

Abstract
RNA molecules are able to bind proteins, DNA and other small or long RNAs using information at primary, secondary or tertiary structure level. Recent techniques that use cross-linking and immunoprecipitation of RNAs can detect these interactions and, if followed by high-throughput sequencing, molecules can be analysed to find recurrent elements shared by interactors, such as sequence and/or structure motifs. Many tools are able to find sequence motifs from lists of target RNAs, while others focus on structure using different approaches to find specific interaction elements. In this work, we make a systematic analysis of RBP–RNA and RNA–RNA datasets to better characterize the interaction landscape with information about multi-motifs on the same RNAs. To achieve this goal, we updated our BEAM algorithm to combine both sequence and structure information to create pairs of patterns that model motifs of interaction. This algorithm was applied to several RNA binding proteins and ncRNAs interactors, confirming already known motifs and discovering new ones. This landscape analysis on interaction variability reflects the diversity of target recognition and underlines that often both primary and secondary structure are involved in molecular recognition.
INTRODUCTION
Interactions between molecules in cells can influence myriad of functions, from localization to co- and post- transcriptional regulation, mediated by degradation or stabilizations of transcription products. The scenario of all possible interactions diversifies based on the principal actors of this interplay: protein, RNA and DNA. RNA-binding proteins (RBPs) can be classified by sequence and/or structural preference, often ascribed to specific RNA binding domains (1,2).
The study of RBPs has witnessed an increasing attention in the last decade, mostly due to the rise of high-throughput screening assays like Cross-Linking ImmunoPrecipitation (or CLIP-Seq) and derived techniques (PAR-CLIP, iCLIP, eCLIP), but a plethora of specific techniques emphasizing some aspects over others has been developed (3,4). These assays allow inquiring the landscape of RBP interactions by forcing in vivo covalent bonds between proteins and their RNA targets, followed by antibodies pull-down and high-throughput (HT) sequencing. In this way, it has been possible to shed light on the binding-sites of NOVA1 and NOVA2 in the mouse brain (5), or other splicing factors such as SRSF1 (6), hnRNP C (7) and FMRP (8), as well as a number of other RBPs with different post-transcriptional regulation roles in the cell (2).
Another well studied protein/RNA–RNA interaction involves microRNAs (miRNAs). MiRNAs, small RNA sequences usually composed of 22 nucleotides, are able to bind AGO proteins inducing mRNA silencing of specific genes that have complementary seed sequence. The seed sequence, a short seven nucleotides region at 5′-end of miRNA, is found to be fundamental for target recognition and regulatory responses, but additional recognition sites emerged in several experiments (9–12). Non-canonical seed pairing in CLASH (Crosslinking, Ligation And Sequencing of Hybrids) experiments was 1.7-fold more frequent than perfect base complementarity (10) and binding sites can occur both in coding region and UTRs. These new evidences suggest other crucial features that could reflect binding site accessibility and strengthen the interaction with miRNAs. Some information about hybridization energy and structural context, that is known to contribute to miRNA/target interactions (13–17), is used in the prediction of new possible miRNA targets: these methods take into consideration binding free energy and secondary structure accessibility in order to find suitable binding sites. The limitation of these methods is that they scan only the binding sites and not the entire mRNA target sequence that could have additional recognition sites for the AGO protein complex. MiRNA interaction is one of the most studied RNA–RNA interactions, but there are many long non coding RNAs that are able to bind other RNA molecules and that can be involved in gene regulation being, for example, competing targets of microRNAs (18).
In this scenario, there is a discrepancy between the amount of data available and the accuracy of current algorithms capable of extracting meaningful information, additionally it must be noted that each of the computational methods deputed to information extraction from RBP/miRNA/RNA HT data has a different and often unique approach.
Of particular interest is the motif discovery problem, which presents itself as the search of common elements (in terms of primary sequence or secondary structures) that justify the molecule selection induced by the assay. A detailed review of recent methods developed to address the motif problem has been presented by Morris et al. (19). At primary sequence level, motif models complexity can be classified by different categories: position-specific sequence matrices (PSSMs and multiple PSSMs) (20), base neighbor dependent (diPSSM and HMM) (21,22) that take into account also dependencies between nucleotide positions, and Higher-order dependencies that are able to capture tri-nucleotide or other higher-order interactions (23). At secondary structure level, structural complexity can be described by site-specific structural context (the ensemble of all structural preferences) (24), Position-specific structural context (described by PSSM-like models) (25) or Higher-order structural dependencies (structure or sequence dependent) (26,27). The complexity of the structural alphabet used to describe motifs found with one of these methods increases from site-specific to higher-order structural context, and a motif can be represented by a single representative structure (28) or an ensemble of potential structural conformations (29).
Some interactions are dictated by sequence patterns, while others by structural patterns, that are recognized and bound by the interaction partner. Often, a given RNA molecule contains more than one motif, and the relationships, interactions and, possibly, cooperativity among different motifs is unclear and hard to study, often because of the lack of sufficiently accurate and fast tools for their identification and mapping.
Recently, we developed BEAM (BEAr Motif finder) (27), which exploits the BEAR secondary structure encoding (30) to discover structural interaction motifs by means of a heuristic simulated annealing procedure, which proved to be suited for protein–RNA interaction HT datasets and their typical size. The evidence of novel and accurate techniques to analyze RNA interactions has laid the foundations for finding new features that characterize RBPs and microRNAs interaction with target RNAs using the BEAM algorithm. The variability in RNA binding sites is the main focus of this work, whose purpose is to explore structural and sequence signals in target RNAs that could play a role in RNA and protein recognition. With this in mind, we further developed the BEAM software, which was previously able to only work on structural motifs, by adding a module for the identification of RNA sequence motifs and by improving the overall search strategy in order to find co-occurring sequence and structure motifs. We systematically analyzed the co-occurrence of RNA sequence and structural motifs involved in RBP and miRNA binding, finding relationships that can better explain the modes of recognition. First, we considered known RBP binding preferences, recapitulating and often expanding and clarifying the interaction motifs and their relationships. Then, we tackled the issue more systematically, by the analysis of large protein–RNA interaction datasets, finding novel motifs and general trends and rules. Finally, we considered the interaction between miRNAs or ncRNA and their targets, identifying structural dependencies of the target sites.
