


Abstract
The telomerase holoenzyme responsible for maintaining telomeres in vertebrates requires many components in vivo, including dyskerin. Dyskerin binds and regulates the accumulation of the human telomerase RNA, hTR, as well as other non-coding RNAs that share the conserved H/ACA box motif. The precise mechanism by which dyskerin controls hTR levels is unknown, but is evidenced by defective hTR accumulation caused by substitutions in dyskerin, that are observed in the X-linked telomere biology disorder dyskeratosis congenita (X-DC). To understand the role of dyskerin in hTR accumulation, we analyzed X-DC substitutions K39E and K43E in the poorly characterized dyskerin N-terminus, and A353V within the canonical RNA binding domain (the PUA). These variants exhibited impaired binding to hTR and polyadenylated hTR species, while interactions with other H/ACA RNAs appear largely unperturbed by the N-terminal substitutions. hTR accumulation and telomerase activity defects of dyskerin-deficient cells were rescued by wildtype dyskerin but not the variants. hTR 3′ extended or polyadenylated species did not accumulate, suggesting hTR precursor degradation occurs upstream of mature complex assembly in the absence of dyskerin binding. Our findings demonstrate that the dyskerin-hTR interaction mediated by PUA and N-terminal residues of dyskerin is crucial to prevent unchecked hTR degradation.
INTRODUCTION
The ends of linear chromosomes, known as telomeres are incompletely replicated due to the conventional DNA polymerase requiring a 3′ hydroxyl to initiate DNA synthesis and the semi-conservative nature of DNA replication. This generates a problem for replicated DNA ends: the potential loss of genomic information. Eukaryotic organisms have evolved mechanisms to maintain telomeres, providing a solution to the end replication problem, and serving as protection against the inappropriate recognition of DNA ends as breaks. Telomeric integrity has implications in cellular aging, as the natural occurrence of telomere attrition serves as a key checkpoint in the control of cell proliferation by triggering replicative senescence (1). However, defects in the telomere-synthesizing enzyme telomerase and other telomere maintenance components cause premature aging syndromes like dyskeratosis congenita (DC) due to progressive telomere shortening in dividing cells and subsequent proliferative-block (2,3).
The rare premature aging disease and telomere biology disorder DC is characterized by pathologic presentation in the proliferative tissues of patients, including the classical triad of diagnostic symptoms: oral leukoplakia, hyperpigmentation of the skin, and nail dystrophy. These patients have higher rates of age-related predispositions, including pulmonary disease and malignancies (4). The most common cause of mortality in DC patients is bone marrow failure caused by the depletion of the hematopoietic stem cell compartment. Ultimately, DC-causative mutations leads to impaired maintenance of telomeres and, in turn, cells in regenerative tissues are unable to maintain their highly proliferative capacity, causing the observed pathology in patients (2,3).
X-linked DC (X-DC), caused by mutations in the dkc1 gene encoding dyskerin, is the most common inherited form of DC (5,6). As with all factors implicated in DC to date, dyskerin is important for proper telomere maintenance. The telomerase enzyme is minimally composed of a reverse transcriptase (hTERT in humans) and an RNA template (hTR in humans) that are able to synthesize telomeric DNA in vitro (7–9). However, this holoenzyme requires many other components in vivo, including dyskerin and the other H/ACA ribonucleoprotein (RNP) complex components NHP2, NOP10, and GAR1. Dyskerin binds hTR, as well as other small non-coding RNAs that share a conserved structural motif known as the H/ACA box (10). Dyskerin is integral for accumulation of hTR, biogenesis of the mature telomerase complex and telomerase activity. X-DC patients suffer from hTR accumulation defects and consequent telomere shortening (6). Several recent studies have identified key factors and pathways implicated in the trimming of hTR 3′ extended and/or polyadenylated species, as well as components involved in hTR degradation (11–15). The involvement of various pathways, including the nuclear RRP6-exosome, PABPN1 and PARN, human TRAMP and NEXT complexes, CBCA complex, XRN1/DCP2, and most recently TOE1, demonstrates the complexity of hTR processing. It has been posited as well as demonstrated that dyskerin interacts with components in these pathways (11–13), and in the absence of dyskerin, accelerated hTR decay in HeLa cells can be partially rescued by co-depleting RRP6 (12). However, the role and regulatory mechanisms of dyskerin in hTR processing, telomerase biogenesis and telomere maintenance remain to be fully elucidated.
There are two hotspots within the dyskerin gene for X-DC causative mutations: one hotspot coincides with the poorly characterized eukaryotic N-terminal extension (amino acids 18–47) and dyskeratosis congenita-like domain (DKCLD) (amino acids 48–106) (16), while the other spans a more C-terminal region encompassing the pseudouridine synthase and archaeosine transglycosylase (PUA) domain (amino acids 297–370) and an uncharacterized domain between the PUA and C-terminal nuclear localization signal (17,18) (Figure 1A). Among the affected residues in the N-terminal hotspot are two lysine residues that have been reported to be substituted to glutamate [K39E (19) and K43E (5)] in two unrelated X-DC families. We previously demonstrated that in HEK293 cells depleted of endogenous dyskerin, the expression of FLAG-tagged dyskerin variants harboring arginine substitutions at either K39 or K43 results in reduced telomerase RNA levels, decreased telomerase activity, and subsequent telomere shortening (20). Additionally, the most commonly reported substitution in X-DC [A353V (5)], lies near the second X-DC hotspot within the PUA, which is the putative RNA binding domain of dyskerin based on homology with other pseudouridine synthases, including the archaeal dyskerin homologue Cbf5 (21,22). X-DC patient-derived cells harboring various mutations, including the recurring A353V variant, exhibit reduced telomerase activity driven by impaired hTR accumulation (6,18,23,24).
Figure 1.

N-terminal and PUA domain dyskerin variants localize to Cajal bodies and the nucleolus. (A) A linear schematic of dyskerin domains. The amino acid (aa) ranges corresponding to the N-terminal extension (18–47) and the pseudouridine synthase and archaesoine transglycosylase domain (PUA – 297–370) are denoted below the schematic. Above the schematic, pink arrows indicate K39 and K43, while the purple arrow indicates A353. All reported X-DC substitutions are indicated by vertical black lines along the top of the schematic. Representative images of the co-localization of FLAG-dyskerin (wildtype WT and dyskerin variants K39E, K43E, and A353V – Cy3 shown in red) with (B) Cajal body marker coilin (FITC shown in green) and (C) nucleolar marker fibrillarin (FITC shown in green), as observed by indirect immunofluorescence. No deviation in localization was observed, as all cells expressing nuclear FLAG-tagged dyskerin (wildtype or X-DC variants) displayed expected co-localization with the nucleolar marker fibrillarin or the Cajal body marker coilin (the number of nuclei with expected co-localization and the number of nuclei counted is indicated for each condition in the merged column). FITC signal is not detected by the Cy3 channel using mock HEK293 cells lacking expression of FLAG-tagged dyskerin, nor is nucleolar FLAG-dyskerin detected by the FITC channel when examining co-localization with coilin. In the bottom panel of (B) the ΔCterm dyskerin variant (Cy3 shown in red) was used as a control for mislocalization. This truncation lacking most of the C-terminal nuclear/nucleolar localization sequence has been previously reported to cause cytosolic accumulation of dyskerin. In (B) the co-localization foci for Cajal bodies are indicated by white arrows in the Cy3 and merged columns. The nucleus is indicated by DAPI staining of nuclear DNA (in blue).
