


Abstract
Alternative splicing is facilitated by accessory proteins that guide spliceosome subunits to the primary transcript. Many of these splicing factors recognize the RNA polymerase II tail, but SFPQ is a notable exception even though essential for mammalian RNA processing. This study reveals a novel role for Dido3, one of three Dido gene products, in alternative splicing. Binding of the Dido3 amino terminus to histones and to the polymerase jaw domain was previously reported, and here we show interaction between its carboxy terminus and SFPQ. We generated a mutant that eliminates Dido3 but preserves other Dido gene products, mimicking reduced Dido3 levels in myeloid neoplasms. Dido mutation suppressed SFPQ binding to RNA and increased skipping for a large group of exons. Exons bearing recognition sequences for alternative splicing factors were nonetheless included more efficiently. Reduced SFPQ recruitment may thus account for increased skipping of SFPQ-dependent exons, but could also generate a splicing factor surplus that becomes available to competing splice sites. Taken together, our data indicate that Dido3 is an adaptor that controls SFPQ utilization in RNA splicing. Distributing splicing factor recruitment over parallel pathways provides mammals with a simple mechanism to regulate exon usage while maintaining RNA splicing efficiency.
INTRODUCTION
Gene expression in mammals is a complex process that requires the interplay of many factors to guide the production of primary transcripts and their processing into mature messengers. The majority of primary transcripts undergo additional processing, such as splicing reactions in which exons are joined together and intervening introns are removed. Most RNA polymerase II (RNA pol II) transcripts are processed by the spliceosome, a large complex assembled from five subunits (U1, U2, U4, U5 and U6) each consisting of multiple proteins and an RNA scaffold (1).
RNA splicing involves assembly of the spliceosome on the primary transcript, which is initiated by recruitment of the U1 subunit to the 5′ splice site (SS) and U2 subunit to the branch-point and 3′ SS (2). Successive incorporation of the remaining subunits leads to increased commitment and ultimately, to spliceosome activation (2). Notwithstanding the complexity of spliceosome assembly, most exons in yeast and mammals are processed shortly after emerging from the transcription complex (3–5). The actual splicing reactions thus occur rapidly once a spliceosome is committed and assembled. Fast substrate processing allows for rapid spliceosome recycling, which maintains the reservoir of free subunits (Supplementary Figure S1). Suppression of recycling greatly reduces RNA splicing yield (6,7), showing that a sufficiently large subunit pool is essential for efficiency.
In contrast to constitutive splicing, exons that undergo alternative splicing must expose ancillary SS, and thus have relatively slow kinetics (8). This situation seems to conflict with the need for rapid recycling, as slow splicing can sequester spliceosome subunits. For this reason, organisms that make extensive use of alternative splicing have evolved mechanisms to prevent depletion of the subunit pool. Instead of controlling the core spliceosome, alternative splicing relies on accessory proteins at the stage of initial SS recognition (9). Core spliceosome subunits have thus changed little from yeast to mammals. To move rate-limiting steps away from the core spliceosome, U2 placement in particular is guided by splicing factors (SF) such as U2AF, SFPQ and serine/arginine-rich (SR) proteins (10–12). 5′ SS recognition by U1 however occurs without additional helper proteins (13), and the ability to generate different messengers largely depends on SF-dependent U2 positioning (9,14). A speed-limiting step prior to U2 docking therefore allows relatively slow SS selection in alternative splicing to coexist with rapid processing and subunit recycling.
Given the prominence of SF in alternative splicing, much effort has been made to define the mechanisms that promote their recruitment to the transcription complex. The current point of view ascribes this function to the carboxy-terminal domain (CTD) of the largest RNA pol II subunit (9,15). Partial coupling of splicing to transcription (5), too, suggests a role for the transcription holoenzyme. Previous studies reported that the CTD, a flexible heptapeptide repeat rich in serine and threonine residues, binds protein complexes needed for splicing and polyadenylation (16,17), and that the number of repeats scales with organism complexity (18). Nonetheless, the sequence of each heptapeptide repeat is almost identical in all organisms. Added CTD length in higher metazoans thus seems to increase capacity without functional diversification. In contrast to the conservative development of the CTD, mammals have evolved a wide variety of accessory SF. Some of these—the SRSF family members—can also be found in fungi but others—for example SFPQ—are specific to higher metazoans. SRSF proteins are recognizable as a separate class from which SFPQ is excluded, because the two families have distinct domain architectures (19). The relation of SFPQ to RNA pol II has indeed lead to questioning; whereas SRSF proteins clearly recognize the CTD, direct binding of SFPQ to this domain is dubious (20,21) even though the latter SF are essential for splicing of practically all exons in nuclear extracts (11,22). Among other functions, SFPQ binds to the polypyrimide tract upstream of the 3′ SS, assisting spliceosome assembly and the second catalytic step of splicing (23). The need for SFPQ in mammalian splicing but its atypical lack of CTD binding suggests that additional molecules contribute to recruitment of this SF. Delegating recruitment of some SF could not only lower capacity pressure on the CTD, but also allow for independent modulation of putative adaptors. Adaptor molecules that bridge SF and RNA pol II could thus contribute to alternative splicing in several ways.
To study these aspects of alternative splicing, we focused on Dido3, a vertebrate protein that is homologous to yeast BYE1 but has an expanded domain architecture (24,25). BYE1 contains an amino-terminal PHD domain that recognizes histone H3 trimethylated on lysine 4 (H3K4me3), and a central TFIIS-like domain that interacts with the RNA pol II jaw and funnel (25,26). Through the latter interaction, the protein could carry out a CTD-independent adaptor function. Whereas the yeast BYE1 gene produces a single protein, vertebrate Dido encodes three messengers that share a 5′ region but have unique 3′ terminal exons (27); in addition to an intermediate isoform (Dido2) corresponding to the yeast homolog BYE1, shorter (Dido1) and longer (Dido3) isoforms are produced. Dido gene mutations and changes in isoform ratio are linked to malignant myelopoiesis (27,28), a family of syndromes closely associated with splicing defects (29). The Dido3-specific protein domain, which is dispensable for RNA pol II binding (30), comprises a large unstructured region characteristic of recent evolution (31). This long isoform is found in SFPQ-producing organisms that make extensive use of alternative splicing but not in yeast. Some evidence for a role in RNA splicing have been found in insects, where the Dido homolog pps functions in sex determination by promoting production of female-specific splicing variants (32).
Here, we show a role for the Dido3-specific domain in SFPQ binding, thereby fulfilling the requirements for the adaptor function mentioned above. Dido3 may thus be the principal catalyst for SFPQ recruitment, rather than the RNA pol II CTD. In contrast to known SF deletions (33,34), we were able to delete large portions of the Dido gene while maintaining cell viability, and gain insight into the effects of Dido mutation on alternative splicing. Disruption of Dido3-specific amino acid sequences reduced SFPQ binding to RNA while increasing the influence of CTD-dependent SF on exon inclusion. Our data thus indicate a two-pronged strategy that promotes efficient SF utilization but still allows for alternative splicing.
MATERIALS AND METHODS
Cell culture and reagents
Mouse embryonic fibroblasts, HEK 293T and HeLa cells were cultured in DMEM supplemented with 10% fetal calf serum and antibiotics. RPE-1 cells were cultured in DMEM+F12 (1:1) supplemented with 10% fetal calf serum and antibiotics. Antibodies against the mouse Dido amino-terminal region have been described before (35,36). Antibodies against GST (sc-138, RRID:AB_627677) were from Santa Cruz Biotechnology (Dallas, TX), antibodies against U2AF1 (GTX106854, RRID:AB_1952473) were from Genetex (Irvine, CA) and antibodies against the V5-tag (ab53418, RRID:AB_883403), Tubulin (ab44928, RRID:AB_2241150) SFPQ (ab177149), HNRNPU (ab20666, RRID:AB_732983) and human Dido (ab92868, RRID:AB_10562305) were from Abcam (Cambridge, MA). Actinomycin D (A9415, Sigma, St. Louis, MO) and isoginkgetin (416154, Merck Millipore) were used as described (3,37). Immunoprecipitation and immunofluorescence were carried out according to standard protocols (36) as outlined in the Supplementary Data.
