes of translation and degradation were assumed to have mass action kinetics, where the translation or degradation rates are proportional to the amount of substrate. These parameters were obtained from a combination of literature information and estimated from our experiment results (Supplementary Table S4; Supplementary Figure S5). The nullclines of the model support that the system possesses one unstable equilibrium point and two steady equilibrium points (Supplementary Figure S6). To explore circuit behaviors during switching processes, we performed a stochastic simulation of changes in repressor protein levels during ligand exposures, in which the Gillespie algorithm was used to interpret the pseudo-reaction ODEs. The simulation results (Supplementary Figure S7) illustrate that TetR and RbsR are sufficient for developing a system that switches between two stable equilibrium points when it is exposed to the corresponding ligand for a sufficient period of time.
Guided by the simulation results, the circuit was then built and it was tuned by using different ribosomal binding site (RBS) for the tetR gene in order to achieve the desirable behaviors (Supplementary Figure S3). These RBS were designed with the RBS calculator (47). Similarly, we used native LacI and hybrid repressor MalR-RbsR to build another toggle switch with the LacI repressing the expression of a mCherry gene (Supplementary Figures S8–S10). The feasibility of this circuit design was also evaluated computationally (Supplementary Figures S6 and S7). With this (MalR-RbsR)/LacI bistable toggle switch, IPTG serves as the signaling molecule to switch the engineered cells to the mCherry-ON stable state and ribose serves as the other signal to switch the cells to the mCherry-OFF state.
To develop a MuTMOS system, we integrated the TetR/RbsR and the (MalR-RbsR)/LacI toggle switches together in E. coli cells (Figure 4A). The simulation of MuTMOS system supports that these two toggle switches are interoperable while both of them respond to ribose to switch to a stable OFF state (Figure 4B). In this circuit, native RbsR and MalR-RbsR interact with orthogonal promoters (PLrbsO and PLmalO, respectively) while both of them respond to ribose as a ligand. As a result, the GFP-ON and mCherry-ON states do not interfere with each other because native RbsR and MalR-RbsR specifically repress tetR and lacI expression, respectively, whereas induction with ribose simultaneously switches the circuit to both GFP-OFF and mCherry-OFF because this ligand releases both native RbsR and MalR-RbsR from their corresponding promoter (Figure 4C). By using our hybrid repressors to generate new wirings between a molecular signal and multiple promoters, we realized the MuTMOS circuit design in which each specific activating signal turns on a different biological activity while all these activities are turned off by one universal deactivating signal.
We also demonstrated that the MuTMOS system could be reprogrammed to respond to different small molecules as the master OFF signal. This is a beneficial feature for biological applications because engineered cells used for different purposes may be desired to respond to different signals. By using hybrid repressors with the same DRM but different ESMs, different signaling molecules can be used to regulate the activity of a particular promoter. We harnessed this flexibility to modify our MuTMOS circuit by replacing native RbsR and MalR-RbsR with other hybrid repressors, RbsR-CelR and MalR-CelR, respectively, such that the new MuTMOS responds to cellobiose as the master OFF signal (Supplementary Figure S11). We first developed two toggle switches, (MalR-CelR)/LacI and TetR/(RbsR-CelR). Our simulation also predicted that these toggle switches are sufficient to produce the desirable cellular behaviors (Supplementary Figures S12–S13). Indeed, these two bistable toggle switches were constructed based upon our circuit designs. Similar to engineering the ribose-responding MutMOS, we optimized the cellobiose-responding MuTMOS by regulating repressor genes with a range of RBS designed with the RBS calculator (47) as shown in Supplementary Figures S14–S17. After integrating these two toggle switches to E. coli, the engineered cells responded to ATc and IPTG to switch to a stable GFP-ON and mCherry-On state, respectively, and the cells were switched back to both GFP-OFF and mCherry-OFF by one signaling molecule, cellobiose (Supplementary Figure S11).