Beyond our previous study (20) and the reported hTR accumulation defects in patient cells harboring certain N-terminal hotspot mutations (6,18,23–30), substitutions in the N-terminal extension of dyskerin including K39E and K43E have not been thoroughly characterized. In addition, the precise mechanism by which dyskerin regulates hTR accumulation is unknown. We aimed to better understand the role of the N-terminal K39 and K43 residues in regulating hTR accumulation through examining the effects of X-DC substitutions at these positions. Thus, in the current study, we characterized the K39E and K43E variants alongside the A353V variant previously reported to cause reduced hTR levels. We observed that all three dyskerin variants are defective at binding polyadenylated species and mature hTR. Interactions with other H/ACA RNAs appear largely unperturbed by the K39E or K43E substitutions, consistent with distinct biogenesis pathways for hTR and other H/ACA class RNAs demonstrated by Fu and Collins in 2003, which implies the sensitivity of hTR in particular to X-DC mutations (31). However, the A353V variant displays more substantial H/ACA RNA interaction defects, indicating that the PUA domain might play a more global role in H/ACA RNA interaction and suggesting a potential telomerase-centric role for the eukaryotic N-terminal extension of human dyskerin. Depletion of dyskerin leads to reduced levels of some H/ACA RNAs, which can be rescued by wildtype dyskerin and variants alike. However, reduced hTR levels in dyskerin-depleted cells can only be rescued by wildtype dyskerin expression. Our observation that other H/ACA RNA levels are comparable between dyskerin-depleted cells expressing wildtype dyskerin or dyskerin variants is consistent with our findings that H/ACA RNP assembly and dyskerin subnuclear localization are unaffected for these variants. The interaction between dyskerin and hTR is probably necessary to mediate the accumulation of hTR to levels required for active telomerase, as total hTR levels in dyskerin-depleted cells with or without expression of dyskerin variants cannot sustain telomerase activity comparable to cells expressing wildtype dyskerin. This is likely because disruption of the dyskerin-hTR interaction favors the degradation of hTR precursors upstream of the assembly of the mature telomerase complex, suggesting an early involvement of dyskerin in hTR biogenesis. This role differs from other recently reported processing factors, in that dyskerin depletion reduces the amount of hTR polyadenylated species rather than causing accumulation of these non-functional RNA species. hTR accumulation defects caused by the inability of dyskerin variants to interact with hTR cannot be rescued by depletion of individual processing pathway components PARN or the core exosome component RRP40, designating a requirement for dyskerin in preventing excessive hTR degradation. Our findings demonstrate that the dyskerin-hTR interaction mediated by PUA and N-terminal residues of dyskerin is crucial to prevent unchecked hTR degradation, and highlights mechanistic links between the regulation of hTR trimming and degradation processes, and the telomere biology disorder X-DC.
MATERIALS AND METHODS
Plasmids and site directed mutagenesis
The plasmid pcDNA3.1-FLAG-dyskerinWT from the lab of Dr. François Dragon was used to generate point mutations or the ΔCterm deletion via site directed mutagenesis, as previously described (20). Specifically, primers (Supplementary Table S1) were designed to generate K39E (c. 115A>G), K43E (c. 127A>G), A353V (c. 1058C>T) and K446X (c. 1336A>T). For expression of HA-SHQ1 in human cells, the coding sequence of HA-SHQ1 was cloned into a pcDNA3 backbone using KpnI and EcoRI cut sites and the plasmid pSR38 obtained from Dr. Tom Meier (32). The plasmid pcDNA6/myc-HisC-hTERT from the lab of Dr. Joachim Lingner (33) was used for expression of wildtype human TERT in cells.
Cell culture and transfection
Human embryonic kidney (HEK293) cells were maintained in Dulbecco's modified Eagle's medium DMEM (Wisent) supplemented with 10% fetal bovine serum FBS (Wisent), and antibiotic-antimycotic (Gibco), at 37°C 5% CO2. Polyclonal FLAG-dyskerin stable cells were maintained under selective pressure in G418 (750 μg/ml). Transfection of pcDNA3.1 (empty vector), pcDNA3.1-FLAG-dyskerin constructs, pcDNA3-HA-SHQ1 and/or pcDNA6/myc-HisC-hTERT was performed using Lipofectamine 2000 Transfection Reagent (Invitrogen) according to the reagent protocol. Prior to transfection, media was changed to DMEM with 10% FBS and lacking antibiotic-antimycotic, and 6 h after transfection the media was replaced with DMEM containing both FBS and antibiotic-antimycotic.
Immunofluorescence
In order to assess localization of FLAG-dyskerin to the Cajal bodies, immunofluorescence experiments were performed on HEK293 cells overexpressing FLAG-dyskerinWT, X-DC variants or ΔCterm following a previously described protocol (34). Specifically, to probe for FLAG-dyskerin variants rabbit anti-FLAG (Sigma-Aldrich F7425, 1:500) was used in PBG (1% cold fish water gelatin, 0.5% bovine serum albumin (BSA), in PBS), followed by mouse anti-coilin (from Dr. Michael Terns (35), 1:10 000) in PBG. Nuclei showing FLAG-dyskerin localization to at least one Cajal body were scored out of the number of nuclei with both FLAG and coilin signal detected, and ≥180 nuclei were counted for scoring of localization of each nuclear FLAG-tagged dyskerin variant.
To assess localization of FLAG-dyskerin to the nucleolus, HEK293 cells overexpressing FLAG-dyskerin variants were fixed with 4% formaldehyde–PBS for 10 min at room temperature. The fixing solution was removed and coverslips were briefly rinsed with PBS, followed by permeabilization of cells with 0.1% Triton X-100-PBS for 5 min at 4°C. Permeabilized cells were incubated with 2× SSC–50% formamide for 5 min at room temperature, and then washed with PBS before blocking in 3% BSA–PBS-T for 1 h at room temperature. Cells were probed for FLAG-dyskerin variants with rabbit anti-FLAG (Sigma-Aldrich F7425, 1:500) in PBG, followed by mouse anti-fibrillarin (monoclonal antibody 72B9 obtained from Dr Kenneth Michael Pollard (36), 1:30) as a nucleolar marker in 3% BSA–PBS-T. Coverslips were washed with PBS and immunostained in PBG with secondary antibodies conjugated to fluorescein isothiocyanate (FITC) (donkey anti-mouse IgG; Jackson ImmunoResearch Lab, Inc., 1:125) or Cy3 (donkey anti-rabbit; Jackson ImmunoResearch Lab, Inc., 1:125). Coverslips were washed with PBS and mounted in Vectashield with DAPI (Vector Laboratories). Nuclei showing FLAG-dyskerin localization to the nucleolus were scored out of the number of nuclei with both FLAG and fibrillarin signal detected, and ≥180 nuclei were counted for scoring of localization of each FLAG-tagged dyskerin variant. Images were captured using an Axio Imager M1 (63×; Carl Zeiss, Jena, Germany).
Co-immunoprecipitation for protein–protein interactions and FLAG-dyskerin–H/ACA RNA interactions
With the exception of co-overexpression of HA-SHQ1 or hTERT with FLAG-dyskerin, all protein-protein interactions were assessed by immunoprecipitating FLAG-dyskerin wildtype or X-DC variants from HEK293 cells and immunoblotting for endogenous dyskerin-interacting proteins. Monoclonal M2 mouse anti-FLAG antibody (Sigma-Aldrich F3165) and Protein G Sepharose (GE Healthcare) were used to immunoprecipitate (IP) FLAG-dyskerin wildtype or X-DC variants. The protocols from the laboratory of Dr Steven Artandi for assessing the interaction between FLAG-dyskerin and hTERT (with and without RNase A treatment), TCAB1, or Reptin have all been described in detail (37,38), and was also used to asses co-IP of the negative control TIP60. The protocol used to assess the interaction of FLAG-dyskerin with HA-SHQ1, the pre-H/ACA RNP components NAF1, NHP2 and NOP10, or mature H/ACA RNP complex component GAR1 was the same used to analyze the interaction between FLAG-dyskerin and H/ACA box RNAs including hTR, and has been described for another hTR-interacting protein (39). For protein-protein interactions, elution from Protein G Sepharose was performed with Laemmli buffer and boiling. For protein–RNA interactions, elution was performed with TRIzol reagent (Invitrogen), followed by chloroform extraction and reverse transcription. Inputs (10% of lysate volume used for IP) were collected prior to IP, and treated with either Laemmli buffer and boiled, or with TRIzol reagent.
Immunoblotting and antibodies
Analysis of protein expression and IP experiments was performed by resolving proteins by SDS-PAGE, transfer to PVDF and immunoblotting. Primary antibodies used for immunoblotting were: anti-FLAG (Proteintech, 20543-1-AP, 1:4000), anti-HA (Cell Signaling, 2367, 6E2, 1:1125), anti-NAF1 (Abcam, ab157106, 1:1000), anti-NHP2 (Proteintech, 15128–1-AP, 1:5000), anti-NOP10 (Abcam, ab134902, 1:500), anti-TCAB1 (Novus, NB100–68252, 1:2000), anti-reptin (Abcam, ab51500, 1:5000), anti-hTERT (Santa Cruz, sc7215, C-20, 1:500), anti-dyskerin (Santa Cruz, sc-373956, H-3, 1:1500), anti-PARN (Abcam, ab154214, 1:500), anti-RRP40 (Bethyl, A303–909A-T, 1/1500), anti-TIP60 (Santa Cruz, sc5725, N-17, 1/1000), anti-hGAR1 (from Dr Witold Filipowicz (40), 1/2000) and anti-actin (Chemicon MAB1501, 1:5000).