GST pull-downs and protein identification
To construct the GST fusion, a fragment corresponding to amino acids 530–2240 from human Dido3 was cloned into pEBG (38). GST fusions were purified as described (35,39), separated by sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and detected by Coomassie Brilliant Blue staining. For protein identification, all bands detected were excised and washed with distilled water. Gel segments corresponding to the same molecular weight were excised from the non-fused GST lane and used as negative controls. Protein identification was carried out by mass spectrometry as described (35). Detailed conditions are given in the Supplementary Data.
Cells lacking Dido exon 16
Mouse embryonic fibroblasts (MEF) lacking exon 16 were derived from embryos that expressed a UBC-Cre-ERT2 transgene and were homozygous for Dido exon 16 flanked by two loxP sites. MEF were derived from individual embryos and immortalized as described (40). After treatment with 1 μM hydroxytamoxifen for two passages, individual clones were selected and exon 16 deletion was confirmed by polymerase chain reaction (PCR) genotyping. Details are provided in the Supplementary Data.
Proximity ligation assay
Intracellular interaction between SFPQ and Dido3 was detected using Duolink®In Situ Orange Starter Kit Mouse/Rabbit (DUO92102, Sigma), according to the manufacturer’s instructions. In brief, the protocol used was as follows. MEF were cultured on glass coverslips until 50% confluent. Cells were washed once in phosphate buffered saline (PBS), fixed with 3.7% paraformaldehyde (10 min) and washed again in PBS. Cells were permeabilized with 0.5% NP-40 (5 min) and blocked using the provided blocking solution (1 h, 37°C). After incubation with primary antibodies against the Dido amino-terminal region and SFPQ for 1 h at room temperature, two proximity ligation probes were added and incubated (1 h, 37°C). DNA was amplified by adding polymerase and complementary fluorescent nucleotides (90 min, 37°C). Coverslips were mounted on Superfrost ultra plus microscope slides (Thermo Fisher Scientific, Waltham, MA) in the provided mounting medium and visualized by immunofluorescence.
RNA crosslinking and immunoprecipitation
Photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) was carried out using mouse embryonic fibroblasts as described (41). In brief, cells were treated with 100 μM 4-Thiouridine and crosslinked with UV light. RNA-bound protein complexes were captured on oligo(dT)25 magnetic beads (NE Biolabs) and washed extensively. RNA–protein complexes were then eluted by RNAse A treatment and analyzed by SDS-PAGE and western blot. Negative controls were RNAse A treated before oligo(dT) capture. Details are given in the Supplementary Data.
RNA crosslinking and immunoprecipitation (CLIP) were performed as described (42) with minor modifications. SFPQ was precipitated with polyclonal antibodies (RN106PW, MBL International, Woburn, MA). A corresponding isotype antibody was used as negative control. After protein digestion with proteinase K, RNA was extracted with the SPLIT RNA extraction kit (Lexogen, Austria) using the manufacturer’s protocol. RNA length was measured on a bioanalyzer (Agilent). Finally, RT-PCR amplifications were performed using a Verso One-step RT-PCR Kit (ThermoFisher) with a pair of primers designed for GAS2L1 gene detection (FW: TCACTGTCCCCAGAGTAGCG; RV: CCGCGCCAGTGATACACATC).
RNA sequencing
Mouse embryonic fibroblasts were grown to 80% confluence, washed once in PBS and lifted by trypsinization. After washing again with PBS, total RNA was isolated from approximately 5 × 106 cells with a SPLIT RNA extraction kit (Lexogen, Austria) using the manufacturer’s protocol. For each mutant and the WT control, biological triplicates were prepared. Subsequent steps were outsourced to on-campus facilities (Parque Científico de Madrid). A Bioanalyzer (Agilent) was used to measure RNA quality and all samples used had RNA integrity numbers of 9 or higher. Libraries were prepared from 1 μg total RNA by the TruSeq RNA v2 method (Illumina) and at least 50 million paired-end (2 × 100) reads per sample were obtained on a HiSeq 2000 system (Illumina).
mRNA sequence analysis
Sequencing adaptors were removed from both ends of the reads. Obtained sequences were aligned with Tophat2 (version 2.1.0) against the Mus musculus mm10 assembly (43), using the corresponding gene annotation model to guide alignment of splice junctions. Initial classification of splicing alterations was performed by Altanalyze software (44). The fragcount script from the RSeQC package (45) was used to determine expression levels. To quantify exon skipping, the number of junctions spanning each exon were counted and divided by the corresponding expression (in FPKM) normalized to gene level (Supplementary Figure S2). Using gene-level expression allows for quantitation of exons that are always skipped in one of the samples. Junctions spanning intergenic regions were ignored. Altered exons were identified as those that were significantly different (P < 0.05) in a two-tailed t-test comparing triplicate Dido mutant samples to the same number of WT controls. For 3′ SS utilization, junctions mapping within three bases of known exon boundaries were analyzed, and normalized according to the total number of junctions sharing the 5′ SS but not coinciding on the same 3′ SS. Altered utilization was defined as a significant difference (P < 0.05) of the ratio between the sampled and alternative 3′ SS, in a two-tailed Welch’s t-test comparing WT and mutant triplicates. Since the t-test is intrinsically conservative and our data showed a large degree of interdependence, no P-value correction was carried out. Exon skipping of GAS2L1 and FAT1 transcripts was verified by reverse transcriptase PCR amplifications on 1 μg total RNA, using a Verso One-step RT-PCR Kit (Thermo Fisher Scientific Inc.). Details are given in the Supplementary Data.
RESULTS
Dido3 protein interactions indicate a role in RNA metabolism
Whereas binding of BYE1/Dido2/Dido3 to H3K4Me3 and the RNA pol II jaw domain has been described (24,25,30), the function of the mammalian-specific Dido3 extension (Figure 1A) remains uncertain. To elucidate a possible function for this part of Dido3, we performed co-purification experiments of glutathione-S-transferase (GST) fusion proteins. In brief, a fusion between GST and Dido3 without histone-binding domain was expressed in HEK293T cells and purified by glutathione-affinity chromatography as described before (35). A construct bearing GST alone was used as negative control. Gel electrophoresis of the co-purified fraction yielded a major band of approximately 100 kDa and several additional proteins (Figure 1B). Subsequent mass spectrometry and confirmation by western blotting (WB) showed that the main interacting protein was SFPQ (Figure 1C), initially identified as an essential SF in mammals (11,22). Mass spectrometry identified several other proteins involved in splicing (DHX9, SRSF7), transcription (HNRNPU) and some predominantly nucleolar proteins (NCL, NPM). The protein repertoire that interacts with Dido3 thus links the Dido gene to aspects of RNA metabolism related to transcription and splicing.
Figure 1.

Interaction between Dido3 and splicing factors. (A) Scheme of Saccharomyces cerevisae BYE1 and Mus musculus Dido proteins, indicating protein interactions (above) and deletions used in this study (below). Colored areas indicate known domain structures. Proteins are drawn to scale. (B) Pull-down experiments using glutathione-S-transferase fused to Dido3. GST and GST-D3 were purified from HEK293T cells and analyzed by SDS-PAGE and Coomassie staining. Interacting proteins identified by proteomics analysis are indicated on the right. (C) WB with indicated antibodies confirmed co-purification. Molecular weight of markers is indicated in kDa (left in B; right in C). (D) RPE-1 cells were seeded on glass coverslips, treated with vehicle only (top) or actinomycin D (bottom), and analyzed by immunofluorescence. Boxed areas show magnification of single nuclei; scale bar: 10 μm.