DISCUSSION
Mix and match of DNA recognition and allosteric response properties of LacI homologs
LacI family repressors have highly conserved protein structure, with an N-terminal helix-turn-helix (HTH) motif for recognizing DNA sequence of the operator, a C-terminal regulatory domain for interacting with the effector molecule, and a hinge-helix motif in between for facilitating the propagation of allosteric effect from the regulatory domain to the HTH motif (54). These domains and motifs are structurally distinct and each plays a well-defined molecular role, which provides us the fundamental basis to design hybrid repressors with a module swapping strategy (20). We defined the HTH motif (LacI residues 1–47 and homologous regions in other family members) as the DRM and the rest of the protein as the ESM. By swapping these modules among LacI homologs, the allosteric response property of a repressor can be matched with the DNA recognition property of another family member. In our studies, we mixed and matched 5 ESMs with 8 DRMs to generate a set of 40 engineered repressors (including 5 native proteins). The characterization of these repressors demonstrated the feasibility of module swapping in all LacI homologs involved in our studies, as all of them can be used to construct hybrid repressors that provide significant activities in allosterically regulated expression induction (Figure 2A). Intriguingly, for some repressors, swapping the DRM leads to a hybrid repressor system with increased dynamic range upon the same induction condition. For instance, the fold induction of the native GalR system is significantly low compared to those involving LacI-GalR or ScrR-GalR (8-fold versus above 200-fold; Figure 2A). Thus, this module swapping strategy also provides a means to engineer the dynamic range of a signal response. In contrast, some DRMs and ESMs are incompatible to construct functional repressors and the resulting hybrid proteins either possess poor ability in DNA binding or allosteric response. These results led us to use a computational approach to study module compatibility among LacI homologs, aiming to advance our ability to predict the behavior of these hybrid repressors. Additionally, a directed evolution strategy is promising for enhancing the performance of these hybrid repressors, in which Meyer et al. recently used rounds of positive and negative selections to screen for repressor mutants that provide expression systems with low background, high dynamic range, high ligand sensitivity, and low crosstalk with other expression systems (55). This approach can be a sufficient way to rescue poorly performing hybrid repressors for further expanding the toolset in the near future.
Compatibility model facilitates the screening of potential hybrids
Due to the fact that not all DRMs and ESMs are compatible with each other (Figure 2A), one major challenge in our module swapping strategy is to identify combinations of these modules that can lead to hybrid repressors with high performance. A computational and statistical approach based on coevolution is well suited for tackling this problem by harnessing the large amount of sequence information of the LacI family, provided that >70 000 LacI homologous sequences have been deposited in Uniprot. We developed a compatibility model to predict the biological performance of hybrid repressors based on coevolutionary analysis, in which module-module allosteric dynamics should involve specific residue-residue associations, which are under evolutionary pressure.
Our model computes a compatibility score for particular DRM-ESM combinations, which serves as a metric to accurately predict whether the resulting repressor is capable of providing an induction dynamic range above a fixed threshold (20-fold induction; Figure 3). This prediction accuracy supports our hypothesis that key residues play an important role in the preservation of module compatibility. Additionally, we can gain confidence in the prediction by performing quantitative comparisons of the compatibility scores from repressors with the same DRM or ESM. In each group of repressors that share a common module, repressors with a top compatibility score have a high probability to perform well (above 80% in all cases). Therefore, to design a new circuit component with a specific DRM or ESM, we may look at the relative compatibility scores to select a promising counterpart for module swapping. Moreover, the compatibility score is useful to guide parameter determination in the simulation of the circuit. In the simulation of four circuits built in this study, the DNA binding affinity parameter θ, of repressors consists of specific DRM and different ESMs, is lower when the compatibility score is favorable, indicating higher binding affinity (Supplementary Tables S3 and S4). Potentially, prediction accuracy can be further improved by feeding the model with additional protein sequences from the ever-growing list of homologs. We may also include sequences of high-performance hybrid repressors in our model, broadening the database for capturing significant module-module interactions. The coevolutionary model provides an efficient and accurate framework to select the modules for functional hybrid repressors without experimental exploration of a large amount of possible combinations when more LacI family transcription factors are involved.
In this model, native repressors possess the top compatibility score among repressors sharing the same ESM (Figure 2B), with PurR-RbsR as the only exception. This is consistent with the result from a LacI family protein phylogenetic tree analysis, where PurR exists as an excluded clade from the branch of RbsR subfamily (56). While the PurR DRM remains highly similar to those in the RbsR subfamily, the DRM of E. coli RbsR used in our study was evolved to become significantly different from other RbsR subfamily members. This also explains the more favorable value of predicted compatibility between PurR DRM and RbsR ESM from our coevolutionary model. In contrast, the ESM of PurR has deviated from its ancestral protein to gain the ability to sense purine instead of ribose and these analyses support our model that module swapping is highly feasible among LacI family members. Additionally, hybrids constructed from LacI ESM show the lowest C(S) scores among all 40 repressors computationally indicating that LacI ESM possesses higher requirement for specificity with DRM and less coevolutionary couplings with other DRM. This result is consistent with the experiment where the seven LacI ESM-containing hybrids exhibit low induction activity with none of them >20-fold change threshold and only two of them are more than 10. Overall, both absolute C(S) scores and relative scores serve as valuable references in designing hybrid repressors using the module swapping approach.