RNA extraction and RT-qPCR
RNA was extracted using TRIzol reagent (Invitrogen), according to the reagent protocol. Reverse transcription was performed with SuperScript II Reverse Transcriptase (Invitrogen) according to the user protocol, either with hexameric random primers for total RNA or oligo-d(T) primer for polyadenylated RNA. PerfeCTa SYBR Green FastMix with Low ROX (Quanta) was used for all qPCR analyses, in a 7500FAST real-time PCR system (ABI) as previously described (20). The comparative ΔΔCT method was used to compare RNA enrichment between samples. For analysis of protein–RNA interactions, 5 μl of RNA from input and 5 μl of RNA from IP fractions were reverse transcribed into cDNA and subjected to qPCR using specific primers for target RNAs (Supplementary Table S1). The ΔΔCT was calculated between the mean CT of the IP and the mean CT of the input for each sample. For analysis of RNA levels, 1 μg of RNA per condition was reverse transcribed into cDNA and subjected to qPCR using specific primers for target RNAs (Supplementary Table S1). The ΔΔCT was calculated between the mean CT of the target and the mean CT of GAPDH for each sample.
siRNA
Transfection of siRNA was performed with Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the user protocol. For dyskerin depletion, two siRNAs targeting the 3′ UTR (24 nM, 72 h treatment, sidkc1.A or sidkc1.B) were tested for depletion of endogenous dyskerin (Supplementary Table S1). The siRNA sequences targeting the 3′ UTR were previously described (41). A mock transfection (no siRNA) and transfection of a scramble siRNA (CCUAAGGUUAAGUCGCCCUCGCUC) were used as negative controls in each experiment. For double depletion experiments, siRNA targeting PARN, or human RRP40 (20 nM, two 48 h treatments) was combined with sidkc1.B (24 nM, 48 h treatment followed by 12 nM, 48 h treatment). The siRNA sequences targeting PARN, or RRP40 (Supplementary Table S1) were previously described (13). The siRNA targeting MTR4 was kindly provided by the laboratory of Dr. François Bachand (11). All other siRNAs were ordered through ThermoFisher Scientific.
RT-PCR analysis of hTR 3′ extended species
Analysis of 3′ extended hTR species was performed as previously described (11). Primers were as follows: F1 forward primer annealing 325 nt upstream of hTR transcription start site (TSS), F2 forward primer annealing 323 nt after hTR TSS, R1 reverse primer annealing 610 nt after the hTR TSS (159 nt after mature hTR end), and GAPDH Forward and Reverse (Supplementary Table S1). The PCR conditions were as follows: 95°C for 3 min; 30 cycles of 95°C for 30 s, 55°C for 40 s, 72°C for 1 min; final extension at 72°C for 8 min. PCR products were analyzed by electrophoresis on 1.5% agarose–TAE gels stained with ethidium bromide.
Q-TRAP
Quantitative analysis of telomerase activity was done using the Q-TRAP protocol previously described (42). Briefly, HEK293 cells with or without expression of FLAG-dyskerin variants were treated with scramble siRNA or siRNA to deplete endogenous dyskerin for 72 h prior to harvesting by scraping and lysis in NP-40 lysis buffer. A standard curve was generated with a serial dilution of mock lysate (HEK293 cells untreated with siRNA and not expressing FLAG-dyskerin) for each experimental replicate (n = 3), with 1, 0.2, 0.04, 0.008 and 0.0016 μg of total protein. For comparison of telomerase activity between conditions, 0.2 μg of total protein was used for each sample.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 7. Unpaired t-tests (P < 0.01) were used to compare RNA enrichment/levels in qPCR experiments, and for comparison of relative telomerase activity (RTA) in Q-TRAP experiments. For analysis of RNA interaction and levels, the enrichment of each RNA target was separately compared to the enrichment of the target in the FLAG-dyskerin wildtype condition. For analysis of telomerase activity, the RTA percentage of each condition was separately compared to the scramble siRNA RTA percentage. Each experiment was performed in triplicate, and error bars represent the standard error of the mean between experimental replicates.
RESULTS
N-terminal or PUA domain variants of dyskerin localize to Cajal bodies and the nucleolus, and do not disrupt pre-H/ACA RNP assembly, or telomerase associated protein-protein interactions
Correct dyskerin localization is essential for its functions both in telomerase and H/ACA RNP biology. Conventionally, dyskerin localizes to both the nucleolus and Cajal bodies, important sites of telomerase trafficking, assembly and activity (43). Additionally, H/ACA RNP complex-guided pseudouridylation for rRNA and snRNA by dyskerin takes place within these subnuclear organelles (44). In order to examine the effect of the K39E, K43E and A353V substitutions on dyskerin localization, immunofluorescence experiments were performed in HEK293 cells transiently expressing FLAG-tagged dyskerin (wildtype and dyskerin variants). The expected localization of dyskerin to Cajal bodies (Figure 1B) and the nucleolus (Figure 1C) was observed for wildtype and disease variants, as indicated by co-localization of FLAG-dyskerin/coilin and FLAG-dyskerin/fibrillarin foci, respectively. A variant of dyskerin lacking a complete C-terminal N/NoLS that has been previously reported to display reduced nuclear import was used as a positive control for mislocalization (45). Indeed, this C-terminal deletion variant accumulated outside of the nucleus as expected (Figure 1B, bottom row). We conclude that the K39E, K43E, and A353V variants of dyskerin localize to the nuclear sites of maturity which are essential for both H/ACA complex and telomerase assembly.
Although these dyskerin variants are able to localize to Cajal bodies and the nucleolus, this does not exclude the possibility that dyskerin is dysfunctional at these sites. For instance, the assembly of dyskerin with its known interacting partners could be disrupted. To test this hypothesis, we immunoprecipitated FLAG-tagged dyskerin (wildtype or variants) and assessed interactions of endogenous H/ACA RNP components with FLAG-tagged dyskerin by immunoblotting. We analyzed NAF1, found only in the pre-RNP; and NOP10 and NHP2, found in both the pre- and mature RNP. We observed that dyskerin variants efficiently associated with H/ACA pre-RNP and RNP components as compared to wildtype (Figure 2A). The observation that the A353V variant is able to interact with these components is consistent with what has been reported for the H/ACA pre-RNP and RNP complex components in vitro (46). Additionally, these variants do not display disruptions in the interactions between dyskerin and other associated proteins important for both H/ACA RNP and telomerase assembly such as SHQ1 (using exogenously expressed HA-SHQ1) (Figure 2B), TCAB1 (Figure 2C), and reptin (Figure 2D). The mature H/ACA complex factor GAR1 was also observed to interact with FLAG-dyskerin variants, as well as wildtype (Figure 2E). Finally, none of the three variants display a defect in interaction with the telomerase reverse transcriptase hTERT when it is exogenously over-expressed in this assay (Figure 2F). This interaction is reduced for all FLAG-tagged dyskerin variants by RNase A treatment during immunoprecipitation (IP), consistent with the interaction between dyskerin and hTERT being dependent on hTR (37) (Supplementary Figure S1A). However, the telomerase activity levels are reduced upon dyskerin depletion with siRNA, and can only be recovered by stable expression of wildtype dyskerin, not X-DC variants (Figure 2G, Supplementary Figure S1B). To further confirm the specificity of the co-IP assay, we assessed whether or not the nuclear chromatin-associated histone acetyltransferase TIP60 could be observed in the FLAG-dyskerin IP fraction. TIP60 assembles with reptin and its AAA+ ATPase partner pontin in a complex independent of dyskerin (47), and as such should not be found to interact with dyskerin. Indeed, we did not observe TIP60 in the FLAG-dyskerin IP fractions (Figure 2H). We conclude that neither the substitutions in the N-terminal extension nor the PUA of dyskerin hinder the assembly of dyskerin into the pre- or mature H/ACA RNP complex, but impede endogenous telomerase complex function.
Figure 2.