Most accessory SF have a function in positioning spliceosome subunits on the primary transcript, but are recycled immediately after initiation of the splicing reactions (9,46). As a consequence, only a small proportion of two different SF co-localizes when splicing is allowed to proceed normally. Actinomycin D, a compound that suppresses topoisomerases and transcription elongation (47), is widely used to interfere with SF localization (48). To further evaluate interaction between Dido3 and components of the splicing machinery, we compared their distribution in the absence and presence of actinomycin D. Briefly, retinal pigment epithelium (RPE-1) cells were co-labeled with antibodies against Dido and SFPQ, and examined by confocal scanning laser microscopy (Figure 1D and Supplementary Figure S3). While a few bright spots showed signal overlap in untreated cells (arrows in top panels), actinomycin D treatment lead to SFPQ and Dido3 accumulation in typical subnuclear domains (bottom panels). This behavior resembles that of known splicing factors (37,48), in agreement with the role of the Dido3-interacting proteins.
Since actinomycin D interferes with transcription and could thus affect RNA pol II recycling (49), isoginkgetin (IGK) was used to inhibit splicing. IGK interferes with recruitment of the U4/U5/U6 subunits, thereby stabilizing the preceding assembly state (the A-Complex) and preventing U1/U2 subunit recycling (3,50). In RPE-1 cells, IGK incubation indeed led to a characteristic Dido3 accumulation in large subnuclear domains, which contained U2AF1 and SFPQ but not HNRNPU (Supplementary Figures S4 and S5). Together, these data suggest a role for Dido3 in the early steps of RNA splicing.
Dido mutation alters SFPQ distribution
Yeast, an organism in which no SFPQ homolog is found, produces the equivalent of Dido2 but not of Dido3 (51). We thus explored the relationship between individual Dido isoforms and SFPQ. Co-immunoprecipitation showed SFPQ binding to Dido3, the isoform that has evolved in vertebrates, but not to the other isoforms (Figure 2A).
Figure 2.

Dido mutation modulates SFPQ behavior (A) Dido exon 16 sequences control SFPQ binding. Individual Dido isoforms were V5-tagged, transiently expressed and immunoprecipitated. Empty vector was used as negative control. WB showed specific co-precipitation of Dido3 and SFPQ. (B and C) Proximity ligation assay detects significant interaction between Dido and SFPQ in WT but not in E16 MEF. E3+4 lacks the necessary epitope. (C) Quantitation showed significant loss of interaction in E3+4 and E16. Error bars represent standard deviation (***P > 0.001, n.s. P > 0.05). (D and E) WT and Dido mutant MEF were seeded on glass coverslips, treated with 40 μM IGK and analyzed by immunofluorescence labeling of SFPQ. (D) IGK treatment produced fewer supramolecular (>2.5 μm2) structures in Dido mutants as compared to WT controls. (E) Representative images of control (top row) and treated (bottom row) samples. (F) WB confirmed equal quantities of SFPQ in WT and Dido mutant MEF. Tubulin was used as loading control; scale bars: 10 μm.
As SFPQ co-immunoprecipitation was specific for Dido3, the single corresponding exon was targeted for deletion (Supplementary Figure S6). Previous attempts to generate constitutive mutants without Dido3 resulted in an embryonic lethal phenotype (30,52). A conditional strategy was therefore used to remove Dido exon 16 (Supplementary Figure S6A and B). In brief, a mouse strain bearing an exon 16 flanked by loxP sites was generated, interbred with Cre-ERT2-producing mice and embryonic fibroblasts were isolated from the second-generation offspring. After 10 days of treatment with 4-hydroxytamoxifen, mouse embryonic fibroblasts (MEF) without Dido exon 16 (E16) were obtained. E16 retains the ability to express Dido1 and Dido2 but lacks the Dido3 carboxy-terminal region (Supplementary Figure S6C and D). A mutant without Dido exons 3 and 4 (E3+4), which leads to amino-terminal truncation of the Dido3 protein (35), was used for comparison. E3+4 has lost the capacity to bind H3K4Me3 but is otherwise intact.
The E3+4 and E16 mutants were used to evaluate Dido3-SFPQ binding by proximity ligation (PLA), an assay that identifies protein interactions in physiological conditions (53). Negative controls in which a single antibody was used or the DIDO NT epitope was removed (E3+4) produced a low number of scattered PLA signals that were considered background (Figure 2B). A combination of anti-SFPQ and -DIDO NT antibodies showed substantial interaction in WT MEF but not in the E16 mutant, even though antibody recognition is not altered in the latter. Quantification confirmed that the number of PLA signals was significantly reduced in E16 compared to WT controls (Figure 2C). The loss of PLA signals underscores the requirement of the Dido3-specific domain for SFPQ binding.
The contribution of Dido to SFPQ incorporation into splicing complexes was further evaluated by inhibiting splicing with IGK. Immunofluorescence detection of SFPQ in untreated WT controls (Figure 2D and E) produced a speckled pattern and a few larger domains, corresponding to its normal nuclear distribution (37). As observed for RPE-1, IGK caused SFQP redistribution into subnuclear domains measuring several square micrometers in MEF. Compared to WT controls, only a small proportion of SFPQ localized to large domains in untreated Dido mutants. In E3+4 and particularly in E16, IGK had little effect on SFPQ redistribution, and most of the protein remained in small speckles. WB showed no significant changes in SFPQ protein levels (Figure 2F). Since IGK causes A-Complex stabilization (50), lack of SFPQ redistribution in the Dido mutants denotes a role for Dido3 in an upstream recruitment step (Supplementary Figure S1).
Widespread splicing alterations in Dido mutant MEF
For a more detailed picture of the role of Dido in RNA metabolism, E3+4 and E16 MEF were subjected to RNA sequencing. Triplicate samples, each containing at least 50 million paired reads, were analyzed and compared to WT controls. Inspection of raw alignments confirmed the absence of aligned reads in the genomic regions targeted for deletion (Supplementary Figure S7).
As several of the proteins that interacted with Dido3 have a role in SS selection and thus could give rise to diverse outcomes, we performed a general classification of alternative splicing events with AltAnalyze software (44). This analysis identified a large number of splicing events that differed between the Dido mutants and WT controls (Figure 3A). Whereas the majority of alterations involved exon-level changes such as inclusion or skipping, we also detected a smaller group of events that gave rise to alternative 5′ or 3′ SS. Quantitative RT-PCR on a subset of targets confirmed splicing alterations identified by massive sequencing (Supplementary Figure S8). Dido mutation in human cells confirmed the combination of reduced SFPQ accretion and exon skipping (Supplementary Figures S9–S11). The effects of this mutation were reverted by exogenic Dido3 expression, which showed that the observations in mouse cells apply to a wide range of mammals and excluded gene targeting artifacts.
Figure 3.

Dido mutation alters exon skipping. (A) General classification of altered splicing events in E3+4 and E16. The majority of events (>60%) corresponded to exon-level alterations. Events are classified as novel when there are not annotated in the AltAnalyze database. (B) Principle component analysis of altered splicing events distinguished WT, E3+4 and E16 groups. (C) Volcano plots of exon skipping events. While E3+4 produced a large group of exons undergoing less skipping (left), E16 resulted in a major population of exons undergoing increased skipping compared to WT controls (right). Similar trends are observed for exons undergoing significant (P < 0.05) and non-significant changes. Only significant events were used for further calculations. (D) Venn analysis confirmed similarities but also differences between Dido mutants. (E) The change in skipping frequencies for exons significantly altered in at least one mutant showed extensive correlation between E3+4 and E16 (r = 0.73). Note the lack of exons that are skipped more in E3+4 but less in E16 (upper left quadrant). Examples provided in the Supplementary Figures are indicated by name.