Another intriguing observation is that among the five repressors that we predicted inaccurately, four of them involve modules from GalR. It is particularly unexpected that the native GalR possesses a relatively low induction activity (8-fold increase upon induction) while its C(S) score is among one of the strongest. The unexpected behavior of GalR may be caused by its unconventional interactions with its promoter and ligand. Previous studies showed that the dissociation constant (Kd) of GalR and its operator is ∼4 nM (57), which is relatively weak compared to the DNA binding affinity of other homologs, such as LacI (Kd ∼10 pM) (58). This potentially explains the relatively high basal GFP expression in the native GalR system (Figure 2A). Additionally, Chatterjee et al. demonstrated that the presence of its ligand, galactose, only reduces the GalR-DNA operator binding affinity by ∼7-fold and does not completely eliminate the GalR-DNA interactions (57). This implies that GalR may still influence the promoter activity in the presence of galactose, leading to a relatively small dynamic range of expression. In this report, our co-evolutionary model focuses in using module-module interactions to understand module compatibility. However, we may also expand our model by also considering interactions of DNA-protein and protein-ligand to further enhance our prediction accuracy.
The model is promising in predicting the potential effects of a residue substitution on the inter-domain compatibility and the intra-domain functionality of hybrid repressors. This methodology can help navigate favorable mutations, reforming the repressors with specific signal-response linkages to exhibit improved compatibility between ESM and DRM while maintaining the functionality of each domain. Furthermore, we anticipate the application of this computational strategy to test compatibility of module swapping in other prokaryotic one-component transcription regulator families consisting of linked input (ESM) and output (DRM) domains, such as the TetR family and the Crp family, expanding the array of signal-response linkages in genetic systems. There are several other prokaryotic one-component signal transduction systems, with similar properties and architectures as the ones studied here. These are additional potential candidates for extensibility of the model. This methodology might be limited by the total number of available sequences and the requirement of having structured independent-folding units that interact. Extension to two-component signal transduction systems to rewire the signal-response linkage while keeping specificity between histidine kinases and response regulators is possible but challenging since they contain similar but differently located input and output domains (59).
Modular repressors open up the possibility of developing MuTMOS systems
By using native repressors, one specific molecular signal can only be connected to a specific promoter for transcriptional regulation. Such rigidity constrains the ways of wiring transcription networks for signal processing and thus, integration of multiple environmental signals for controlling cellular behavior often requires multiple layers of gene expression. For instance, Moon et al. used a layered transcription approach to link four small-molecule inputs for controlling an AND logic gate, in which the output from one layer of transcription is the input of a downstream layer (60). With recent advances in dCas9 technologies, this layered transcription approach is highly programmable and robust for engineering logic-based operations (61,62). However, this approach requires a large amount of biological components to connect multiple inputs and outputs and an increase in the number of circuit parts and circuit complexity also elevates the difficulties in construction of biological systems. This requirement poses limitations to circuit design for biological signal processing. With our set of hybrid repressors, multiple signals can be directly wired to a promoter and by harnessing this property, we previously developed a passcode kill switch that requires the presence of multiple environmental signals to repress the expression of a lethal gene (20). With the new hybrid repressors developed in this study, the signal complexity of the passcode circuit can potentially be further expanded, creating more specific environments for biocontaining engineered organisms. Vice versa, these repressors also provide the opportunities to use one environmental signal for controlling the activities of multiple promoters, which allows us to develop the MuTMOS system. Generally, these engineered repressors provide a convenient means to integrate multiple signals for controlling cellular functions, which renders new possibilities in circuit topologies for engineering biological outcomes. In this study, the MuTMOS detects two ON signals, in which each of the two independent input signals switches a different biological activity to the ON state while one master OFF signal switches off both functions. The feasibility of this engineered cellular behavior depends on these modular repressors, in which they respond to the same molecular signal but regulate different promoters. As shown in Figure 4A, the MutMOS system involves two toggle switches that are constructed with four repressors. These repressors recognize orthogonal promoter and thus, each repressor only regulates the expression of one specific repressor gene in the circuit. Specifically, for the (MalR-RbsR)/LacI toggle switch, LacI represses malR-rbsR expression and also, MalR-RbsR only represses lacI but not tetR. Similarly, for the TetR/RbsR toggle switch, TetR and RbsR reciprocally represses the expression of each other and both of them do not affect the expression of genes in the (MalR-RbsR)/LacI circuit. These two toggle switches do not cross-interact on the repressor-promoter level and thereby, switching any one of them to the ON state does not affect the other toggle switch. This circuit topology cannot be realized by using only native repressors because they only connect a signal to a specific promoter, in which it is not possible to avoid strong interference between the two toggle switches if a master OFF signal is used. Additionally, we showed that the signal for programming cellular behavior can be altered easily with these hybrid repressors. By replacing the RbsR ESM with the CelR ESM, we then changed the master OFF signal from ribose to cellobiose (Supplementary Figure S11). Together, the MuTMOS system reveals the capabilities of these modular repressors in engineering new genetic behavior that were previously infeasible.