Dyskerin variants co-immunoprecipitate (co-IP) H/ACA RNP assembly factors comparable to wildtype dyskerin, but result in reduced telomerase activity in cells depleted of endogenous dyskerin. Interactions of FLAG-dyskerin WT and variants with factors needed for assembly of the H/ACA ribonucleoprotein complex and telomerase were assessed by co-immunoprecipitation from HEK293 cell lysates. Assembly of the (A) H/ACA pre-RNP complex involving NAF1 (input protein not detectable), NHP2, and NOP10 was investigated by immunoblotting for the endogenous H/ACA pre-RNP components and FLAG-dyskerin proteins. Interaction with the cytosolic chaperone and RNA mimic (B) SHQ1 was assessed by co-expressing HA-tagged SHQ1 and FLAG-tagged dyskerin, and immunoblotting for HA and FLAG. Similar to the H/ACA pre-RNP complex, the interaction between dyskerin and nuclear RNP assembly factors (C) TCAB1 and (D) reptin and mature H/ACA complex component (E). GAR1 was examined by immunoblotting for endogenous assembly factors and FLAG-dyskerin. (F) The interaction between dyskerin and the telomerase reverse transcriptase hTERT was assessed by co-expressing hTERT and FLAG-tagged dyskerin, and immunoblotting for hTERT and FLAG. (G) Q-TRAP was repeated in experimental replicate n = 3, and quality of telomeric repeat amplification products were visually assessed on 10% non-denaturing acrylamide gel (see Supplementary Figure S1B for representative image). Statistically significant reductions in relative telomerase activity are indicated by * (P value < 0.01). Error bars represent SEM. (H) The nuclear chromatin-associated histone acetyltransferase TIP60 could not be observed in the FLAG-dyskerin IP fraction, though the expected 55 kDa protein band was observed in the input fractions (note that the upper band in the input panel represents a non-specific band that is expected based on the antibody datasheet). Immunoblotting targets are indicated to the right of each panel as ‘WB: α target’, and a list of antibodies can be found in the materials and methods section. In the IP panel of (E) the GAR1-specific band is indicated by an arrow, while immunoglobulin light chain is the strong band present in all IP fractions indicated by the asterisk. Each co-IP and immunoblotting was performed in experimental replicate a minimum of n = 2, representative blots are shown.
N-terminal dyskerin variants have disrupted interactions with telomerase RNA
Although the K39E and K43E variants have not been well characterized to date, there has been a great deal of focus on the A353V variant. A353 is located in the putative RNA binding domain of dyskerin, and mouse models and X-DC patient cells harboring the A353V substitution exhibit decreased hTR levels (18,26,30,48). Additionally, this substitution has been reported to disrupt the interaction between hTR and dyskerin in vitro (46,49). In vitro and in X-DC patient-derived cell studies, several other X-DC mutations have also been reported to disrupt the interaction with hTR (29,49), and reduced levels of hTR are commonly reported in patients with X-DC (6,18,23–30,50). Concomitant with this and our previous observation of reduced hTR levels in dyskerin knockdown cells expressing K39R and K43R variants (20), we hypothesized that the interaction between hTR and the K39E and/or K43E variants may also be disrupted. To test this hypothesis, the RNA interaction of dyskerin variants was examined by qPCR after FLAG IP. We first established that the interaction between wildtype dyskerin and hTR could be reliably analyzed by this method, as past reports of this interaction have used mainly northern blotting. FLAG-tagged wildtype dyskerin IP fractions displayed enrichment of hTR cDNA as expected, without enrichment of the C/D box RNA U3 indicating successful and specific IP of the dyskerin-hTR interaction (Supplementary Figure S2A). However, IP fractions for FLAG-K39E and K43E dyskerin variants showed less enrichment of hTR relative to wildtype (≤50% of wildtype), demonstrating a reduced ability of these variants to interact with hTR (Figure 3A, black bars). Similarly, and in agreement with what has been previously reported in vitro (46), the A353V variant also displayed reduced hTR enrichment relative to wildtype.
Figure 3.
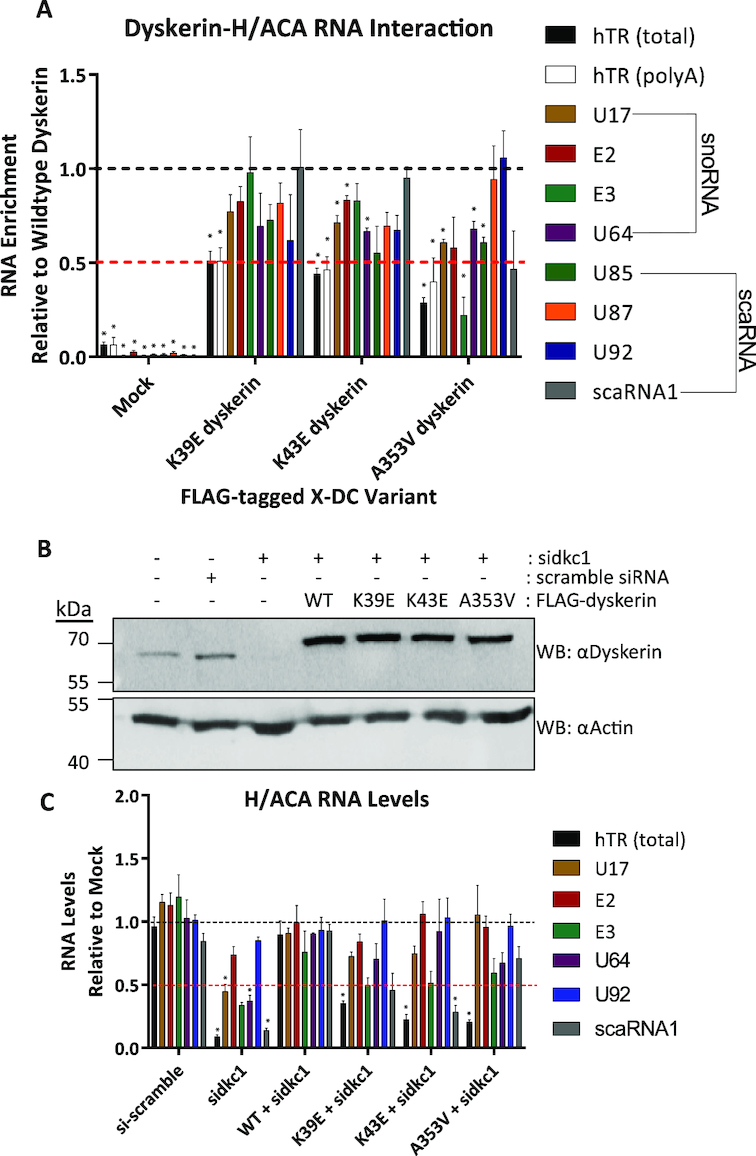
N-terminal dyskerin variants disrupt the dyskerin–RNA interaction and lead to reduced hTR accumulation in cells depleted for endogenous dyskerin. Dyskerin-RNA interactions were assessed by IP of FLAG-tagged dyskerin followed by RNA extraction and qPCR. Relative to wildtype IP fractions (indicated by black dashed line), dyskerin variants K39E, K43E, and A353V display (A) reduced enrichment of hTR following IP (approximately at or below 50% of wildtype, indicated by the red dashed line). Black bars indicate total hTR (cDNA primed with random hexamer) and white bars outlined in black indicate precursor hTR with poly(A) tails (cDNA primed with oligo dTs). Other H/ACA RNAs examined are indicated by coloured bars, and listed in the figure legend to the right of the graph. HEK293 cells lacking FLAG-tagged dyskerin (indicated as mock) were used as a negative control for RNA binding to the FLAG antibody and/or Protein G Sepharose. Mock cells were subject to the same IP protocol detailed for fractions containing FLAG-tagged dyskerin. These data represent experimental replicates of n = 3. Statistically significant reductions in enrichment relative to wildtype are indicated by * (P value < 0.01). Error bars represent SEM. (B) Depletion of endogenous dyskerin with siRNA (sidkc1.B), as well as expression of FLAG-tagged constructs (the higher molecular weight band detected by αDyskerin) was assessed by immunoblotting. (C) RNA levels were assessed from HEK293 cells with stable expression of FLAG-tagged wildtype or variants of dyskerin following 72h of siRNA targeting endogenous dyskerin (sidkc1.B), relative to empty vector HEK293 cells (mock) lacking FLAG-tagged dyskerin and untreated with siRNA. Mock cell RNA levels are indicated by the black dashed line, while the red dashed line indicates 50% of mock cell RNA levels. Empty vector HEK293 cells treated with scramble siRNA were used to control for possible effects of siRNA transfection. These data represent experimental replicates of n = 3. Statistically significant reductions in RNA enrichment compared to WT + sidkc1 treated cells are indicated by * (P value < 0.01). Error bars represent SEM.
