


Abstract
Hepatitis C virus (HCV) is a positive-sense RNA virus that interacts with the liver-specific microRNA, miR-122. miR-122 binds to two sites in the 5′ untranslated region (UTR) and this interaction promotes HCV RNA accumulation, although the precise role of miR-122 in the HCV life cycle remains unclear. Using biophysical analyses and Selective 2′ Hydroxyl Acylation analyzed by Primer Extension (SHAPE) we investigated miR-122 interactions with the 5′ UTR. Our data suggests that miR-122 binding results in alteration of nucleotides 1–117 to suppress an alternative secondary structure and promote functional internal ribosomal entry site (IRES) formation. Furthermore, we demonstrate that two hAgo2:miR-122 complexes are able to bind to the HCV 5′ terminus simultaneously and SHAPE analyses revealed further alterations to the structure of the 5′ UTR to accommodate these complexes. Finally, we present a computational model of the hAgo2:miR-122:HCV RNA complex at the 5′ terminus of the viral genome as well as hAgo2:miR-122 interactions with the IRES–40S complex that suggest hAgo2 is likely to form additional interactions with SLII which may further stabilize the HCV IRES. Taken together, our results support a model whereby hAgo2:miR-122 complexes alter the structure of the viral 5′ terminus and promote formation of the HCV IRES.
INTRODUCTION
Hepatitis C virus (HCV) infects ∼71 million people worldwide, with the majority of patients developing a persistent infection that can lead to chronic hepatitis, cirrhosis and hepatocellular carcinoma (1–3). HCV is an enveloped, positive-sense RNA virus of ∼9.6 kb that encodes a single open reading frame (4), flanked by highly structured 5′ and 3′ untranslated regions (UTRs) (5,6). As a positive-sense RNA virus, the HCV genome itself is used as a template for translation, replication, and packaging (6,7). Therefore, the viral RNA must be a dynamic structure able to readily accommodate unwinding, elongation, and exposure of different regions of the RNA to various cellular and viral proteins in order to mediate the different stages of the viral life cycle. The 5′ and 3′ UTRs contain cis-acting RNA elements that play key roles in the viral life cycle, including an internal ribosomal entry site (IRES) that drives translation of the viral polyprotein located in the 5′ UTR. Specifically, stem loops (SL) II-IV make up the viral IRES required for translation and stem–loop structures and sequences in both the 5′ and 3′ UTRs have been shown to be required for viral replication (6–11). In addition, the 5′ terminus of the viral genome interacts with an abundant, liver-specific human microRNA (miRNA), called miR-122 (12–14).
miR-122 is highly expressed in the liver where it constitutes ∼70% of liver miRNAs, with ∼66 000 copies/hepatocyte (13,15). Although miRNAs typically interact with the 3′ UTR of target mRNAs to downregulate gene expression, miR-122 binds to two sites in the 5′ UTR of the HCV genome and this interaction promotes HCV RNA accumulation (Figure 1A) (13,14,16,17). While sequestration of miR-122 using antisense inhibitors results in impairment of HCV RNA accumulation in both cell culture and the livers of HCV-infected patients, the precise mechanism(s) of miR-122-mediated viral RNA accumulation remain incompletely understood (13,18). Recent studies have suggested two major mechanisms for miR-122′s promotion of viral RNA accumulation: (i) protection of the 5′ triphosphate moiety of the HCV genome from recognition by 5′ pyrophosphatases and subsequent decay by 5′-3′ exoribonucleases (19–22); and (ii) an RNA chaperone-like mechanism whereby miR-122 binding alters the structure of the viral RNA to promote functional IRES formation (23,24).
Figure 1.

miR-122 binds to the HCV 5′ terminus with a 2:1 stoichiometry. (A) Model of the interaction of miR-122 (purple) and nts 1–42 of the HCV genome (black). (B) Non-denaturing gel electrophoretic mobility shift assay (EMSA) of 1–42 nt HCV RNA and increasing amounts of miR-122. (C) Thermogram and resulting binding curve of the titration of 1–42 nt HCV RNA with miR-122. Affinities of each binding site, Kd1 (Site 1) and Kd2 (Site 2), are indicated. All data are representative of at least three independent replicates and Kd values are an average of three measurements (±) error propagated from individual fits.
Although miR-122 is important for HCV RNA accumulation, there is evidence of mutations in the viral 5′ terminus that are less reliant on miR-122 and allow HCV to replicate to low levels in the absence of the miRNA. One of these mutants, G28A, was isolated from patients who underwent miR-122 inhibitor-based therapy after viral rebound and was subsequently demonstrated to allow low levels of viral RNA accumulation in miR-122 knock-out (KO) cells (25,26). Although the mechanism(s) underlying miR-122-independent viral RNA accumulation remain unclear, it is hypothesized that the G28A mutant can take on a conformation that favours viral RNA accumulation.
Several studies have suggested that miR-122 binding to the HCV genome can alter the structure of the HCV 5′ UTR. Two previous in vitro Selective 2′ Hydroxyl Acylation analyzed by Primer Extension (SHAPE) studies using HCV genotype 1b (Con1b) indicate that miR-122 binding alters the secondary structure of the HCV 5′ UTR, specifically at the two miR-122 binding sites, but also in the HCV IRES region (22,27). In agreement with this finding, an Argonaute (Ago) high-throughput sequencing and crosslinking immunoprecipitation (HITS-CLIP) study suggests that Ago interacts with the HCV 5′ UTR at the two miR-122 binding sites, but also with regions in the HCV IRES that do not align to known human miRNAs (17). Furthermore, computational prediction analyses suggested that the HCV 5′ UTR may be able to take on an alternative conformation that could be influenced by miR-122 interactions with the viral genome (28). More recently, two studies have suggested that miR-122 binding to the HCV genome suppresses a more energetically favorable alternative secondary structure in the SLII region of the HCV 5′ UTR (termed SLIIalt), thereby promoting the folding of a functional IRES (SLII-IV) that drives HCV translation (23,24). This is further supported by the observation that mutations in the 5′ UTR are predicted to favor functional IRES formation and are able to accumulate in the absence of miR-122 (23,24). Thus, we sought to further investigate this model using biophysical analyses and provide insight into miR-122:HCV RNA interactions as well as the contribution of Ago in positioning this complex on the HCV 5′ UTR.
Herein, we performed biophysical characterization of miR-122:HCV RNA interactions in vitro using isothermal titration calorimetry (29), electrophoretic mobility shift assays (EMSA), and structural analysis of the wild-type (WT) and G28A HCV 5′ UTR in the presence and absence of miR-122. We also performed in vitro binding studies using hAgo2:miR-122 and we provide a computational prediction of the three-dimensional (3D) structure of the hAgo2:miR-122 complexes at the 5′ terminus of the HCV genome as well as a model of the IRES–40S complex to help interpret the role Ago plays in stabilizing the bent IRES SLII conformation required for activation of HCV translation. Taken together, our results suggests that two hAgo2:miR-122 complexes can simultaneously bind to the HCV 5′ terminus and we provide a more comprehensive biophysical analysis of alterations to the 5′ UTR that occur upon miR-122 and hAgo2:miR-122 binding. Our results suggest that miR-122 plays three roles in the viral life cycle, which include: (i) an RNA chaperone-like mechanism whereby hAgo2:miR-122 recruitment suppresses formation of an alternative secondary structure (SLIIalt) and promotes functional IRES formation; (ii) protection of the 5′ terminus from pyrophosphatase activity and subsequent exoribonuclease-mediated decay; and (iii) direct promotion of HCV translation through contacts between hAgo2 at Site 2 and SLII of the HCV IRES.
MATERIALS AND METHODS
Viral RNA and microRNAs
A plasmid containing the full-length HCV genotype 2a, JFH-1 isolate with three cell culture-adapted mutations that increase overall viral replication (JFH-1T) was provided by Rodney Russell, Memorial University (30). A DNA template of 419 nucleotides (nts) of the 5′ UTR of HCV was prepared using PCR amplification. The 419 nt PCR product was gel extracted from a 1% agarose gel, which was then used for in vitro transcription with the T7 RiboMAX Express kit (Promega) according the manufacturer's instructions. Full-length in vitro transcribed RNA was gel purified on a 6% polyacrylamide gel (29:1 acrylamide:bis-acrylamide, 1X TBE) and the RNA was eluted in 400 μl elution buffer (500 mM NH4OAc, 10 mM EDTA and 0.1% sodium dodecylsulphate) overnight at room temperature (RT) with gentle agitation. The purified RNA was precipitated as previously described and stored at −20°C until use. The same procedure was followed for 1–371 nt, 1–117 nt of the 5′ UTR (WT and G28A) as well as 1–117 nt containing point mutations in positions 5 and 6 at Site 2 (A38U and C39G).
Wild-type (WT) HCV 1–42 (5′-ACC UGC CCC UAA UAG GGG CGA CAC UCC GCC AUG AAU CAC UCC-3′), HCV Site 2p3,4 1–42 (5′- ACC UGC CCC UAA UAG GGG CGA CAC UCC GCC AUG AAU CAC AGC -3′), and the individual sites, Site 1 (5′-ACC UGC CCC UAA UAG GGG CGA CAC UCC-3′) and Site 2 (5′-GCC AUG AAU CAC UCC-3′) as well as single-stranded guide strand miR-122 (5′-UGG AGU GUG ACA AUG GUG UUU GU-3′) and miR-124 (5′-UAA GGC ACG CGG UGA AUG CC-3′), were synthesized by IDT. Viral RNAs were gel purified as described above.
Isothermal titration calorimetry (ITC)
HCV RNAs were resuspended in ITC buffer (100 mM HEPES pH 7.5, 100 mM KCl, 5 mM MgCl2, filter-sterilized). Five-hundred microliters of 5 μM HCV RNA were heated to 65°C for 10 min and then snap-cooled on ice for 10 min then centrifuged for 1 min at 13 000 rpm for ITC analyses. The temperature was set at 37°C and the reference power was 3 μcal/s. A total of 20 injections of 2 μl were performed with 180 s spacing and 4 s per injection of a 100 μM stock of single-stranded guide strand miR-122 prepared in ITC buffer (22). ITC was carried out on the ITC200 and data analysis using the Origin 7 software (MicroCal), where the first injection was removed. The same procedure was performed for the individual sites (Site 1 and Site 2). The data was fitted to a two sequential binding model, except for the individual Sites 1 and 2, where the data was fitted to a one-site binding model.
Electrophoretic mobility shift assay (EMSA)
2.8 μM stocks of 1–42 nt WT and Site2p3,4 HCV RNA were prepared by diluting in RNase-free water. The RNA was re-folded by incubating at 65°C for 5 min and then incubated at 37°C for 1 h. Dilutions of miR-122 or miR-124 (control) were prepared from serial dilutions from a 4:1 ratio of miRNA: HCV RNA in folding buffer (100 mM HEPES (pH 7.5), 100 mM KCl and 5 mM MgCl2). The different dilutions of miR-122 were added to 2.8 μM of HCV RNA and incubated at 37°C for 30 min (total volume of 7 μl). An equal volume of loading dye was added (30% glycerol, 0.5× TBE, 6 mM MgCl2, 3 μl bromophenol blue and 3 μl xylene cyanol). Five microliters of the sample were separated on a 15% non-denaturing polyacrylamide gel (29:1 acrylamide:bis-acrylamide, 0.5× TBE, 6 mM MgCl2) for 2 h at 8 W at 4°C. The band shifts were visualized with SYBR Gold (Invitrogen) staining (22).
RNA structure prediction
RNA structure predictions were carried out using the RNA prediction software RNAstructure available from the Matthews lab at https://rna.urmc.rochester.edu/index.html (31). Briefly, the RNA sequence was loaded into the RNA structure software using the ‘Fold RNA Single Strand’ command (31). The results were saved as dot bracket files, which were used to generate the predicted structures in VARNA (32).