Clustering of altered events by principal component analysis produced three distinct sample groups corresponding to WT, E3+4 and E16 (Figure 3B). Compared to WT controls, both E3+4 and E16 were displaced along one axis (PC1) but only E3+4 shifted along the second axis (PC2). This indicates that the two mutants share some alterations but also produce unique splicing events. Gene expression analysis followed by clustering produced the same groups (Supplementary Figure S12), and gene ontology showed coordinated regulation of interferon-stimulated genes that likely represents a response to aberrant RNAs in the Dido mutants (54).
Since the most common differences produced by Dido mutation were exon-level events, we calculated metrics to directly reflect the corresponding defects. Instead of representing splice isoform distribution, we used a flattened gene model to quantify exon skipping. The calculated parameter represents the total number of times each exon was skipped after correcting for gene expression. Volcano plots to compare expression-corrected exon skipping between WT controls and each mutant again showed marked differences between E3+4 and E16 (Figure 3C). Whereas E3+4 significantly reduced skipping in a large number of exons and increased skipping in a small group, E16 produced a small group of exons that underwent less skipping while increasing skipping for a large number of exons. Most exons that showed altered skipping belonged to genes that were expressed moderately in both WT and Dido mutant MEF (Supplementary Figure S13). Venn analysis summarized the differences between E3+4 and E16, validating population sizes of exons that underwent altered skipping (Figure 3D).
We next calculated the ratio of exon skipping relative to WT controls in each Dido mutant. To establish a correlation between E3+4 and E16, all exons significantly altered in at least one mutant were analyzed, and the data for each mutant were plotted on its own axis (Figure 3E). This analysis showed substantial correlation between E3+4 and E16; the majority of exons that underwent significantly altered inclusion or skipping in one mutant showed the same behavior in the other mutant, although changes were not always significant in both conditions. In accordance with the prior analysis, a large group of exons underwent more efficient inclusion in E3+4 only (Figure 3E, bottom quadrants) while E16 produced a typical population of exons that were skipped more (Figure 3E, right quadrants). We found hardly any exons that underwent more skipping in E3+4 but less in E16. Since increased exon skipping was the most common alteration in general, this finding suggests a partial conservation of function in E3+4 that is lacking in E16.
Dido mutation reduces SFPQ binding to RNA
SFPQ binds to the polypyrimide tract upstream of the 3′ SS to assist spliceosome assembly (23). A compromised interaction between SFPQ and RNA might thus account for the splicing defects in the Dido mutants. To test this hypothesis, SFPQ binding to RNA was evaluated by PAR-CLIP (41). Photocrosslinking followed by RNA pull-down allowed for efficient SFPQ recovery from WT cells while negative controls showed a minimal yield (Figure 4A). SFPQ recovery was markedly reduced in the E16 mutant, and to a lesser degree in E3+4. Quantitation of three independent experiments revealed that Dido mutation, in particular E16 deletion, significantly reduced overall SFPQ binding to RNA (Figure 4B). Altered SFPQ binding to GAS2L1 RNA was confirmed by CLIP and RT-PCR (42). After photocrosslinking, SFPQ was immunoprecipitated and the associated RNA amplified by RT-PCR (Figure 4C). Inputs showed comparable GAS2L1 expression in all samples. After immunoprecipitation, a band of the correct size was produced in WT MEF, but no amplification was seen in E3+4 or E16. These data confirmed SFPQ binding to exons undergoing alternative splicing and the role of Dido3 in this interaction.
Figure 4.

Dido mutation reduces SFPQ binding to RNA. (A) SFPQ binding to RNA was assayed by PAR-CLIP. While robust binding was observed in WT MEF, SFPQ signal was partially lost in the Dido mutants. Inputs showed comparable SFPQ expression levels in all samples. (B) Quantitation of three independent experiments showed significant loss of SFPQ binding in E16 and a similar tendency in E3+4. Error bars represent standard deviation (**P < 0.01). (C) iCLIP followed by RT-PCR confirmed lack of SFPQ binding to the GAS2L1 target, in which exon skipping increased. Positions of SFPQ binding and RT-PCR primers are indicated (right). Next, SFPQ binding to the 5′ end of exons was quantified from iCLIP data (iCLIP IP) and corrected for expression (iCLIP CTL). (D) Average SFPQ binding to alternatively used exons was significantly higher than SFPQ binding in a random control group. Error bars represent standard error of the mean (**P < 0.01). (E) SFPQ binding levels of individual exons in WT cells correlate positively with increased skipping in E16 and E3+4, indicating high SFPQ dependency of affected exons.
To further evaluate the Dido3-SFPQ axis, we tested whether altered exon utilization in E3+4 and E16 correlates with SFPQ binding in WT cells. SFPQ occupancy on the 5′ end of alternatively spliced exons was derived from available CLIP-SEQ data (34) and compared to a random control population. Exons that underwent altered splicing in E3+4 or E16 showed significantly higher SFPQ occupancy than the random subset of unchanged exons (Figure 4D). Furthermore, SFPQ occupancy correlated positively with exon skipping (Figure 4E); low SFPQ binding marked exons prone to inclusion whereas high SFPQ binding corresponded to exons prone to skipping. Exons of moderate SFPQ occupancy showed increased inclusion in E3+4 but were skipped more frequently in E16. Lack of Dido3 thus reduces overall SFPQ binding to messenger RNA, which primarily affects a group of exons that is highly SFPQ-dependent.
Dido3 maintains an advantage for upstream 3′ SS
As SFPQ is involved in early 3′ SS recognition (22), we focused on this sequence feature. The combination of all 3′ SS that share a common 5′ SS was considered a splicing system, where the presence of multiple 3′ SS denotes alternative splicing (Supplementary Figure S14A). For each splicing system, we determined 3′ SS utilization relative to alternative sites; utilization thus represents the contribution of a single 3′ SS (between 0 and 100%) to a splicing system. To reflect the sequential nature of transcription, 3′ SS were grouped by distance relative to alternative sites (measured from the common 5′ SS).
We first applied the utilization parameter to define the 3′ SS that respond to Dido mutation (Figure 5A). In a random set of unaffected splicing systems, upstream 3′ SS contributed on average 80% of splicing events and downstream sites just 10%. Most genes undergoing alternative splicing thus produced only a small proportion of transcripts that skip exons. We then calculated the same parameter for 3′ SS whose utilization was altered significantly to determine whether 3′ SS utilization in WT MEF defines their behavior in the mutants. Compared to control sites, 3′ SS that were affected by Dido mutation already showed different utilization in WT cells. For 3′ SS affected by Dido mutation, we observed a comparatively low utilization of upstream 3′ SS and high utilization of downstream 3′ SS. Both Dido mutants thus act on splicing systems with a high intrinsic tendency to skip exons in WT cells. Statistical analysis showed that differences were significant (Supplementary Figure S14B).
Figure 5.

Dido3 maintains an advantage for upstream 3′ SS. (A) 3′ SS utilization in WT cells predicts their behavior in Dido mutants. Unaffected controls are indicated by a dotted line, and splice sites that will change utilization in Dido mutants are indicated with colors. Upstream 3′ SS affected by Dido mutation are used less than random controls in WT cells, while downstream 3′ SS affected by Dido mutation are used more than controls. (B and C) Unaffected controls are indicated by a black dotted line. Low WT utilization characterized upstream 3′ SS while high WT utilization characterized downstream 3′ SS undergoing significantly altered use in the mutants (continuous lines), indicating susceptibility to exon skipping in this group. (B) E3+4 increases utilization of relatively strong upstream 3′ SS at the cost of weak sites. No notable changes in downstream 3′ SS utilization were found. (C) E16 decreased the utilization of naturally strong sites, but increased the use of a subset of weak sites. (D) Frequency distribution of the number of exons skipped by each splice junction shows that disruption of SFPQ recruitment leads to excessive exon skipping. Error bars represent standard error of the mean. (E) The number of skipped exons was compared to the number of alternative 3′ SS used. Skipping of multiple exons at once is not accompanied by increased complexity of the splicing systems. Instead, splicing is simply displaced to a single 3′ SS far downstream. Error bars show standard deviation.