In some analyses of mouse and X-DC patient cells, certain X-DC variants (including A353V but not K39E or K43E) have also been reported to reduce the accumulation of other H/ACA RNAs (24,26,28,48). Although the interaction of A353V with other H/ACA RNAs has not been looked at directly in cells or in vivo, it is possible that this substitution and others could disrupt the interaction between dyskerin and H/ACA RNAs other than hTR. We therefore wanted to consider whether the variants tested in this study display a general H/ACA RNA interaction defect, using the same assay. For the N-terminal hotspot variants K39E and K43E, the observed RNA interaction defect appears to be most substantial for hTR compared to other H/ACA RNAs. For instance, other H/ACA RNAs examined did not display significant differences in enrichment after IP of the dyskerin variant K39E compared to the wildtype dyskerin (Figure 3A, colored bars). In contrast, the K43E variant displayed a significant reduction in enrichment for three of the four H/ACA snoRNAs examined (with the exception of E3) >50% of wildtype (Figure 3A, brown, red and purple bars), and no significant defects in scaRNA interactions when compared to wildtype dyskerin (Figure 3A, green, orange, blue and gray bars). As such, we propose that in the case of the K43E variant, the interaction defect for hTR is the most severe of the H/ACA RNAs considered here, though this does not exclude potential functional defects related to the other H/ACA RNAs with reduced enrichment.
The A353V variant displayed reduced enrichment of several H/ACA RNAs including the U85 scaRNA and three of the four H/ACA snoRNAs (with the exception of E2) (Figure 3A, green, brown, teal, and purple bars). Among the H/ACA RNAs with significantly reduced enrichment, the E3 snoRNA falls below 50% of the levels enriched by wildtype dyskerin (Figure 3A, teal bar). These notable interaction defects with H/ACA RNAs other than hTR are consistent with the position of A353 in the PUA domain, supporting the notion that this region is the conserved RNA-binding domain of dyskerin as a member of the pseudouridine synthase family of proteins. As such, we propose that substitutions within the PUA domain, like A353V, might cause more global H/ACA RNA interaction defects than those within the N-terminal X-DC hotspot. Our findings also suggest a potential telomerase-centric role for the eukaryotic N-terminal extension of dyskerin (16), containing the N-terminal X-DC hotspot.
Dyskerin variants are unable to maintain hTR levels during depletion of endogenous dyskerin
Depleting endogenous dyskerin has been previously reported to decrease the levels of several H/ACA RNAs, including hTR and polyadenylated hTR (12). We therefore examined whether dyskerin variants also have an impact on RNA levels. Towards this end, we performed siRNA targeting the 3′ UTR of dyskerin mRNA in HEK293 cells stably expressing FLAG-tagged wildtype or variants of dyskerin (Figure 3B). While dyskerin-depleted HEK293 cells rescued with wildtype dyskerin were able to partially recover levels of hTR, none of the variants significantly rescued hTR levels (Figure 3C, black bars). The levels of several other H/ACA RNAs (E2 P value 0.155, E3 P value 0.0165, and U92 P value 0.485—Figure 3C, red, teal and blue bars) were not significantly affected by depletion of endogenous dyskerin, compared to knockdown cells expressing FLAG-tagged wildtype dyskerin. Interestingly, the levels of certain H/ACA RNAs (U17, U64 and scaRNA1) that were significantly reduced by dyskerin depletion were rescued by expression of wildtype dyskerin (Figure 3C, brown, purple and gray bars). Following dyskerin depletion in empty vector cells, the level of U17, U64 and scaRNA1 were significantly reduced compared to dyskerin-depleted cells expressing FLAG-wildtype dyskerin. The recovered level of each of these H/ACA RNAs is comparable between cells expressing variants and wildtype dyskerin, with the exception of scaRNA1 that remained significantly reduced in K43E cells. Furthermore, with a second siRNA targeting endogenous dyskerin, an even more modest effect on H/ACA RNA levels was observed compared to knockdown cells expressing FLAG-wildtype dyskerin (Supplementary Figures S2B and C). As such, we speculate that, of the H/ACA RNAs examined, hTR is one of the most sensitive to the dyskerin–RNA interaction defect. However, we do not rule out potential effects of these mutations on rRNA pseudouridylation, or functional consequences on the ribosome.
Substitutions in the N-terminus or PUA domain of dyskerin disrupt interactions with polyadenylated hTR species and favour hTR degradation over processing
Consistent with reports indicating an important role for dyskerin in hTR processing and maturation (11–13,51,52), wildtype dyskerin interacts with polyadenylated hTR species, as assessed by enrichment of hTR cDNA species primed with oligo d(T)s (Supplementary Figure S2A, white bars). Furthermore, all three variants displayed a reduced interaction with polyadenylated hTR species relative to wildtype dyskerin, at or below 50% of enrichment with wildtype, similarly to what was observed for total hTR species (Figure 3A, white bars). Given this observation, we postulate that it is likely that these interaction defects have implications upstream of mature hTR function during an early assembly step with nascent or precursor hTR species (see model in Figure 5).
Figure 5.

Summary model. Under wildtype conditions, dyskerin binds to newly transcribed hTR, protecting species with 3′ extensions and/or poly(A) tails from degradation. This ensures the trimming of hTR 3′ extended and/or polyadenylated species into mature hTR can take place, followed by assembly of the functional telomerase complex, and telomere maintenance by hTERT. In the context of X-DC, dyskerin variants with defects in binding to newly transcribed hTR result in deprotection of the H/ACA box of hTR, leading to misregulated degradation of 3′ extended and polyadenylated species by a variety of reported RNA processing and degradation complexes (i.e. PABPN1/PARN, NEXT/TRAMP/exosome, DCP2/XRN1, and TOE1). Ultimately, this causes a reduced amount of functional telomerase and a lack of telomere maintenance, as has been observed in X-DC patients. Two copies of the H/ACA ribonucleoprotein complex have been reported to be assembled with the H/ACA box of hTR in active telomerase, with the 5′ stem loop dyskerin likely being anchored to the complex via the 3′ stem loop-bound dyskerin, which may be disrupted by amino acid substitutions in the N-terminal extension or DKCLD X-DC hotspot.
Unlike the reported accumulation of polyadenylated hTR species resulting from the knockdown of some RNA processing components recently identified in hTR maturation—such as the poly(A) ribonuclease PARN, the exosome complex components, recruitment factors involved in these pathways, or TOE1 (11–15)—knockdown of dyskerin leads to a reduction of polyadenylated hTR species as well as total hTR species (Figure 4A). Additionally, we observed no accumulation of hTR 3′ extended precursors by semi-quantitative RT-PCR following dyskerin knockdown (Figure 4B), contrasting what has been observed upon knockdown of components in the aforementioned processing pathways. There was also no accumulation of hTR 3′ extended precursors observed in cells depleted of endogenous dyskerin while stably expressing either wildtype or variants of dyskerin (Figure 4B). Based on these data, we posited that hTR precursors that are not bound by dyskerin variants are rapidly degraded, causing the hTR accumulation defect, and not leading to defects in hTR processing or trimming per se.
Figure 4.

Deprotection of hTR results in degradation and not accumulation of poly(A) or 3′ extended hTR species. Following siRNA targeting of dyskerin (sidkc1.A and sidkc1.B), qPCR analysis revealed that (A) total and poly(A) hTR levels are both reduced relative to untreated HEK293 cells. The levels of the C/D RNA U3 are not reduced by depletion of dyskerin. This has been performed in experimental replicate n = 3. Additionally, (B) the accumulation of 3′ extended hTR precursors was examined by PCR following knockdown of endogenous dyskerin (sidkc1.B), or the NEXT/hTRAMP complex component MTR4 (siMTR4 - a positive control for defective processing of hTR). The expected 288 nt product observed with cDNA from HEK293 cells depleted of MTR4 (indicated by an asterisk) was not observed in cells depleted of dyskerin, with or without exogenous WT and dyskerin variants (middle panel: F2 + R1). GAPDH amplification was used as a positive control (bottom panel), and no amplification of genomic contamination was observed (top panel: F1 + R1), indicating that the amplified product is a 3′ extended hTR transcript. This was repeated in experimental replicate n = 2, and a representative image is shown. Double depletion of endogenous dyskerin and (C) PARN or (D) RRP40 from HEK293 cells with siRNA (sidkc1.B and siPARN, or sidkc1.B and siRRP40), as well as expression of FLAG-tagged constructs (the higher molecular weight band detected by αDyskerin) was assessed by immunoblotting following 96h of siRNA treatment. Total hTR levels were assessed from HEK293 cells with stable expression of FLAG-tagged wildtype or variants of dyskerin by qPCR following 96 h of siRNA treatment, relative to empty vector HEK293 cells (mock) lacking FLAG-tagged dyskerin and untreated with siRNA. Mock cell hTR level is indicated by the black dashed line, while the red dashed line indicates 50% of mock cell hTR level. Empty vector HEK293 cells treated with scramble siRNA were used to control for possible effects of siRNA transfection. These data represent experimental replicates of n = 3. No statistically significant reductions in RNA enrichment compared to WT + siPARN + sidkc1 were observed. Statistically significant reductions in RNA enrichment compared to WT + siRRP40 + sidkc1 treated cells are indicated by * (P value < 0.01). Error bars represent SEM.