Synthesis of NAI-N3
Synthesis of NAI-N3 has been previously described (33). Briefly, methyl 2-methylnicotinate was dissolved in anhydrous dichloromethane, followed by the addition of trichloroisocyanuric acid with overnight stirring at RT. Anhydrous N,N-dimethylformamide and sodium azide were added to the reaction. The product of this reaction (methyl 2-(azidomethyl)nicotinate) was stirred in a solution of 1:1 methanol and aqueous sodium hydroxide (NaOH). Water was added and the solution was acidified to pH 4 by the addition of HCl and extracted by ethyl acetate. This became the acid precursor of NAI-N3, 2-(azidomethyl)nicotinic acid. Fresh carboyldiimidazole (CDI) and anhydrous DMSO were stirred until homogenous. 2-(azidomethyl)nicotinic acid was also mixed with anhydrous DMSO until homogenous. CDI with DMSO were then added to the acid precursor in a drop-wise manner. The reaction proceeded for 1 h at RT while stirring. This resulted in a 2 M NAI-N3 stock that was stored at −80°C.
In vitro SHAPE analysis
Five picomoles of the HCV 5′ UTR RNA were re-folded and incubated with 10 μM of single-stranded, guide strand miR-122 or miR-124 (control) for 30 min at 37°C in SHAPE buffer (333 mM HEPES, pH 8.0, 20 mM MgCl2, 333 mM NaCl), followed by treatment with 0.1 M (gel electrophoresis) or 0.01 M (capillary electrophoresis) NAI-N3 or DMSO (control) at 37°C for 5 min. Labeled RNA was then extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. RNA was precipitated and stored at −80°C. The same procedure was followed for G28A. For in vitro SHAPE with hAgo2:miR-122 complexes, 25 pmol of hAgo2 loaded with miR-122 was incubated with 5 pmol of the refolded HCV 5′ UTR for 45 min at 37°C followed by treatment with NAI-N3 and DMSO followed by capillary electrophoresis (as described below).
SHAPE analysis by gel electrophoresis
32P-end-labeled oligonucleotides (0.1 pmol/μl) were annealed to 500 fmol of acylated HCV 5′ UTR RNA by incubating at 95°C for 2 min, followed by step-down cool to 4°C for 2 min. Subsequently, 1× First-Strand Buffer, 5 mM DTT, 0.5 mM dNTPs and SUPERaseIn RNase Inhibitor (Life Technologies) were added to the reaction. One millimolar ddCTP and ddTTP were used in sequencing ladder reactions. The reactions were incubated at 52°C for 1 min, followed by the addition of SuperScript III Reverse Transcriptase (2 U/μl) followed by incubation at 52°C for 30 min. One microliter of 2 M NaOH was then added to each reaction at 95°C for 5 min and reactions were stopped by addition of 2 μl formamide loading dye (95% formamide, 0.01M EDTA, bromophenol blue and xylene cyanol). The cDNA extensions were loaded and run out on a 12% polyacrylamide gel (29:1 acrylamide:bis-acrylamide, 1× TBE). Gels were dried at 80°C for 3 h in a gel dryer (Bio Rad, Model 583) and cDNA fragments were visualized by phosphoimager (Personal Molecular Imager, BioRad) and analyzed using SAFA software (34,35). A normalized reactivity of 1.0 was defined as the average intensity of the top 10% most reactive peaks, where the most reactive 2% of all intensities were removed from the pool. The intensities of the next 8% most reactive peaks are averaged and all reactivities are divided by this average value, as previously described (36,37).
SHAPE analysis by capillary electrophoresis
For SHAPE analysis by capillary electrophoresis, 1 pmol of 6-FAM-labeled oligonucleotide (5′-6-FAM-CGC CCG GGA ACT TAA CGT CTT-3′) was annealed to 5 pmol of modified HCV RNA. The same primer extension buffers as for gel electrophoresis were used, with the addition of 2 μl DMSO per reaction sample. One picomole of NED-labeled olignonucleotide (5′ NED-CGC CCG GGA ACT TAA CGT CTT-3′) was used for sequencing ladders with either 0.5 mM ddGTP or 1 mM ddCTP. Capillary electrophoresis was performed at Plate-forme d’Analyses Génomiques de l’Université Laval on an ABI 3100 Genetic Analyzer. Raw fluorescence data was analyzed using QuSHAPE software (38). A normalized reactivity of 1.0 was defined as the average intensity of the top 10% most reactive peaks, where the most reactive 2% of all intensities were removed from the pool. The intensities of the next 8% most reactive peaks were averaged and all reactivities are divided by this average value, as previously described (39). The Wilcoxon rank test was done between the SHAPE data of HCV RNA with hAgo2:miR-122 and that of HCV RNA alone to determine if there is a statistical difference between the SHAPE reactivities, as previously described (40). The Wilcoxon test determined there are highly significant differences in SHAPE reactivity between both groups (p<0.0001). The baseline value for ΔSHAPE (miR-122 reactivity – miR-124 reactivity or hAgo2:miR-122 reactivity – HCV RNA reactivity) was determined by finding the average of the absolute ΔSHAPE values. ΔSHAPE values above baseline were considered significant increases and below negative baseline value were considered significant decreases in SHAPE reactivity.
Purification of recombinant human Ago2 loaded with miR-122
Human Ago2 was expressed using a baculovirus system and purified with homogeneously loaded miR-122 according to a previously published protocol (41). Briefly, Sf9 cells were infected with baculovirus encoding His-tagged hAgo2. Cells were then lysed and hAgo2 was purified by Ni-NTA affinity chromatography. The His-tag was removed using Tobacco etch virus (TEV) protease and hAgo2 was loaded with synthetic miR-122 bearing a 5′-phosphate (IDT). Homogeneously loaded hAgo2:miR-122 was captured using an antisense oligonucleotide (IDT), eluted, and purified by size exclusion chromatography using an ÄKTA FPLC (GE Healthcare Life Science). Protein concentrations in purified Ago2:miR-122 complex samples were determined by absorption at 280 using an extinction coefficient of 198 370 M−1 cm−1 and by Bradford assay using BSA as standard.
Electrophoretic mobility shift assays with hAgo2:miR-122
Binding reactions were prepared in reaction buffer (30 mM Tris pH 8.0, 100 mM KOAc, 2 mM Mg(OAc)2, 0.5 mM TCEP, 0.005% NP-40) with a final volume of 20 μl, and a final concentration of the labeled RNAs of 0.1 nM, and of hAgo2:miR-122 ranging from 0 to 5 nM. Reactions were incubated for 45 min at room temperature and then run on a 15% acrylamide native gel in 0.5× TBE (45 mM Tris-borate, 1 mM EDTA, pH 8.3). The gels were used to expose a phosphorscreen overnight, which was imaged on a Typhoon scanner (GE Healthcare Life Sciences).
To calculate the Kd, equilibrium binding data of triplicate electrophoretic mobility shift assays were fit to following equation:
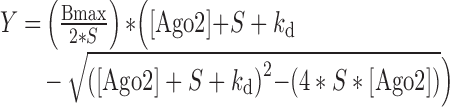 |
where Y is the fraction of target RNA bound, Bmax is the calculated value of maximum binding, [AGO2] is the total concentration of the AGO2:miR-122 complex, S is the concentration of RNA (0.1 nM) and Kd is the calculated dissociation constant, obtained with Prism (GraphPad Software). For the wildtype data, Bmax was normalized to 1: the total bound sites at each concentration were calculated as (2*[double shift intensity] + [single shift intensity]), and the free sites were calculated as ([single shift intensity] + 2*[unbound RNA intensity]). These numbers were then used to calculate the total signal and the bound fraction.
Computational modeling
We modeled Ago2:miR-122:HCV RNA ternary complexes, using experimentally determined secondary RNA-target duplexes and solved human Ago structures (42,43). Our model building is based on an algorithm we developed previously to construct C. elegans and human RISC structures (44). To construct RISC structures, we generated up to 1000 3D duplex conformations for each miRNA-target duplex using the MC-Sym algorithm (45); sampled 1000 Ago conformations using an elastic network model theory; docked each duplex conformation to each Ago conformation using a solved T. thermophilus RISC structure (PDB code: 3HJF); and screened for favorable RISC structures with minimal steric clashes, followed by structure refinement using energy minimization. Throughout, we used human Ago2 (4OLA) to model RISC structures. Complexes of hAgo2 bound simultaneously to adjacent recognition sites were assembled by joining two separate RISC structures, as described previously (46). We defined a 3-base linker (CGC) in HCV RNA between the miR-122 binding sites. From experimentally solved RNA structures, we obtained 1542 CGC fragments (downloaded from RNA FRABASE 2.0). These fragments sampled many relative orientations between adjacent RISC structures. Viable RISC–RISC complexes with minimal steric clashes (<100 atomic overlaps) were then refined using energy minimization.
A similar strategy was used to the assemble RISC–RISC–IRES–40S complexes. Using the cryo-EM structure of human 40S-IRES (5A2Q) and a modeled RISC–RISC complex, we searched for complexes free of steric clashes by sampling ∼2000 CC fragments (HCV RNA nucleotides 42–43) linking the duplexes in IRES SLIIa and adjacent Site 2 RISC. Since the IRES in 5A2Q structure contains nucleotides 40–42 (5′CUC) that are known to form base pairs with the seed region of miR-122 at Site 2, we removed these three nucleotides in the solved structure. Given the large size of the 40S ribosomal subunit, we simplified computational assessment of steric clashes by considering only components of IRES–40S in the vicinity of the RISC–RISC complex, which include IRES and a subset of ribosomal proteins.
RESULTS
Two miR-122 molecules can bind to the HCV 5′ terminus simultaneously with different binding affinities
The HCV genome has two miR-122 binding sites in its 5′ UTR, which are both required for optimal viral RNA accumulation (Figure 1A) (14). It was previously reported that two molecules of miR-122 can bind to the Con1b genotype of HCV RNA and alter the secondary structure of the viral terminus (22,27). As such, we first sought to confirm the stoichiometry of miR-122:HCV RNA interactions with the JFH-1 isolate (genotype 2a). To investigate miR-122 binding to the wild-type (WT) HCV 5′ UTR, we performed EMSA using nts 1–42 of the HCV genome with increasing concentrations of miR-122 (Figure 1B). With miR-122 addition, at low molar ratios, the HCV RNA began to show retarded mobility (Figure 1B), while at higher concentrations, the viral RNA was further retarded and began to become saturated at a 2:1 molar ratio of miR-122:HCV RNA, indicating that both miR-122 binding sites are able to bind simultaneously to the HCV genome (Figure 1B). Importantly, EMSAs carried out with an unrelated miRNA, miR-124, did not cause retardation of the viral RNA (Supplementary Figure S1A). This suggests that miR-122 can bind to the 5′ UTR of the HCV JFH-1 isolate RNA with a 2:1 stoichiometry.
To further investigate the miR-122:HCV RNA interactions, we used isothermal titration calorimetry (29) to measure the affinity of miR-122 for the viral RNA (Figure 1C). In agreement with our EMSA results, we found that the ITC binding curve was best fit with a two-site binding model, with equilibrium dissociation constants (Kds) of Kd1 = 20.45 ± 11.8 nM and Kd2 = 186.08 ± 57.76 nM (Figure 1C and Table 1). We observed no binding of miR-124 to the HCV RNA by ITC analysis (Supplementary Figure S1B). These results suggest that the two miR-122 binding sites have different binding affinities; and although they have different dissociation constants, the interactions were similar in enthalpy and entropy (Table 1). Both interactions were highly exothermic (ΔH1 = –73.28 kcal/mol and ΔH2 = –57.67 kcal/mol) with entropy changes of ΔS1 = –200.67 cal/mol/deg and ΔS2 = –155.33 cal/mol/deg and their Gibbs free energy values were calculated to be ΔG1 = –11.04 kcal/mol and ΔG2 = –9.49 kcal/mol. Thus, based on the EMSA and ITC results, two miR-122 molecules are able to interact with the HCV RNA simultaneously in vitro, albeit with differing binding affinities (Table 1).
Table 1.