The effect of each Dido mutation was assayed by separating 3′ SS used significantly more from those used significantly less in the mutants (Figure 5B and C). E3+4 increased utilization of upstream 3′ SS that were used at a frequency close to unaffected controls in WT MEF, and suppressed utilization of upstream 3′ SS used infrequently in the WT (Figure 5B). Downstream 3′ SS showed variable behavior in E3+4. WT utilization was predictive of the use of upstream 3′ SS in E3+4, but not of the use of downstream 3′ SS (Supplementary Figure S14C and D). The effect of E16 on 3′ SS was fundamentally different from E3+4. In E16, 3′ SS used frequently in WT controls lost their natural advantage (Figure 5C). At the same time, E16 showed increased utilization of 3′ SS that were rarely used in WT MEF irrespective of upstream or downstream location. The effect on both up- and downstream sites was significant in this mutant (Supplementary Figure S14C and D). Given the role of Dido3, these data indicate that efficient SFPQ recruitment sustains an advantage for high-utilization 3′ SS. Since most high-utilization 3′ SS are located upstream in WT cells, increased exon skipping is the typical outcome in the E16 mutant.
The E16 effect was particularly notable for 3′ SS located far up- and downstream (log2 distance ratio >2.5), which might correspond to skipping of multiple exons. We thus classified 5′-3′ SS junctions according to the number of exons being skipped. The majority of splice junctions traversed no exons, and this fraction decreased by an order of magnitude for each additional skipped exon (Figure 5D). Whereas the proportion of junctions skipping multiple exons decreased for all categories but one in E3+4, they increased in E16. After normalization, the increase in skipping of multiple exons was more evident in E16 while a decrease was found in E3+4 (Supplementary Figure S14E). As transcription across exons exposes alternative 3′ SS to the splicing machinery, skipping of multiple exons could involve numerous 3′ SS. We thus compared the number of exons skipped in each splicing system to the number of alternative 3′ SS actually used (Figure 5E). The mean number of alternative 3′SS used increased slightly for splicing systems that involved multiple exons, but rapidly reached a ceiling. The distributions for WT and Dido mutant MEF were comparable. Since the number of alternative 3′SS does not scale with the number of skipped exons, excessive skipping entails simple slippage toward downstream 3′SS without increasing splicing complexity.
Sequence motifs modulate exon skipping
Earlier studies reported that sequences surrounding the actual splice sites might control splicing in higher vertebrates; both regional AT content and short SF recognition motifs can impose regulation (55,56). To test whether a composition bias modulates splicing in E3+4 and E16, we analyzed the sequences surrounding alternatively used exons (Supplementary Figure S15A). A random selection was used as control. High AT content of the surrounding introns promoted exon inclusion in both mutants, with E3+4 showing a greater effect than E16. Only marginal differences in AT content were found within exons. We next examined the presence of short AT-rich sequence motifs, which have been linked to pausing of transcription elongation complexes (57). To extract short motifs, we calculated the frequency of all combinations of 5-mers downstream of significantly altered exons (Figure 6A). In accordance with the published RNA pol II behavior, AT-rich sequence motifs located immediately downstream (<200 basepairs) promoted exon inclusion in Dido mutants. In contrast to introns, exon bodies did not adhere to a simple composition bias. Here, a few specific motifs (GAGGAG and CAGCAG) favored exon inclusion (Figure 6B). The motifs identified support the upstream 3′ SS when recognized by SRSF proteins such as SRSF1 (58–60). Downstream AT-rich sequences showed a more pronounced contribution to 3′ SS selection in E3+4, whereas putative SRSF binding motifs had a greater effect in E16. Calculated 3′ SS entropy (61) contributed little, if anything, to 3′ SS selection (Supplementary Figure S15B–E).
Figure 6.

Identification of sequence motifs associated with altered splicing. (A) Scatter plots showing frequency of 5-mer sequences in the first 200 bases downstream of exons skipped significantly less (horizontal axis) or more (vertical axis) in E3+4 (left) or E16 (right). (B) Scatter plots showing frequency of 5-mer sequences located in exons as in (A), for E3+4 (left) or E16 (right). Diagonal dashed lines indicate no change in exon usage. A subset of 5-mers undergoing considerable changes is marked with sequences. Error bars (subset) represent standard deviation and are derived from a random selection of 5 × 1000 5-mers based on the flattened gene model.
DISCUSSION
Although the basic steps that catalyze the fusion of exons within precursor RNA are conserved in all eukaryotes, alternative splicing in vertebrates requires additional layers of regulation. Because the most common forms of alternative splicing depend on alternative 3′ SS, and the U2 spliceosome subunit is modulated by a variety of proteins (12,62), the acceptor site seems a versatile point at which to intercede in the splicing reactions.
In this study, we show that the Dido gene, a mammalian homolog of yeast BYE1 (25,51), encodes a hitherto unknown 3′ SS modulator. The Dido gene produces smaller isoforms that are present throughout evolution, and a single large isoform found only in vertebrates. The yeast BYE1 acts in transcription (25), and we now established a role for the vertebrate-specific isoform (Dido3) in RNA splicing. Dido has apparently acquired additional sequences that correspond to particular requirements in alternative splicing. The acquired genomic sequences comprise a single exon in all vertebrates, which suggests that the Dido3-specific protein domain forms a discrete functional unit. We show that this domain interacts with SFPQ in vitro and in vivo. In contrast to other accessory SF, SFPQ does not seem to be recruited directly by the RNA pol II CTD in vivo (20). In addition, the combination of SRSF and RNA pol II CTD can be found in lower eukaryotes but SFPQ only has homologs in chordates and arthropods. These data suggest that the Dido3 carboxy-terminus and SFPQ have evolved together and that their combination allows for an extra level of regulation in alternative splicing.
Practical approaches that address SFPQ function have met with difficulties because its mutation compromises cell viability (34). SFPQ has a role in several processes (23), including recognition of the polypyrimidine tract prior to spliceosome assembly (11,22). Immunodepletion indeed indicates that RNA splicing in mammals cannot proceed without this protein (11,22). Efficient SFPQ elimination might thus be impossible. Our data show that splicing inhibition leads to a combined accumulation of SFPQ and Dido3 in enlarged nuclear compartments. Both SFPQ accumulation and its RNA binding were reduced in Dido mutants, particularly in the mutant lacking the SFPQ-interacting region. The combination of these observations shows a role for Dido3 in the recruitment of SFPQ to precursor RNA.
In contrast to the lethality that is caused by SFPQ deletion (11,22), we obtained viable cell lines without the Dido3 isoform. Dido3 apparently promotes SFPQ recruitment without taking part in splicing. We thus envision that Dido3 forms a bridge between RNA pol II and SFPQ, which it delivers to nascent transcripts. The Dido3 amino-terminal half comprises PHD, TFIIS-like and SPOC domains (51) and is represented in yeast by BYE1. A structural study of the transcription holoenzyme established that Dido/BYE1 competes with TFIIS for occupation of RNA pol II (25). Since TFIIS can be recruited after transcription initiation and has a specific role in transcription across DNA lesions (63), BYE1/Dido could be the transcription factor normally bound to RNA pol II. In contrast to the Dido3 amino-terminus, which contains a series of well-defined domains, bioinformatics predict that its carboxy-terminus is largely unstructured. Such lack of structure is a common feature in recently evolved splicing-related proteins (19,64). The Dido3-specific protein region (65) and SFPQ itself (66) however include coiled-coil domains suitable for transient interactions; coiled coils are able to shift between different oligomerization phases in a concentration-dependent manner (67), which allows a dynamic transition between free forms, dimers and oligomers. Overexpression of an isolated Dido3 coiled-coil, which interfered with isoginkgetin-induced SFPQ accumulation (Supplementary Figure S9), corroborated this idea.
