

Abstract
Breast cancer is the most common malignancy among women, with the highest incidence rate worldwide. Dysregulation of long noncoding RNAs during the preliminary stages of breast carcinogenesis is poorly understood. In this study, we performed RNA sequencing to identify long noncoding RNA expression profiles associated with early‐stage breast cancer. RNA sequencing was performed on six invasive ductal carcinoma (IDC) tissues along with paired normal tissue samples, seven ductal carcinoma in situ tissues, and five apparently normal breast tissues. We identified 375 differentially expressed lncRNAs (DElncRNAs) in IDC tissues compared to paired normal tissues. Antisense transcripts (~ 58%) were the largest subtype among DElncRNAs. About 20% of the 375 DElncRNAs were supported by typical split readings leveraging their detection confidence. Validation was performed in n = 52 IDC and paired normal tissue by qRT‐PCR for the identified targets (ADAMTS9‐AS2, EPB41L4A‐AS1, WDFY3‐AS2, RP11‐295M3.4, RP11‐161M6.2, RP11‐490M8.1, CTB‐92J24.3, and FAM83H‐AS1). We evaluated the prognostic significance of DElncRNAs based on TCGA datasets and report that overexpression of FAM83H‐AS1 was associated with patient poor survival. We confirmed that the downregulation of ADAMTS9‐AS2 in breast cancer was due to promoter hypermethylation through in vitro silencing experiments and pyrosequencing.
Keywords: ADAMTS9‐AS2, breast cancer, FAM83H‐AS1, long noncoding RNAs, ncRNAs, RNA sequencing
Abbreviations
- DCIS
ductal carcinoma in situ
- IDC
invasive ductal carcinoma
- lincRNA
long intergenic noncoding RNA
- lncRNA
long noncoding RNAs
- PCA
principal component analysis
- PCC
Pearson′s correlation coefficient
- TNBC
triple‐negative breast cancers
1. Introduction
Breast cancer is the most common cancer among women (ASR‐43.1) with highest mortality rates (Ferlay et al., 2013). Breast cancer is broadly classified into noninvasive ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC). Understanding the mechanism of breast carcinogenesis at genetic and transcriptional level can aid in characterization of DCIS or early‐stage IDC tumors. Gene expression signatures are used to classify IDC subtypes of hormone receptor‐positive (estrogen and progesterone receptors), that is, luminal A and B, and hormone receptor‐negative HER2 and basal‐like (Perou et al., 2000; Sorlie et al., 2001) breast cancer subtypes. Next‐generation sequencing has enabled global profiling of mRNAs and noncoding RNAs (ncRNAs) including long ncRNAs (lncRNAs) and microRNAs. lncRNAs have gained immense importance in gene regulation and are known to play an important role in cancer development and prognosis (Huarte, 2015; Prensner and Chinnaiyan, 2011; Rao et al., 2017). Understanding the divergent expression of lncRNAs in early‐stage breast tumors can help elucidate its functional role in carcinogenesis.
Specific lncRNAs signatures are known to be associated with different molecular subtypes of breast cancer. DSCAM‐AS1 was identified specifically in ER‐positive breast tumors and shown to increase aggression and drug resistance (Niknafs et al., 2016). Similarly, AFAP1‐AS1 was predominantly found to be dysregulated in HER2 and triple‐negative breast cancers (TNBC) (Shen et al., 2015; Yang et al., 2016a). H19 was identified to be overexpressed in ER/PR‐positive breast adenomas, and BC200 was implicated to be distinctly elevated in benign tumors and not in invasive subtypes and hence is of prognostic significance (Adriaenssens et al., 1998; Iacoangeli et al., 2004). HOTAIR was demonstrated to gain activity in BRCA1‐mutated tumors. In a normal cell, BRCA1 competes with HOTAIR in binding to EZH2 of PRC2 (Wang et al., 2013). The functional characteristics of certain lncRNAs, such as UCA1, GAS5, and XIST, have established them as breast cancer‐associated tumor suppressors, while HOTAIR, TINCR, and DSCAM‐AS1 are known as oncogenic lncRNAs (Wang et al., 2017; Xu et al., 2017). Support vector machine‐based prediction of breast cancer intrinsic subtype using lncRNA expression profile and PAM50 gene signature using TCGA datasets was recently proposed as an improved prediction model (Zhang et al., 2018).