ITC analysis of miR-122 binding to HCV RNAs
| K d (nM) | ||||
|---|---|---|---|---|
| HCV RNA (nt) | ΔH (kcal/mol) | ΔS (cal/mol/deg) | Site 1 | Site 2 |
| HCV (1-42) | −73.28 ± 2.50 | −200.67 | 20.45 ± 11.8 | |
| −57.67 ± 3.53 | −155.33 | 186.08 ± 57.76 | ||
| HCV S2p3,4 (1-42) | −75.70 ± 1.04 | −209.00 | 25.80 ± 9.40 | |
| −20.30 ± 1.94 | −38.53 | 1315.24 ± 416.06 | ||
| Site 1 (1-27) | −55.53 ± 0.53 | −143.67 | 18.80 ± 3 | |
| Site 2 (29-42) | −128.23 ± 6.54 | −372.67 | 228.34 ± 65.41 | |
| HCV WT (1-117) | −101.09 ± 2.51 | −294.67 | 132.55 ± 35.43 | |
| −20.82 ± 4.67 | −44.83 | 13296.58 ± 7518.62 | ||
| HCV S2p5,6 (1-117) | −58.87 ± 2.26 | −160 | 319.3 ± 86.01 | |
| −14.33 ± 6.06 | −23.93 | 13596.11 ± 5024.61 | ||
| G28A (1-117) | −102.18 ± 2.36 | −294.33 | 21.24 ± 8.08 | |
| −39.31 ± 5.94 | −101.17 | 2590.99 ± 863.68 | ||
| HCV WT (1-371) | −104.36 ± 11.32 | −308 | 514.27 ± 229.88 | |
| −41.85 ± 14.72 | −108.2 | 1426.17 ± 517.93 | ||
aValues reported are an average of three measurements (±) error propagated from individual fits.
To confirm whether Kd1 represents binding of miR-122 to Site 1 or Site 2, we performed EMSA and ITC analyses on an HCV RNA that contained mutations in positions 3 and 4 of the seed sequence of Site 2 (S2p3,4) (Supplementary Figure S1C-E). The results show that the first binding site had an affinity of 25.80 ± 9.40 nM, similar to Site 1 of WT HCV (Figure 1C and Table 1), while the binding affinity of the second site was 1315.24 ± 416.06 nM, as a result of the mutations in the seed sequence of Site 2 (Supplementary Figure S1E). This suggests that when nts 1–42 of the HCV genome are present, Site 1 is bound first in vitro. Additionally, the enthalpy, entropy and Gibbs free energy of the first binding site of HCV S2p3,4 had very similar to the first binding site of the WT HCV RNA (1–42 nt) (Table 1). However, the second binding site had a higher enthalpy and entropy (ΔH = –20.30 ± 1.94 kcal/mol and ΔS = –38.53 cal/mol/deg, respectively) with ΔG = –8.35 kcal/mol, suggesting that this reaction is not as spontaneous at 37°C compared to the first binding site of WT HCV (Table 1). These results were confirmed qualitatively by performing EMSA on HCV S2p3,4 where the first binding event occurred quickly, while the second binding event did not occur until a 4:1 ratio of miR-122 to HCV S2p3,4 was reached (Supplementary Figure S1D). These results suggest that when only the two sites are present (1–42 nt HCV RNA), miR-122 binds to Site 1 with a higher affinity in vitro.
To investigate the specific binding sites further, we tested miR-122 binding to each individual site by ITC analysis using Site 1 (nt 1–27) or Site 2 (nt 29–42) RNAs. We found that miR-122 bound to Site 1 (Kd = 18.80 ± 3 nM) with a stronger affinity than to Site 2 (Kd = 228.34 ± 65.41 nM) (Table 1 and Supplementary Figure S2). More specifically, we found that miR-122 binding to Site 1 was exothermic, with ΔH = –55.53 kcal and an entropy of ΔS = –143.67 cal/mol/deg (Table 1 and Supplementary Figure S2A). These results are in good agreement with computational binding predictions, where miR-122 interactions with Site 1 are predicted to have ΔH = –69.5 kcal/mol and ΔS = –192.2 cal/mol/deg. Additionally, we found that Site 2 had a lower affinity for miR-122 with a Kd = 228.34 ± 65.41 nM; however, miR-122 binding was more exothermic, with ΔH = –128.23 kcal/mol and an entropy of ΔS = –372.67 cal/mol/deg, again in good agreement with computational binding predictions (ΔH = –109 kcal/mol and ΔS = –306.6 cal/mol/deg) (Table 1 and Supplementary Figure S2B). This suggests that the individual miR-122 binding sites, have a different rearrangement and organization compared to the 1–42 nt HCV RNA based on enthalpy and entropy. However, the Kd of the individual sites are similar to those values obtained in the context of the 1–42 nt HCV RNA. Thus, at least in vitro, miR-122 has a higher affinity for Site 1 than for Site 2 on the 1–42 nt HCV RNA.
miR-122 binding alters the secondary structure of SLII of the HCV 5′ UTR
To investigate structural alterations mediated by miR-122 binding to the HCV 5′ UTR, we performed in vitro SHAPE analysis on the full HCV 5′ UTR (nts 1–371) in the presence and absence of miR-122 using both capillary (Figure 2 and Supplementary Figure S3) and gel electrophoresis (Supplementary Figure S4). As recent studies and computational predictions suggest that the WT HCV 5′ UTR favours a non-canonical alternative SLII structure (SLIIalt) in the absence of miR-122 (23,24), we used SHAPE analysis to map the reactivity of the first 117 nts (including SLI and II) of the HCV RNA in the presence or absence of miR-122 (Figure 2).
Figure 2.

miR-122 binding suppresses an alternative structure (SLIIalt) and promotes functional folding of SLII. (A) Normalized SHAPE reactivities of HCV RNA (nts 1–117) with no miRNA (top) or in the presence of miR-122 (bottom). Nucleotides 1–5 and 14–19 were omitted due to high background reactivity. Nucleotides with high (≥0.85, red), intermediate (between 0.4 and 0.85, orange), low (between 0.2 and 0.4, blue) and very low (≤0.2, black) SHAPE reactivity are indicated. (B) Predictions of the lowest free energy structures formed by the first 117 nts of HCV 5′ UTR and their predicted free energies (ΔG, kcal/mol). (C) SHAPE data from (A) superimposed onto two of the predicted structures of HCV RNA alone or (D) HCV RNA + miR-122, where the predicted structure and free energy values are indicated. The SHAPE data represents the results of six independent replicates and error bars indicate the standard error of the mean (SEM).
Reactivity information was obtained for almost every nucleotide in the RNA; however, we were not able to discern the SHAPE reactivity at nts 1–5 due to the high reactivity at the 5′ end of the transcript and nts 14–19 due to the high background observed in this region, in line with previous studies (22,27). In the absence of miR-122, the loop of SLI (11–13 nts) and Site 2 (30–39 nts) were found to be relatively unstructured/flexible, as reported previously (Figure 2A and Supplementary Figures S3 and S4) (22,24,27). However, Site 1 (consisting of nts 20–27) had a relatively low SHAPE reactivity, suggesting that this region is constrained in the context of the 5′ UTR (Figure 2A).
When SHAPE was performed on the HCV 5′ UTR in the presence of miR-122, we observed a reproducible reduction in SHAPE reactivity, particularly in the seed region of Site 2, consistent with previous models of miR-122:HCV RNA interactions (Figure 2A and Supplementary Figure S3) (22,24,27). We also observed a reproducible decrease in the SHAPE reactivity in the stem region of SLI, suggesting that miR-122 binding may further stabilize this stem–loop structure (Figure 2A and Supplementary Figure S3). In addition, there was a reduction in SHAPE reactivity at positions A33, G34, and U36 on the HCV RNA (14,22). This is in agreement with previous studies and suggests that miR-122 binding may increase rigidity in this region and that the U at position 36 may be able to engage in a wobble base pair with the G at position 9 on Site 2-bound miR-122 (22,24,27). Moreover, we observed a lower SHAPE reactivity in SLIIa (nts 52–59) in the presence of miR-122 when compared to the HCV RNA alone (Figure 2A).
Structural predictions of nts 1–117 of the HCV genome with and without miR-122 suggest that the most energetically favourable structure(s) of the WT HCV RNA in the absence of miR-122 is an alternative non-canonical SLII structure from nts 20–104, previously termed SLIIalt (Figure 2B) (23,24). The functional SLII structure is only predicted to be the fifth most energetically favourable structure, with a ΔG of –37.8 kcal/mol, compared to the free energies of the SLIIalt structures, which range from –40.8 to –38.2 kcal/mol (Figure 2B). When we analyzed our SHAPE data with respect to the non-canonical SLIIalt and SLII structures, we found that the reactivity agrees well with both of these predicted structures (Figure 2C). Specifically, there are high overall SHAPE reactivities in nts 30–39, 53–59 and 80–85 which are all either single-stranded or in bulge regions in both structures (Figure 2C). However, nts 20–29 had relatively low SHAPE reactivities in the absence of miR-122, which is more consistent with the SLIIalt structure, since these nucleotides are base-paired versus single-stranded in the functional SLII structure (Figure 2C). Conversely, SHAPE analysis in the presence of miR-122 is consistent with formation of the canonical SLII structure, predicted to be the most energetically favorable structure in the presence of miR-122 (ΔG = –66.8 kcal/mol, Figure 2A and D). Thus, although our data provides an averaged view of the equilibrium of several of the most energetically favorable structures, the quantitative SHAPE results suggest that in the absence of miR-122, the SLIIalt is favoured, while interactions with miR-122 promote functional SLII formation, which would support HCV IRES-mediated translation (24,47–49).
To further validate these findings, we predicted that if SLIIalt is the preferred conformation of the viral RNA, then in the context of nts 1–117, Site 2 should have a greater miR-122 binding affinity than Site 1, as Site 2 is in a more open (single-stranded) conformation, while Site 1 is in a closed (double-stranded) conformation (Figure 2C). Thus, we performed ITC analyses to measure the binding affinity of miR-122 for the 1–117 nt and 1–371 nt HCV RNA. Both binding sites had weaker affinities when compared to the 1–42 nt HCV RNA, with Kd1 (Site 2) = 132.55 ± 35.43 nM (ΔG = –79.2 kcal/mol) and Kd2 (Site 1) = 13296.58 ± 7518.62 nM (ΔG = –17.5 kcal/mol) for 1–117 nt HCV RNA, and Kd1 (Site 2) = 514.27 ± 229.88 nM (ΔG = –81.53 kcal/mol) and Kd2 (Site 1) = 1426.17 ± 517.93 nM (ΔG = –33.83 kcal/mol) for 1–371 nt HCV RNA, respectively (Table 1 and Supplementary Figure S5). This is not unexpected since the longer RNAs (1–117 and 1–371 nt) are likely able to sample more conformations than the 1–42 nt RNA. In order to discern whether Kd1 represents binding of miR-122 to Site 1 or Site 2 on the 1–117 nt RNA, we performed ITC analyses on a Site 2 mutant (S2p5,6), whereby positions 5 and 6 of the Site 2 seed sequence were mutated to their respective Watson-Crick base (Table 1). These mutations are predicted to reduce binding of Site 2, without significantly altering the predicted conformation or miR-122 interactions with Site 1. As we predicted, a reduction in binding affinity was only observed for Kd1 (Site 2) = 319.3 ± 86.01 nM (ΔG = –47 kcal/mol), while Kd2 (Site 1) = 13596.11 ± 5024.61 nM (ΔG = –12.6 kcal/mol) remained unchanged (Table 1 and Supplementary Figure S5). Thus, in contrast to what we observed with nts 1–42, in the context of nts 1–117, Site 2 has a higher affinity, while Site 1 has a significantly reduced affinity for miR-122. These results are in agreement with formation of the energetically more favorable SLIIalt, where Site 2 is predicted to be in an open and accessible conformation, while Site 1 is predicted to be in a closed conformation.
