


Abstract
The complex role of linker histone (LH) on chromatin compaction regulation has been highlighted by recent discoveries of the effect of LH binding variability and isoforms on genome structure and function. Here we examine the effect of two LH variants and variable binding modes on the structure of chromatin fibers. Our mesoscale modeling considers oligonucleosomes with H1C and H1E, bound in three different on and off-dyad modes, and spanning different LH densities (0.5–1.6 per nucleosome), over a wide range of physiologically relevant nucleosome repeat lengths (NRLs). Our studies reveal an LH-variant and binding-mode dependent heterogeneous ensemble of fiber structures with variable packing ratios, sedimentation coefficients, and persistence lengths. For maximal compaction, besides dominantly interacting with parental DNA, LHs must have strong interactions with nonparental DNA and promote tail/nonparental core interactions. An off-dyad binding of H1E enables both; others compromise compaction for bendability. We also find that an increase of LH density beyond 1 is best accommodated in chromatosomes with one on-dyad and one off-dyad LH. We suggest that variable LH binding modes and concentrations are advantageous, allowing tunable levels of chromatin condensation and DNA accessibility/interactions. Thus, LHs add another level of epigenetic regulation of chromatin.
INTRODUCTION
The physical structure of our genome and its epigenetic regulation by the complex cellular machinery represent important open problems today. Along with histone tail modifications and remodeling proteins, the organization of the chromatin fiber in eukaryotic nuclei itself is recognized a key epigenetic regulator of gene expression. The chromatin fiber is made of DNA and specialized nucleosome particles that help compact the 2 m of DNA (in human) into a micrometer sized nucleus (1). Specifically, the nucleosome particle contains DNA wrapped 1.75 times around the eight histone proteins (two copies of each of the highly conserved H2A, H2B, H3 and H4 proteins), forming a disk-like structure (2). The highly conserved core histone proteins, with their flexible tails, interact with neighboring nucleosomes and help bend parental DNA linkers entering/exiting the nucleosome to direct chromatin compaction. However, to produce a highly compacted fiber with repressed DNA, additional proteins, most prominently linker histones (LHs), are also recruited (3–5). The LH proteins not only help condense the chromatin fiber (3,6,7), but are also involved in DNA replication, genome stability, and DNA repair (5, 8). Although less conserved than the core histone proteins, LHs are present in all higher organisms in multiple subtypes (9). Higher eukaryotes have multiple variants variably expressed throughout cell cycle (4,10). For instance, there are 11 known LH variants in humans and mice, with 7 present dominantly in normal body tissues (H1a, H1b, H1c, H1d, H1e, H10, H1x) and four dominant in sperm and eggs cells (H1tm, H1T2, HILS1m, H1oo) (5, 11). The H1 subtypes are expressed and differentially regulated at all stages of cell cycle, with the somatic subtypes mostly expressed during DNA replication (11).
Besides different LH variants (12), variable LH stoichiometric ratios (13–16), multiple LH post-translational modifications (5), and variable on- and off-dyad modes are also recognized (9,17). Namely, biochemical (10,18–22), structural (NMR, crystallographic, cryo-EM) (17,23–26) and modeling studies (23–26) suggest heterogeneous structural ensembles of chromatosome configurations and variable LH binding modes. These ensembles also depend on the DNA sequence, DNA linker length, post-translational modifications, and environmental conditions (8,9).
In the symmetric on-dyad binding mode, the LH globular head (GH), positioned on the nucleosome dyad, interacts with both linker DNA arms, while in the asymmetric off-dyad binding mode, the GH is shifted and interacts with only one linker DNA (26). In this way, the GH’s own asymmetry contributes to the asymmetry of LH’s interactions with parental DNA linkers (27,28). Furthermore, the LH-parental DNA interaction depends on the nucleosomal environment (27,29) and the salt concentration (30). Modeling has shown that in low-salt environments, LH bound on-dyad primarily interacts with one linker DNA, but at physiological concentrations the interaction becomes more symmetric (30).
The types of asymmetric, off-dyad LH binding modes also vary. For example, the GH may bind the nucleosome adjacent to the dyad axis, but interact only with one linker DNA (23,25,31,32). Alternatively, the GH can bind the nucleosome in an off-dyad mode, with the CTD interacting with both DNA linkers (31,33,34). In a recent cryo-EM study (33), an off-dyad binding mode has been observed in spirally twisted 12-nucleosome arrays with NRL = 177 and 187 bp. This asymmetric, off-dyad LH binding may be favorable for stabilizing a tightly packed zigzag array that defines a left-handed superhelix: it facilitates interactions between GHs of LHs in parallelly stacked nucleosomes, keeps DNA linkers straight, and promotes tail-to-tail interactions between tetranucleosomal units for additional packing and twisting (33). Whether these observations in crystal hold for large fiber arrays in crowded environment in cell nucleus remains to be seen.
How such different LH binding modes are related to different levels of chromatin condensation is still unknown. Experimental and modeling studies have shown that the LH conformation depends on the chromatin environment (27,29), but the influence of the LH type and binding mode on the chromatin structure still lacks a full account. For example, Zhou et al. (17) present measurements of sedimentation coefficients that are higher for on-dyad binding, while Bednar et al. (35) associate on-dyad binding with uncondensed nucleosomes and Song et al. (33) show condensed nucleosome arrays with off-dyad binding. The speculation that the off-dyad mode could be a consequence of cross-linking effects (35), complicates interpretations as glutaraldehyde is known to perturb nucleosome array conformations (36). Furthermore, how more than one LH is bound in a chromatosome is not known, though densities >1 have been observed (13,14,16). Here we examine the influence of on-dyad and two off-dyad binding modes (+/– 20° with respect to dyad axis) on chromatin folding for two LH variants (H1C or H1.2, and H1E or H1.4) in combination with different LH densities (from 0.5 to 1.6) and linker lengths with our mesoscale chromatin model (Figure 1) (30,37–46). See description of covered systems in Table 1.
Figure 1.