Dido3 is probably bound to RNA pol II via its TFIIS domain (25,30), which indicates that the two move together during RNA synthesis (26,30). Due to RNA pol II processivity, Dido3 has a limited timeframe in which to promote SFPQ recruitment. The role of Dido3 matches a kinetic competition model, which has gained much importance for understanding mammalian splicing. Recent refinement of this model suggests that splice site utilization depends on a flexible window of opportunity that is subject to stochastic modulation and recycling (68,69). The proximity ligation assay, which requires close interaction of two proteins, reflected this limited timeframe by labeling a small but significant proportion of total SFPQ. These findings are easily integrated into a general model (Supplementary Figure S1) in which SF are recruited from a common reservoir and recycled after splicing (6).
In addition to protein distribution, the RNA sequencing data support a common pool from which individual SF molecules are recruited. For example, suppression of ribosomal protein genes during yeast meiosis alleviates the burden on splicing of non-ribosomal genes (70). Our data show that a comparable trans-competition governs the subtle changes in mammalian alternative splicing. RNA sequencing indicated that both Dido mutations cause increased skipping in a subset of exons. Reduced utilization of 3′ SS, typical SFPQ targets, explains the increased skipping of exons that were already susceptible to skipping in WT controls. A complementary 3′ SS group showed contrasting behavior, however, and was used more frequently. In a model in which individual SF units are recruited from a shared but restricted pool, reduced utilization in one 3′ SS subset automatically increases SF availability for other transcripts. The same dynamics explain observations related to pharmacological suppression of RNA pol II (71). Here, too, slowing down RNA pol II promotes inclusion of some exons while skipping others.
In contrast to generalized suppression of RNA pol II by pharmacological inhibition (71), local sequence composition is an important physiological modulator of transcription (55). For example, several studies show that AT-rich sequences can stall RNA polymerase for several minutes at a time (57,72,73). In our study, these AT-rich sequences allowed for functional discrimination between the two Dido mutants, and were particularly dominant in E3+4. The lack of chromatin binding (24) implies that Dido3 is no longer enriched at the 5′ end of active genes in E3+4, but instead resides free in the nucleoplasm (35). RNA sequencing nonetheless indicates that paused RNA pol II can recruit both Dido3 (without the amino-terminus) and SFPQ from a soluble state and thereby promote exon inclusion. The E16 mutant does not involve a Dido3 population shift but comprises an SFPQ recruitment defect that cannot be compensated. As a result, this mutation leads to global excessive exon skipping. In E16, exons using alternative recruitment pathways such as SRSF (74,75) benefit from reduced U2 utilization by the Dido3-SFPQ axis. Dido mutant behavior reproduces the modular architecture of Dido3, important for its ability to consecutively bind chromatin, RNA pol II and SFPQ (Figure 7).
Figure 7.
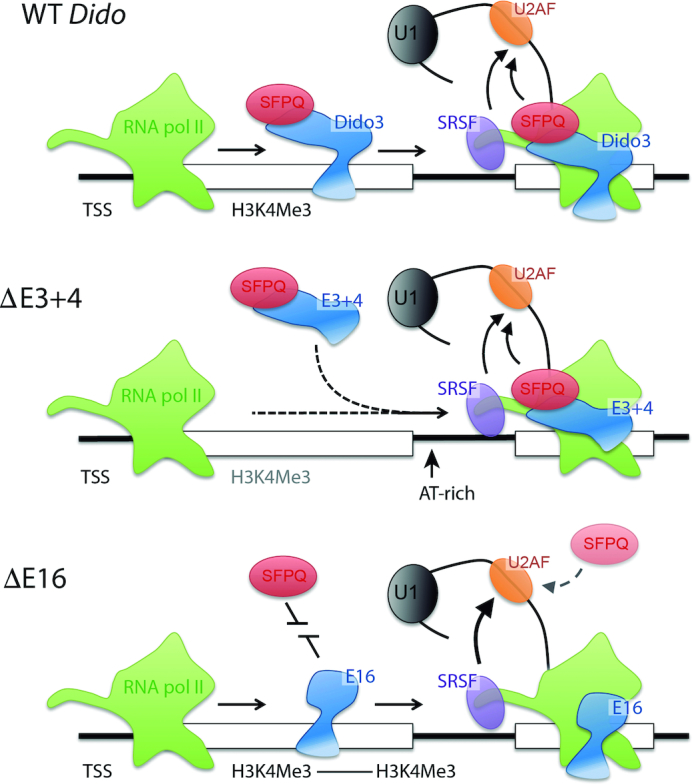
Model for Dido3 function and parallel SF recruitment in WT conditions (top), the Dido3 PHD domain anchors the protein to the 5′ end of active genes, where it can be picked up by the RNA pol II jaw domain (25). Along the gene body, Dido3 catalyzes SFPQ recruitment to promote exon inclusion. The E3+4 mutant (center) lacks enrichment on active genes but is otherwise intact. RNA pol II pausing could thus induce recruitment in two steps, initially of Dido3 without the amino-terminus and subsequently of SFPQ. The E16 mutant (bottom) no longer promotes SFPQ recruitment. In this case, SFPQ might rely on an inefficient direct interaction with RNA to catalyze 3′SS recognition. The Dido3-SFPQ axis acts in parallel to the RNA pol II CTD, known to favor SRSF recruitment (16,17). RNA sequencing showed that impairment of the Dido3-SFPQ axis promotes the use of the CTD-dependent SRSF pathway.
Our model of a common SF pool requires no arbitration between adjacent 3′ SS to explain alternative RNA splicing. Exon skipping can be a simple result of reduced utilization in an upstream 3′ SS, and utilization of downstream sites increases because it is the next available alternative. Absence of arbitration is suggested by the sequential skipping of multiple exons, where intermediate 3′ SS are not considered (Figure 5E). If arbitration between all exposed 3′ SS would occur, skipping of multiple exons would encompass increased complexity that was absent from our data. Since only putative pause sites located nearby (within approximately 0.5 kb) stimulate the upstream 3′ SS (Supplementary Figures S16–S18), our data support a narrow SF recruitment window. Such a narrow window explains why no arbitration at a distance takes place. The long introns in mammalian genes imply that splice sites move easily out of range as transcription proceeds.
Together, our data indicate that independent yet interconnected pathways affect 3′ SS selection, revealing a two-pronged strategy to maintain RNA splicing efficiency. Dido3 contributes to this process by facilitating SFPQ recruitment within a limited window of opportunity. Since the RNA pol II CTD has expanded during evolution but has acquired hardly any additional sequence variation, enrollment of Dido3 as an independent linker might reduce competition between recruitment pathways that ultimately converge on the U2 spliceosome subunit. The use of parallel pathways thus provides a high-capacity mechanism to promote efficient RNA splicing in mammals, reminiscent of transcriptional coupling in yeast (76), but still allowing for alternative splicing.
DATA AVAILABILITY
Sequencing data are deposited in the NCBI sequence read archive under accession number SRP150516 and PRJNA476070. SFPQ iCLIP data were obtained from PRJNA378774.