The G28A mutation favours formation of SLII even in the absence of miR-122
The G28A mutation was isolated from chronic HCV patients that had undergone miR-122 inhibitor-based therapy after viral rebound and was subsequently shown to be able to accumulate to low levels in miR-122 knockout (KO) cells (25,50). RNA structural predictions suggest that in contrast to WT HCV RNA, the G28A mutation is predicted to adopt only two main conformations with very similar free energies, SLIIalt (ΔG = –37.9 kcal/mol) and SLII (ΔG = –37.8 kcal/mol), even in the absence of miR-122 (Figure 3). SHAPE analysis of G28A suggests that the seed region of Site 1 (nts 20–27) has an overall higher SHAPE reactivity when compared with WT HCV RNA (Figure 2A versus 3A). Importantly, this region represents the most significant difference between the SLIIalt and SLII structures in that it is predicted to be base-paired in the former and single-stranded in the latter (Figure 3B). When we superimposed the SHAPE data onto the two G28A predictions, the data is more consistent with formation of SLII, where nts in the Site 1 seed region (nts 20–27), predicted to be single-stranded in this conformation, have a high overall SHAPE reactivity (Figure 3). We also observed high overall SHAPE reactivities in the loop regions of SLII (nts 52–59 and 80–85), but these are largely consistent with both structural conformations (Figure 3). With respect to the remaining IRES structure (i.e. SLIII-IV), the G28A mutant demonstrated similar SHAPE reactivity to the WT HCV RNA (Supplementary Figure S6). To further confirm these findings, we performed ITC analysis on the 1–117 nt G28A mutant (Table 1 and Supplementary Figure S6). In agreement with our SHAPE data, G28A had higher affinities for miR-122 than WT, with Kd1 (Site 1) = 21.24 ± 8.08 nM (ΔG = –80.36 kcal/mol) and Kd2 (Site 2) = 2590.99 ± 863.68 nM (ΔG = –31.81 kcal/mol) (Table 1). Thus, RNA structural predictions, SHAPE, and ITC are consistent with the G28A mutation favoring formation of SLII even in the absence of miR-122, while the WT HCV RNA is more likely to adopt an alternative conformation(s) (i.e. SLIIalt, Figure 2B–C) in the absence of miR-122.
Figure 3.

The G28A mutation favours SLII formation in the absence of miR-122. (A) Normalized SHAPE reactivities of G28A (nts 1–117). Nucleotides 1–5 and 14–19 were omitted due to high background reactivity. Nucleotides with high (≥0.85, red), intermediate (between 0.4 and 0.85, orange), low (between 0.2 and 0.4, blue) and very low (≤0.2, black) SHAPE reactivity are indicated. (B) SHAPE data from (A) superimposed on the predicted lowest free energy structures formed by the first 117 nts of the G28A 5′ UTR (SLIIalt and SLII) and their free energy values (ΔG, kcal/mol). The SHAPE data represents the results of four independent replicates and error bars indicate the standard error of the mean (SEM).
Two hAgo2:miR-122 complexes can bind to the HCV RNA 5′ UTR simultaneously
It is widely accepted that miR-122 interacts with the HCV genome, at least initially, in the context of a hAgo protein (14,51–54). Previous studies suggest that Ago proteins are required for miR-122 duplex unwinding, miRNA-induced silencing complex (miRISC)-loading, and recognition of the HCV RNA binding sites; however, it is unclear whether Ago remains bound to the viral RNA to participate in events leading to promotion of HCV RNA accumulation (14,51–55). Due to the close proximity of the miR-122 binding sites on the HCV genome (Figure 1A), we sought to test whether two hAgo2:miR-122 complexes are physically able to occupy the viral RNA simultaneously. To this end, we prepared purified samples of hAgo2:miR-122 in complex and performed gel shift assays with the WT (1–42 nt) and S2p3,4 HCV RNAs (Figure 4A). Using radiolabeled HCV S2p3,4 RNA, where the Site 2 sequence is mutated to disrupt pairing to the miR-122 seed region, we observed a single shift in samples containing hAgo2:miR-122, indicating a binding event occurred (Figure 4A, left side). Using WT HCV RNA, two shifts were observed, indicating two binding events occurred on the RNA. Importantly, the fraction of the WT HCV RNA in the supershifted band increased as hAgo2-miR-122 concentrations were increased, demonstrating a dose-dependent response (Figure 4A, right side).
Figure 4.
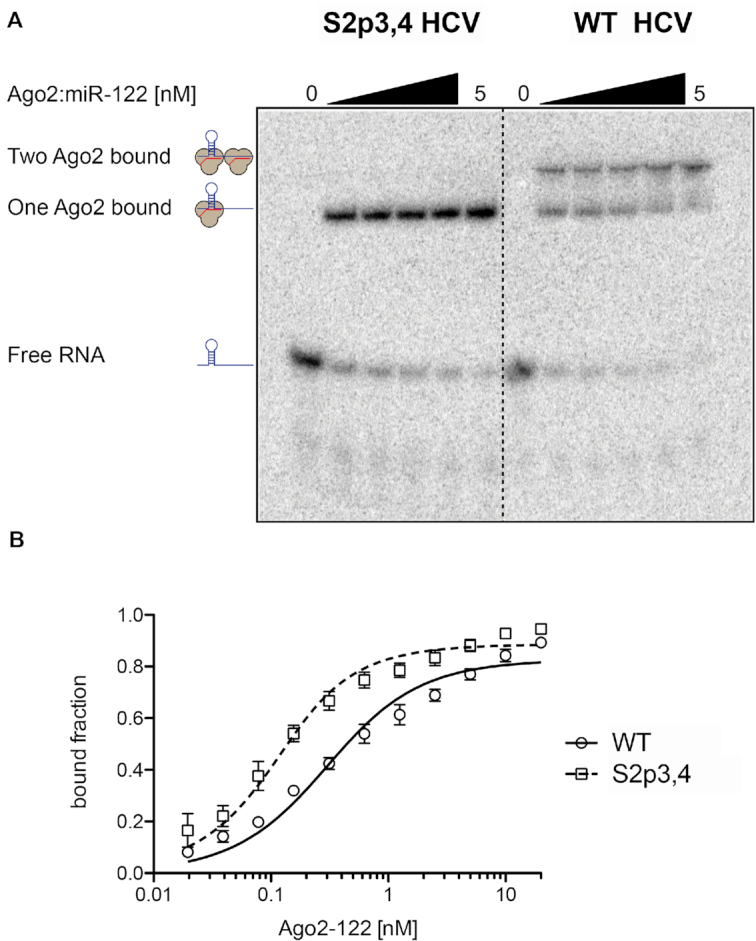
Two Ago2:miR-122 complexes can bind simultaneously to the 5′ terminus of the HCV genome. (A) Recombinantly expressed hAgo2 loaded with miR-122 (final conentration: 0, 0.3125, 0.625, 1.25, 2.5 and 5 nM) was incubated with radiolabeled viral RNA comprising nts 1–42. Incubation with HCV S2p3,4 (left side) resulted in a single shift, whereas incubation with WT HCV RNA (right side) yielded an additional shift corresponding to the complex containing two hAgo2:miR-122 complexes. The depicted gel is one of five independent replicates. (B) Equilibrium binding data for Ago2:miR-122 and HCV WT and S2p3,4, based on gel shift assays (Supplementary Figure S7A). Data points are means of three replicates with standard error. The Kd ± SE for S2p3,4 is 63 ± 10 pM, the Kd apparent for WT HCV is 250 ± 31 pM.
Equilibrium binding data using a broader range of hAgo2 concentrations (Figure 4B and Supplementary Figure S7) revealed a Kd of 63 pM for the complex with HCV S2p3,4, and a Kd apparent of 250 pM for WT HCV. Under the assumption of independent binding, binding to Site 2 can be calculated by subtraction of the S2p3,4 data from the WT data (Supplementary Figure S7B). Fitting the calculated Site 2 data to the binding model yields a Kd = 1.13 nM, while previous experiments place the Kd for such a target in the low pM range for miR-122 (56). This discrepancy suggests that, although Site 1 and Site 2 can be simultaneously occupied by two Ago2:miR-122 complexes, the two binding events are negatively affected by each other in the system. This conclusion is in agreement with the increased SHAPE reactivity measured for the nucleotides spanning the 5′ end of Site 1 and the 3′ end of Site 2 on the HCV genome (Supplementary Figure S3).
hAgo2:miR-122 binding alters the secondary structure of the HCV 5′ UTR
To determine how hAgo2:miR-122 complex binding to the HCV 5′ UTR alters the secondary structure of the viral RNA, we performed in vitro SHAPE analysis in the presence of purified hAgo2:miR-122 (Figure 5 and Supplementary Figures S3 and S8). When focusing on the 5′ terminus of the HCV RNA (nts 1–42), we observed an overall increase in SHAPE reactivity at nts C27, C29, A31 and G33 in the presence of hAgo2:miR-122 (Figure 5). The increase in SHAPE reactivity at C27 was unexpected since this is the first nucleotide of the seed region of miR-122 bound to Site 1; however, the increase in reactivity observed may be due to the close proximity of the two hAgo2:miR-122 complexes which could force this base-pair to become unpaired (particularly since the 5′ end of the miR-122 molecule is anchored in the pocket of the Mid domain). The increase in SHAPE reactivity at C29 was observed with both miR-122 alone (Figure 2) and hAgo2:miR-122 (Figure 5), suggesting an alteration in the structure such that this nucleotide is more flexible, independently of the presence of hAgo2. Like C27, the significant increase in SHAPE reactivity at C31 in the presence of hAgo2:miR-122 suggests that the secondary structure of the viral RNA may be further altered in order to accommodate two hAgo2:miR-122 complexes simultaneously (Figure 5). We also observed a change in reactivity at G33 (in the bulge region of Site 2), which could suggest that it is further exposed in the presence of hAgo2:miR-122 due to lack of base pairing at this position. Similarly, we observed an increase in SHAPE reactivity in the loop region of SLI, which may be due to SLI being forced out of the hAgo2:miR-122 complex at Site 1. Beyond nts 1–42, we also observed changes in the SHAPE reactivity of SLII in the presence of hAgo2:miR-122, particularly in the bulge region of SLIIa (nts 52–58) as well as a few nucleotides on the 3′ arm of SLII opposite SLIIa (104–109), which suggests further stabilization of this region (Figure 5).
Figure 5.
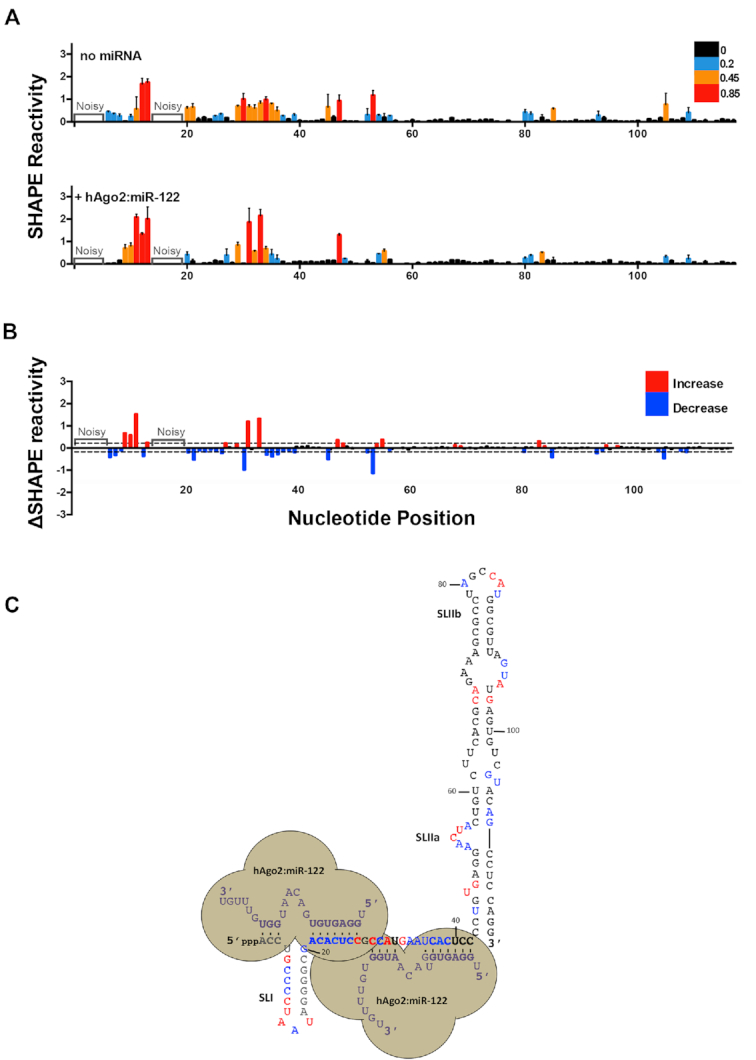
hAgo2:miR-122 complexes alter the secondary structure of the 5′ terminus and SLII of the HCV genome. (A) Normalized SHAPE reactivities of 1–117 nt HCV RNA in the absence (top) or presence of hAgo2:miR-122 (bottom). Nucleotides 1–5 and 14–19 were omitted due to high background reactivity. (B) Difference plot showing changes in SHAPE reactivity upon miR-122 binding, ΔSHAPE = hAgo2:miR-122:HCV RNA reactivity – HCV RNA (no miRNA) reactivity. Significant increases in reactivity (above baseline, red) and decreases in reactivity (below baseline, blue) are indicated. (C) Changes in reactivity upon hAgo2:miR-122 binding are superimposed onto the hAgo2:miR-122:HCV RNA secondary structure model (coloured as in (B)). The data represents the results of four independent replicates and error bars represent the standard error of the mean (SEM).