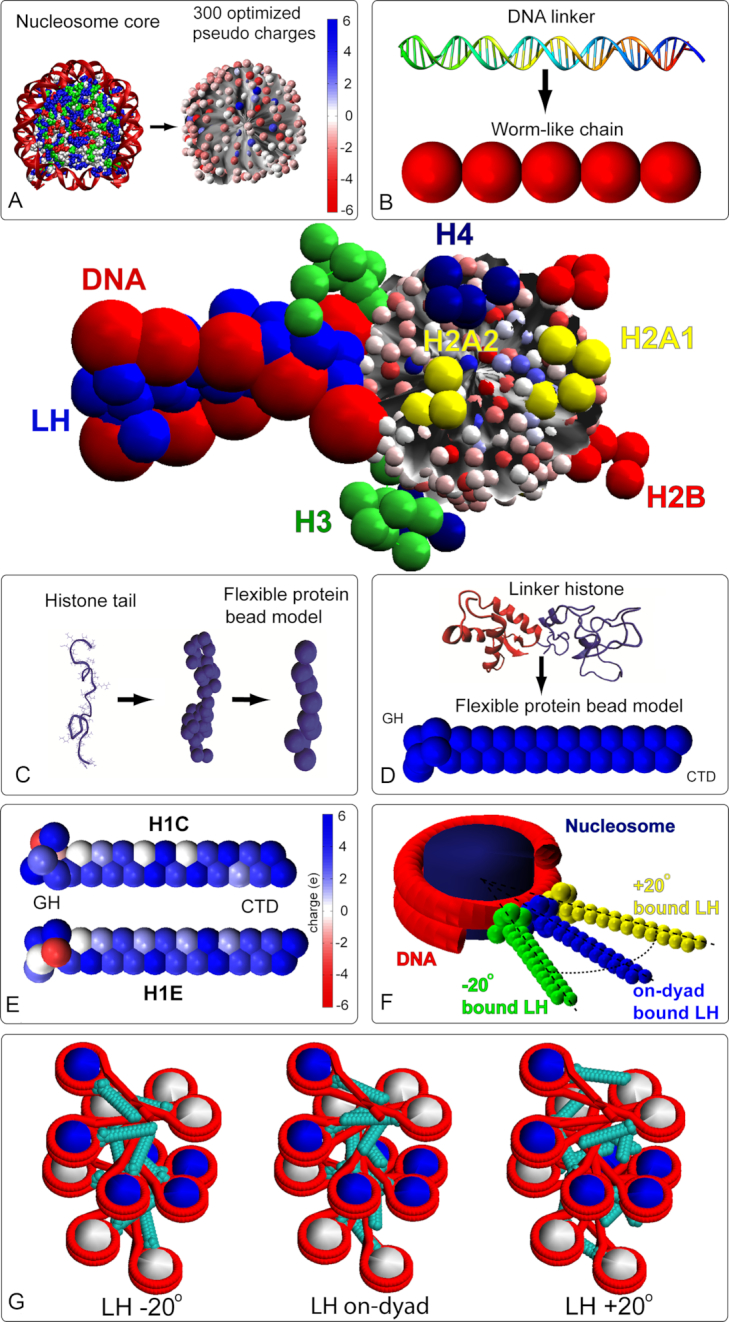
Coarse grained model with its components. (A) nucleosome core modeling with color coded 300 charges; (B) Worm-like chain model of DNA linker made of polymer segments, with two segments connected by a spherical bead; each segment represents 9 bp. (C) Histone tails modeling where each bead corresponds to five amino acids; (D) linker histone modeling; (E) two LH models (H1C and H1E) with color coded charges; For H1C, six beads for the Globular Head (GH) and 21 bead for the C-terminal domain (CTD) are used. For H1E we use 6 GH + 22 CTD beads respectively; (F) on and off-dyad binding modes; and (G) starting configuration with three LH binding modes.
Table 1.
Fiber systems considered in this study
| LH density | LH type | LH distribution | NRL (bp) | LH binding mode |
|---|---|---|---|---|
| 0.5 | H1C, H1E | 1 LH per 2 nucleosomes | 182, 191, 200, 209, 218 | on-dyad, –20°, or +20° |
| 1 | H1C, H1E | 1 LH per nucleosome | 182, 191, 200, 209, 218 | on-dyad, –20°, or +20° |
| 1.3 or 1.6 | H1C, H1E | 1 LH or 2 LH per nucleosome | 200, 209 | 1 LH chromatosome: on-dyad, –20°, or +20° |
| 2 LH chromatosome: H1C(on-dyad, –20°, or +20°) + H1E(on-dyad, –20°, or +20°) or 2 H1C (on-dyad, –20°, or +20°) |
We consider chromatosomes with zero, one, or two LHs bound. Densities (ρ) >1 have been associated with heterochromatin in the center of mature rod photoreceptors nuclei (ratio of 1.3 LH per nucleosome) (16), chicken erythrocyte nuclei (1.3 LH per nucleosome) (13), and glia cells (1.04 LH per nucleosome) (14). When two LHs bind to the same chromatosome, we also explore which combinations are favorable for nucleosome packing (on-dyad + off-dyad, or two off-dyads, both in various combinations with both LH types).
Overall, we find that fiber structures, sedimentation rates, packing ratios, and bending propensities depend sensitively on the LH type and binding mode. Some variations produce highly compact, but rigid fibers, while others decrease compaction, but enhance long-range interactions via increased bending, thereby promoting looping. Importantly, the LH binding modes that reduce fiber compaction also expose nucleosomes to the environment, thus making them more accessible to the cellular machinery.
Our results show that the intensity of interaction between LH and parental and nonparental DNA, as well as tail interactions with nonparental cores are the strongest factors that dictate the chromatin geometry. Specifically, H1E in the –20° off-dyad binding mode produces the highest chromatin compaction and sedimentation rates over a wide range of NRLs, namely 191–218 bp.
With ρ = 0.5, the off-dyad binding mode that produces highest packing (–20o with H1E) has significantly reduced packing. Other modes are more robust to sub-saturated levels of LH.
An LH density (ρ) >1 LH per nucleosome increases bending and reduces sedimentation and packing ratios, compared to highly compact fibers with H1E bound off-dyad. However, a couple of LH binding setups with ρ > 1, when H1C and H1E are bound together in one nucleosome (primarily in an off-dyad/on-dyad combinations), produce both bendable and compact fibers.
Overall, our results indicate that variable LH types and binding modes have physiological roles. Thus, while H1E in bound –20° produces highly compact, though rigid fibers, other binding modes decrease nucleosome packing, but allow fiber bending and thereby encourage hierarchical looping inside the cell nucleus (47). We propose that the LH subtypes and variability of their binding modes can add an additional layer of epigenetic regulation, complementary to the histone code (48), by controlling accessibility to the genome template.