MATERIALS AND METHODS
BEAM sequence module and statistical tests
We improved the BEAM algorithm with the addition of a primary sequence motif discovery module. In particular, now BEAM2.0 can search for primary sequence motifs during a standard secondary structure motif-seeking run. Since the nature of the problem is separable, the simultaneous sequence and secondary structure motifs runs are split into different threads using java. The heuristic strategy for the primary sequence motifs discovery is the same we followed for the original BEAM formulation (27), but with a match/mismatch 3/-2 substitution matrix for nucleotides and no restraints on the motif length, to allow for short sequence motifs.
For both sequence and structure motif discovery, the significance of a motif model is assessed by rejecting the null hypothesis that the medians of the alignment score distributions induced by the molecules involved in the creation of the motif and the background are equal. This is accomplished by means of a Mann-Whitney-test (MW): this non-parametric test is used to compare the medians of the scores distribution, in particular the scores of the RNA sequences containing the motif aligned against the motif model versus the background scores, estimated by sampling Rfam sequences having similar length and fraction of predicted paired nucleotides (see the (27) for more details on Rfam binning).
On top of the core software, an additional python module has been added to post-process results in order to better describe the motif landscape in terms of co-occurrence of primary sequence and secondary structure motifs. To address this, for each run, now BEAM reports the distance distribution between pairs of motifs present on the same RNAs, as well as percentages of co-occurrence and motif mutual information. An estimation of the significance of both co-presence and mutual distance is made respectively with an Hypergeometric (HG)-test and a Kolmogorov–Smirnov (KS)-test.
The HG-test is computed by considering the presence or absence of a motif in a certain RNA and testing if the two motifs are co-present more than by chance alone. The problem is restated as a conditioned sampling problem (more details in the Supplementary Materials).
The KS is used here to assess if two motifs have a significantly enriched distance. For each motif we build a distribution based on the starting positions on involved RNAs. We state that two motifs have an enriched distance if the distribution given by the difference of the two starting positions (on each RNA containing both) has a definite peak, detectable by the algorithm encoded in the peakutils python library. Since the random distribution given by the difference of two uniform random variables is a triangular curve, we use a KS with the null hypothesis that our difference distribution has a triangular distribution with suitable parameters (Supplementary Materials).
In this work, all the experiments were conducted with the following parameters:
three random starting points for the simulated annealing for each motif (-R 3)
three motifs in sequence and three motifs in structure (-M 3 -N t)
The (N)ucleotide parameter is the new addition (a full list of the parameters can be found in (27)): a run with the (N) flag activated searches for (M) motifs in structure and (M) motifs in sequence. The choice of (R) = 3 is due to optimal results obtained in the artificial testing done in the original paper. Moreover, stability tests done with the data presented in this work have shown satisfactory results (Supplementary Materials).
DoRiNA and eCLIP dataset
All high-throughput data involving RNA interacting with RBPs was downloaded from the DoRiNa database (31) and from the ENCODE project database (32,33). From DoRiNa, we collected 104 HITS-CLIP/PAR-CLIP experiments, both from human (hg19) and mouse (mm9). From ENCODE, we collected 223 eCLIP experiments only from human (hg19), with 150 unique RBPs spanning two different cell types (K562 and HepG2). Then, for both DoRiNA and ENCODE datasets, we extended the RBP-binding region 100 nt upstream and downstream using Bedtools utilities (34) to facilitate the RNA folding prediction with RNAfold (33). Then, we divided every dataset in two different sub-datasets by mapping every sequence to its genomic region (coding regions-CDS and Untranslated Regions UTR) by means of the Gencode provided annotation file; for those RBP known to act in the nucleus on unspliced RNA (mostly splicing factors SRSF1, HNRNPL, NOVA, WTAP) we kept the fragments mapping on introns too. The structure for each RNA was obtained using RNAfold (35) with default parameters (results presented are, however, applicable to RNAstructure MFE predictions too, see Supplementary Materials, Table S2). Sequences longer than 500 nt were filtered out to avoid misleading structure prediction that can occur with very long primary sequences (36,37). In this way, we filtered out about 90k sequences out of ∼26 millions in a total of 71 datasets.
The datasets we downloaded had already been processed and are to be considered post peak-calling, we use all the available data. The only available filter that we decided to ignore is the one on the experiment score (e.g. CLIP score). There are methods available which make use for e.g. the first 1000 intervals with high score (25), but we deliberately used each interval after the robustness tests done in the original paper (where we show how BEAM can handle up to 80% of noise) in order not to cut out any positive information. The only limitation refers to sequence length in order to obtain a more accurate prediction.
Every RNA structure was then translated into the BEAR encoding (30) (the structural alphabet) to be BEAM-ready.
Co-variation analysis
In order to analyze the role of the structure with respect to the sequence motif, we have tested a two-way approach: We have simplified the structural alphabet using a 7-characters encoding, and plotted a logo of the structure underlying the sequence motif (Supplementary Table S2).
The underlying sequence co-variation in structural motifs can instead be used as an information to select a motif among many alternatives. We have used an information-based approach to perform the sequence covariation analysis within the structure. For each motif found, we have calculated the per-position Shannon sequence entropy on the relative model Position Frequency Matrix, normalized from 0 to 1, 0 being assigned to fully conserved columns. A single value estimation E(PFM) of the sequence conservation underlying the structural motif is done by taking the mean of all the involved positions:
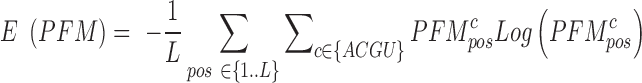 |
where L is the length of the motif model.
miRNA and RNA–RNA interaction data set
The miRNA dataset was composed of miRNA target data from Helwak and collaborators (10) containing information about chromosome coordinates of the mRNA targets. 13 905 molecules formed our collection of mRNAs. For each of these sequences, we chose the first nucleotide at the 5′ end of the longest stem predicted within each chimeric CLASH target (hybridization data provided by Helwak et al., 2013) as reference nucleotide. From this specific nucleotide, as for DoRiNa RBPs datasets, the collections of target RNAs were extended by 100 nucleotides upstream and downstream and then folded using RNAfold (35).