To determine if hAgo2:miR-122 alters the structure of HCV RNA beyond SLII, we mapped SHAPE reactivities of the entire HCV 5′ UTR (nts 1–371) in the presence of miR-122, hAgo2:miR-122, and controls (Supplementary Figures S3 and S8). In vitro SHAPE analysis of the HCV 5′ UTR alone (nts 1–371) agreed well with previous models of SLIII and IV of the HCV IRES (Supplementary Figure S8) (22,27). Additionally, we found that in the presence of hAgo2:miR-122, a few nts in the apical loops SLIIIa-e displayed a modest but reproducible decrease in overall SHAPE reactivity. Moreover, we observed significant changes in SHAPE reactivity in several nts of SLIV, in close proximity to the pseudoknot and AUG start codon. Taken together, our data suggest that hAgo2:miR-122 binding results in alteration of the structure of the viral RNA at least in vitro, both at the 5′ terminus (where the miR-122 seed and auxiliary binding sites are located), but also in the IRES region (SLII-IV), in a manner which may promote functional IRES activity.
Computational modeling of hAgo2:miR-122 and HCV RNA
As we established that two hAgo2:miR-122 molecules were able to bind to the 5′ terminus of the HCV genome (nts 1–42) simultaneously, we sought to model these interactions using the crystal structure of hAgo2. Of the ∼106 possible complexes at each site from modeled hAgo2 and RNA duplex conformations, we considered an increasing number of constructed complexes until sterically favorable conformations were found. Site 1 required ∼150 000 complexes, whereas Site 2 needed only ∼30 000 structures. The much greater number of considered complexes at Site 1 reflects the wider conformational search needed to fit a double-stranded RNA structure with a three-stem junction into a hAgo2 conformation; in contrast, the RNA structure at Site 2 has an unbranched conformation (see Figure 6 displaying viable complexes at each site).
Figure 6.
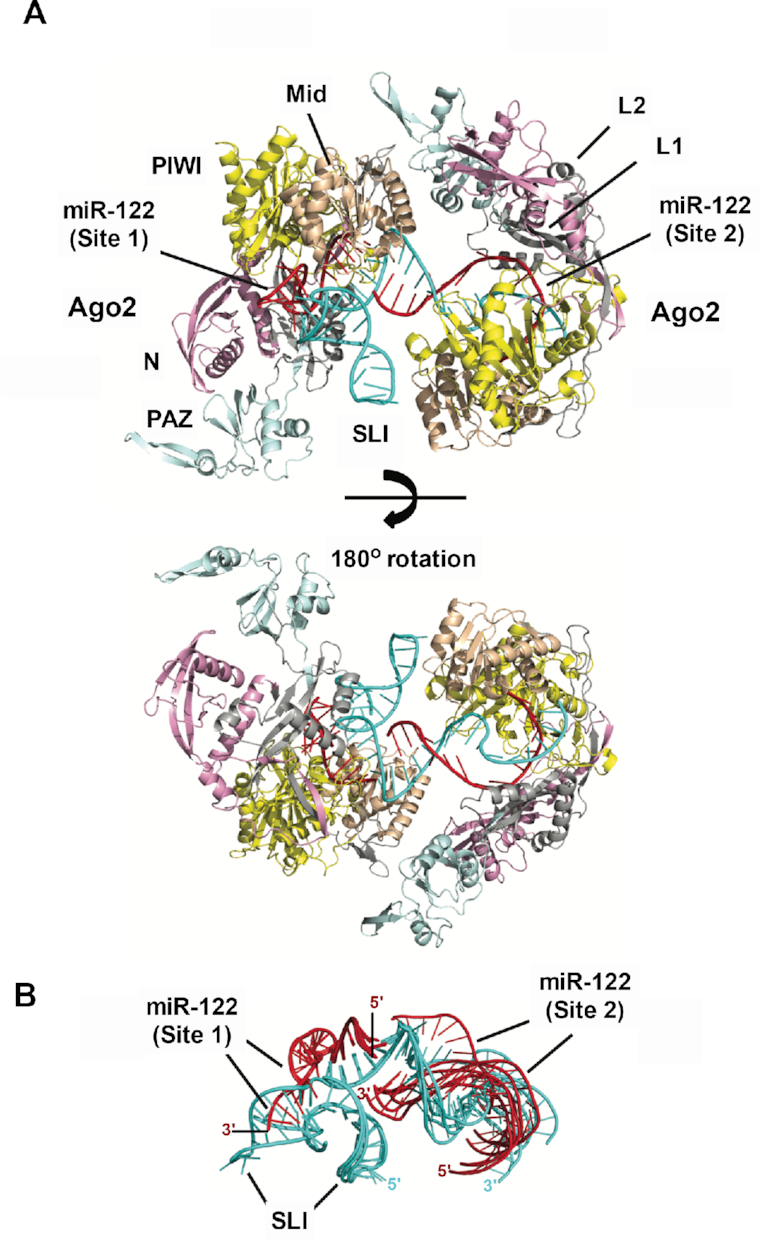
Model of hAgo2:miR-122:HCV RNA interactions at Sites 1 and 2. (A) A modeled RISC–RISC complex at Site 1 and Site 2 on the HCV genome and (B) miR-122:HCV RNA conformations of different complexes. (A) Two views of the RISC–RISC complex are shown: top structure shows the RNA-binding channel of Site 1 with the protruding SLI structure and the RNA bridge between the adjacent hAgo2 structures; bottom structure (180° rotated view) highlights the RNA conformation at Site 2 that enables simultaneous occupation of both hAgo2 binding sites. (B) Superimposed miR-122:HCV RNA conformations from six RISC–RISC complexes show conformational variability. In particular, SLI-3′ stem structure in Site 1 has significant conformational flexibility; while the Site 2 RNA exhibits multiple orientations with respect to Site 1 RNA, indicating that the adjacent hAgo2 structures can adopt distinct conformations. The HCV RNA (cyan), miR-122 (red), and hAgo2 N terminal (pink), L1 (gray), PAZ (cyan), L2 (gray), mid (wheat) and PIWI domain (yellow) are indicated.
Computational modeling produced multiple RNA-induced silencing complex (RISC) structures at Site 1 and Site 2 that are free of steric clashes. Assembly of these structures generated viable RISC–RISC complexes where the 3′ end duplex of miR-122 at Site 2 forms a solvent-exposed bridge between the adjacent RISC structures (Figure 6A). Modeling trials showed that only 3′ duplexes protruding from the RNA-binding channel at Site 2 were capable of assembling into RISC–RISC complexes; straight duplex conformations generated collisions between the Mid and N-terminal domains of adjacent hAgo2 structures. This suggests that Site 2 duplex conformations are distorted in the internal loop region, resulting in the duplex protruding out of the hAgo2 RNA-binding channel (Figure 6A, lower panel). Alternatively, the auxiliary interactions with Site 2 of the HCV genome may become unpaired as the hAgo2:miR-122 at Site 2 adjusts to accommodate Site 1-bound hAgo2:miR-122. This latter hypothesis is further supported by our SHAPE data and the hAgo2:miR-122 gel shifts which suggest higher SHAPE reactivity in this region and a reduction in binding affinities when both Sites are occupied by hAgo2:miR-122, respectively.
Model RISC–RISC complexes exhibit conformational variability that is captured by their RNA conformations (Figure 6B). At Site 1, the SLI structure shows significant conformational flexibility. The Site 2 RNA has multiple orientations that are consistent with the RISC–RISC complex, implying that different Ago-Ago conformations are feasible. The adjacent hAgo2 structures display Mid/N-terminal and PAZ/L2 contacts. However, further computational/experimental analysis will be needed to determine whether a specific Ago–Ago conformation is favored.
To probe the RISC–RISC complex further, we examined the two RISC subunits separately. At Site 1, the shape of the predicted hAgo2 structure determined the allowed RNA conformations (Figure 6B). The dsRNA forms a 3-stem junction with a 7 base pair stem in the seed region (seed stem), a 7 bp stem–loop (SLI), and a 3 bp stem in the 3′ end (3′ stem). The seed stem is in an expected orientation for a recognized mRNA target. All allowed RNA structures show that the SLI-3′ stem forms an L-shaped conformation, which adopts multiple orientations (Figure 6B) with respect to the hAgo2 structure. The source of the L-shaped structure's orientations is the miR-122′s six unpaired bases (GACAAU) at the 3-stem junction. These bases also produce a sharp turn (nearly 90°) at the end of the seed stem to allow the L-shaped structure to protrude out of the RNA-binding channel; thus, avoiding steric clashes with hAgo2′s PIWI and N-terminal domains. At Site 2, the assembled RISC structures show that the RNA can adopt straight and distorted duplex conformations. Duplex distortions occur at the internal loop just beyond the seed stem region, generating nonstandard RNA backbone conformations. The straight duplexes fit within the RNA-binding channel, whereas distorted duplexes protrude out of the RNA-binding channel. As noted above, only the distorted duplex conformations that protrude out of the RNA-binding channel are consistent with the overall RISC–RISC complex, which may lend support to the hypothesis that the auxiliary interactions with miR-122 at Site 2 may become unpaired when both sites are occupied by hAgo2:miR-122.
Computational modeling of RISC–IRES–40S complex
The two RISC complexes form immediately upstream of SLII of the HCV IRES. Cryo-EM structures of IRES–40S show that SLII can adopt an active, bent conformation that maintains the 40S subunit in an open conformation to allow mRNA loading (57,58). An experimental characterization of miR-122-HCV IRES interactions suggests that the RISC Ago proteins play a role in activation of HCV translation (51,59). To help further interpret Ago's role, we used structural modeling to characterize the RISC–IRES–40S interactions.
We assembled the RISC–IRES–40S complex by using the cryo-EM of the IRES–40S (PDB: 5A2Q) and our modeled RISC–RISC structures (see Materials and Methods). We found many viable RISC–IRES–40S complexes with only slight differences in the orientation between the IRES–40S and RISC–RISC structures. The RISC–RISC structure is oriented away from the IRES–40S complex, and it only interacts with the IRES via the hAgo2 at Site 2 (Figure 7). Both single- and double-stranded regions of the IRES interact with Ago surfaces opposite the RNA-binding channel. The IRES-Ago interface is extremely tight fitting with a strong shape complementarity (Figure 7, right panels). Specifically, the Ago Mid and L2 domains interact with the backbone of the single-stranded IRES region (ACCC, nts 118–121) between SLII and III. An unstructured loop in the PIWI domain (residues 719–728, DKNERVGKSG; bold residues conserved among hAgo 1–3) interacts with the bulge nucleotides in SLIIa where SLII adopts a bent conformation. Interestingly, benzimidazole, an inhibitor of HCV IRES-driven translation initiation, binds to the same bulged IRES region to lock SLII in an extended, inactive conformation (PDB 3TZR) (60). Lastly, we found that the SLI structure does not interact with the IRES–40S complex, a result consistent with the experimental finding that deletion of SLI does not influence HCV IRES-mediated translation (59).