MODEL AND METHODS
Mesoscale model
Our mesoscale model (Figure 1) interprets chromatin as a polymer chain composed of four types of components, linker DNAs, nucleosome particles, flexible histone tails and flexible linker histones. The Supplementary material summarizes details of our mesoscale model with LH (modeled after H1E) that has evolved over 18 years and has been validated against various structural, thermodynamic and dynamic data. There we summarize each model component and associated energy calculations. See also recent reviews (48–50). Below we describe the H1C modeling (instead of H1E used before) and the setups specific to this work.
H1C modeling
To add a coarse-grained LH H1C model, we performed a similar procedure as with H1E (30). The rigid H1C GH was modeled using homology modeling principles, as above. The initial full-atom model of GH was created using the Modeller package (51), based on the GH from the nucleosome crystal structure 5NL0. The model was equilibrated for 4 ns using the NAMD molecular dynamics package (52). The 6 beads were extracted using VMD (53), and the DiSCO charge optimization program (39) was applied to extract the corresponding bead charges. The 6-bead GH model was spatially aligned with the GH in the 5NL0 crystal structure and positioned at the corresponding on-dyad position of the nucleosome model using the Tcl/Tk and Matlab programming languages and the matrix manipulation routines embedded in VMD (53).
The N-terminal domain (NTD) was not modeled as it was demonstrated that its elimination does not affect the LH binding or higher-order structures of chromatin (30). The flexible CTD tail was modeled analogously to the H1E tail (see Supplementary Information of (30)). Namely, the 105 amino acids of the unfolded H1C CTD tail were divided into Twenty-one 5-amino acids segments with each segment represented by a charged bead. Beads charges were calculated using the approach described in (30). The same bending and stretching coefficients were used as in the H1E model, based on the parameters used for the nucleosome tails (30). See Figure 1 for the final H1C model and Supplementary Figure S1 for a sequence alignment of both LHs. While H1E formed our initial LH model (30), the H1C model was developed due to interesting experiments showing importance of LH roles in chromatin structure and vision disorders (16,54).
Linker histone off-dyad modeling
The modeled binding modes, which are unchanged during the simulations, are generated by rigidly rotating the on-dyad LH models (GH and CTD) around the axis perpendicular to the nucleosome plane, passing through the nucleosome center. The LH was rotated around that axis, –20° or +20°. The –20° mode was chosen by measuring the off-dyad orientation of GH in the experimentally generated cryo-EM image of tetranucleosome (33). The +20° mode was chosen as its symmetric analog, following the modeling studies by the Wade group (26,55). See Figure 1 for illustration.
Modeling LH concentrations above 1 LH/nucleosome
To produce fibers with LH concentrations above 1 LH/nucleosome (ρ > 1) we developed a nucleosome particle model with two LHs. The two LHs both can be H1C or H1E, or one of each. They can be bound in any mode defined for a single LH, namely, on-dyad, –20° or +20° (see Table 1). Both LHs cannot be bound in the same mode, but all other combinations are allowed (one on-dyad, one off-, or two off-, in various LH types). The LH concentration is regulated by the number of nucleosomes with two LH. For instance, to set up a 100-core oligonucleosome array with ρ = 1.3, 70 nucleosomes have one LH (H1E), and 30 nucleosomes include two LHs, for a total of 130 LHs. Nucleosomes with one and two LH are distributed randomly. However, we require that for fibers with ρ = 1.3 two sequentially adjacent nucleosomes do not have two LHs. For fibers with ρ = 1.6, 40 nucleosomes have one LH (H1E), and 60 nucleosomes include 2 LHs, for a total of 160 LHs. For ρ = 0.5 every other nucleosome has a bound LH.
The binding modes were tested at physiological salt concentrations (150 mM NaCl) on 24 and 100-core oligonucleosomes with ρ = 0.5, 1, 1.3 and 1.6 LH/nucleosome densities. We examined 5 nucleosome repeat lengths (NRL) (182, 191, 200, 209 and 218 bp), as shown in Table 1. With ρ =1.3 and 1.6, we covered two NRLs, 200 and 209 bp due to their closeness to the NRL = 206 bp observed in chromatin with the above stoichiometric ratio of LH (ρ =1.3 in mature mouse photoreceptors (16)). For each fiber system in Table 1 we sampled the equilibrium ensemble through three independent MC trajectories using three different random seeds, each trajectory having 40–60 million MC steps to ensure convergence of global and local properties (44); analyses were performed on the last 5 million MC steps per trajectory. The starting configurations (shared among all LH binding modes) are depicted in Supplementary Figure S2.
For details of the Monte Carlo algorithm, energy terms, fiber packing, persistence length and contact patterns calculations see the Supplementary material.
RESULTS
As we are interested in the dependence of chromatin condensation on the LH subtype, binding mode, and density, we assess overall fiber topologies and measure packing ratios, sedimentation coefficients, and persistence lengths of the simulated chromatin arrays for all the fiber systems explored (Table 1). We also analyze the influence of binding modes on the chromatin compaction via the energetic contributions of various chromatin building blocks (DNA linkers, nucleosome cores, other LHs, histone tails).
Off-dyad LH binding modes produce the highest nucleosome packing
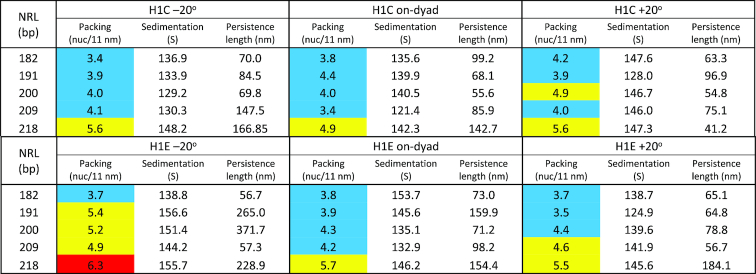
Quantitatively, the analysis of packing ratios reveals that fibers with ρ = 1 and H1E bound –20° off-dyad produce overall highest packing ratio for all NRLs and all LH densities, except the shortest NRL (182 bp) (Table 2, color coded by packing). On the other hand, fibers with ρ = 1 and H1E bound +20° have lower values. Fibers with ρ = 1 and H1E bound on dyad have generally lowest packing densities. The H1C LH achieves lower densities regardless of the LH binding mode, consistent with its association as a weak chromatin condenser (54). The observed packing densities for ρ = 1 correlate with NRL, as observed previously (3,6,44,56,57). Only arrays with NRL = 209 bp slightly deviate from this trend; this can be explained by relatively strong electrostatic interactions between LH and parental DNA and weak interactions with nonparental DNA (discussed below).