In order to extend the landscape of RNA interaction motifs, we chose the RISE interaction database, that collects data from different HT experiments in which both interactors are RNA molecules. We downloaded the 87 474 human interactions and selected only the ones involving non-coding RNAs and coding RNAs, discarding the coding-coding interactions and the miRNAs ones (that were previously analyzed). From the 39 249 total interactions, we have applied the BEAM2 algorithm to the 39 ncRNA that had more than 50 target interactors. Also in this case we extended the binding site region 100 nucleotides upstream and downstream and we folded the sequences using RNAfold.
Structuration and sequence analysis workflow for miRNAs
To globally analyze the structural trends for miRNA target sequences, we have converted each target sequence from dot-bracket notation to a string of 1s and 0s standing for paired and unpaired nucleotides, respectively. Each RNA has been centered at the reference nucleotide (as described in the previous paragraph) to obtain a superposition, and structuration was represented as a binary vector S:
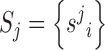 |
where s ji = 0 if NT in position i of RNA j is unpaired, 1 otherwise.
For each position of the aligned sequences, the position-specific mean of Si has been measured, and denoted as the Position Specific Structuration Score (PSSS).
13 905 random nucleotide sequences (each generated sequence has a uniform nucleotide sampling probability for each position) with the same length were generated, folded with RNAfold and compared to target nucleotide sequences from our dataset using TwoSampleLogo (38) to create a nucleotide composition logo. The logo was obtained from 25 nucleotides upstream and downstream with respect to the central position of the sequences as described in dataset preparation. Another set of random sequences with the same GC content of the experimental target sequences was generated and folded with RNAfold. In order to build this second random set we have evaluated the GC content frequency for each position in the experimental dataset and generated random sequences with the same nucleotides frequency in each position. PSSS was measured and compared to the CLASH experimental data.
RESULTS
RNA binding proteins benchmark
We applied BEAM to 104 CLIP (HITS-CLIP and PAR-CLIP) and 223 eCLIP experiments corresponding to 45 (CLIP) and 150 (eCLIP) different proteins in vivo, with 15 proteins in common between the two classes of techniques. The full list of results is reported in the Supplementary Materials. The CLIP datasets are both from human (hg19, 74 experiments, 13 different cell lines, principally HEK293 cells) and mouse (mm9, 30 experiments, 7 different cell lines, mostly brain cells and ESC), while eCLIP data is from human chronic myelogenous leukemia K562 and Hepatocellular Carcinoma HepG2 cells.
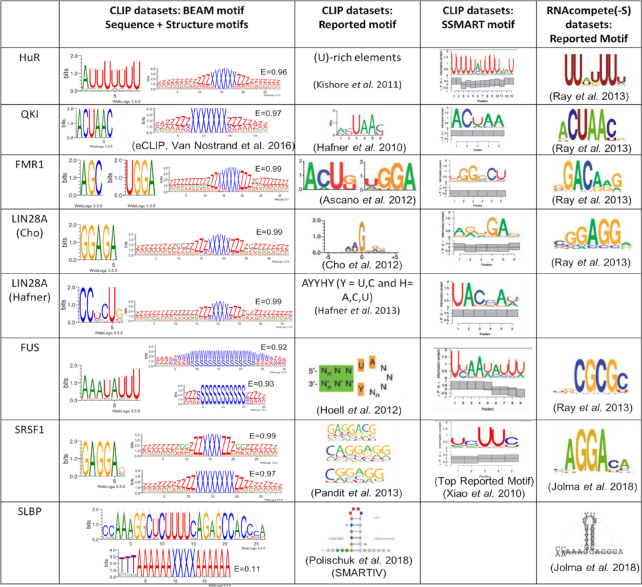
We obtained sequence and structure motifs in order to find pairs of motifs that act concurrently in the determination of the interaction. For each dataset we took the best 3 motifs in sequence and structure, respectively, and analysed their co-coverage in pairs, which is the fraction of RNAs in the dataset that contain both motifs, and their reciprocal localization. First we compared sequence and structure motifs found by BEAM with the reported motifs from corresponding original works, RNAcompete and HTR SELEX (39), SSMART and SMARTIV (25,40), when available (Table 1). Since eCLIP suffers from less experimental noise than CLIP (41), for proteins of which we had both assays we reported the eCLIP only if BEAM was not able to retrieve the signal in the CLIP datasets.
Table 1.
Comparison with known motifs from various sources. The first column shows the BEAM results with sequence and structural motif logos. Alongside the structural logos, the mean Shannon Entropy (E) of the underlying sequence. Structural logos are displayed in qBEAR (Supplementary Materials Table S5); the second column shows the motifs reported in the original papers, when available (exception: SLBP interaction motif has a better representation in the SMARTIV paper); the third column contains the top reported motifs by SSMART along the pairing probability found in the structural context of the sequence motif; the last column contains the motifs reported both by RNAcompete and HTR SELEX
|
Results show strong agreement between our predictions and the known motifs (both in sequence and structure, when available) for the reported RBPs.
Moreover, for each of the presented motifs, we calculated the mean Shannon Entropy value. As we could expect from the absence of sequence motifs superposed to structural ones, the entropy is high (∼0.9) for all the motifs except for SLBP that has an entropy score of 0.1. These results suggest that sequence covariation would be an excellent guide to choose among different structural motifs, yet it cannot be considered a conditio sine qua non to determine the validity of a structural motif, since this tool has originally been primarily designed to tackle the problem of conserved structure/non conserved sequence motifs.