To test this hypothesis, we performed a double knockdown of dyskerin and components of processing or degradation pathways; PARN or the human exosome core component RRP40 were depleted along with endogenous dyskerin using siRNA, in cells expressing wildtype or variant FLAG-tagged dyskerin. In the context of dyskerin depletion, neither siRNA targeting of PARN nor RRP40 completely recovers hTR levels in HEK293 cells, with or without expression of FLAG-tagged variants (Figure 4C and D). Importantly, following PARN knockdown in dyskerin depleted cells expressing FLAG-tagged wildtype dyskerin, hTR levels are also lower than mock and scramble siRNA treated cells. This is consistent with previous reports that PARN knockdown and PARN mutations result in reduced mature hTR levels due to hTR processing defects which generate 3′ extended species that undergo degradation, likely by the human exosome and/or cytosolic RNA decay machinery (11–13). Additionally, the partial rescue of hTR levels that we observe upon knockdown of RRP40 in dyskerin depleted cells is comparable to the partial rescue observed upon knockdown of RRP6 by Shukla et al. (12), consistent with these two factors functioning in the same exosome complex acting on hTR in dyskerin depleted cells. We speculate that various RNA degradation pathways may function redundantly with respect to hTR degradation in this context, consistent with reports of many different nucleases acting during the hTR trimming and degradation processes, including the previously reported role of the cytoplasmic RNA decay machinery acting on a subset of hTR species exported to the cytoplasm upon dyskerin depletion (11–14).
DISCUSSION
Defective accumulation of hTR is a common consequence of X-DC mutations, reported in both cell culture studies and in patients with this premature aging disease (18,23–29,50,53). Here, we show that for the K39E, K43E and A353V dyskerin variants, reduced hTR accumulation is driven by an interaction defect between dyskerin and precursor hTR species. Interaction defects have been previously reported in vitro for A353V, as well as studies done in vitro or in patient-derived cells for substitutions G402E, T49M and ΔL37 (29,46,49). While none of these previous studies examined precursors of hTR, it seems likely that the interaction defects reported for other variants would affect precursor hTR species that have an H/ACA box available for dyskerin binding. As such, we propose that X-DC variants that display hTR interaction defects cause reduced accumulation of hTR and telomerase activity by deprotecting hTR precursors.
Recent studies examining the processing and degradation pathways for hTR have shed light on roles for many different complexes, including the nuclear exosome, PABPN1 and PARN, human TRAMP and NEXT complexes, CBCA complex, XRN1/DCP2 and most recently TOE1 (11–15). While the balance between degradation and trimming of hTR precursors into mature functional species is clearly a tightly regulated and complex process, much remains uncertain regarding the redundancies in these pathways and how the balance between them is coordinated. Our findings suggest that in a context of dysfunctional H/ACA complex interactions with hTR, targeting only one of these pathways is insufficient to rescue hTR accumulation defects in cells. Furthermore, 3′ extended hTR precursors do not accumulate in cells depleted of endogenous dyskerin, regardless of whether or not exogenous dyskerin (wildtype or variant) is expressed. These findings lead us to the conclusion that the interaction between dyskerin and hTR is one of the earliest steps in hTR biogenesis that prevents hTR degradation, as has been proposed previously by Tseng et al. (13) for the H/ACA RNP complex as a whole. However, in contrast to what has been observed for other components that regulate the correct processing of hTR, depletion of dyskerin ultimately leads to a reduction of both polyadenylated and mature hTR species, which cannot be rescued by targeting individual degradation pathways. We propose that the interaction between dyskerin and hTR is essential for blocking canonical RNA degradation pathways, as well as preventing hTR degradation by components that would canonically function as trimming or processing factors. In the X-DC context, we speculate that dyskerin-hTR interaction defects could promote misregulated PARN trimming leading to hTR degradation, as well as formation of 3′ extended hTR precursors which rapidly undergo degradation by the exosome rather than being properly trimmed into mature hTR species. This is consistent with a recent report detailing a role for dyskerin in the regulation of short 3′ extended hTR trimming by PARN, as well as preventing the stabilization of long 3′ extended hTR products which are targeted for degradation by the RRP6-exosome (52). More recently still, it has been demonstrated that targeting the exosome via depletion of RRP40 can partially rescue total hTR levels in human embryonic stem cells (hESCs) that were genetically engineered by CRISPR/Cas9 to harbor the A353V variant of dyskerin, as well as RRP40 depletion partially restoring telomerase activity, telomere length, and reducing DNA damage signaling. However, targeting the non-canonical poly(A) RNA polymerase PAPD5 in the A353V hESCs was able to rescue hTR levels to a greater extent than downregulation of RRP40, as well as uniquely rescue hematopoiesis defects comparable to rescue by overexpression of hTR in these hESCs (54). This is consistent with polyadenylation of hTR being upstream of the recruitment of various nucleases, including the exosome and PARN. Furthermore, as has been demonstrated by Shukla et al. (12), it is likely that the cytoplasmic RNA decay machinery also contributes to the inability to fully rescue hTR levels by solely targeting the exosome or PARN, with DCP2 and XRN1 acting on cyTER (cytoplasmic telomerase RNA) species which are exported from the nucleus following dyskerin depletion.
While the predicted secondary structure of H/ACA box RNAs is likely comparable between specific RNA molecules, many differences between hTR and other H/ACA class RNAs have been reported in the literature. Indeed, biogenesis and structural differences among H/ACA class RNAs including hTR have been demonstrated (31,55–57), as well as variations in hairpin size, for instance as predicted for members of the AluACA class RNAs, the E2 snoRNA, and hTR (58). Similarly, structural elements within the H/ACA RNA stem loops such as the BIO box motif present in hTR (31,56), or the CAB box found in scaRNAs (55,57) also provide variations in protein–RNA complex assembly due to differences in RNA structure and/or folding. RNA structural variations, as well as differences in H/ACA RNA biogenesis may offer an explanation for the evident variability of dyskerin–RNA interactions, and the downstream consequences. For instance, although reduced total 18S rRNA pseudouridine content has been observed in X-DC patient lymphocytes expressing the A353V variant, several studies have reported normal processing and accumulation of mature 18S and 28S rRNA species in patient cells with this variant (18,26,28,59). We speculate that the reduction in pseudouridine content may be caused by an impaired interaction of dyskerin with select H/ACA RNAs, such as the interaction defect that we observed between the A353V variant and the E3 snoRNA, but this reduction does not substantially impede mature rRNA function. One explanation for this could be that H/ACA RNAs that interact poorly with A353V still accumulate to sufficient levels to maintain a functional amount of pseudouridine synthesis. Indeed, though the interaction between A353V and E3 is defective, this X-DC variant can still at least partially rescue total levels of E3 snoRNA in cells depleted of endogenous dyskerin. In contrast, hTR accumulation cannot be recovered by any of the variants in this study, and the amount of hTR that remains in cells depleted of endogenous dyskerin while expressing X-DC variants is not sufficient for telomerase function. As such, it is tempting to speculate that X-DC presents as a telomere biology disorder because hTR is particularly sensitive to any disruptions in dyskerin–RNA interaction, as has also been previously proposed based on differential biogenesis requirements between hTR and other H/ACA class RNAs (31).
Importantly, while some X-DC patient cells have presented with lower dyskerin protein levels than wildtype controls (26,28,60), in the context of this study we did not observe notable differences in the levels of expression for wildtype, K39E, K43E and A353V dyskerin constructs, suggesting that these variants are not subject to more protein degradation than wildtype dyskerin. All three variants that we examined are capable of forming H/ACA RNP complexes and localizing to the subnuclear compartments where mature H/ACA RNP complexes are found. This lends further support to the notion that not all H/ACA RNAs are disrupted by these mutations, as complex formation and function is largely dependent on protein–RNA interactions (61). One striking observation that we made was that all three of these variants are able to interact with exogenously expressed hTERT. The interaction between dyskerin and hTERT is dependent on hTR, and so we expected to observe a decreased co-IP of hTERT with the dyskerin variants correlating with the amount of hTR in X-DC variant IP fractions. However, hTR stability can be improved upon hTERT overexpression alone (62), and it has been reported that X-DC variants are able to co-IP with active telomerase in the context of hTERT and hTR overexpression (18). As such, we postulate that overexpression of hTERT likely masks possible interaction defects between hTERT and the variants examined in this study. Indeed, given that cells depleted of endogenous dyskerin suffer a reduction in telomerase activity that cannot be rescued by these X-DC variants, we postulate that telomerase assembly is defective in this context, despite our inability to detect a defective interaction between these variants and hTERT by co-IP.