Table 2.
Fiber properties (packing densities in nucleosomes per 11 nm, sedimentation coefficients in S units, and persistence lengths in nm) for 100-nucleosome arrays fully saturated with 1 LH (H1C or H1E) per nucleosome. The values are color coded by nucleosome packing ranges: blue - low packing (<4.5 nuc/11 nm), yellow – medium (4.5 type=“Other"> nuc/11 nm < 6), and red – high (> 6 nuc/11 nm)
|
Corresponding data for ρ > 1 (Supplementary Table S1) show that such fibers cannot reach high packing achieved with ρ = 1 and –20° off-dyad binding with H1E, but some combinations with ρ = 1.3 and ρ = 1.6 (particularly in which H1E is bound either on-dyad or +20°, and chromatosomes with two LHs have LHs bound +20°/on-dyad) also approach the density of 5 nuc/11 nm. This also holds for fibers with ρ = 1.3 in which H1E is bound –20°. However, fibers with ρ = 1.6, although approaching 5 nuc/11 nm with some LH binding setups, generally produce less consistent packing, because they also have more LH binding configurations with packing densities below 4 nuc/11 nm, compared to fibers with ρ = 1.3. Arrays with ρ = 1.3 achieve higher packing when chromatosomes with two LH have H1E + H1C, instead of two H1C, with H1E off-dyad.
Generally, the highest packing densities we measure are close to the nucleosome packing (6 nuc/11 nm) estimated experimentally in chicken erythrocytes (58,59), and by numerical models with very simple LH model and without explicit tails, for NRL = 200 bp (Stehr et al. (7) (∼5.5 nuc/11 nm, and Wedemann and Langowski ∼6.1 nuc/11 nm (56)).
Differences in fiber topologies for these variable binding modes emerge from our models shown in Figure 2, where LHs in chromatosomes with one LH are colored as their parental histone core (white or blue), and yellow for chromatosomes with 2 LHs (with yellow histone core, in fibers with ρ > 1). The –20° binding mode with H1E emerges as the best condenser of nucleosome arrays with nucleosome density of ∼5.2 nuc/11 nm. In that mode LHs naturally avoid steric clashes with nonparental nucleosomes and their LHs (primarily ± 2 contacts) and allow nucleosome stacking (see images in Figures 1G and 2C, D). With the on-dyad mode, on the other hand, nucleosomes have to rotate around the dyad as well as nucleosome axes to avoid nonparental nucleosomes and their LHs (particularly ± 2 contacts), resulting in reduced packing. With the +20° mode, LHs have limited interactions with parental DNA and produce weaker stems with linker DNA.
Figure 2.

Packing ratios and fiber images for various array setups for NRL = 200 bp. Six arrays are depicted: high packing, fully saturated with H1E bound -20° off-dyad; high packing with ρ = 1.6; high packing with ρ = 1.3; with ρ = 1, and H1E bound +20° off-dyad; with ρ = 1, and H1E bound on-dyad; and a low packing array with 1.6 LH/nucleosome. (A) packing ratios; (B) fibers’ images and (C) detailed images of side views of short segments of fibers; (D) top fiber views of the same segments. Alternating nucleosomes are colored blue and white, while nucleosomes with two LHs are colored yellow. LHs are colored as their parental cores (blue, white or yellow). Tails are omitted for clarity.
Our measured sedimentation values closely mirror the corresponding packing ratios of the fibers (Table 2). In low packing arrays (3.6–4.5 nuc/11 nm), the sedimentation rates are between ∼125 S and ∼140 S. The range of sedimentation values for high packing arrays (4.5–6.7 nuc/11 nm) ranges from 140 S to 160 S, close to the sedimentation values measured by Kepper et al. (6) for cross-linked 100-core arrays using much simpler chromatin models with implicit LH and tails.
For ρ = 0.5, our data for 24-nucleosome systems (Figure 3 and Supplementary Table S2) show that lower LH density reduces packing for all LH binding modes in comparison to fully saturated arrays (ρ = 1). The high packing LH binding modes we identified are most sensitive to reduced LH density (–20° mode with H1E, see Supplementary Tables S2A and S2B). Surprisingly, the +20° mode with H1E is rather robust to changes in LH density.
Figure 3.

Packing densities for 24 core arrays with 1 LH per nucleosome and 0.5 LH per nucleosome, as functions of NRL for various binding modes and LH densities, for H1C (top, A) and H1E (bottom, B). The fiber images correspond to fibers with highest and lowest packing ration for the corresponding NRL and LH type.
Increased LH density bends chromatin but maintains compactness
Our analysis of persistence length in arrays with ρ = 1 shows correlation with the packing density. Compact arrays with packing ratios over 5 nuc/11 nm have longer persistence lengths (see Table 2), reaching over 300 nm. In most cases those values correspond to the –20° LH binding mode. The other two modes produce more bendable fibers, with +20° mode having generally the shortest persistence lengths. Interestingly, with the high packing H1E, the shortest persistence lengths for medium to long NRLs (191–218 bp) are obtained with NRL = 209 bp. These fibers have strong electrostatic interactions between LH and parental DNA linkers, and weak interactions between LH and nonparental linkers and between tails and cores.
In arrays with ρ > 1, persistence lengths decrease (for NRL = 200 bp), but packing is preserved. Namely, while in arrays with ρ = 1 and H1E in the –20° mode, fibers with NRL = 200 bp have much longer persistence lengths than fibers with NRL = 209 bp compared to values when ρ > 1 (see Table 2 and Supplementary Table S1).
Figure 4 illustrates the dependence of the persistence length on the LH binding mode using the same fiber examples as in Figure 2. Figure 4 shows that the LH binding mode with highest packing (ρ = 1, H1E –20°, NRL = 200 bp, packing ∼ 5.2 nuc/11 nm) has significantly longer persistence length than other LH binding modes (with ρ = 1, and ρ > 1, see Figure 4B).
Figure 4.