Despite known association of lncRNA expression with molecular subtype, recently reported lncRNAs have emerging role in relevant signaling or druggable pathways. lncRNA CYTOR was reported to be associated with breast cancer progression through EGFR signaling pathway (Van Grembergen et al., 2016). NKILA was observed to promote heterotrimeric complex formation (p50/p60/IκB) and inhibit IκB phosphorylation, thus regulating NF‐κB signaling (Liu et al., 2015). LINK‐A was reported to aid in stabilizing HIF1α in normoxic conditions of TNBC. Through BRK/PTK6 activation and phosphorylation of HIF1α, LINK‐A substantiates its kinase activation and cancer signaling potential (Lin et al., 2016). Alternatively, breast cancer‐associated lncRNAs important in drug targeting pathways can also be useful prognostic biomarkers. In the present study, we have done RNA sequencing in early‐stage tumors (stage I–IIA IDC, DCIS) and noncancerous breast tissue samples to identify lncRNAs that play a role in early‐stage breast cancer. We speculate that aberrant expression of lncRNAs could be an early event in breast cancer development, and hence, the study was aimed to identify dysregulated lncRNAs and the mechanism of dysregulation in breast cancer.
2. Materials and methods
2.1. Study population and sample classification
The study cohort includes patients diagnosed and treated for breast cancer at Cancer Institute (WIA), Chennai, Tamil Nadu, India. These patients were histologically confirmed of invasive ductal carcinoma (IDC—stage I–II A) and DCIS. Apparently, normal breast tissues were obtained from patients undergoing surgery for breast conditions other than malignancy. Samples having > 70% for cancer cells following histopathological examination were included in the study. Paired normal and apparently normal tissues completely free of tumor cells were selected and kept frozen (−80 °C) until further processing. Total RNA sequencing was done for 24 samples, that is, tumor (n = 6), paired normal (matched normal; n = 6), DCIS (n = 7), and apparently normal (n = 5). Validation cohort of IDC (n = 52) and corresponding paired normal tissue were used to gauge candidate lncRNAs. The clinicopathological features of patients in the discovery and validation cohort are detailed in Table S1. All patients were informed about the study, and their written consent for participation was obtained. The Institutional Ethical Committee approved the study and the protocol duly conforming the guidelines set by the Declaration of Helsinki.
2.2. RNA isolation and library preparation
Total RNA was isolated from frozen tissues using TRIzol method and purification by NucleoSpin RNA Isolation Kit (Macherey‐Nagel, Düren, Germany), which includes an on‐column DNase treatment. The quality and quantity of total RNA was evaluated through 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Ribosomal RNA was depleted (EpiGentek, USA), and cDNA library was prepared using Illumina TruSeq Stranded Total RNA Library Prep Kit. The library profile was verified using 2100 Bioanalyzer (Agilent Technologies). Subsequent RNA sequencing of cDNA libraries with paired‐end reads (2 × 100 bps reads) was performed according to the standard Illumina protocol using HiSeq 2500 sequencing platform.
2.3. RNA sequencing and data analysis
Raw reads were assessed for Phred quality using fastqc (Andrews, 2010), and low bases and adaptor sequences were trimmed off using Fqtrim (Pertea, 2015) retaining reads of length ≥ 75 bases. Clean reads were aligned against human reference genome (GRCh38 assembly) with Gencode V24 annotation using hisat2 (Baruzzo et al., 2017) with default parameters. Exon centric read counts were obtained from binary alignment map using HTSeq (Anders et al., 2015) using the script ‘htseq count’ for all samples independently. lncRNAs identified with ≥ 15 reads in at least three samples per cohort, that is, IDC, paired normal, DCIS, and apparent normal were further investigated for differential expression using DESeq (Anders and Huber, 2010). Read counts obtained from HTSeq were normalized using ‘estimateSizeFactors’ variance and were modeled using ‘estimateDispersions’. The differentially expressed genes were computed using ‘nbinomTest’ functions of DESeq. Significant differential expression was defined if |log2 (fold‐change)| > 1 and q‐value (Bonferroni and Benjamini–Hochberg adjusted P value) < 0.1. Expression profile of lncRNAs from TCGA breast cancer dataset (TCGA‐BRCA; n = 837 invasive tumors and n = 105 normal samples) was used for survival analysis (Li et al., 2015). Kaplan–Meier plots for differentially expressed lncRNAs (DElncRNAs) were generated for tumor stages as well as molecular subtypes and evaluated using log‐rank test.