Figure 7.

Model of RISC-HCV IRES–40S interactions. A modeled RISC–IRES–40S complex (top left panel) and detailed views of hAgo2:IRES interactions (other panels). Front and back views of the space-filling representation show a tight fit with shape complementarity between hAgo2 and IRES SLIIa (right panels). hAgo2 Mid, L2 and PIWI domains interact with single- and double-stranded regions of the HCV IRES SLIIa (bottom left). The IRES (cyan), ribosomal RNA (yellow), ribosomal proteins (gray), hAgo2s (pink and blue), and miR-122s (orange) are indicated.
DISCUSSION
Two miR-122 molecules are able to bind to the HCV 5′ UTR simultaneously in vitro
Biophysical analysis of miR-122:HCV RNA interactions revealed a 2:1 stoichiometry in agreement with previous studies using Con1b (genotype 1b) (22). In the context of nts 1–42, we found that miR-122 binds to Site 1 with a stronger affinity than to Site 2; and miR-122 binding to Site 1 was observed to be more exothermic with a decreased entropy, when compared with Site 2. This effect could be due to different local conformational changes/rearrangements required for Site 1 to bind with miR-122, when compared with Site 2. Individual analysis of Sites 1 and 2 alone revealed that Site 1 has a higher overall binding affinity than Site 2; however, miR-122 binding to Site 2 was more exothermic and entropically favoured suggesting that in this isolated system, Site 2 binding to miR-122 brings the system to a lower energy ordered state (Table 1). This is in agreement with previous results with a J6/JFH-1 (genotype 2a) isolate (52); but is in contrast to studies with Con 1b (genotype 1b), where miR-122 binding to Site 2 had a stronger binding affinity than Site 1 (22). Although both ITC experiments were done similarly, it is possible that our results vary due to differences in the sequences between these two genotypes as well as the specific lengths used for binding experiments of the individual sites, since the tail of miR-122 bound to Site 2 would be predicted to have extended binding into the Site 1 seed region in the absence of Site 1-bound miR-122 (22). In contrast, when we performed ITC analyses using 1–117 nt HCV RNA, we observed that Site 2 had a much greater affinity than Site 1. This was confirmed using S2p5,6 which contains a mutation in the Site 2 seed region that resulted in a reduction in Site 2 binding, but had no effect on Site 1. This is likely due to the more energetically favorable SLIIalt structure that the 1–117 nt HCV RNA is able to adopt. More specifically, in the SLIIalt conformation, Site 2 is predicted to be in a single-stranded and accessible loop region, while Site 1 is in a closed conformation. Thus, our biophysical analyses suggest that two miR-122 molecules are able to bind simultaneously to the HCV 5′ terminus in vitro, with binding to Site 2 occurring with a higher affinity than Site 1 in the context of nts 1–117 of the HCV genome.
Binding of miR-122 to the HCV genome alters the structure of the 5′ UTR
In vitro SHAPE analysis in the presence of miR-122 revealed decreases in the overall SHAPE reactivity across the predicted miR-122 seed sequences, consistent with miR-122 interactions reducing the flexibility of the viral RNA in this region (22,27). Moreover, we observed an overall decrease in SHAPE reactivity at G33 and A34 of the HCV genome, which are not predicted to bind to miR-122, suggesting that miR-122 binding imparts rigidity to this region of the viral RNA. Furthermore, the decrease in SHAPE reactivity at U36 is consistent with formation of a G:U wobble pair between miR-122 and the viral RNA at this position, allowing us to further refine the model of miR-122:HCV RNA interactions at Site 2 (22,24,27). In addition to the seed regions, we also observed some subtle differences in the SHAPE reactivity in SLI, with an overall decrease in the stem and slight increase in the loop region. These changes in reactivity suggest that miR-122 binding may stabilize SLI. However, changes in reactivity of the loop region are not likely to have biological significance because the sequence identity of the loop is poorly conserved across HCV genotypes (14,61).
In agreement with recent studies that suggest that the HCV 5′ UTR is able to adopt a more energetically favorable alternative (SLIIalt) structure (23,24), our SHAPE analyses support this model, whereby miR-122 binding results in a RNA chaperone-like switch in conformation to SLII, which would promote functional IRES formation by positioning the AUG start codon within the 40S ribosomal subunit for translation initiation (47,48). However, as our SHAPE data is an average of the structures that form in solution, and most of the loop regions in SLIIalt and SLII are overlapping, we could not definitively rule out formation of SLII and the functional IRES conformation in the absence of miR-122 for WT HCV RNA. However, this model is further supported by our ITC analyses which suggest Site 2 has a greater binding affinity in the context of 1–117 nt WT HCV RNA. Thus, our data is consistent with formation of SLIIalt in the absence of miR-122, and miR-122 interactions promote functional folding of SLII at least in vitro.
G28A favours the canonical SLII structure
The G28A mutation was first isolated from patients who had undergone therapy with a miR-122 antagonist and this mutation was subsequently demonstrated to reduce HCV’s reliance on miR-122 in cell culture (13,18). As RNA structural predictions of the G28A mutant suggested that this mutation results in a reduced number of energetically favorable alternative structures and formation of the functional SLII structure with similar free energy to the SLIIalt structure, we sought to verify this using SHAPE analyses and ITC (23). Our SHAPE analysis indeed suggests that SLII is more preferentially formed by G28A when compared to WT HCV RNA, as reflected by higher SHAPE reactivities in the seed sequence of Site 1, which is in a more open (single-stranded) conformation in the SLII structure. Specifically, in several of the lowest free energy structures of WT HCV RNA, the G28 position is found in a G-C Watson-Crick pair and thus the G28A mutation would be predicted to disrupt formation of several of the predicted alternative structures. Moreover, nts 20–24 have a much higher SHAPE reactivity in G28A when compared with WT HCV RNA, suggesting that these nucleotides are in a more open (single-stranded) conformation in the presence of the G28A mutation. This is further supported by our ITC analyses, which suggests a higher affinity for both sites when compared with WT HCV RNA. Taken together, this data suggests that the G28A mutation is able to more favorably form the SLII conformation, even in the absence of miR-122; but presumably, miR-122 is still required to protect the 5′ terminus from pyrophosphatase activity and subsequent exonuclease-mediated decay by Xrn1 and 2 (19–22). Interestingly, most HCV genotypes (1a, 1b, 3a, 4a, and 5a) already carry an A at position 28, which may indicate that these genotypes have a greater propensity to form SLII than the JFH-1 isolate used herein (6,7). This suggests that these HCV genotypes may be less reliant on miR-122 to form the SLII structure; although we cannot rule out the possibility that additional nucleotide polymorphisms preclude formation of SLII. Notably, downstream of SLII, the WT and G28A HCV RNAs had similar SHAPE reactivities consistent with formation of SLIII-IV of the HCV IRES.
Two hAgo2:miR-122 complexes can bind to the 5′ UTR simultaneously and their interactions with the 5′ terminus further alter the structure of the 5′ UTR
Previous studies have suggested that HCV RNA accumulation requires miRNA biogenesis components, including Dicer, TRBP and the four Ago proteins (14,52–55,62,63). However, it is unclear whether Ago participates beyond miR-122 loading, as sequences in miR-122 dispensable for canonical silencing as well as downstream mediators of silencing do not appear to be required for miR-122′s promotion of HCV RNA accumulation (14,64–66). Moreover, the two miR-122 sites in the 5′ UTR are in very close proximity (separated by a single base) and hence, we were curious whether two hAgo2:miR-122 complexes were able to bind simultaneously to the 5′ terminus of the HCV genome. To this end, we purified hAgo2 loaded with the miR-122 guide strand, and performed gel shift assays. Indeed, we observed that two hAgo2:miR-122 complexes can bind to the 5′ terminus of the HCV genome simultaneously, despite the close proximity of the miR-122 sites; however, the two binding events were negatively affected by one another. Previous studies have suggested that, at least for canonical RNA silencing, seed sites separated by 13–35 nt (from the seed start) may act synergistically (67). As the two miR-122 binding sites on the HCV genome are separated by 15 nt (from seed to seed), this may allow them to have a synergistic effect on HCV RNA accumulation. Alternatively, it is also possible that binding to one site (presumably Site 2) promotes changes to the conformation of the viral RNA that promote hAgo2:miR-122 interactions with the second site (Site 1). Moreover, we observed significant affinity enhancement in the context of hAgo2:miR-122 (pM range) when compared with miR-122 alone (nM range). This is not altogether surprising since the hAgo protein holds the miRNA is a conformation that is conducive to target RNA binding. Additionally, it suggests that binding affinity predictions based solely on miRNA-RNA interactions may significantly underestimate binding affinity.
Our in vitro SHAPE analysis of the HCV RNA in the presence of hAgo2:miR-122 demonstrated that additional structural changes occur at the miR-122 recognition sites at the 5′ terminus in the presence of hAgo2. Specifically, we observed higher overall SHAPE reactivities in the region predicted to form the Site 2 auxiliary interactions with nts 13–16 of Site 2-bound miR-122. This suggests that in order to accommodate both hAgo2:miR-122 complexes, these nucleotides may become unpaired and are therefore available for acylation. Alternatively, since SHAPE reactivities represent an average of the HCV RNA conformations present in solution, the increase in reactivity at these sites could also be due to the contribution of viral RNA bound by hAgo2:miR-122 at one site and not the other, because of the differences in the availability and/or the affinity between the two recognition sites.
Our SHAPE analyses on the entire 5′ UTR during miR-122 or hAgo2:miR-122 binding were very similar; however, we did observe some further changes in SHAPE reactivity when hAgo2:miR-122 was present. Specifically, we observed an overall decrease in SHAPE reactivity in nts 52–58 of SLIIa and nts 104–109 in the 3′ arm of SLII that form base-pair interactions adjacent to SLIIa. Although this was unexpected, our computational prediction of the hAgo2:miR-122:HCV IRES complex suggest that the PIWI domain of hAgo2 at Site 2 may make contact with this region of the viral RNA, which could further stabilize this structure. This is not unprecedented as during Enterovirus 71 infection, Ago2 has been demonstrated to promote viral replication through interactions with the IRES region of the viral RNA, and hence this may point to a related mechanism during HCV infection (68). Interestingly, SLII was previously shown to fold independently of the IRES (47), and it acts as an initiator for HCV translation by taking on an L-shaped conformation that interacts with the 40S subunit, holding the AUG start codon in the P-site of the ribosome until the translation machinery is properly assembled (9,24,48,49,69,70). Our SHAPE results support this model and suggest that hAgo2:miR-122 binding may not only promote translation through suppression of an alternative secondary structure (SLIIalt), but may further stabilize the SLII structure through additional contacts between hAgo2 bound to Site 2 and the viral IRES.
In addition to SLII, SLIIIa and d have been shown to make important contacts with the 40S ribosomal subunit, while SLIIIb-c makes contact with the eukaryotic translation initiation factor 3 (eIF3), to promote HCV RNA translation (9,71–73). We also observed some changes in SHAPE reactivity in the presence of hAgo2:miR-122 in SLIV at nts 337–345 and 350–371, where the 40S and 60S subunits bind (71). In further support of this finding, SHAPE analysis performed on the HCV IRES (nts 42–371) in the presence of the 40S ribosomal subunit demonstrated similar changes in reactivity to what we observed here in the presence of hAgo2:miR-122 (71). In both cases, an overall decrease in SHAPE reactivity was observed in the apical loops of SLIII, with a decrease in SHAPE reactivity directly upstream, and an increase in SHAPE reactivity at a few nts immediately downstream, of the start codon. This suggests that, beyond the 5′ terminus (nts 1–42), miR-122 binding might promote a conformation of the HCV IRES that is similar to the ternary complex with eIF3 and the 40S ribosomal subunit. This is further supported by an Ago HITS-CLIP study during HCV infection which demonstrated miR-122-dependent HITS-CLIP reads in the IRES region (17). Thus, hAgo2:miR-122 interactions may alter the structure of the HCV 5′ UTR that both promote formation of SLII (IRES folding) as well as additional structural changes in SLIII and IV that may promote association with the ribosome and eIF3. However, future work will be needed to confirm these findings in the context of viral replication.