Persistence lengths and fiber images for various array setups for NRL = 200 bp. Six setups are depicted, as shown below vertical bars. The fibers are schematic representations with only nucleosomes (green), with superhelix axis (blue), as calculated by the analysis routine (see the Persistence length calculation routine in the Supplementary material).
A comparison to experimental measurements shows that persistence lengths (Table 2 and Supplementary Table S1) are in close agreement to estimates for human fibroblasts based on the Flory statistical approach (100–140 nm) (56,60,61), or based on the Porod-Kratky worm-like chain model (70–110 nm) (56,62) (see green and yellow vertical lines in Figure 4A). Our values cover the theoretical ranges measured by Rippe (30–220 nm) (63), Wedemann and Langowski (30–265 nm) (56), and Kepper et al. (26–390 nm) (6).
Contact patterns reveal the effects of off-dyad LH binding
Our analysis of internucleosome contact patterns (Figure 5, Supplementary Figures S3 and S4) reveals that the LH binding mode has a strong effect on the fiber architecture, particularly for arrays with NRL = 200 and 209 bp. The LH binding orientation has the strongest effect on ±2, ±3 and ±5 internucleosome contacts. The on-dyad binding mode promotes ±2 (or zigzag) contacts, while the –20° binding mode reduces them, but increases ±3 and ±5 contacts, particularly for NRL = 200 bp and LH = H1E. This corresponds to a tighter packing, as revealed above. The +20° LH binding mode is a strong promoter of dominant ±3 contacts, at the expense of the reduced ±2 contacts. It orients LHs against the natural twisting of the left-handed fiber helix (see Figure 1G), and favors contacts with nonparental over parental DNA (see Figure 5 and Table 3). This produces weaker nucleosome stems, bent linkers, and reduced persistence lengths (the +20° mode has generally lowest persistence lengths).
Figure 5.

Internucleosome contact patterns for 100-core arrays fully saturated with H1C or H1E. Dashed lines correspond to arrays with H1C and full lines to arrays with H1E. (A) Contact patterns for NRL = 182 bp; (B) Contact patterns for NRL = 191 bp; (C) Contact patterns for NRL = 200 bp; (D) Contact patterns for NRL = 209 bp; (E) Contact patterns for NRL = 218 bp.
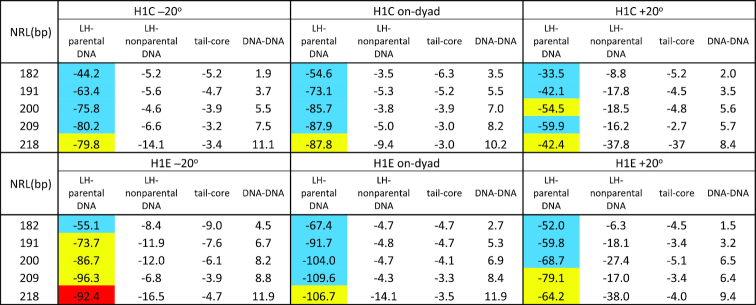
Table 3.
Electrostatic energies (in kcal/mol), between linker histone and parental DNA strands, between linker histone and nonparental DNA strands, between tails and nonparental cores, and between DNA linkers, for 100-nucleosome arrays fully saturated with H1C (upper half) or H1E (lower half). The values are color coded by nucleosome packing: blue - low packing (<4.5 nuc/11 nm), yellow – medium (4.5 > nuc/11 nm < 6), and red - high (>6 nuc/11 nm).
|
In arrays with ρ = 1.3 (Supplementary Figure S3), the nucleosome contacts follow the trends described above, but are softer. Namely, the ±3 contacts are still increased for the dominant +20° binding mode, compared to arrays with dominant on-dyad and –20° binding modes, but they are similar to the ±2 contacts. Overall, these trends associate increased fiber bending and reduced ±5 contacts with lower packing (compare Figures S3 and S4 for NRL = 200 bp). In arrays with ρ = 1.6 (Supplementary Figure S4), the dominant contacts for every setup are generally decreased, compared to ρ = 1.3.
Energetic analysis reveals importance of LH/nonparental DNA and tail/core interactions for fiber compaction
Our analysis of electrostatic energies, in particular LH/parental DNA interactions, LH/nonparental DNA interactions, tail/core interactions, and DNA/DNA interactions (Table 3 and Supplementary Figure S3, and Figure 6), help explain the compaction and bending patterns described above by showing that an optimal balance among these interactions results in high fiber packing. Attractive interactions between LHs and parental DNA linkers (Figure 6A) are always strongest among these four interactions, but intense attraction between LHs and nonparental DNA (Figure 6B), as well between tails and cores (Figure 6C) are necessary for high compaction, such as in fibers with ρ = 1 in which H1E is bound –20° off-dyad. However, dominant electrostatic attraction between LH and parental DNA, at the expense of weak interactions with nonparental DNA (as in setups 5 and 6) cannot produce highly compact fibers. This is the case in the low-packing arrays saturated with H1E bound on-dyad and the ρ = 1.6 case in the last setup. On the other hand, in fibers with ρ = 1 in which H1E is bound +20° off-dyad (setup 4), strong LH interactions with nonparental DNA cannot compensate for the weaker LH/parental DNA interactions (see Supplementary Table S3 and Figure 6). Interestingly, in fibers with ρ > 1, the LH binding modes that individually lead to low packing increase packing when combined, e.g. when fibers with dominant on-dyad LH binding mode have additional LHs bound +20° off-dyad, or vice versa (see Supplementary Table S3 and Figure 6).
Figure 6.

Electrostatic energies (in kcal/mol) for the same setups as in Figure 2 and 4. Electrostatic energy for: (A) linker histones and parental DNA strands; (B) linker histones and nonparental DNA strands; (C) tails and nonparental cores; (D) DNA linkers; (E) Sum.
Interactions between tails and nonparental cores are also important for optimal condensation. In compact arrays, for example with ρ = 1, saturated with H1E bound –20°, tail/nonparental core interactions are 30–50% stronger compared to arrays with H1E bound on-dyad or +20°.
Repulsive interactions between DNA linkers reflect the nucleosome packing level (Figure 6D). The strongest repulsion occurs in arrays with H1E bound on-dyad and –20° where linkers are tightly packed in nucleosome stems or in a zigzag, left-handed superhelix arrangements and LHs intensely interact with both parental and nonparental DNA (setup 1 of Figure 2).