Python 2.7 scripts used to generate data are available from https://github.com/kvanwely/DIDO-LAB.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Alfredo Castello in Oxford for sharing PAR-CLIP techniques and cell lines. The authors thank Catherine Mark for editorial assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Economics, Industry, and Competitiveness [SAF2016-75456-R, AEI/FEDER, EU]; Comunidad Autónoma de Madrid [B2017/BMD-3703 MITIC]; Foundation Alfonso Martin Escudero (to C.M.A.); CSIC predoctoral fellow FPI grant [BES-2014-068580 to C.M.G.]. Funding for open access charge: Project grants (SAF2016 and B2017) as listed under Funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Will C.L., Luhrmann R.. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011; 3:a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoskins A.A., Moore M.J.. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem. Sci. 2012; 37:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huranova M., Ivani I., Benda A., Poser I., Brody Y., Hof M., Shav-Tal Y., Neugebauer K.M., Stanek D.. The differential interaction of snRNPs with pre-mRNA reveals splicing kinetics in living cells. J. Cell Biol. 2010; 191:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oesterreich F.C., Herzel L., Straube K., Hujer K., Howard J., Neugebauer K.M.. Splicing of nascent RNA coincides with intron exit from RNA polymerase II. Cell. 2016; 165:372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tilgner H., Knowles D.G., Johnson R., Davis C.A., Chakrabortty S., Djebali S., Curado J., Snyder M., Gingeras T.R., Guigo R.. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012; 22:1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Medenbach J., Schreiner S., Liu S., Luhrmann R., Bindereif A.. Human U4/U6 snRNP recycling factor p110: mutational analysis reveals the function of the tetratricopeptide repeat domain in recycling. Mol. Cell Biol. 2004; 24:7392–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raghunathan P.L., Guthrie C.. A spliceosomal recycling factor that reanneals U4 and U6 small nuclear ribonucleoprotein particles. Science. 1998; 279:857–860. [DOI] [PubMed] [Google Scholar]
- 8. Khodor Y.L., Menet J.S., Tolan M., Rosbash M.. Cotranscriptional splicing efficiency differs dramatically between Drosophila and mouse. RNA. 2012; 18:2174–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kornblihtt A.R., Schor I.E., Allo M., Dujardin G., Petrillo E., Munoz M.J.. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013; 14:153–165. [DOI] [PubMed] [Google Scholar]
- 10. House A.E., Lynch K.W.. Regulation of alternative splicing: more than just the ABCs. J. Biol. Chem. 2008; 283:1217–1221. [DOI] [PubMed] [Google Scholar]
- 11. Patton J.G., Porro E.B., Galceran J., Tempst P., Nadal-Ginard B.. Cloning and characterization of PSF, a novel pre-mRNA splicing factor. Genes Dev. 1993; 7:393–406. [DOI] [PubMed] [Google Scholar]
- 12. Ruskin B., Zamore P.D., Green M.R.. A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell. 1988; 52:207–219. [DOI] [PubMed] [Google Scholar]
- 13. Spiluttini B., Gu B., Belagal P., Smirnova A.S., Nguyen V.T., Hebert C., Schmidt U., Bertrand E., Darzacq X., Bensaude O.. Splicing-independent recruitment of U1 snRNP to a transcription unit in living cells. J. Cell Sci. 2010; 123:2085–2093. [DOI] [PubMed] [Google Scholar]
- 14. Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J.. Deciphering the splicing code. Nature. 2010; 465:53–59. [DOI] [PubMed] [Google Scholar]
- 15. Saldi T., Cortazar M.A., Sheridan R.M., Bentley D.L.. Coupling of RNA polymerase II transcription elongation with Pre-mRNA splicing. J. Mol. Biol. 2016; 428:2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCracken S., Fong N., Yankulov K., Ballantyne S., Pan G., Greenblatt J., Patterson S.D., Wickens M., Bentley D.L.. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997; 385:357–361. [DOI] [PubMed] [Google Scholar]
- 17. Pineda G., Shen Z., de Albuquerque C.P., Reynoso E., Chen J., Tu C.C., Tang W., Briggs S., Zhou H., Wang J.Y.. Proteomics studies of the interactome of RNA polymerase II C-terminal repeated domain. BMC Res. Notes. 2015; 8:616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jeronimo C., Collin P., Robert F.. The RNA polymerase II CTD: the increasing complexity of a low-complexity protein domain. J. Mol. Biol. 2016; 428:2607–2622. [DOI] [PubMed] [Google Scholar]
- 19. Haynes C., Iakoucheva L.M.. Serine/arginine-rich splicing factors belong to a class of intrinsically disordered proteins. Nucleic Acids Res. 2006; 34:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das R., Yu J., Zhang Z., Gygi M.P., Krainer A.R., Gygi S.P., Reed R.. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell. 2007; 26:867–881. [DOI] [PubMed] [Google Scholar]
- 21. Kameoka S., Duque P., Konarska M.M.. p54(nrb) associates with the 5′ splice site within large transcription/splicing complexes. EMBO J. 2004; 23:1782–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gozani O., Patton J.G., Reed R.. A novel set of spliceosome-associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. EMBO J. 1994; 13:3356–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yarosh C.A., Iacona J.R., Lutz C.S., Lynch K.W.. PSF: nuclear busy-body or nuclear facilitator. Wiley Interdiscip. Rev. RNA. 2015; 6:351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gatchalian J., Futterer A., Rothbart S.B., Tong Q., Rincon-Arano H., Sanchez de Diego A., Groudine M., Strahl B.D., Martinez A.C., van Wely K.H. et al.. Dido3 PHD modulates cell differentiation and division. Cell Rep. 2013; 4:148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kinkelin K., Wozniak G.G., Rothbart S.B., Lidschreiber M., Strahl B.D., Cramer P.. Structures of RNA polymerase II complexes with Bye1, a chromatin-binding PHF3/DIDO homologue. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:15277–15282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pinskaya M., Ghavi-Helm Y., Mariotte-Labarre S., Morillon A., Soutourina J., Werner M.. PHD and TFIIS-Like domains of the Bye1 transcription factor determine its multivalent genomic distribution. PLoS One. 2014; 9:e102464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Futterer A., Campanero M.R., Leonardo E., Criado L.M., Flores J.M., Hernandez J.M., San Miguel J.F., Martinez A.C.. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J. Clin. Invest. 2005; 115:2351–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berzoti-Coelho M.G., Ferreira A.F., de Souza Nunes N., Pinto M.T., Junior M.C., Simoes B.P., Martinez A.C., Souto E.X., Panepucci R.A., Covas D.T. et al.. The expression of death inducer-obliterator (DIDO) variants in myeloproliferative neoplasms. Blood Cells Mol. Dis. 2016; 59:25–30. [DOI] [PubMed] [Google Scholar]
- 29. Wong A.C.H., Rasko J.E.J., Wong J.J.. We skip to work: alternative splicing in normal and malignant myelopoiesis. Leukemia. 2018; 32:1081–1093. [DOI] [PubMed] [Google Scholar]
- 30. Futterer A., de Celis J., Navajas R., Almonacid L., Gutierrez J., Talavera-Gutierrez A., Pacios-Bras C., Bernascone I., Martin-Belmonte F., Martinez A.C.. DIDO as a switchboard that regulates Self-Renewal and differentiation in embryonic stem cells. Stem Cell Rep. 2017; 8:1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jemth P., Karlsson E., Vogeli B., Guzovsky B., Andersson E., Hultqvist G., Dogan J., Guntert P., Riek R., Chi C.N.. Structure and dynamics conspire in the evolution of affinity between intrinsically disordered proteins. Sci. Adv. 2018; 4:eaau4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson M.L., Nagengast A.A., Salz H.K.. PPS, a large multidomain protein, functions with sex-lethal to regulate alternative splicing in Drosophila. PLoS Genet. 2010; 6:e1000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng Y., Luo C., Wu W., Xie Z., Fu X., Feng Y.. Liver-Specific deletion of SRSF2 caused acute liver failure and early death in mice. Mol. Cell Biol. 2016; 36:1628–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takeuchi A., Iida K., Tsubota T., Hosokawa M., Denawa M., Brown J.B., Ninomiya K., Ito M., Kimura H., Abe T. et al.. Loss of Sfpq causes Long-Gene transcriptopathy in the brain. Cell Rep. 2018; 23:1326–1341. [DOI] [PubMed] [Google Scholar]