We additionally report structural motifs found in the same datasets, and relative positions between the motifs are calculated (see Supplementary Table S1 for coverages and distances between motif pairs).
The RNA motifs identified as binding the ELAVL1 (HuR) protein (42) show agreement with the known U-rich sequence motif present in 50% of the data. We also identified a hairpin structure with a 6 nt loop in the 36% of the RNAs. An overall 18% of the molecules share both the sequence and the structural motif and the distribution of the distance between the two shows a peak at 15nt from the start of the structural motif. The evidence however is not statistically significant (KS test against random distance distribution P-value >0.05, Supplementary Materials Table S1) to prove both the localization of the sequence motif in the hairpin loop and the mutual presence on the same molecules (HG test pval > 0.05).
RNA molecules binding the quaking (QKI) protein (9,41) are characterized by a strong sequence motif on which all methods agree (ACUAA). BEAM could not find a structure motif in the CLIP dataset, thus we reported the eCLIP results on the same RBP where a structure motif was found in 52% of the RNA molecules. A 7nt long hairpin loop is reported in the 35% of the structures, and both are shared by the 19% of the whole dataset (HG pval < 0.001). No preferred distance between the two significantly emerges (KS pval > 0.05).
The fragile X-mental retardation 1 (FMR1) protein (9) has multiple RNA-binding domains. The original paper reports two sequence motifs, ACUK and WGGA, which distinctly interact with KH and RGG domains. Our results show the WGGA motif, but fail to retrieve the ACUK one, in favour of a 3nt enriched 3-mer (AGC). The two sequence motifs are respectively found in 77% and 96% of the RNAs, shared in the 74% (HG pval < 0.05) and have a significantly enriched distance peak of 14 nucleotides (KS pval < 0.01). The structure motif is found in 33% of the RNAs and is a hairpin structure with a 5–6 nt long loop and small internal loop 3′ of the main loop, the co-occurrence between the structure and the two sequence motifs however is not statistically relevant (HG pval > 0.05).
LIN28A (43,44) has a zinc-finger domain CCHC that has been hypothesized to bind an internal loop in the hairpin containing the sequence motif, at variable distances (45,46). The sequence motif that BEAM identifies is similar to the one found by other methods: the motif found in the data from Cho and colleagues is found in 38% of the RNA, and the structure motif shows an internal loop structure in the hairpin, in 37% of RNAs. Both occur in 14% of the RNA (HG pval < 0.001). The data from Hafner and colleagues led to similar results: the sequence motif is found in 58% of the RNA and the structural hairpin with an internal loop is found in the 40%, with both occurring in the 23% of the molecules.
FUS (47), an RNA binding protein whose mutation can generate two incurable neurological diseases (ALS—Amyotrophic Lateral Sclerosis and FTLD—Frontotemporal Lobar Degeneration), has a Zinc finger domain (RanBP2-type) that recognizes a variable length hairpin loop that is found by BEAM (it finds different structural motifs with variable loop lengths) and a sequence motif that is in agreement with the top motif reported by SSMART (HG pval < 0.01).
SRSF1 (6,48) has a role in preventing exon skipping and it controls the accuracy of splicing and alternative splicing. This protein interacts by RS domains with other spliceosomal components and it is known to bind purine-rich RNA sequences. It recognizes three reported motifs in the original paper, all composed of poly-purinic stretches. Both the predictions by HT SELEX and by the original paper are in agreement with our sequence motif. The structural motifs found are two, one with a symmetrical internal loop and a 4 nt hairpin loop and the other with a longer 7 nt hairpin loop. The signal is found respectively in the 26% and 29% of the data, shared by the 19% (HG pval < 0.001) and have an enriched distance of 63 nt (KS pval < 0.01) between the two structures.
The RNAs binding the SLBP protein (48,49), involved in histone pre-mRNA processing, is known to bind stem-loop structure. BEAM results within an eCLIP dataset, shows that SLBP targets are enriched in a long sequence motif in perfect agreement with the data reported by SMARTIV and HT SELEX, included in a short stem with a 4 nt hairpin loop. However the data is not sufficient to statistically consider the two motif to be co-localized significantly (KS pval > 0.05). The sequence motif is present in 66% of the RNAs while the structure motif is in 59%. Both are present in 51% of the RNAs (HG pval < 0.001).
RNA binding proteins landscape
We studied the pairs of motifs generated in each experiment dataset to better characterize the landscape of interaction. We divided the results in 3 classes, characterized by the motif implicated: sequence-sequence pairs, sequence-structure pairs, structure–structure pairs. A global analysis on all the motif pairs, conducted by plotting the distribution of motif pairs co-coverage for each dataset (CLIP and eCLIP), shows a general trend of low co-occurrence of motifs on the RNAs (Figure 1). This is probably due to the inherent noise in the CLIP techniques, since the co-localization distribution moves to higher values for the eCLIP datasets (t-test P-values < 0.001 for all three classes in both datasets).
Figure 1.

Absolute frequency of co-localized pairs of motifs in the CLIP (left) and eCLIP (right) datasets. The majority of sequence-structural pairs are centered around 19% with extreme cases of ∼40% (orange line). More cases of co-localization on RNAs are present in the sequence-sequence pairs while the structure-structure pairs are relatively low, with a mean of 10%. eCLIP results show a generalized higher co-localization because of the lower experimental noise produced by the assay.
We analyzed the top sequence and structure pairs in eCLIP datasets (ranked by co-coverage) and reported some interesting results (for a full list of results, see Supplementary Materials).