SUMMARY AND DISCUSSION
Recent discoveries of the existence of various LH binding modes have opened new possibilities for the interpretation of the chromatin structure puzzle (9,10,17–22,31,33,34). Together with various LH subtypes and their multiple posttranslational modifications, linker histones offer an additional epigenetic mechanism for gene expression control, besides histone tails post-translational modifications (48). Such regulation controls development and disease progress. For example, changes in LH density in rod photoreceptor cells (16) affect vision disorders. Photoreceptors, as well as erythroid and cells in the nerve system, have LH densities beyond one LH per nucleosome (ρ = 1.3) (13–16), this density can help DNA inside photoreceptor nuclei to be compact and mostly repressed.
Here we have systematically examined by mesoscale modeling the effects of various off and on-dyad LH binding modes and LH densities, over a range of NRLs usually encountered in nature. Specifically, we covered a wide range of DNA lengths (NRL = 182–218 bp), two LH types (H1C and H1E), one on-dyad and two off-dyad binding modes, and LH densities ranging from 0.5 to 1.6 LH/nucleosome for oligonucleosomes with 24 and 100 cores.
Our results help interpret experimental data (6,56,57,60–63) and offer a structural interpretation of the existence of different LH binding modes in chromatin and their synergetic mechanism for producing optimal fiber compaction. Overall, we find that the –20° LH binding mode with H1E achieves highest packing levels (generally above 5 nuc/11 nm) at the cost of significantly increased persistence length. Such fibers, although highly condensed, have limited long-range interactions and serve as strong barriers to transcription. This agrees with the study of Bednar et al. that associates on-dyad binding with uncondensed nucleosomes (35), while Song et al. show condensed nucleosome arrays with LH bound off-dyad (33). Along the same line, Garcia-Saez et al. suggest that a change from fully to partially condensed fiber is associated with a shift from off- to on-dyad binding (64). The on-dyad and +20° modes offer smaller packing ratios but produce more bendable fibers with increased long-range interactions, necessary for looping and folding. We propose that all binding modes are concurrently employed inside chromatin. The simultaneous presence of LHs bound with different modes offers a balance between highly compact and bendable fiber segments. We also show that H1C is a weak condenser of chromatin, as observed experimentally (54).
When ρ > 1, fibers have decreased packing but are generally more bendable. Although the precise LH density in vivo is not known, changes in density affect cell development and disease progression (4,5,10,11). However, some combinations with ρ > 1 produce bendable fibers with relatively high packing ratios as well. In fibers with ρ = 1.3, arrays with dominant –20o LH binding mode are both relatively high packed, as well as flexible. Surprisingly, some LH binding combinations that individually do not produce highly compact fibers reach packing densities of 5 nuc/11 nm when present together. Because LH binding is dynamic (65–68), the binding of the second LH might be transient. Additional LHs might act as backups for the detached LHs and help keep chromatin fiber in a compact and repressed state.
For LH densities below 1 (0.5 LH per nucleosome), the high packing –20° mode with H1E show decreased packing relative to ρ = 1, but other modes are less sensitive to ρ changes. This may be biologically advantageous as LHs diffuse more or less freely through chromatin, so fiber segments robust to the fluctuations of ρ may help maintain the overall chromatin fold. In our work on metaphase chromatin (47), we showed how an LH density decrease from one to zero can control zigzag fiber self-association and enhance long-range interactions by hierarchical looping.
We also showed here that NRL values affect fiber sensitivity to the type of LH, binding mode, and density. While highly compact fibers with NRL = 191 or 200 bp condensed with H1E are sensitive to changes in these parameters, the less compact fibers with NRL = 209 bp are more robust to the LH binding mode and density. This is significant because NRL = 200 bp corresponds to the linker length most often found in human cell lines (55 bp ∼ NRL = 202 bp) (69,70), and belongs to the 10n + 5 series of linker lengths (n = 0, 1, 2, 3 …) generally recognized as repressors of gene expression (70–72). The bendable NRL = 209 bp is close to NRL = 207 bp, which belongs to the 10n series of linker lengths that produces most stable fibers in vitro, when compacted with divalent Mg2+ ions (57,71). Therefore, NRL values of 191 and 200 bp are advantageous for organisms because they offer efficient mechanisms for the control of chromatin compaction (open versus closed chromatin).
Our electrostatic analysis reveals that interactions between LH and parental and nonparental DNA, and between tails and nonparental cores are essential for optimal compaction. The LH binding modes that overemphasize either LH interactions with parental or nonparental DNA (on-dyad or +20°) generally cannot reach maximal compaction because their tail/core interactions are compromised. In arrays with ρ > 1, LH binding modes that individually emphasize one type of LH-DNA interactions, when present together, combine to lead to optimal packing and increased bendability and elasticity (Figure 4 and Supplementary Tables S1 and S3). Additional LHs increase fiber elasticity through increased attractive forces between LH and DNA linkers to enhance fiber folding.
We thus propose that various LH subtypes, their binding modes, and posttranslational modifications (48) form an additional layer of epigenetic control besides modifications of histone tails and DNA methylation. Through the manipulation of nucleosome packing, LHs regulate the local accessibility of DNA to cellular machinery and, through control of fiber bending, determine which segments of chromatin are exposed, and which interact through looping and other long-range contacts.
Taken together, our studies show how LH binding modes and densities can control chromatin structure and act as strong epigenetic regulators. Further studies on the dynamic aspects of the LH binding/unbinding events and interchange among different binding modes for a range of variants will advance our understanding of LH’s role in chromatin structure and function.