Potential biological applications of the MuTMOS system
We have constructed a genetic MuTMOS circuit that controls the switch of multiple cellular activities to the stable ON state with each triggered by a different signal, while all activities return to the stable OFF state in response to the same OFF signal. For electric circuits, it is a common practice to use a master signal for resetting multiple states in an electrical device, as this strategy effectively reduces signal complexity, which improves the robustness and efficiency of the device in many situations. In synthetic biology, the MuTMOS signal response behavior may facilitate the development of living diagnostics for in vivo monitoring of pathological conditions. A recent study has constructed a toggle switch system to serve as a biosensor for probing bacterial growth in a host organism (63). It has been proposed that an array of these biosensing systems can be integrated into a strain of engineered cells for monitoring multiple parameters in host environments (64). For diagnostic cells designed for monitoring patients for a long duration, those biosensors may need to be reset to the OFF state after a certain period of time. With the MuTMOS system, resetting multiple sensors can be performed by the injection of one chemical signal to the host, instead of using a different signal for each sensor. This is advantageous for in vivo monitoring because with reduced number of chemical signals used in the host, there is a reduced risk of triggering unfavorable physiological responses by these chemicals, such as immune response and changes in metabolic activities. We also showed that the master OFF signal can be substituted by integrating different ESMs of the modular repressors; the ease of changing the response pathway enhances the utility of this design in situations that require different signaling molecules. Furthermore, our genetic MuTMOS system may be potentially used for other environmental, industrial and biomedical applications, given that the MuTMOS behavior has been shown to be extensively implemented in various mechanical and electrical devices.
DATA AVAILABILITY
The sequence of all plasmids will be deposited to GenBank:
TetR-RbsR_toggle_switch_plasmid: MK753225
(MalR-RbsR)-LacI_toggle_switch_plasmid: MK753226
TetR-(RbsR-CelR)_toggle_switch_plasmid: MK753227
(MalR-CelR)-LacI_toggle_switch_plasmid: MK753228
The datasets related to the computational model is accessible in datadryad with the DOI: 10.5061/dryad.cf604dn. Scripts and code can be found in http://morcoslaboratory.org/?page_id=385.
The authors declare that all other data supporting the findings of this study are available within the paper and/or the associated supplementary files. All custom scripts are available from the corresponding author upon request.
Supplementary Material
ACKNOWLEDGEMENTS
Authors contributions: R.P.D. and C.T.Y.C. designed and performed the biological experiments. X.J. and F.M. designed and conducted the compatibility model. X.J., J.A.P. and F.M. conducted the ODEs model and stochastic simulation. X.J., F.M., C.T.Y.C. wrote the paper. F.M. and C.T.Y.C. designed the overall research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
R.P.D. and C.T.Y.C. acknowledge funding from the University of Texas System Rising STARs Program [802-1053-T000674F], the University of Texas at Tyler New Faculty Grants Program, and the Welch Foundation [Grant # BP-0037]. X.J., J.A.P, F.M. acknowledge funding from the University of Texas at Dallas. Funding for open access charge: University of Texas at Tyler.
Conflict of interest statement. None declared.