RBM5, a component of spliceosomal A complex that is able to regulate alternative splicing of target mRNAs, contains two zinc finger domains. Results show the sequence motif ‘GGGAGGUGG’ in 70% of the dataset and the structure motif (in qBEAR notation, a reduced version of the alphabet with 19 characters instead of 83, details in Supplementary Materials) ‘z**zzzzzccczzzxxxxxzzzzcczzz****zz’ in 69% of sequences. Together these two are present in 45% of the molecules (HG pval < 0.01). The structural motif shows a symmetrical internal loop situated in a short hairpin loop of about 5nt, and located outside the binding site (Figure 2). This is consistent with previous findings about other ZF domains (e.g. LIN28A Zinc Finger), that is known to guide the protein to the interaction hairpin loop and has a similar structural motif (11). A similar structural motif is found in the LIN28A dataset, with which RBM5 share a ZF domain (Supplementary Materials, Figure S2).
Figure 2.

RBM5 interaction landscape. Top: sequence and structure motifs shared by 45% of the RNA molecules. Bottom: location of sequence (left) and structure (right) motifs; the residue in position 0 corresponds to the start of the binding site reported by the experiment. Lower right: score distributions of input sequences and background sequences with respect to the motif model.
TIAL1 (a protein involved in translational control, splicing and apoptosis) shows preference for U-rich sequence motifs with a sequence motif present in 65% of targets and a structure motif ‘zzzzzcc*zzzzxxxxzzzzzz*zzz’ that is shared by 68% of mRNAs (HG pval < 0.01). Together these are present in 45% of the molecules. This protein has three recognition motifs and is known to bind adenine and uridine elements. In the TIAL1 eCLIP dataset, the sequence motif is located in the binding site, while the structural motif has no enriched position (Figure 3), yet it has significantly higher scores than the background (Mann-Whitney P-value < 0.001) within the BEAM scoring model. A similar sequence motif is found in the ELAVL1 dataset, with which TIAL1 share a common RRM domain (Supplementary Materials, Figure S2).
Figure 3.

TIAL1 interaction landscape. Top: sequence and structure motifs shared by 49% of the sequences. Bottom: location of sequence (left) and structure (center) motifs; the residue in position 0 corresponds to the start of the binding site reported by the experiment. Lower right: score distributions of input sequences and background sequences with respect to the motif model.
miRNA sequence and structure motifs
Sequence analysis of target RNAs captured by CLASH binding miRNAs has shown an enrichment in GC content along the target region interacting with microRNA. The local increase in this composition is consistent with the GC percentage in microRNA seed sequences, reflecting the complementarity of the two RNA strands. The sequence information obtained aligning the target sequences along the interaction sites have confirmed previous analyses about the importance of GC content in miRNA recognition (11) together with a novel signal about how structure can influence microRNA interaction. In fact, a global analysis of structuration trends reveals a preference in more structured nucleotides in the position corresponding to the microRNA interaction site as in Figure 4.
Figure 4.
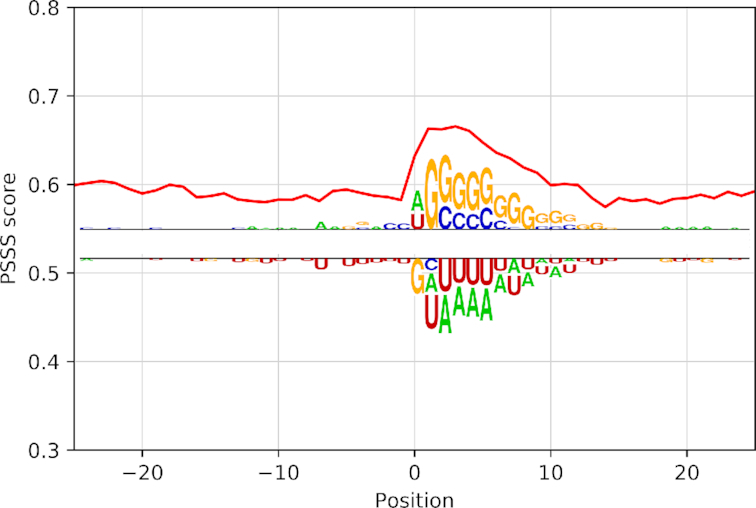
Structural trend and sequence motifs for miRNA targets. Red lines correspond to PSSS mean score; the weblogo shows the sequence motif identified (top) in that specific regions compared to background (bottom). Here, position 0 corresponds to miRNA binding site as described in methods.
The overlapping of the structure trend and sequence preference in GC content appear to be localized in the same position, corresponding to miRNA binding sites suggesting that, before the interaction, target transcripts are more structured in these sites compared to flanking regions. Results do not change if we take into account 10 sub-optimal secondary structure predictions for each RNA sequence (Supplementary Materials, Figures S3 and S4).
In order to validate this result with respect to a background, we compared this data with an equal number of randomly generated sequences of the same length with the same nucleotide composition for each aligned position (see Materials and Methods) and then folded them using RNAfold. Results of the comparison of real targets and control sequences reveal the same preference for structured regions when GC content increases. This same structuration trend decreases in the real data when the GC percentage of nucleotide composition decreases under 0.5 in the binding site region (Supplementary Figure S1). This data reveals that primary sequence specific composition influences structure conformation, leading to an increase of paired nucleotides produced by variations in nucleotide composition related to guanine and cytosine abundance in miRNA targets across the miRNA binding site. This variation in nucleotide composition is important for the interaction and it also generates structural preferences. In fact, the signal is not specific for certain groups of targets (Supplementary Figure S2), defined as specific miRNAs’ target sets. Even if miRNAs have different unique seed sequences, the displacement in nucleotide composition generates a structure trend shared by the vast majority of target RNAs.
Common structure interaction motifs
We have shown that an enrichment in GC content generates a typical structure trend, but this structure could be part of a specific structure motif. To investigate further, we used the BEAM2.0 algorithm to discover common motifs, shared by all the miRNA targets as a whole. Motif discovery on the whole dataset revealed that some specific motifs are shared by ∼30% of target genes. Since the found structural motifs seem not to be specific for all targets under miRNA control (Supplementary Table S2), we looked for the presence of motifs in groups of mRNAs that interact with the same miRNA. The results of motif finding are schematized in Figure 5, with the some specific examples highlighted with red circles, and listed in Table 2.