Supplementary Material
ACKNOWLEDGEMENTS
Computing was performed on the New York University High Performance Computing cluster Prince and our group's High Performance Computing cluster Schulten.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences [R35GM122562]; Philip Morris and Philip Morris International (to T. S.). Funding for open access charge: National Institute of General Medical Sciences (to T.S.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Kornberg R.D. Chromatin structure: a repeating unit of histones and DNA. Science. 1974; 184:868–871. [DOI] [PubMed] [Google Scholar]
- 2. Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 3. Routh A., Sandin S., Rhodes D.. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hergeth S.P., Schneider R.. The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 2015; 16:1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fyodorov D.V., Zhou B.R., Skoultchi A.I., Bai Y.. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018; 19:192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kepper N., Foethke D., Stehr R., Wedemann G., Rippe K.. Nucleosome geometry and internucleosomal interactions control the chromatin fiber conformation. Biophys. J. 2008; 95:3692–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stehr R., Kepper N., Rippe K., Wedemann G.. The effect of internucleosomal interaction on folding of the chromatin fiber. Biophys. J. 2008; 95:3677–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Öztürk M.A., Cojocaru V., Wade R.C.. Toward an ensemble view of chromatosome structure: a paradigm shift from one to many. Structure. 2018; 26:1050–1057. [DOI] [PubMed] [Google Scholar]
- 9. Cutter A.R., Hayes J.J.. Linker histones: novel insights into structure-specific recognition of he nucleosome. Biochem. Cell Biol. 2017; 95:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Widom J. Chromatin structure: Linking structure to function with histone H1. Curr. Biol. 1998; 8:R788–R791. [DOI] [PubMed] [Google Scholar]
- 11. Pan C., Fan Y.. Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim. Biophys. Acta. 2016; 1859:496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Happel N., Doenecke D.. Histone H1 and its isoforms: contribution to chromatin structure and function. Gene. 2009; 431:1–12. [DOI] [PubMed] [Google Scholar]
- 13. Bates D.L., Thomas J.O.. Histones H1 and H5: one or two molecules per nucleosome?. Nucleic Acids Res. 1981; 9:5853–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pearson E.C., Bates D.L., Prospero T.D., Thomas J.O.. Neuronal nuclei and glial nuclei from mammalian cerebral cortex. Nucleosome repeat lengths, DNA contents and H1 contents. Eur. J. Biochem. 1984; 144:353–360. [DOI] [PubMed] [Google Scholar]
- 15. Woodcock C.L., Skoultchi A.I., Fan Y.. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006; 14:17–25. [DOI] [PubMed] [Google Scholar]
- 16. Popova E.Y., Grigoryev S.A., Fan U., Skoultchi A.I., Zhang S.S., Barnstable C.J.. Developmentally regulated linker histone H1c promotes heterochromatin condensation and mediates structural integrity of rod photoreceptors in mouse retina. J. Biol. Chem. 2013; 288:17895–17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou B.R., Jiang J., Feng H., Ghirlando R., Xiao T.S., Bai Y.. Structural mechanisms of nucleosome recognition by linker histones. Mol. Cell. 2015; 59:628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pruss D., Bartholomew B., Persinger J., Hayes J., Arents G., Moudrianakis E.N., Wolffe A.P.. An asymmetric model for the nucleosome: a binding site for linker histones inside the DNA gyres. Science. 1996; 274:614–617. [DOI] [PubMed] [Google Scholar]
- 19. Hayes J.J. Site-Directed Cleavage of dna by a linker histone-Fe(II) EDTA conjugate: localization of a globular domain binding site within a nucleosome. Biochemistry. 1996; 35:11931–11937. [DOI] [PubMed] [Google Scholar]
- 20. Hayes J.J., Kaplan R., Ura K., Pruss D., Wolffe A.. A putative DNA binding surface in the globular domain of a linker histone is not essential for specific binding to the nucleosome. J. Biol. Chem. 1996; 271:25817–25822. [DOI] [PubMed] [Google Scholar]
- 21. An W., Leuba S.H., van Holde K., Zlatanova J.. Linker histone protects linker DNA on only one side of the core particle and in a sequence-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:3396–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. White A.E., Hieb A.R., Luger K.. A quantitative investigation of linker histone interactions with nucleosomes and chromatin. Sci. Rep. 2016; 6:19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cui F., Zhurkin V.B.. Distinctive sequence patterns in metazoan and yeast nucleosomes: implications for linker histone binding to AT-rich and methylated DNA. Nucleic Acids Res. 2009; 37:2818–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown D.T., Izard T., Misteli T.. Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat. Struct. Mol. Biol. 2006; 13:250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pachov G.V., Gabdoulline R.R., Wade R.C.. On the Structure and Dynamics of the Complex of the Nucleosome and the Linker Histone. Nucleic Acids Res. 2011; 39:5255–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Öztürk M.A., Pachov G.V., Wade R.C., Cojocaru V.. Conformational selection and dynamic adaptation upon linker histone binding to the nucleosome. Nucleic Acids Res. 2016; 44:6599–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fang H., Wei S., Lee T.H., Hayes J.J.. Chromatin structure-dependent conformations of the H1 CTD. Nucleic Acids Res. 2016; 44:9131–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nikitina T., Wang D., Gomberg M., Grigoryev S., Zhurkin V.. Combined micrococcal nuclease and exonuclease III digestion reveals precise positions of the nucleosome core/linker junctions: implications for high-resolution nucleosome mapping. J. Mol. Biol. 2013; 425:1946–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perišić O., Schlick T.. Dependence of the linker histone and chromatin condensation on the nucleosome environment. J. Phys. Chem. B. 2017; 121:7823–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luque, Collepardo-Guevara R., Grigoryev S., Schlick T.. Dynamic condensation of linker histone C-terminal domain regulates chromatin Structure. Nucleic Acids Res. 2014; 42:7553–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou B.R., Feng H., Kato H., Dai L., Yang Y., Zhou Y., Bai Y.. Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou Y.B., Gerchman S.E., Ramakrishnan V., Travers A., Muyldermans S.. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 1998; 395:402–405. [DOI] [PubMed] [Google Scholar]
- 33. Song F., Chen P., Sun D., Wang M., Dong L., Liang D., Xu R.M., Zhu P., Li G.. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014; 344:376–380. [DOI] [PubMed] [Google Scholar]