Residues in the N-terminal extension of dyskerin are highly evolutionarily conserved among eukaryotes, but the exact function of this region in humans has been largely uncharacterized to date. Our findings support structural studies done in yeast indicating that this region, along with the PUA, plays an important RNA-interacting role (63). The recently solved cryo-EM structure of human telomerase also demonstrates that this region is in close proximity to both the PUA and hTR itself (64), strongly supporting the possibility that the function of the N-terminal extension and DKCLD may be as an RNA-binding domain cooperating with the PUA to stabilize the interaction of dyskerin with the H/ACA domain of H/ACA class RNAs. In addition to the interaction defects that we observe for K39E and K43E, other X-DC substitutions in the N-terminal hotspot have also been reported to disrupt the interaction between dyskerin and hTR in vitro (T49M) (49) and in patient-derived cell studies (ΔL37) (29). In concordance with what was proposed by Nguyen et al. (64) based on the human telomerase cryo-EM structure, we speculate that hTR is particularly sensitive to substitutions in the N-terminus of dyskerin due to the 5′ hairpin of the H/ACA domain in hTR which is unique amongst other H/ACA box RNAs. Given that the interaction of dyskerin with this 5′ hairpin is largely dependent on dyskerin interfacing with the 3′ hairpin-bound dyskerin (64), it is possible that substitutions in the N-terminal hotspot disrupt this interface and therefore the interaction between dyskerin and the 5′ hairpin in hTR without affecting other H/ACA RNA interactions as substantially. Additionally, this may explain why K39E and K43E present an hTR interaction of approximately 50% of wildtype dyskerin, as the 3′ hairpin dyskerin–RNA interaction is stabilized by other H/ACA protein–RNA contacts which could remain intact based on the co-IP data analyzing H/ACA RNP complex assembly. We suggest that protection of only the 3′ hairpin in the H/ACA domain of hTR may be insufficient to regulate the balance between trimming and degradation of hTR precursors. Further analyses will be needed to test this hypothesis, and to assess whether other X-DC substitutions within the N-terminal extension and DKCLD present a similar disruption of the dyskerin-hTR interaction.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Tom Meier, Dr. Joachim Lingner and Dr. François Dragon for providing us with pSR38 (SHQ1), pcDNA6/myc-HisC-hTERT and pcDNA3.1-FLAG-dyskerinWT plasmids, respectively. We thank Dr. Michael Terns, Dr. Kenneth Michael Pollard and Dr. Witold Filipowicz for providing us with mouse anti-coilin, mouse anti-fibrillarin and rabbit anti-hGAR1 antibodies. We thank Dr. François Bachand for providing us with siRNA targeting human MTR4, as well as experimental advice regarding RNA processing pathways. We thank Marc Fabian and Jian Qin for discussion and comments.
Author contributions: D.E.M. and C.A. designed research; D.E.M. and P.L.-L. performed experiments. D.E.M. and C.A. wrote the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research (CIHR) [MOP-133449 to C.A.]; Canada Graduate Scholarships-Master's Program (CIHR), Cole Foundation Fellowship and CIHR Doctoral Research Award (to D.E.M.). Funding for open access charge: Canadian Institutes of Health [MOP-133449].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lopez-Otin C., Blasco M.A., Partridge L., Serrano M., Kroemer G.. The hallmarks of aging. Cell. 2013; 153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bertuch A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016; 13:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wegman-Ostrosky T., Savage S.A.. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br. J. Haematol. 2017; 177:526–542. [DOI] [PubMed] [Google Scholar]
- 4. Alter B.P., Giri N., Savage S.A., Rosenberg P.S.. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018; 103:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knight S.W., Heiss N.S., Vulliamy T.J., Greschner S., Stavrides G., Pai G.S., Lestringant G., Varma N., Mason P.J., Dokal I. et al.. X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am. J. Hum. Genet. 1999; 65:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mitchell J.R., Wood E., Collins K.. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999; 402:551–555. [DOI] [PubMed] [Google Scholar]
- 7. Meyerson M., Counter C.M., Eaton E.N., Ellisen L.W., Steiner P., Caddle S.D., Ziaugra L., Beijersbergen R.L., Davidoff M.J., Liu Q. et al.. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997; 90:785–795. [DOI] [PubMed] [Google Scholar]
- 8. Feng J., Funk W.D., Wang S.S., Weinrich S.L., Avilion A.A., Chiu C.P., Adams R.R., Chang E., Allsopp R.C., Yu J. et al.. The RNA component of human telomerase. Science. 1995; 269:1236–1241. [DOI] [PubMed] [Google Scholar]
- 9. Nakamura T.M., Morin G.B., Chapman K.B., Weinrich S.L., Andrews W.H., Lingner J., Harley C.B., Cech T.R.. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997; 277:955–959. [DOI] [PubMed] [Google Scholar]
- 10. Lafontaine D.L., Bousquet-Antonelli C., Henry Y., Caizergues-Ferrer M., Tollervey D.. The box H + ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev. 1998; 12:527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nguyen D., Grenier St-Sauveur V., Bergeron D., Dupuis-Sandoval F., Scott M.S., Bachand F.. A polyadenylation-dependent 3′ end maturation pathway is required for the synthesis of the human telomerase RNA. Cell Rep. 2015; 13:2244–2257. [DOI] [PubMed] [Google Scholar]
- 12. Shukla S., Schmidt J.C., Goldfarb K.C., Cech T.R., Parker R.. Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat. Struct. Mol. Biol. 2016; 23:286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tseng C.K., Wang H.F., Burns A.M., Schroeder M.R., Gaspari M., Baumann P.. Human telomerase RNA processing and quality control. Cell Rep. 2015; 13:2232–2243. [DOI] [PubMed] [Google Scholar]
- 14. Deng T., Huang Y., Weng K., Lin S., Li Y., Shi G., Chen Y., Huang J., Liu D., Ma W. et al.. TOE1 acts as a 3′ exonuclease for telomerase RNA and regulates telomere maintenance. Nucleic Acids Res. 2018; 47:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Son A., Park J.E., Kim V.N.. PARN and TOE1 constitute a 3′ end maturation module for nuclear Non-coding RNAs. Cell Rep. 2018; 23:888–898. [DOI] [PubMed] [Google Scholar]
- 16. Cerrudo C.S., Ghiringhelli P.D., Gomez D.E.. Protein universe containing a PUA RNA-binding domain. FEBS J. 2014; 281:74–87. [DOI] [PubMed] [Google Scholar]
- 17. Podlevsky J.D., Bley C.J., Omana R.V., Qi X., Chen J.J.. The telomerase database. Nucleic Acids Res. 2008; 36:D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zeng X.L., Thumati N.R., Fleisig H.B., Hukezalie K.R., Savage S.A., Giri N., Alter B.P., Wong J.M.. The accumulation and not the specific activity of telomerase ribonucleoprotein determines telomere maintenance deficiency in X-linked dyskeratosis congenita. Hum. Mol. Genet. 2012; 21:721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heiss N.S., Megarbane A., Klauck S.M., Kreuz F.R., Makhoul E., Majewski F., Poustka A.. One novel and two recurrent missense DKC1 mutations in patients with dyskeratosis congenita (DKC). Genet Couns. 2001; 12:129–136. [PubMed] [Google Scholar]
- 20. Brault M.E., Lauzon C., Autexier C.. Dyskeratosis congenita mutations in dyskerin SUMOylation consensus sites lead to impaired telomerase RNA accumulation and telomere defects. Hum. Mol. Genet. 2013; 22:3498–3507. [DOI] [PubMed] [Google Scholar]
- 21. Li L., Ye K.. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature. 2006; 443:302–307. [DOI] [PubMed] [Google Scholar]
- 22. Duan J., Li L., Lu J., Wang W., Ye K.. Structural mechanism of substrate RNA recruitment in H/ACA RNA-guided pseudouridine synthase. Mol. Cell. 2009; 34:427–439. [DOI] [PubMed] [Google Scholar]
- 23. Wong J.M., Collins K.. Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev. 2006; 20:2848–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong J.M., Kyasa M.J., Hutchins L., Collins K.. Telomerase RNA deficiency in peripheral blood mononuclear cells in X-linked dyskeratosis congenita. Hum. Genet. 2004; 115:448–455. [DOI] [PubMed] [Google Scholar]