Figure 5.
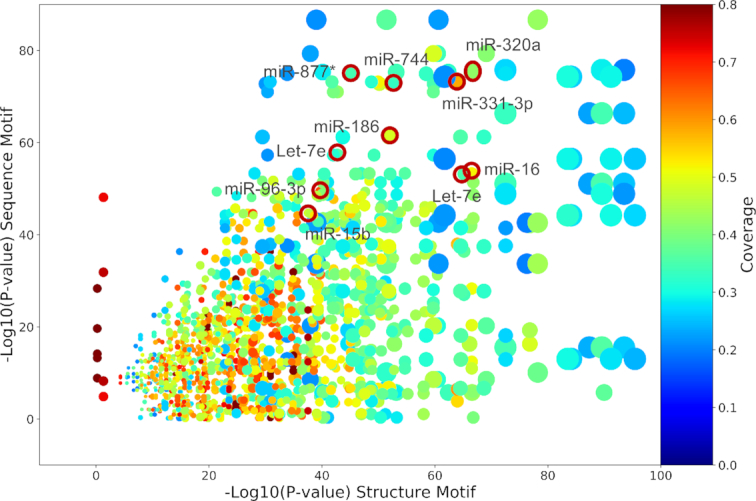
Overview of motifs results for miRNA targets. Each spot is a sequence-structure motif pair found in one of the 383 miRNA set of targets. Ten representative pairs are shown in red circles. The size of the spot is proportional to the size of the dataset and the color represents the co-coverage, the position along x- and y- axes are respectively the reverse log10P-value for structure and sequence motifs.
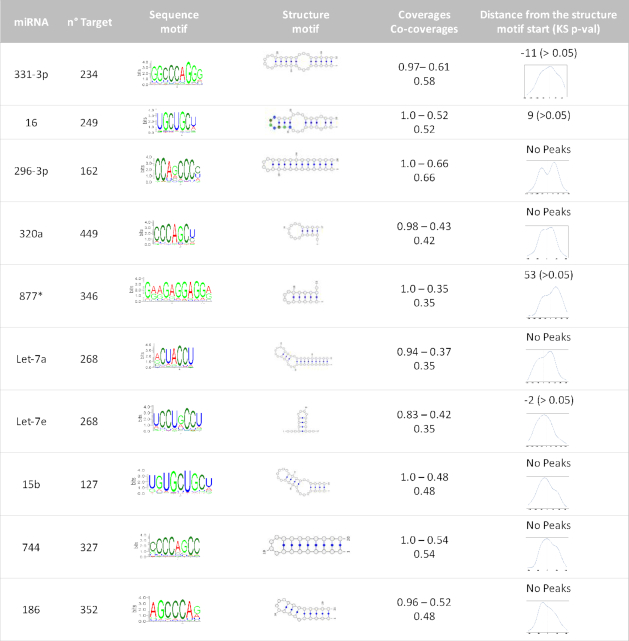
Table 2.
Ten representative sequence-structure motif pairs. From the first column: miRNAs, the number of interactors for each miRNA, the web logo of the sequence motif found, the structure motif best representative, sequence motif coverage, structure motif coverage and co-coverage of the pair. Last column contains an estimate of the enriched distance (if present) between sequence and structure motif. If the two motifs overlap, the model structure in the fourth column as a color-coded sequence motif, otherwise a KDE is shown in the last column (in nucleotide units)
|
The motifs found by BEAM are, at the structural level, all similar and are in general hairpin structures, some perfectly paired, some presenting internal loops or bulges. At the sequence level, they are all complementary to seed sequences (this is consistent with the high specificity of the miRNA/mRNA interaction for target recognition). The sequence motifs confirm also the GC enrichment found in the global analysis of target genes and the structural motifs obtained, all similar, are in agreement with the structuration trend.
RNA motifs in RNA–RNA interactions
The analysis of sequence and structure motifs in RNA–RNA interactions has shown some interesting motifs. The Terminal differentiation-INduced Non Coding RNA TINCR is known to be involved in cancer as tumor suppressor and it regulates prostate cancer cell proliferation, migration and invasion (50). It is a 3.7 kb long non-coding RNAs that in normal condition is able to control human epidermal differentiation (51). Our data shows that 48% of the 258 target RNAs share a C-rich sequence motif ‘CCCCUCC’ and 40% of molecules have a specific structure in the binding site region ‘zzzzzzzz*cczzzzxxxxxxzzzzz*zzzzzzz’. The sequence and structure motifs co-exist in only 16% of the targets and that is not sufficient to reject the hypothesis that it is due to chance (HG pval > 0.05) (Figure 6).
Figure 6.

TINCR interaction landscape. Top: sequence and structure motifs shared by 16% of the sequences. Bottom: location of sequence (left) and structure (center) motifs; the residue in position #0 corresponds to the start of the binding site reported by the experiment. Lower right: score distributions of input sequences and background sequences with respect to the motif model.
Another important ncRNA, XIST, is located in the nucleus and it is involved in X chromosome silencing in mammalian females to provide equal dosage distribution between male and female (52). Motif discovery with BEAM2 shows that 73% of XIST targets share the ‘UCUGAG’ sequence motif and 57% of the molecules have the structure motif ‘**zzz*czc*zzzzxxxxxxzzzz**zz****zzz’ mostly located in the binding site region. The two motifs co-exist in 42% of the molecules but the data is not sufficient to reject the hypothesis that their coupling is due to chance (HG pval > 0.05) (Figure 7).
Figure 7.

XIST interaction landscape. Top: sequence and structure motifs shared by 42% of the sequences. Bottom: location of sequence (left) and structure (center) motifs; the residue in position 0 corresponds to the start of the binding site reported by the experiment. Lower right: score distributions of input sequences and background sequences with respect to the motif model.