Finally, we used computational prediction to model the 3D structure of the hAgo2:miR-122:HCV RNA ternary and higher-order complexes at the 5′ terminus of the HCV genome. Computational prediction of the ternary complex at Site 1 required sampling of approximately 5 times the conformations needed to model Site 2. This was expected as there is an unusually large stem–loop structure (SLI, nts 4–20) between the seed site (nts 2–8) and the 3′ supplemental interactions (nts 15–17) with Site 1-bound miR-122 that can adopt numerous possible conformations. At Site 1, the dsRNA forms a 3-stem junction which is predicted to force nts 1–3 of the HCV genome into an L-shaped conformation to avoid steric clashes with hAgo2′s PIWI and N terminal domains, while the seed region appears to take on an orientation expected for canonical mRNA targets. The Site 2 duplex can adopt either straight or distorted conformations, but only distorted conformations that protrude out of the RNA-binding channel, which are induced by the unpaired bases in the internal loop region, allow simultaneous hAgo2 occupation of the recognition sites. Multiple RISC–RISC structural conformations can be assembled, suggesting that the higher-order complex with adjacent hAgo2 structures likely exhibits some degree of conformational variability in addition to the flexibility of the SLI structure at Site 1. Specifically, the two hAgo2 structures are orientated differently to avoid steric clashes. Formation of the RISC–RISC complex forces adoption of a specific set of duplex conformations at Site 2, implying that hAgo2 occupation of Site 1 influences the RISC structure at Site 2. Interestingly, in this model the auxiliary interactions between the HCV RNA at Site 2 and miR-122 appears to protrude from hAgo2 (Figure 6A, lower panel). While this may be the case, it seems more likely that miR-122 is retained in the hAgo2-binding channel and that these auxiliary interactions with nts 13–16 of miR-122 at Site 2 become unpaired to accommodate Ago2:miR-122 binding to Site 1 (particularly since the end of the miRNA is likely to be tightly bound by the 3′ nucleotide-binding pocket of the PAZ domain) (41). This is further supported by our biophysical analyses which suggest interference between the two sites, and our SHAPE data which indicates a higher SHAPE reactivity in this region in the presence of hAgo2:miR-122. Thus, it is possible that the auxiliary interactions between Site 2-bound miR-122 and nts 29–32 of the HCV genome are required for efficient initial recruitment of hAgo2:miR-122 to the HCV genome, but that these interactions become unpaired in order to accommodate hAgo2:miR-122 interactions with Site 1.
Assembly of the larger RISC–IRES–40S complex demonstrates that the interface between the hAgo2 at Site 2 and the IRES SLIIa exhibits a tight fit with structural complementarity. Given that the RISC Ago may aid in miR-122-mediated activation of HCV translation (59,74), a plausible interpretation of our modeled complex is that Ago binding to SLIIa helps to stabilize the flexible IRES SLII in the active, bent conformation. These interactions may also help to stabilize the hAgo2:miR-122 complex at Site 2 after recruitment of Site 1, to accommodate both hAgo2:miR-122 complexes on the viral RNA. Moreover, the close proximity between Ago at Site 2 and the viral IRES may preclude interactions between Ago and the downstream RNA silencing machinery, which could help explain how the viral RNA is able to escape canonical RNA silencing. However, we note that the hAgo2 residues known to form contacts with GW182 appear to be accessible in our model (16,75). Thus, further experimental work will be needed to verify the details of these predicted RISC–RISC and RISC–IRES–40S conformations in live cells and to clarify how HCV escapes canonical RNA silencing.
miR-122 has at least three roles in the HCV life cycle
Taken together, our biophysical, SHAPE and computational prediction analyses suggest a new model for hAgo2:miR-122 interactions with the HCV genome (Figure 8). Specifically, we predict that the HCV genome initially adopts an alternative conformation (SLIIalt) that allows recruitment of hAgo2:miR-122 to Site 2 of the 5′ terminus. Binding to Site 2 acts in an RNA-chaperone like manner to convert the 5′ terminus into SLII, allowing functional formation of the HCV IRES. This SLII conformation then allows recruitment of hAgo2:miR-122 to Site 1, which protects the 5′ triphosphate moiety from pyrophosphatase activity and subsequent viral RNA decay by Xrn1 and 2 (19–22). In order to accommodate the hAgo2:miR-122 at Site 1, the auxiliary interactions between the 3′ tail of the miR-122 molecule at Site 2 are likely destabilized, but the hAgo2:miR-122 complex at Site 2 is likely further stabilized by interactions with the HCV IRES. This model is consistent with our biophysical and SHAPE analyses as well as previous mutational analyses where mutation of nts 17–18 of the Site 2-bound miR-122 (predicted to reduce auxiliary interactions with the HCV genome between Sites 1 and 2) led to an ∼1.5-fold increase in viral RNA accumulation (13). This model may also explain the sequence conservation and requirement for the auxiliary interactions with Site 2 (13), since they are likely required for the efficient initial recruitment of hAgo2:miR-122 to this site, even if they are weakened upon hAgo2:miR-122 binding to Site 1. In agreement with this, a recent study that used cooperative-immunoprecipitation of Ago complexes in HCV-infected cells suggests that Site 1 has a higher affinity for Ago:miR-122 than Site 2 in live cells (52). Finally, the highly conserved spacing between the two miR-122 binding sites, which appears to result in interference between the two Ago proteins, is likely retained across HCV isolates because of the requirements for these sequences to form secondary structures (SL-I' and SL-IIz') in the 3′ end of the negative-strand intermediate which constitutes the promoter for positive-strand viral RNA synthesis (76).
Figure 8.

Model for Ago2:miR-122 interactions with the HCV 5′ UTR. Schematic representation of Ago2:miR-122 binding to the HCV genome and the effects on the secondary structure of the viral RNA. The HCV 5′ UTR initially takes on the most energetically favorable conformation (SLIIalt), which allows recruitment of hAgo2:miR-122 to Site 2 of the HCV genome (black). This results in a chaperone-like switch in conformation, resulting in formation of SLII, part of the HCV IRES (SLII-IV). This change in conformation allows recruitment of hAgo2:miR-122 to Site 1, which protects the 5′ triphosphate moiety from pyrophosphatase activity and subsequent exonuclease-mediated decay. In order to accommodate hAgo2:miR-122 interactions at Site 1, the auxiliary interactions with Site 2-bound hAgo2:miR-122 are weakened; however, Site 2-bound hAgo2:miR-122 is further stabilized by interactions with the HCV IRES (in SLIIa and the region between SLII-III). The miR-122 seed and auxiliary binding sites on the HCV genome are indicated (red).
In conclusion, our results suggest that miR-122 is likely to play at least three roles in the HCV life cycle. First, the initial recruitment of hAgo2:miR-122 to Site 2 results in a chaperone-like switch in conformation which suppresses an alternative secondary structure (SLIIalt) and promotes SLII formation (23,24). Second, recruitment of hAgo2:miR-122 to Site 1 protects the 5′ terminus from pyrophosphatase activity and subsequent exonuclease-mediated decay (14,19–21,53,77). And lastly, the hAgo2:miR-122 complex at Site 2 may further stabilize the HCV IRES through interactions between the PIWI and Mid domains of Ago and SLII of the HCV IRES to promote viral translation (59,74). This provides new insight into the role of miR-122 in the HCV life cycle and future work will focus on clarifying this model in the context of infection and further dissecting the contribution of each of these miR-122 activities.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge Charlie Rice (University of Rockefeller) for providing Huh-7.5 cells and Rodney Russell (Memorial University) for providing JFH-1T. We would also like to thank Julie Magnus (McGill University) for technical support, Veronique Sauvé (McGill University) for assistance with ITC analysis, Tamara Giguère and Jean-Pierre Perreault (University of Sherbrooke) for assistance with the SHAPE experiments and data analysis, and Zamboni Chem Solutions Inc. for assistance in synthesis of NAI-N3. 3D modeling was done using High Performance Computing at NYU.
Author Contributions: J.C. and S.M.S. conceptualized and designed the study. J.C. generated and analyzed the data with help from E.C. for ITC analysis. L.F.R.G. and I.J.M. performed hAgo2 purification and generated and analyzed hAgo2:miR-122:HCV RNA gel shift assays. H.H.G. and K.G. performed computational modeling, analysis and interpretation of the modeling results. J.C. and S.M.S. drafted the manuscript and all authors contributed to editing the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Gerald Clavet Fellowship from McGill University (to J.C.); McGill University as well as grants from the NSERC Discovery grant program [RGPIN-2014-05907 to S.M.S.]; US National Institutes of Health grants [R01-GM104475, R01-GM115649, R35-GM127090 to I.J.M.]; Swiss National Science Foundation [Advanced Postdoc Mobility Fellowship P300PA_177860 to L.F.R.G.]; NYU Abu Dhabi (to H.H.G. and K.C.G.); and this research was undertaken, in part, thanks to the Canada Research Chairs program (to S.M.S.). Funding for open access charge: NSERC discovery grant.
Conflict of interest statement. None declared.