REFERENCES
- 1. Nielsen A.A., Segall-Shapiro T.H., Voigt C.A.. Advances in genetic circuit design: novel biochemistries, deep part mining, and precision gene expression. Curr. Opin. Chem. Biol. 2013; 17:878–892. [DOI] [PubMed] [Google Scholar]
- 2. Arkin A.P. A wise consistency: engineering biology for conformity, reliability, predictability. Curr. Opin. Chem. Biol. 2013; 17:893–901. [DOI] [PubMed] [Google Scholar]
- 3. Purnick P.E., Weiss R.. The second wave of synthetic biology: from modules to systems. Nat. Rev. Mol. Cell Biol. 2009; 10:410–422. [DOI] [PubMed] [Google Scholar]
- 4. Lu T.K., Khalil A.S., Collins J.J.. Next-generation synthetic gene networks. Nat. Biotechnol. 2009; 27:1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., Lee K.J.. The genetic design of signaling cascades to record receptor activation. PNAS. 2008; 105:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H.. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011; 365:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitaker W.R., Davis S.A., Arkin A.P., Dueber J.E.. Engineering robust control of two-component system phosphotransfer using modular scaffolds. PNAS. 2012; 109:18090–18095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morsut L., Roybal K.T., Xiong X., Gordley R.M., Coyle S.M., Thomson M., Lim W.A.. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016; 164:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwarz K.A., Daringer N.M., Dolberg T.B., Leonard J.N.. Rewiring human cellular input-output using modular extracellular sensors. Nat. Chem. Biol. 2017; 13:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baeumler T.A., Ahmed A.A., Fulga T.A.. Engineering synthetic signaling pathways with programmable dCas9-Based chimeric receptors. Cell Rep. 2017; 20:2639–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roybal K.T., Williams J.Z., Morsut L., Rupp L.J., Kolinko I., Choe J.H., Walker W.J., McNally K.A., Lim W.A.. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. 2016; 167:419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Porter D.L., Kalos M., Zheng Z., Levine B., June C.. Chimeric antigen receptor therapy for B-cell malignancies. J. Cancer. 2011; 2:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazumder M., McMillen D.R.. Design and characterization of a dual-mode promoter with activation and repression capability for tuning gene expression in yeast. Nucleic Acids Res. 2014; 42:9514–9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomez-Martinez M., Schmitz D., Hergovich A.. Generation of stable human cell lines with Tetracycline-inducible (Tet-on) shRNA or cDNA expression. J.Vis.Exp.: JoVE. 2013; 73:50171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ulmasov B., Capone J., Folk W.. Regulated expression of plant tRNA genes by the prokaryotic tet and lac repressors. Plant Mol. Biol. 1997; 35:417–424. [DOI] [PubMed] [Google Scholar]
- 16. Cronin C.A., Gluba W., Scrable H.. The lac operator-repressor system is functional in the mouse. Genes Dev. 2001; 15:1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Licht A., Preis S., Brantl S.. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in Bacillus subtilis. Mol. Microbiol. 2005; 58:189–206. [DOI] [PubMed] [Google Scholar]
- 18. Tungtur S., Egan S.M., Swint-Kruse L.. Functional consequences of exchanging domains between LacI and PurR are mediated by the intervening linker sequence. Proteins. 2007; 68:375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meinhardt S., Manley M.W. Jr, Becker N.A., Hessman J.A., Maher L.J. 3rd, Swint-Kruse L.. Novel insights from hybrid LacI/GalR proteins: family-wide functional attributes and biologically significant variation in transcription repression. Nucleic Acids Res. 2012; 40:11139–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan C.T., Lee J.W., Cameron D.E., Bashor C.J., Collins J.J.. ‘Deadman’ and ‘Passcode’ microbial kill switches for bacterial containment. Nat. Chem. Biol. 2016; 12:82–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bick M.J., Greisen P.J., Morey K.J., Antunes M.S., La D., Sankaran B., Reymond L., Johnsson K., Medford J.I., Baker D.. Computational design of environmental sensors for the potent opioid fentanyl. Elife. 2017; 6:e28909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thyme S.B., Boissel S.J., Arshiya Quadri S., Nolan T., Baker D.A., Park R.U., Kusak L., Ashworth J., Baker D.. Reprogramming homing endonuclease specificity through computational design and directed evolution. Nucleic Acids Res. 2014; 42:2564–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Risso V.A., Sanchez-Ruiz J.M., Ozkan S.B.. Biotechnological and protein-engineering implications of ancestral protein resurrection. Curr. Opin. Struct. Biol. 2018; 51:106–115. [DOI] [PubMed] [Google Scholar]