DISCUSSION
We conducted a systematic analysis over 104 CLIP and 223 eCLIP datasets to uncover a broad landscape of interactions between RNA-binding Proteins/RNAs and their targets. Given the current lack of tools able to simultaneously and efficiently detect RNA sequence and secondary structure patterns, we implemented a sequence module for BEAM, extending the breadth at which it describes an interaction landscape. BEAM has been developed to optimize execution time when analyzing high-throughput datasets (CLIP-Seq and similar assays), and now adds a sequence characterization to the predicted interaction motif. This module acts independently from the main unit, which looks for conserved structural motifs. Results with CLIP-Seq and eCLIP data are in agreement with recent findings (represented by HT SELEX and SSMART), and add a structural information layer which may be disjointed from the sequence motif. Although the BEAM software has been demonstrated to work well with noisy datasets (up to 80% of unrelated data), CLIP-Seq data may be difficult to analyse, given the possibly large proportion of unspecific RNAs that the method can detect amidst those that are really able to interact with the immunoprecipitated RBP; however, the eCLIP assay has proven a better technique and overcomes CLIP-Seq limitations by reporting less unspecific targets (33,48). In few cases, we have been able to identify conserved structural motifs that are found at specific distances from the sequence motifs, or pairs of sequence-sequence or structure-structure motifs that have also enriched distances. Some words should be spent about the aspect of modularity, which has been recently reviewed by (1) and (53): The presence of multiple binding domains on RBP alters the landscape of interaction. Dominguez and colleagues have shown how same singular domains do not guarantee the same signal structure. Our dataset was not sufficient to extensively test the modularity of the binding interaction. Yet we can see from our results how similar proteins sharing one or two domains may have similar binding partners (domains shown in Supplementary Materials).
Moving to the landscape analysis with the focus on structural aspects of miRNA target recognition we show novel structural characteristics in target genes that could be important for the specificity of the interaction. Even if the seed sequence is fundamental for target recognition, also structure could play a role in the complex mechanism of gene regulation. Our results show a trend to a more structured folding in the target region bound by miRNA. This structuration composition reflects a nucleotide variation in the region with an increment in GC content localized in the same position. GC enrichment do not change with different types of interaction and it is localized in the target region complementary to the seed, suggesting a role in the stability of the miRNA–target hybrid. Even if more investigations about hybrid stability are necessary to confirm our data, these results could open new scenarios about the importance of structure in miRNA interactions, allowing a better understanding of the recognition mechanisms and, in principle, a better target prediction.
The RNA–RNA interaction analysis have revealed some interesting motifs that are enriched in the protein-coding mRNA targets of a selected number of ncRNA, especially long non coding RNAs, and their protein-coding targets. These results may be the starting point to highlight the mechanism by which these molecules interact with their targets and how this can affect their regulatory function.
Summarizing, we presented a survey of the landscape of interaction motifs, using an update of our BEAM method, by analyzing both sequence and structure motifs independently and then combining the information obtained to better characterize RBP–RNA and miRNA/RNA interactions.
DATA AVAILABILITY
The full list of results and BEAM2.0 is available on the BEAM web server (http://beam.uniroma2.it/data).
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
EPIGEN flagship project MIUR-CNR [to MHC]. Funding for open access charge: AIRC project [to MHC] (grant number IG 16895).
Conflict of interest statement. None declared.
REFERENCES
- 1. Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., Bazile C., Lambert N.J., Van Nostrand E.L., Pratt G.A. et al.. Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell. 2018; 70:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hannigan M.M., Zagore L.L., Licatalosi D.D.. Mapping transcriptome-wide protein–RNA interactions to elucidate RNA regulatory programs. Quant. Biol. 2018; 6:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferrè F., Colantoni A., Helmer-Citterich M.. Revealing protein–lncRNA interaction. Brief. Bioinform. 2016; 17:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Licatalosi D.D., Mele A., Fak J.J., Ule J., Kayikci M., Chi S.W., Clark T.A., Schweitzer A.C., Blume J.E., Wang X. et al.. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008; 456:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sanford J.R., Wang X., Mort M., Vanduyn N., Cooper D.N., Mooney S.D., Edenberg H.J., Liu Y.. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009; 19:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B.. Europe PMC Funders Group iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nature structural & molecular biology. 2011; 17:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y.S., Mele A., Fraser C.E., Stone E.F., Chen C., Fak J.J., Chi S.W. et al.. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011; 146:247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jungkamp A.-C., Munschauer M. et al.. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics. 2014; 30:1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seok H., Ham J., Jang E.-S., Chi S.W.. MicroRNA target Recognition: Insights from Transcriptome-Wide Non-Canonical interactions. Mol. Cells. 2016; 39:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ding J., Li X., Hu H.. MicroRNA modules prefer to bind weak and unconventional target sites. Bioinformatics. 2015; 31:1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding J., Li X., Hu H.. TarPmiR: a new approach for microRNA target site prediction. Bioinformatics. 2016; 32:2768–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Enright A.J., John B., Gaul U., Tuschl T., Sander C., Marks D.S.. MicroRNA targets in Drosophila. Genome Biol. 2003; 5:R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lewis B.P., Shih I., Jones-Rhoades M.W., Bartel D.P., Burge C.B.. Prediction of mammalian MicroRNA targets. Cell. 2003; 115:787–798. [DOI] [PubMed] [Google Scholar]
- 17. Lewis B.P., Burge C.B., Bartel D.P.. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are MicroRNA targets. Cell. 2005; 120:15–20. [DOI] [PubMed] [Google Scholar]
- 18. Guil S., Esteller M.. RNA–RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem. Sci. 2015; 40:248–256. [DOI] [PubMed] [Google Scholar]