- 35. Prieto I., Kouznetsova A., Futterer A., Trachana V., Leonardo E., Alonso Guerrero A., Cano Gamero M., Pacios-Bras C., Leh H., Buckle M. et al.. Synaptonemal complex assembly and H3K4Me3 demethylation determine DIDO3 localization in meiosis. Chromosoma. 2009; 118:617–632. [DOI] [PubMed] [Google Scholar]
- 36. Sanchez de Diego A., Alonso Guerrero A., Martinez A.C., van Wely K.H.. Dido3-dependent HDAC6 targeting controls cilium size. Nat. Commun. 2014; 5:3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shav-Tal Y., Blechman J., Darzacq X., Montagna C., Dye B.T., Patton J.G., Singer R.H., Zipori D.. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell. 2005; 16:2395–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheng D., Cote J., Shaaban S., Bedford M.T.. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell. 2007; 25:71–83. [DOI] [PubMed] [Google Scholar]
- 39. Rachez C., Suldan Z., Ward J., Chang C.P., Burakov D., Erdjument-Bromage H., Tempst P., Freedman L.P.. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell- free system. Genes Dev. 1998; 12:1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen S., Choo A., Chin A., Oh S.K.. TGF-beta2 allows pluripotent human embryonic stem cell proliferation on E6/E7 immortalized mouse embryonic fibroblasts. J. Biotechnol. 2006; 122:341–361. [DOI] [PubMed] [Google Scholar]
- 41. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr, Jungkamp A.C., Munschauer M. et al.. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B.. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003; 302:1212–1215. [DOI] [PubMed] [Google Scholar]
- 43. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emig D., Salomonis N., Baumbach J., Lengauer T., Conklin B.R., Albrecht M.. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 2010; 38:W755–W762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang L., Wang S., Li W.. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012; 28:2184–2185. [DOI] [PubMed] [Google Scholar]
- 46. Chen M., Manley J.L.. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009; 10:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sobell H.M. Actinomycin and DNA transcription. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:5328–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carmo-Fonseca M., Pepperkok R., Sproat B.S., Ansorge W., Swanson M.S., Lamond A.I.. In vivo detection of snRNP-rich organelles in the nuclei of mammalian cells. EMBO J. 1991; 10:1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shandilya J., Roberts S.G.. The transcription cycle in eukaryotes: from productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta. 2012; 1819:391–400. [DOI] [PubMed] [Google Scholar]
- 50. O’Brien K., Matlin A.J., Lowell A.M., Moore M.J.. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 2008; 283:33147–33154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rojas A.M., Sanchez-Pulido L., Futterer A., van Wely K.H., Martinez A.C., Valencia A.. Death inducer obliterator protein 1 in the context of DNA regulation. Sequence analyses of distant homologues point to a novel functional role. FEBS J. 2005; 272:3505–3511. [DOI] [PubMed] [Google Scholar]
- 52. Futterer A., Raya A., Llorente M., Izpisua-Belmonte J.C., de la Pompa J.L., Klatt P., Martinez A.C.. Ablation of Dido3 compromises lineage commitment of stem cells in vitro and during early embryonic development. Cell Death Differ. 2012; 19:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Weibrecht I., Leuchowius K.J., Clausson C.M., Conze T., Jarvius M., Howell W.M., Kamali-Moghaddam M., Soderberg O.. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteom. 2010; 7:401–409. [DOI] [PubMed] [Google Scholar]
- 54. Zevini A., Olagnier D., Hiscott J.. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017; 38:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Amit M., Donyo M., Hollander D., Goren A., Kim E., Gelfman S., Lev-Maor G., Burstein D., Schwartz S., Postolsky B. et al.. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Rep. 2012; 1:543–556. [DOI] [PubMed] [Google Scholar]
- 56. Goren A., Ram O., Amit M., Keren H., Lev-Maor G., Vig I., Pupko T., Ast G.. Comparative analysis identifies exonic splicing regulatory sequences–The complex definition of enhancers and silencers. Mol. Cell. 2006; 22:769–781. [DOI] [PubMed] [Google Scholar]
- 57. Palangat M., Hittinger C.T., Landick R.. Downstream DNA selectively affects a paused conformation of human RNA polymerase II. J. Mol. Biol. 2004; 341:429–442. [DOI] [PubMed] [Google Scholar]
- 58. Cavaloc Y., Bourgeois C.F., Kister L., Stevenin J.. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999; 5:468–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jobbins A.M., Reichenbach L.F., Lucas C.M., Hudson A.J., Burley G.A., Eperon I.C.. The mechanisms of a mammalian splicing enhancer. Nucleic Acids Res. 2018; 46:2145–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schaal T.D., Maniatis T.. Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein-specific enhancer sequences. Mol. Cell Biol. 1999; 19:1705–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yeo G., Burge C.B.. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004; 11:377–394. [DOI] [PubMed] [Google Scholar]
- 62. Bai Y., Lee D., Yu T., Chasin L.A.. Control of 3′ splice site choice in vivo by ASF/SF2 and hnRNP A1. Nucleic Acids Res. 1999; 27:1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kalogeraki V.S., Tornaletti S., Cooper P.K., Hanawalt P.C.. Comparative TFIIS-mediated transcript cleavage by mammalian RNA polymerase II arrested at a lesion in different transcription systems. DNA Repair. 2005; 4:1075–1087. [DOI] [PubMed] [Google Scholar]
- 64. Jarvelin A.I., Noerenberg M., Davis I., Castello A.. The new (dis)order in RNA regulation. Cell Commun. Signal. 2016; 14:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Trachana V., van Wely K.H., Guerrero A.A., Futterer A., Martinez A.C.. Dido disruption leads to centrosome amplification and mitotic checkpoint defects compromising chromosome stability. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:2691–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee M., Sadowska A., Bekere I., Ho D., Gully B.S., Lu Y., Iyer K.S., Trewhella J., Fox A.H., Bond C.S.. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Res. 2015; 43:3826–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Groger K., Gavins G., Seitz O.. Strand displacement in coiled-coil structures: controlled induction and reversal of proximity. Angew. Chem. Int. Ed. Engl. 2017; 56:14217–14221. [DOI] [PubMed] [Google Scholar]
- 68. Coulon A., Ferguson M.L., de Turris V., Palangat M., Chow C.C., Larson D.R.. Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife. 2014; 3:03939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rino J., Carvalho T., Braga J., Desterro J.M., Luhrmann R., Carmo-Fonseca M.. A stochastic view of spliceosome assembly and recycling in the nucleus. PLoS Comput. Biol. 2007; 3:2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Munding E.M., Shiue L., Katzman S., Donohue J.P., Ares M. Jr. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol. Cell. 2013; 51:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fong N., Kim H., Zhou Y., Ji X., Qiu J., Saldi T., Diener K., Jones K., Fu X.D., Bentley D.L.. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 2014; 28:2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Artsimovitch I., Landick R.. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Henriques T., Gilchrist D.A., Nechaev S., Bern M., Muse G.W., Burkholder A., Fargo D.C., Adelman K.. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol. Cell. 2013; 52:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pandit S., Zhou Y., Shiue L., Coutinho-Mansfield G., Li H., Qiu J., Huang J., Yeo G.W., Ares M. Jr, Fu X.D.. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell. 2013; 50:223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tian H., Kole R.. Strong RNA splicing enhancers identified by a modified method of cycled selection interact with SR protein. J. Biol. Chem. 2001; 276:33833–33839. [DOI] [PubMed] [Google Scholar]
- 76. Herzel L., Ottoz D.S.M., Alpert T., Neugebauer K.M.. Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017; 18:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are deposited in the NCBI sequence read archive under accession number SRP150516 and PRJNA476070. SFPQ iCLIP data were obtained from PRJNA378774.
Python 2.7 scripts used to generate data are available from https://github.com/kvanwely/DIDO-LAB.