- 34. Zhou B.R., Feng H., Ghirlando R., Li S., Schwieters C.D., Bai Y.. A small number of residues can determine if linker histones are bound on or off dyad in the chromatosome. J. Mol. Biol. 2016; 428:3948–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bednar J., Garcia-Saez I., Boopathi R., Cutter A.R., Papai G., Reymer A., Syed S.H., Lone I.N., Tonchev O., Crucifix C. et al.. Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol. Cell. 2017; 66:384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou B.R., Jiang J., Ghirlando R., Norouzi D., Sathish Yadav K.N., Feng H., Wang R., Zhang P., Zhurkin V., Bai Y.. Revisit of reconstituted 30-nm nucleosome arrays reveals an ensemble of dynamic structures. J. Mol. Biol. 2018; 430:3093–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlick T., Li B., Olson W.K.. The Influence of Salt on the Structure and Energetics of Supercoiled DNA. Biophys. J. 1994; 67:2146–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beard D., Schlick T.. Modeling salt-mediated electrostatics of macromolecules: the discrete surface charge optimization algorithm and its application to the nucleosome. Biopolymers. 2001; 58:106–115. [DOI] [PubMed] [Google Scholar]
- 39. Zhang Q., Beard D., Schlick T.. Constructing irregular surfaces to enclose macromolecular complexes for mesoscale modeling using the discrete surface charge optimization (DISCO) algorithm. J. Comput. Chem. 2003; 24:2063–2074. [DOI] [PubMed] [Google Scholar]
- 40. Arya G., Schlick T.. Role of histone tails in chromatin folding revealed by a new mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arya G., Schlick T.. A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A. 2009; 113:4045–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grigoryev S., Arya G., Correll S., Woodcock C., Schlick T.. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:13317–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schlick T., Perišić O.. Mesoscale simulations of two nucleosome-repeat length oligonucleosomes. Phys. Chem. Chem. Phys. 2009; 11:10729–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perišić O., Collepardo-Guevara R., Schlick T.. Modeling studies of chromatin fiber structure as a function of DNA linker length. J. Mol. Biol. 2010; 403:777–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Collepardo-Guevara R., Schlick T.. Crucial role of dynamic linker histone binding and divalent ions for DNA Accessibility and gene regulation revealed by mesoscale modeling of oligonucleosomes. Nucleic Acids Res. 2012; 40:8803–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Collepardo-Guevara R., Schlick T.. Chromatin fiber polymorphism triggered by variations of DNA linker lengths. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:8061–8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grigoryev S.A., Bascom G., Buckwalter J.M., Schubert M.B., Woodcock C.L., Schlick T.. Hierarchical looping of zigzag nucleosome chains in metaphase chromosomes. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perišić O., Schlick T.. Computational strategies to address chromatin structure problems. Phys. Biol. 2016; 13:035006. [DOI] [PubMed] [Google Scholar]
- 49. Ozer G., Luque A., Schlick T.. The chromatin fiber: multiscale problems and approaches. Curr. Opin. Struct. Biol. 2015; 31:124–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bascom G.D., Schlick T.. Mesoscale modeling of chromatin fibers. Nuclear Archit. Dyn.- Transl.Epigenet. 2018; 2:123–147. [Google Scholar]
- 51. Eswar N., Marti-Renom M.A., Webb B., Madhusudhan M.S., Eramian D., Shen M., Pieper U., Sali A.. Comparative protein structure modeling With MODELLER. Curr.Protoc.Bioinformatics. 2006; 15:5561–5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Humphrey W., Dalke A., Schulten K.. VMD - Visual Molecular Dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 54. Clausell J., Happel N., Hale T.K., Doenecke D., Beato M.. Histone H1 subtypes differentially modulate chromatin condensation without preventing ATP-Dependent remodeling by SWI/SNF or NURF. PLoS One. 2009; 4:e0007243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Öztürk M.A., Cojocaru V., Wade R.C.. Dependence of chromatosome structure on linker histone sequence and posttranslational modification. Biophys J. 2018; 114:2363–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wedemann G., Langowski J.. Computer simulation of the 30-nanometer chromatin fiber. Biophys. J. 2002; 82:2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Robinson P., Fairall L., Huynh V., Rhodes D.. EM measurements define the dimensions of the “30-nm" chromatin fiber: evidence for a compact, interdigitated structure. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:6506–6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gerchman S.E., Ramakrishnan V.. Chromatin higher-order structure studied by neutron scattering and scanning transmission electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:7802–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bednar J., Horowitz R. A., Grigoryev S.A., Carruthers L.M., Hansen J.C., Koster A.J., Woodcock C.L.. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14173–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van den Engh G., Sachs R., Trask B.J.. Estimating genomic distance from DNA sequence location in cell nuclei by a random walk model. Science. 1992; 257:1410–1412. [DOI] [PubMed] [Google Scholar]
- 61. Yokota H., van den Engh G., Hearst J.E., Sachs R.K., Trask B.J.. Evidence for the organization of chromatin in megabase pair-sized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J. Cell. Biol. 1995; 130:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mehring C. Modellierung der Dynamik von Interphasechromosomen. 1998; Universita¨t Heidelberg: Diplomarbeit. [Google Scholar]
- 63. Rippe K. Making contacts on a nucleic acid polymer. Trends Biochem. Sci. 2001; 26:733–740. [DOI] [PubMed] [Google Scholar]
- 64. Garcia-Saez I., Menoni H., Boopathi R., Shukla M.S., Soueidan L., Noirclerc-Savoye M., Le Roy A., Skoufias D.A., Bednar J., Hamiche A. et al.. Structure of an H1-Bound 6-Nucleosome array reveals an untwisted two-start chromatin fiber conformation. Mol. Cell. 2018; 72:1–14. [DOI] [PubMed] [Google Scholar]
- 65. Misteli T., Gunjan A., Brown D.T.. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000; 408:877–881. [DOI] [PubMed] [Google Scholar]
- 66. Lever M.A., Th’ng J.P., Hendzel M.J.. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature. 2000; 408:873–876. [DOI] [PubMed] [Google Scholar]
- 67. Stasevich T.J., Mueller F., McNally J.G.. Dissecting the binding mechanism of the linker histone in live cells: an integrated FRAP analysis. EMBO J. 2010; 29:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Collepardo-Guevara R., Schlick T.. The effect of linker histone's nucleosome binding affinity on chromatin unfolding mechanisms. Biophys. J. 2011; 101:1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schones D.E., Cui K., Cuddapah S., Roh T.-Y., Barski A., Wang Z., Wei G., Zhao K.. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008; 5:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bascom G.D., Kim T., Schlick T.. Kilobase pair chromatin fiber contacts promoted by living-system-like DNA linker length distributions and nucleosome depletion. J. Phys. Chem. B. 2017; 121:3882–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Norouzi D., Katebi A., Cui F., Zhurkin V.B.. Topological diversity of chromatin fibers: Interplay between nucleosome repeat length, DNA linking number and the level of transcription. AIMS Biophys. 2017; 2:613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Voong L.N., Xi L., Sebeson A.C., Xiong B., Wang J.-P., Wang X.. Insights into nucleosome organization in mouse embryonic stem cells through chemical mapping. Cell. 2016; 167:1555–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.