- 24. Chakrabarti S., Panchenko A.R.. Coevolution in defining the functional specificity. Proteins. 2009; 75:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar A., Glembo T.J., Ozkan S.B.. The Role of Conformational Dynamics and Allostery in the Disease Development of Human Ferritin. Biophys. J. 2015; 109:1273–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gobel U., Sander C., Schneider R., Valencia A.. Correlated mutations and residue contacts in proteins. Proteins. 1994; 18:309–317. [DOI] [PubMed] [Google Scholar]
- 27. Morcos F., Pagnani A., Lunt B., Bertolino A., Marks D.S., Sander C., Zecchina R., Onuchic J.N., Hwa T., Weigt M.. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kass I., Horovitz A.. Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations. Proteins. 2002; 48:611–617. [DOI] [PubMed] [Google Scholar]
- 29. Suel G.M., Lockless S.W., Wall M.A., Ranganathan R.. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat. Struct. Biol. 2003; 10:59–69. [DOI] [PubMed] [Google Scholar]
- 30. Oliveira L., Paiva A.C.M., Vriend G.. Correlated mutation analyses on very large sequence families. ChemBioChem. 2002; 3:1010–1017. [DOI] [PubMed] [Google Scholar]
- 31. Tillier E.R.M., Lui T.W.H.. Using multiple interdependency to separate functional from phylogenetic correlations in protein alignments. Bioinformatics. 2003; 19:750–755. [DOI] [PubMed] [Google Scholar]
- 32. dos Santos R.N., Ferrari A.J.R., de Jesus H.C.R., Gozzo F.C., Morcos F., Martinez L.. Enhancing protein fold determination by exploring the complementary information of chemical cross-linking and coevolutionary signals. Bioinformatics. 2018; 34:2201–2208. [DOI] [PubMed] [Google Scholar]
- 33. Sulkowska J.I., Morcos F., Weigt M., Hwa T., Onuchic J.N.. Genomics-aided structure prediction. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:10340–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marks D.S., Colwell L.J., Sheridan R., Hopf T.A., Pagnani A., Zecchina R., Sander C.. Protein 3D structure computed from evolutionary sequence variation. PLoS One. 2011; 6:e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morcos F., Jana B., Hwa T., Onuchic J.N.. Coevolutionary signals across protein lineages help capture multiple protein conformations. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:20533–20538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karml S.H., Holt S.H., Song L.H., Tamir S., Luo Y.T., Bai F., Adenwalla A., Darash-Yahana M., Sohn Y.S., Jennings P.A. et al.. Interactions between mitoNEET and NAF-1 in cells. PLoS One. 2017; 12:e0175796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malinverni D., Jost Lopez A., De Los Rios P., Hummer G., Barducci A.. Modeling Hsp70/Hsp40 interaction by multi-scale molecular simulations and coevolutionary sequence analysis. Elife. 2017; 6:e23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang X.L., Martinez-Ledesma E., Morcos F.. Revealing protein networks and gene-drug connectivity in cancer from direct information. Sci. Rep. 2017; 7:3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sanchez-Ibarra H.E., Reyes-Cortes L.M., Jiang X.L., Luna-Aguirre C.M., Aguirre-Trevino D., Morales-Alvarado I.A., Leon-Cachon R.B., Lavalle-Gonzalez F., Morcos F., Barrera-Saldana H.A.. Genotypic and phenotypic factors influencing drug response in mexican patients with type 2 diabetes mellitus. Front. Pharmacol. 2018; 9:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou Q., Kunder N., De la Paz J.A., Lasley A.E., Bhat V.D., Morcos F., Campbell Z.T.. Global pairwise RNA interaction landscapes reveal core features of protein recognition. Nat. Commun. 2018; 9:2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng R.R., Morcos F., Levine H., Onuchic J.N.. Toward rationally redesigning bacterial two-component signaling systems using coevolutionary information. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E563–E571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng R.R., Nordesjo O., Hayes R.L., Levine H., Flores S.C., Onuchic J.N., Morcos F.. Connecting the Sequence-Space of bacterial signaling proteins to phenotypes using coevolutionary landscapes. Mol. Biol. Evol. 2016; 33:3054–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boyd J.S., Cheng R.R., Paddock M.L., Sancar C., Morcos F., Golden S.S.. A combined computational and genetic approach uncovers network interactions of the cyanobacterial circadian clock. J. Bacteriol. 