2.4. lncRNA‐mRNA co‐expression network analysis
Pearson′s correlation coefficient (PCC) was used to determine linear correlation between mRNA and lncRNA expression profiles using r package. DElncRNAs DElncRNAs ‐mRNA pairs with |PCC| ≥ 0.9 were considered for network analysis using string v10 (Szklarczyk et al., 2015) with organism ‘Human’ as backend database and Cytoscape (Shannon et al., 2003).
2.5. Real‐time quantitative PCR
Total RNA of 500 ng was used for preparing cDNA libraries using QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Gene expression was estimated by QuantStudio 12K Flex Real‐Time PCR System using TaqMan™ gene expression assays (Applied Biosystems, Thermo Fisher Scientific, Waltham, Massachusetts, USA) containing primers and probes specific for lncRNA and GAPDH. The expression values were calculated using the method (ΔC t = ΔC t target gene‐ΔC t reference gene).
2.6. siRNA‐mediated knock‐down of DNMT1
Expression of ADAMTS9‐AS2 was evaluated in MDAMB‐231 and MCF7 cells. The cells were cultured in Dulbecco's modified Eagle's medium with 10% FBS at 37 °C. Knock‐down was carried out using Lipofectamine 3000 (Life Technologies, Carlsbad, CA, USA), siRNA targeting DNMT1 (Ambion, USA) with cells maintained in Opti‐MEM (Life Technologies) during and after transfection. Transfected cells were collected after 48 and 72 h for total RNA and DNA isolation.
2.7. DNA extraction, Bisulfite treatment, and pyrosequencing
Genomic DNA was extracted from tissues and cultured MDAMB‐231 and MCF7 cells using NucleoSpin Kit (Macherey‐Nagel, GmbH). About 500 ng of DNA was used for bisulfite treatment following manufacturer's protocol of EZ DNA Methylation‐Gold Kit (Zymo Research, Irvine, CA, USA). Bisulfite‐treated DNA was amplified using inventoried PyroMark CpG assay Hs_AC132007.1_01_PM (Qiagen, GmbH) with primers spanning ADAMTS9‐AS2 promoter region. The amplified fragment was sequenced using pyromark Q48 Autoprep (Qiagen, GmbH) and analyzed by pyromark q24 software v 2.0.7.
2.8. Statistical analyses
graphpad prism7 (GraphPad Prism Software Inc., San Diego, CA, USA) was used for evaluating qRT‐PCR gene expression data. Student's t‐test was used for pairwise analysis of tumor and paired normal samples. Welch correction was done if significant difference in variance was observed and Wilcoxon rank sum test was applied whenever non‐Gaussian distribution was followed.
3. Results
3.1. Expression profile of lncRNAs in ductal carcinoma in situ and invasive ductal carcinoma
RNA sequencing resulted in generation of ~ 89 million reads per sample with ~ 87.24% alignment against human genome build Hg38. We identified ~ 2 689 lncRNAs and ~ 18 132 mRNAs with ≥ 15 reads in at least three samples per cohort (Table 1, Table S2). In agreement with previous reports, lncRNAs were expressed at comparatively lower levels than mRNAs (Fig. S1A–D). Principal component analysis (PCA) plots based on lncRNA quantification showed distinct segregation of tumors (IDC and DCIS) from paired and apparent normal samples reflecting the characteristic variation of lncRNA expression profile (Fig. 1A, Fig. S1E). Differential expression analysis was performed between IDC, DCIS, and control samples in four categories, that is, IDC vs. paired normal (TN), IDC vs. apparent normal (TA), DCIS vs. apparent normal (DA), and IDC vs. DCIS (TD); (Fig 1B–D, Fig S2, Table S3‐S6).
Table 1.
Number of DElncRNAs in DCIS and early‐stage breast cancer
| Comparison set | lncRNA | |||
|---|---|---|---|---|
| Overexpressed | Downregulated | Total | Split reads | |
| IDC vs. paired normal | 195 | 180 | 375 | 96 |
| IDC vs. apparent normal | 38 | 56 | 94 | 25 |
| DCIS vs. apparent normal | 29 | 40 | 69 | 24 |
| IDC vs. DCIS | 5 | 7 | 12 | 3 |
Figure 1.