- 19. Sasse A., Laverty K.U., Hughes T.R., Morris Q.D.. Motif models for RNA-binding proteins. Curr. Opin. Struct. Biol. 2018; 53:115–123. [DOI] [PubMed] [Google Scholar]
- 20. Kazan H., Ray D., Chan E.T., Hughes T.R., Morris Q.. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput. Biol. 2010; 6:e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frith M.C., Saunders N.F.W., Kobe B., Bailey T.L.. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput. Biol. 2008; 4:e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao Z., Weinberg Z., Ruzzo W.L.. CMfinder–a covariance model based RNA motif finding algorithm. Bioinformatics. 2006; 22:445–452. [DOI] [PubMed] [Google Scholar]
- 23. Maticzka D., Lange S.J., Costa F., Backofen R.. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 2014; 15:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hiller M., Pudimat R., Busch A., Backofen R.. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res. 2006; 34:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polishchuk M., Paz I., Yakhini Z., Mandel-Gutfreund Y.. SMARTIV: combined sequence and structure de-novo motif discovery for in-vivo RNA binding data. Nucleic Acids Res. 2018; 46:W221–W228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cook K.B., Vembu S., Ha K.C.H., Zheng H., Laverty K.U., Hughes T.R., Ray D., Morris Q.D.. RNAcompete-S: Combined RNA sequence/structure preferences for RNA binding proteins derived from a single-step in vitro selection. Methods. 2017; 126:18–28. [DOI] [PubMed] [Google Scholar]
- 27. Pietrosanto M., Mattei E., Helmer-Citterich M., Ferrè F.. A novel method for the identification of conserved structural patterns in RNA: from small scale to high-throughput applications. Nucleic Acids Res. 2016; 44:8600–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heller D., Krestel R., Ohler U., Vingron M., Marsico A.. ssHMM: extracting intuitive sequence-structure motifs from high-throughput RNA-binding protein data. Nucleic Acids Res. 2017; 45:11004–11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rabani M., Kertesz M., Segal E.. Computational prediction of RNA structural motifs involved in post-transcriptional regulatory processes. Methods Mol. Biol. 2011; 714:467–479. [DOI] [PubMed] [Google Scholar]
- 30. Mattei E., Ausiello G., Ferrè F., Helmer-Citterich M.. A novel approach to represent and compare RNA secondary structures. Nucleic Acids Res. 2014; 42:6146–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blin K., Dieterich C., Wurmus R., Rajewsky N., Landthaler M., Akalin A.. DoRiNA 2.0—upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2015; 43:D160–D167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Advani S.H., Lee T.S., Dean R.H., Pak C.K., Avasthi J.M.. Consequences of fluid lag in three-dimensional hydraulic fractures. Int. J. Numer. Anal. Methods Geomech. 1997; 21:229–240. [Google Scholar]
- 33. Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., Bazile C., Lambert N.J., Van Nostrand E.L., Pratt G.A. et al.. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Molecular Cell. 2018; 70:854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lorenz R., Bernhart S.H., Höner Zu, Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gardner P.P., Giegerich R.. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics. 2004; 5:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lange S.J., Maticzka D., Möhl M., Gagnon J.N., Brown C.M., Backofen R.. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012; 40:5215–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vacic V., Iakoucheva L.M., Radivojac P.. Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics. 2006; 22:1536–1537. [DOI] [PubMed] [Google Scholar]
- 39. Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X., Gueroussov S., Albu M., Zheng H., Yang A. et al.. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013; 499:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Munteanu A., Mukherjee N., Ohler U.. SSMART: sequence-structure motif identification for RNA-binding proteins. Bioinformatics. 2018; 34:3990–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin- C., Fang M.Y., Sundararaman B., Blue S.M., Thai B., Surka C., Elkins K. et al.. Robust transcriptome-wide discovery of RNA binding protein binding sites with enhanced CLIP. Nat. Methods. 2016; 13:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kishore S., Jaskiewicz L., Burger L., Hausser J., Khorshid M., Zavolan M.. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods. 2011; 8:559–564. [DOI] [PubMed] [Google Scholar]
- 43. Cho J., Chang H., Kwon S.C., Kim B., Kim Y., Choe J., Ha M., Kim Y.K., Kim V.N.. LIN28A is a suppressor of ER-associated translation in embryonic stem cells. Cell. 2012; 151:765–777. [DOI] [PubMed] [Google Scholar]
- 44. Ascano M., Hafner M., Cekan P., Gerstberger S., Tuschl T.. Identification of RNA–protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA. 2012; 3:159–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pietrosanto M., Adinolfi M., Casula R., Ausiello G., Ferrè F., Manuela H.-C.. BEAM web server: a tool for structural RNA motif discovery. Bioinformatics. 2018; 34:1058–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Warrander F., Faas L., Kovalevskiy O., Peters D., Coles M., Antson A.A., Genever P., Isaacs H. V.. Lin28 proteins promote expression of 17∼92 family miRNAs during amphibian development. Dev. Dyn. 2016; 245:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakaya T., Alexiou P., Maragkakis M., Chang A., Mourelatos Z.. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. RNA. 2013; 19:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin- C., Fang M.Y., Sundararaman B., Blue S.M., Thai B., Surka C., Elkins K. et al.. Robust transcriptome-wide discovery of RNA binding protein binding sites with enhanced CLIP. Nat. Methods. 2016; 13:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brooks L., Lyons S.M., Mahoney J.M., Welch J.D., Liu Z., Marzluff W.F., Whitfield M.L.. A multiprotein occupancy map of the mRNP on the 3′ end of histone mRNAs. RNA. 2015; 21:1943–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dong L., Ding H., Li Y., Xue D., Liu Y.. LncRNA TINCR is associated with clinical progression and serves as tumor suppressive role in prostate cancer. Cancer Manag. Res. 2018; 10:2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kretz M., Siprashvili Z., Chu C., Webster D.E., Zehnder A., Qu K., Lee C.S., Flockhart R.J., Groff A.F., Chow J.. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013; 493:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmitt A.M., Chang H.Y.. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016; 29:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lunde B.M., Moore C., Varani G.. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007; 8:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full list of results and BEAM2.0 is available on the BEAM web server (http://beam.uniroma2.it/data).