2016; 198:2439–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol.Syst.Biol. 2006; 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lutz R., Bujard H.. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997; 25:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Litcofsky K.D., Afeyan R.B., Krom R.J., Khalil A.S., Collins J.J.. Iterative plug-and-play methodology for constructing and modifying synthetic gene networks. Nat. Methods. 2012; 9:1077–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Salis H.M., Mirsky E.A., Voigt C.A.. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009; 27:946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gillespie D.T. Exact stochastic simulation of coupled Chemical-Reactions. J. Phys. Chem. 1977; 81:2340–2361. [Google Scholar]
- 49. Lugagne J.B., Sosa Carrillo S., Kirch M., Kohler A., Batt G., Hersen P.. Balancing a genetic toggle switch by real-time feedback control and periodic forcing. Nat. Commun. 2017; 8:1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. UniProt Consortium, T UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018; 46:2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johnson L.S., Eddy S.R., Portugaly E.. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010; 11:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gardner T.S., Cantor C.R., Collins J.J.. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000; 403:339–342. [DOI] [PubMed] [Google Scholar]
- 54. Lewis M., Chang G., Horton N.C., Kercher M.A., Pace H.C., Schumacher M.A., Brennan R.G., Lu P.. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 1996; 271:1247–1254. [DOI] [PubMed] [Google Scholar]
- 55. Meyer A.J., Segall-Shapiro T.H., Glassey E., Zhang J., Voigt C.A.. Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 2019; 15:196–204. [DOI] [PubMed] [Google Scholar]
- 56. Ravcheev D.A., Khoroshkin M.S., Laikova O.N., Tsoy O.V., Sernova N.V., Petrova S.A., Rakhmaninova A.B., Novichkov P.S., Gelfand M.S., Rodionov D.A.. Comparative genomics and evolution of regulons of the LacI-family transcription factors. Front. Microbiol. 2014; 5:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chatterjee S., Zhou Y.N., Roy S., Adhya S.. Interaction of Gal repressor with inducer and operator: induction of gal transcription from repressor-bound DNA. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:2957–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oehler S., Alberti S., Muller-Hill B.. Induction of the lac promoter in the absence of DNA loops and the stoichiometry of induction. Nucleic Acids Res. 2006; 34:606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheng R.R., Haglund E., Tiee N.S., Morcos F., Levine H., Adams J.A., Jennings P.A., Onuchic J.N.. Designing bacterial signaling interactions with coevolutionary landscapes. PLoS One. 2018; 13:e0201734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moon T.S., Lou C., Tamsir A., Stanton B.C., Voigt C.A.. Genetic programs constructed from layered logic gates in single cells. Nature. 2012; 491:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nielsen A.A., Voigt C.A.. Multi-input CRISPR/Cas genetic circuits that interface host regulatory networks. Mol. Syst. Biol. 2014; 10:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gander M.W., Vrana J.D., Voje W.E., Carothers J.M., Klavins E.. Digital logic circuits in yeast with CRISPR-dCas9 NOR gates. Nat. Commun. 2017; 8:15459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Certain L.K., Way J.C., Pezone M.J., Collins J.J.. Using engineered bacteria to characterize infection dynamics and antibiotic effects in vivo. Cell Host Microbe. 2017; 22:263–268. [DOI] [PubMed] [Google Scholar]
- 64. Meylan S., Andrews I.W., Collins J.J.. Targeting antibiotic tolerance, pathogen by pathogen. Cell. 2018; 172:1228–1238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence of all plasmids will be deposited to GenBank:
TetR-RbsR_toggle_switch_plasmid: MK753225
(MalR-RbsR)-LacI_toggle_switch_plasmid: MK753226
TetR-(RbsR-CelR)_toggle_switch_plasmid: MK753227
(MalR-CelR)-LacI_toggle_switch_plasmid: MK753228
The datasets related to the computational model is accessible in datadryad with the DOI: 10.5061/dryad.cf604dn. Scripts and code can be found in http://morcoslaboratory.org/?page_id=385.
The authors declare that all other data supporting the findings of this study are available within the paper and/or the associated supplementary files. All custom scripts are available from the corresponding author upon request.