REFERENCES
- 1. World Health Organization Global Hepatitis Report 2017. 2017; Geneva: 1–83. [Google Scholar]
- 2. Hajarizadeh B., Grebely J., Dore G.J.. Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2013; 10:553–562. [DOI] [PubMed] [Google Scholar]
- 3. Messina J.P., Humphreys I., Flaxman A., Brown A., Cooke G.S., Pybus O.G., Barnes E.. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015; 61:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tice J.A., Chahal H.S., Ollendorf D.A.. Comparative clinical effectiveness and value of novel Interferon-Free Combination Therapy for Hepatitis C Genotype 1: Summary of California Technology Assessment Forum Report. JAMA Intern Med. 2015; 175:1559–1560. [DOI] [PubMed] [Google Scholar]
- 5. Wilson J.A., Sagan S.M.. Hepatitis C virus and human miR-122: Insights from the bench to the clinic. Curr. Opin. Virol. 2014; 7:11–18. [DOI] [PubMed] [Google Scholar]
- 6. Tuplin A., Struthers M., Simmonds P., Evans D.J.. A twist in the tail: SHAPE mapping of long-range interactions and structural rearrangements of RNA elements involved in HCV replication. Nucleic Acids Res. 2012; 40:6908–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sagan S.M., Chahal J., Sarnow P.. cis-Acting RNA elements in the hepatitis C virus RNA genome. Virus Res. 2015; 206:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friebe P., Lohmann V., Krieger N., Bartenschlager R.. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J. Virol. 2001; 75:12047–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khawaja A., Vopalensky V., Pospisek M.. Understanding the potential of hepatitis C virus internal ribosome entry site domains to modulate translation initiation via their structure and function. Wiley Interdiscipl. Rev.: RNA. 2015; 6:211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tellinghuisen T.L., Evans M.J., von Hahn T., You S., Rice C.M.. Studying hepatitis C Virus: Making the best of a bad virus. J. Virol. 2007; 81:8853–8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang C., Le S.Y., Ali N., Siddiqui A.. An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region. RNA. 1995; 1:526–537. [PMC free article] [PubMed] [Google Scholar]
- 12. Jopling C.L. Regulation of hepatitis C virus by microRNA-122. Biochem. Soc. Trans. 2008; 36:1220–1223. [DOI] [PubMed] [Google Scholar]
- 13. Jopling C.L., Yi M., Lancaster A.M., Lemon S.M., Sarnow P.. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005; 209:1577–1581. [DOI] [PubMed] [Google Scholar]
- 14. Machlin E.S., Sarnow P., Sagan S.M.. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. PNAS. 2011; 108:3193–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang J., Nicolas E., Marks D., Sander C., Lerro A., Buendia M.A., Xu C., Mason W.S., Moloshok T., Bort R. et al.. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004; 1:106–113. [DOI] [PubMed] [Google Scholar]
- 16. Jopling C.L., Schütz S., Sarnow P.. Position-dependent function for a tandem MicroRNA miR-122-Binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008; 4:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luna J.M., Scheel T.K.H., Danino T., Shaw K.S., Mele A., Fak J.J., Nishiuchi E., Takacs C.N., Catanese M.T., De Jong Y.P. et al.. Hepatitis C virus RNA functionally sequesters miR-122. Cell. 2015; 160:1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Janssen H.L.A., Reesink H.W., Lawitz E.J., Zeuzem S., Rodriguez-Torres M., Patel K., van der Meer A.J., Patick A.K., Chen A., Zhou Y. et al.. Treatment of HCV infection by targeting MicroRNA. N. Engl. J. Med. 2013; 368:1685–1694. [DOI] [PubMed] [Google Scholar]
- 19. Amador-Canizares Y., Bernier A., Wilson J.A., Sagan S.M.. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res. 2018; 46:5139–5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kincaid R.P., Lam V.L., Chirayil R.P., Randall G., Sullivan C.S.. RNA triphosphatase DUSP11 enables exonuclease XRN-mediated restriction of hepatitis C virus. PNAS. 2018; 115:8197–8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Y., Masaki T., Yamane D., McGivern D.R., Lemon S.M.. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. PNAS. 2013; 110:1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mortimer S.A., Doudna J.A.. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res. 2013; 41:4230–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amador-Canizares Y., Panigrahi M., Huys A., Kunden R.D., Adams H.M., Schinold M.J., Wilson J.A.. miR-122, small RNA annealing and sequence mutations alter the predicted structure of the Hepatitis C virus 5′ UTR RNA to stabilize and promote viral RNA accumulation. Nucleic Acids Res. 2018; 46:9776–9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schult P., Roth H., Adams R.L., Mas C., Imbert L., Orlik C., Ruggieri A., Pyle A.M., Lohmann V.. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018; 9:2613–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hopcraft S.E., Azarm K.D., Israelow B., Leveque N., Schwarz M.C., Hsu T.H., Chambers M.T., Sourisseau M., Semler B.L., Evans M.J.. Viral determinants of miR-122 independent hepatitis C virus replication. mSphere. 2016; 1:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ottosen S., Parsley T.B., Yang L., Zeh K., van Doorn L.-J., van der Veer E., Raney A.K., Hodges M.R., Patick A.K.. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti-hepatitis C virus therapeutic targeting the human factor miR-122. Antimicrob. Agents Chemother. 2015; 59:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pang P.S., Pham E.A., Elazar M., Patel S.G., Eckart M.R., Glenn J.S.. Structural map of a microRNA-122: hepatitis C virus complex. J. Virol. 2012; 86:1250–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fricke M., Dünnes N., Zayas M., Bartenschlager R., Niepmann M., Marz M.. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA. 2015; 21:1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiao X., Chang J.H., Kilic T., Tong L., Kiledjian M.. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol. Cell. 2013; 50:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Russell R.S., Meunier J.-C., Takikawa S., Faulk K., Engle R.E., Bukh J., Purcell R.H., Emerson S.U.. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:4370–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reuter J.S., Mathews D.H.. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010; 11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Darty K., Denise A., Ponty Y.. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flynn R.A., Zhang Q.C., Spitale R.C., Lee B., Mumbach M.R., Chang H.Y.. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 2016; 11:273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das R., Laederach A., Pearlman S.M., Herschlag D., Altman R.B.. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA. 2005; 11:344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laederach A., Das R., Vicens Q., Pearlman S.M., Brenowitz M., Herschlag D., Altman R.B.. Semiautomated and rapid quantification of nucleic acid footprinting and structure mapping experiments. Nat. Protoc. 2008; 3:1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rausch J.W., Sztuba-Solinska J., Lusvarghi S., Le Grice S.F.J.. Novel Biochemical Tools for Probing HIV RNA Structure. HIV Protoc. 2016; 1354:91–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spitale R.C., Crisalli P., Flynn R.A., Torre E.A., Kool E.T., Chang H.Y.. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013; 9:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karabiber F., McGinnis J.L., Favorov O.V., Weeks K.M.. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA. 2013; 19:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Low J.T., Weeks K.M.. SHAPE-directed RNA secondary structure prediction. Methods. 2010; 52:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilkinson K.A., Gorelick R.J., Vasa S.M., Guex N., Rein A., Mathews D.H., Giddings M.C., Weeks K.M.. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008; 6:883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schirle N.T., Sheu-Gruttadauria J., MacRae I.J.. Structural basis for microRNA targeting. Science. 2014; 346:608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Faehnle C.R., Elkayam E., Haase A.D., Hannon G.J., Joshua-Tor L.. The making of a slicer: activation of human Argonaute-1. Cell Rep. 2013; 3:1901–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schirle N.T., MacRae I.J.. The crystal structure of human Argonaute2. Science. 2012; 336:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gan H.H., Gunsalus K.C.. Assembly and analysis of eukaryotic Argonaute-RNA complexes in microRNA-target recognition. Nucleic Acids Res. 2015; 43:9613–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parisien M., Major F.. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008; 452:51–55. [DOI] [PubMed] [Google Scholar]
- 46. Flamand M.N., Gan H.H., Mayya V.K., Gunsalus K.C., Duchaine T.F.. A non-canonical site reveals the cooperative mechanisms of microRNA-mediated silencing. Nucleic Acids Res. 2017; 45:7212–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lukavsky P.J., Kim I., Otto G.A., Puglisi J.D.. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Biol. 2003; 10:1033–1038. [DOI] [PubMed] [Google Scholar]
- 48. Lukavsky P.J., Otto G.A., Lancaster A.M., Sarnow P., Puglisi J.D.. Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat. Struct. Biol. 2000; 7:1105–1110. [DOI] [PubMed] [Google Scholar]
- 49. Odreman-Macchioli F., Baralle F.E., Buratti E.. Mutational analysis of the different bulge regions of hepatitis C virus domain II and their influence on internal ribosome entry site translational ability. J. Biol. Chem. 2001; 276:41648–41655. [DOI] [PubMed] [Google Scholar]
- 50. Israelow B., Mullokandov G., Agudo J., Sourisseau M., Bashir A., Maldonado A.Y., Dar A.C., Brown B.D., Evans M.J.. Hepatitis C virus genetics affects miR-122 requirements and response to miR-122 inhibitors. Nat. Commun. 2015; 5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Conrad K.D., Giering F., Erfurth C., Neumann A., Fehr C., Meister G., Niepmann M.. microRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS One. 2013; 8:e56272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nieder-Röhrmann A., Dünnes N., Gerresheim G.K., Shalamova L.A., Herchenröther A., Niepmann M.. Cooperative enhancement of translation by two adjacent microRNA-122/argonaute 2 complexes binding to the 5′ untranslated region of hepatitis C virus RNA. J. Gen. Virol. 2017; 98:212–224. [DOI] [PubMed] [Google Scholar]
- 53. Shimakami T., Yamane D., Jangra R.K., Kempf B.J., Spaniel C., Barton D.J., Lemon S.M.. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. PNAS. 2012; 109:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilson J.A., Zhang C., Huys A., Richardson C.D.. Human Ago2 is required for efficient MicroRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J. Virol. 2011; 85:2342–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Randall G., Panis M., Cooper J.D., Tellinghuisen T.L., Sukhodolets K.E., Pfeffer S., Landthaler M., Landgraf P., Kan S., Lindenbach B.D. et al.. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:12884–12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sheu-Gruttadauria J., Xiao Y., Gebert L.F.R., MacRae I.J.. Beyond the seed: structural basis for supplementary microRNA targeting. bioRxiv. 2018; doi:10.1101-476960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quade N., Boehringer Daniel, Leibundgut Marc, van den Heuvel Joop, Ban Nenad. Cryo-EM structure of Hepatitis C virus IRES bound to the human ribosome at 3.9-Å resolution. Nat. Commun. 2015; 6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yamamoto H., Collier M., Loerke J., Ismer J., Schmidt A., Hilal T., Sprink T., Yamamoto K., Mielke T., Burger J. et al.. Molecular architecture of the ribosome-bound Hepatitis C Virus internal ribosomal entry site RNA. EMBO J. 2015; 34:3042–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roberts A.P., Lewis A.P., Jopling C.L.. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011; 39:7716–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dibrov S.M., Parsons J., Carnevali M., Zhou S., Rynearson K.D., Ding K., Garcia Sega E., Brunn N.D., Boerneke M.A., Castaldi M.P. et al.. Hepatitis C virus translation inhibitors targeting the internal ribosomal entry site. J. Med. Chem. 2014; 57:1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Y.P., Ramirez S., Gottwein J.M., Bukh J.. Non-genotype-specific role of the hepatitis C virus 5′ untranslated region in virus production and in inhibition by interferon. Virology. 2011; 421:222–234. [DOI] [PubMed] [Google Scholar]
- 62. Masaki T., Arend K.C., Li Y., Yamane D., McGivern D.R., Kato T., Wakita T., Moorman N.J., Lemon S.M.. MiR-122 stimulates hepatitis C virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe. 2015; 17:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang C., Huys A., Thibault P.A., Wilson J.A.. Requirements for human Dicer and TRBP in microRNA-122 regulation of HCV translation and RNA abundance. Virology. 2012; 433:479–488. [DOI] [PubMed] [Google Scholar]
- 64. Berezhna S.Y., Supekova L., Sever M.J., Schultz P.G., Deniz A.A.. Dual regulation of hepatitis C viral RNA by cellular RNAi requires partitioning of Ago2 to lipid droplets and P-bodies. RNA. 2011; 17:1831–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pager C.T., Schutz S., Abraham T.M., Luo G., Sarnow P.. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology. 2013; 435:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Roberts A.P., Doidge R., Tarr A.W., Jopling C.L.. The P body protein LSm1 contributes to stimulation of hepatitis C virus translation, but not replication, by microRNA-122. Nucleic Acids Res. 2014; 42:1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saetrom P., Heale B.S., Snove O. Jr, Aagaard L., Alluin J., Rossi J.J.. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 2007; 35:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lin J.Y., Brewer G., Li M.L.. HuR and Ago2 bind the internal ribosome entry site of enterovirus 71 and promote virus translation and replication. PLoS One. 2015; 10:e0140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Berry K.E., Waghray S., Mortimer S.A., Bai Y., Doudna J.A.. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure. 2011; 19:1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lyons a.J., Lytle J.R., Gomez J., Robertson H.D.. Hepatitis C virus internal ribosome entry site RNA contains a tertiary structural element in a functional domain of stem–loop II. Nucleic Acids Res. 2001; 29:2535–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Filbin M.E., Kieft J.S.. HCV IRES domain IIb affects the configuration of coding RNA in the 40S subunit's decoding groove. RNA. 2011; 17:1258–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kieft J.S., Zhou K., Grech A., Jubin R., Doudna J.A.. Crystal structure of an RNA tertiary domain essential to HCV IRES-mediated translation initiation. Nat. Struct. Biol. 2002; 9:370–374. [DOI] [PubMed] [Google Scholar]
- 73. Kieft J.S., Zhou K., Jubin R., Doudna J.A.. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001; 7:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Henke J.I., Goergen D., Zheng J., Song Y., Schüttler C.G., Fehr C., Jünemann C., Niepmann M.. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008; 27:3300–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Elkayam E., Faehnle C.R., Morales M., Sun J., Li H., Joshua-Tor L.. Multivalent recruitment of human argonaute by GW182. Mol. Cell. 2017; 67:646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Friebe P., Bartenschlager R.. Role of RNA structures in genome terminal sequences of the hepatitis C virus for replication and assembly. J. Virol. 2009; 83:11989–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Thibault P.A., Huys A., Amador-Cañizares Y., Gailius J.E., Pinel D.E., Wilson J.A.. Regulation of hepatitis C virus genome replication by Xrn1 and microRNA-122 binding to individual sites in the 5′ untranslated region. J. Virol. 2015; 89:6294–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.