- 25. Alder J.K., Parry E.M., Yegnasubramanian S., Wagner C.L., Lieblich L.M., Auerbach R., Auerbach A.D., Wheelan S.J., Armanios M.. Telomere phenotypes in females with heterozygous mutations in the dyskeratosis congenita 1 (DKC1) gene. Hum. Mutat. 2013; 34:1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gu B.W., Apicella M., Mills J., Fan J.M., Reeves D.A., French D., Podsakoff G.M., Bessler M., Mason P.J.. Impaired telomere maintenance and decreased canonical WNT signaling but normal ribosome biogenesis in induced pluripotent stem cells from X-Linked dyskeratosis congenita patients. PLoS One. 2015; 10:e0127414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parry E.M., Alder J.K., Lee S.S., Phillips J.A., 3rd L., J.E. D., Armanios M.. Decreased dyskerin levels as a mechanism of telomere shortening in X-linked dyskeratosis congenita. J. Med. Genet. 2011; 48:327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bellodi C., McMahon M., Contreras A., Juliano D., Kopmar N., Nakamura T., Maltby D., Burlingame A., Savage S.A., Shimamura A. et al.. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep. 2013; 3:1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batista L.F., Pech M.F., Zhong F.L., Nguyen H.N., Xie K.T., Zaug A.J., Crary S.M., Choi J., Sebastiano V., Cherry A. et al.. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011; 474:399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Machado-Pinilla R., Carrillo J., Manguan-Garcia C., Sastre L., Mentzer A., Gu B.W., Mason P.J., Perona R.. Defects in mTR stability and telomerase activity produced by the Dkc1 A353V mutation in dyskeratosis congenita are rescued by a peptide from the dyskerin TruB domain. Clin. Transl. Oncol. 2012; 14:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fu D., Collins K.. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol. Cell. 2003; 11:1361–1372. [DOI] [PubMed] [Google Scholar]
- 32. Grozdanov P.N., Roy S., Kittur N., Meier U.T.. SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA. 2009; 15:1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cristofari G., Lingner J.. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 2006; 25:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chu T.W., D'Souza Y., Autexier C.. The insertion in fingers domain in human telomerase can mediate enzyme processivity and telomerase recruitment to telomeres in a TPP1-Dependent manner. Mol. Cell Biol. 2016; 36:210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tomlinson R.L., Ziegler T.D., Supakorndej T., Terns R.M., Terns M.P.. Cell cycle-regulated trafficking of human telomerase to telomeres. Mol. Biol. Cell. 2006; 17:955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pogacic V., Dragon F., Filipowicz W.. Human H/ACA small nucleolar RNPs and telomerase share evolutionarily conserved proteins NHP2 and NOP10. Mol. Cell Biol. 2000; 20:9028–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Venteicher A.S., Abreu E.B., Meng Z., McCann K.E., Terns R.M., Veenstra T.D., Terns M.P., Artandi S.E.. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009; 323:644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Venteicher A.S., Meng Z., Mason P.J., Veenstra T.D., Artandi S.E.. Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell. 2008; 132:945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Booy E.P., Meier M., Okun N., Novakowski S.K., Xiong S., Stetefeld J., McKenna S.A.. The RNA helicase RHAU (DHX36) unwinds a G4-quadruplex in human telomerase RNA and promotes the formation of the P1 helix template boundary. Nucleic Acids Res. 2012; 40:4110–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dragon F., Pogacic V., Filipowicz W.. In vitro assembly of human H/ACA small nucleolar RNPs reveals unique features of U17 and telomerase RNAs. Mol Cell Biol. 2000; 20:3037–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin P., Mobasher M.E., Alawi F.. Acute dyskerin depletion triggers cellular senescence and renders osteosarcoma cells resistant to genotoxic stress-induced apoptosis. Biochem. Biophys. Res. Commun. 2014; 446:1268–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herbert B.S., Hochreiter A.E., Wright W.E., Shay J.W.. Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat. Protoc. 2006; 1:1583–1590. [DOI] [PubMed] [Google Scholar]
- 43. MacNeil D.E., Bensoussan H.J., Autexier C.. Telomerase regulation from beginning to the end. Genes (Basel). 2016; 7:E64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu Y.T., Meier U.T.. RNA-guided isomerization of uridine to pseudouridine–pseudouridylation. RNA Biol. 2014; 11:1483–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Heiss N.S., Girod A., Salowsky R., Wiemann S., Pepperkok R., Poustka A.. Dyskerin localizes to the nucleolus and its mislocalization is unlikely to play a role in the pathogenesis of dyskeratosis congenita. Hum. Mol. Genet. 1999; 8:2515–2524. [DOI] [PubMed] [Google Scholar]
- 46. Trahan C., Martel C., Dragon F.. Effects of dyskeratosis congenita mutations in dyskerin, NHP2 and NOP10 on assembly of H/ACA pre-RNPs. Hum. Mol. Genet. 2010; 19:825–836. [DOI] [PubMed] [Google Scholar]
- 47. Ikura T., Ogryzko V.V., Grigoriev M., Groisman R., Wang J., Horikoshi M., Scully R., Qin J., Nakatani Y.. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000; 102:463–473. [DOI] [PubMed] [Google Scholar]
- 48. Mochizuki Y., He J., Kulkarni S., Bessler M., Mason P.J.. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10756–10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ashbridge B., Orte A., Yeoman J.A., Kirwan M., Vulliamy T., Dokal I., Klenerman D., Balasubramanian S.. Single-molecule analysis of the human telomerase RNA.dyskerin interaction and the effect of dyskeratosis congenita mutations. Biochemistry. 2009; 48:10858–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kropski J.A., Mitchell D.B., Markin C., Polosukhin V.V., Choi L., Johnson J.E., Lawson W.E., Phillips J.A. 3rd, Cogan J.D., Blackwell T.S. et al.. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest. 2014; 146:e1–e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ballarino M., Morlando M., Pagano F., Fatica A., Bozzoni I.. The cotranscriptional assembly of snoRNPs controls the biosynthesis of H/ACA snoRNAs in Saccharomyces cerevisiae. Mol. Cell Biol. 2005; 25:5396–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tseng C.K., Wang H.F., Schroeder M.R., Baumann P.. The H/ACA complex disrupts triplex in hTR precursor to permit processing by RRP6 and PARN. Nat. Commun. 2018; 9:5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Agarwal S., Loh Y.H., McLoughlin E.M., Huang J., Park I.H., Miller J.D., Huo H., Okuka M., Dos Reis R.M., Loewer S. et al.. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010; 464:292–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fok W.C., Shukla S., Vessoni A.T., Brenner K.A., Parker R., Sturgeon C.M., Batista L.F.Z.. Posttranscriptional modulation of TERC by PAPD5 inhibition rescues hematopoietic development in dyskeratosis congenita. Blood. 2019; 133:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Theimer C.A., Jady B.E., Chim N., Richard P., Breece K.E., Kiss T., Feigon J.. Structural and functional characterization of human telomerase RNA processing and cajal body localization signals. Mol. Cell. 2007; 27:869–881. [DOI] [PubMed] [Google Scholar]
- 56. Egan E.D., Collins K.. An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol. Cell Biol. 2012; 32:2428–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jady B.E., Bertrand E., Kiss T.. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J. Cell Biol. 2004; 164:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ketele A., Kiss T., Jady B.E.. Human intron-encoded AluACA RNAs and telomerase RNA share a common element promoting RNA accumulation. RNA Biol. 2016; 13:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thumati N.R., Zeng X.L., Au H.H., Jang C.J., Jan E., Wong J.M.. Severity of X-linked dyskeratosis congenita (DKCX) cellular defects is not directly related to dyskerin (DKC1) activity in ribosomal RNA biogenesis or mRNA translation. Hum. Mutat. 2013; 34:1698–1707. [DOI] [PubMed] [Google Scholar]
- 60. Perdigones N., Perin J.C., Schiano I., Nicholas P., Biegel J.A., Mason P.J., Babushok D.V., Bessler M.. Clonal hematopoiesis in patients with dyskeratosis congenita. Am. J. Hematol. 2016; 91:1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Massenet S., Bertrand E., Verheggen C.. Assembly and trafficking of box C/D and H/ACA snoRNPs. RNA Biol. 2017; 14:680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yi X., Tesmer V.M., Savre-Train I., Shay J.W., Wright W.E.. Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol. Cell Biol. 1999; 19:3989–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li S., Duan J., Li D., Yang B., Dong M., Ye K.. Reconstitution and structural analysis of the yeast box H/ACA RNA-guided pseudouridine synthase. Genes Dev. 2011; 25:2409–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nguyen T.H.D., Tam J., Wu R.A., Greber B.J., Toso D., Nogales E., Collins K.. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature. 2018; 557:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.