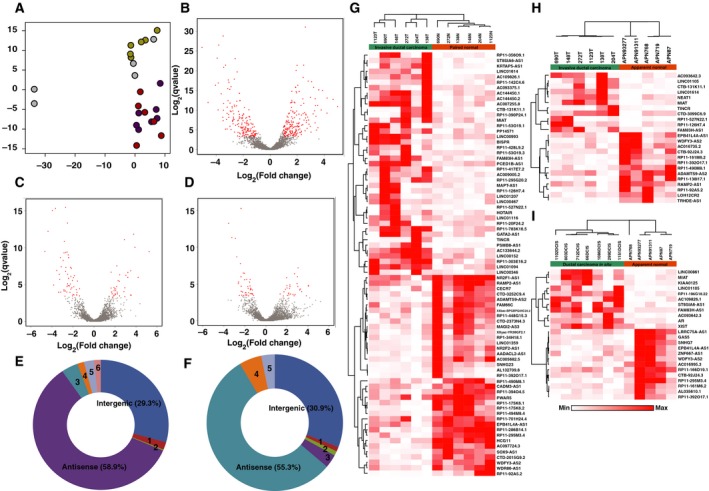
Summary of DElncRNAs identified in DCIS and early‐stage breast cancer. (A) PCA based on lncRNA expression profile to demonstrate distinct segregation of tissues of various pathological types. Color legend: Apparent normal: yellow; DCIS: purple; Paired normal: green; IDC: red samples. (B) Volcano plot represents the expression pattern of lncRNA in IDC vs. paired normal samples. (C) Volcano plot represents the expression pattern of lncRNA in IDC vs. apparent normal samples. (D) Volcano plot represents the expression pattern of lncRNA in DCIS vs. apparent normal samples. (E) Pie chart representing DElncRNA subtypes in IDC vs. paired normal samples [1—intron overlapping (1.9%); 2—noncoding transcript (0.3%); 3—TEC (0.4%); 4—sense overlapping (1.5%); 5—processed transcript (2.4%); 6—completely intronic (1.6%)]. (F)Pie chart representing DElncRNA subtypes in IDC vs. apparent normal samples [1—intron overlapping (1.1%); 2—completely intronic (1.1%); 3—TEC (3.2%); 4—processed transcript (5.3%); 5—sense overlapping (3.2%)]. (G) Heatmap with supervised clustering represents the expression trend of DElncRNAs in IDC vs. paired normal samples. (H) Heatmap with supervised clustering represents the expression trend of DElncRNAs in IDC vs. apparent normal samples. (I) Heatmap with supervised clustering represents the expression trend of DElncRNAs in DCIS vs. apparent normal samples.
We observed antisense RNAs (asRNA) and long intergenic noncoding RNAs (lincRNAs) to be the major lncRNA subtypes differentially expressed among these four groups. asRNAs accounted for 58.9% of total DElncRNAs in IDC compared to paired normal and 55.3% compared to apparently normal samples (Fig. 1E–F). WDR86‐AS1 emerged as a novel antisense lncRNA in our data, whereas ADAMTS9‐AS2 (Li et al., 2017; Peng et al., 2017) and ST8SIA6‐AS1 (Yang et al., 2016a,2016b) have previously been reported in other studies (Fig. 1G–I).
3.2. Identification of novel lncRNAs differentially expressed in breast tumors
Dysregulated lncRNAs with evidence of ≥ 2 junction reads in each comparison groups were further investigated (Fig. S1F–I). We identified 21 lncRNAs (11 overexpressed and 10 downregulated) showing a differential expression pattern (Table 2, Fig. 2). Among them, MIAT, FAM83H‐AS1, EPB41L4A‐AS1, WDFY3‐AS2, and RP11‐392O17.1 were commonly deregulated in TN, TA, and DA comparison groups (Fig. 2). Further, LINC01614, RP11‐490M8.1, and CTB‐92J24.3 were novel DElncRNAs identified in early‐stage breast cancer.
Table 2.
List of DElncRNAs common among various comparison sets
| lncRNA | IDC vs. apparent normal | IDC vs. paired normal | DCIS vs. apparent normal | Expression status |
|---|---|---|---|---|
| MIAT | 2.89 | 1.47 | 2.72 | Overexpressed |
| FAM83H‐AS1 | 1.96 | 1.92 | 2.01 | Overexpressed |
| LINC01614 | 5.24 | 6.1 | – | Overexpressed |
| RP11‐527N22.1 | 4.2 | 3.77 | – | Overexpressed |
| TINCR | 3.22 | 4.22 | – | Overexpressed |
| CTB‐131K11.1 | 2.42 | 1.96 | – | Overexpressed |
| RP11‐126H7.4 | 2.22 | 1.77 | – | Overexpressed |
| LINC01105 | 3.48 | 4.04 | – | Overexpressed |
| AC093642.3 | 2.94 | 3.39 | – | Overexpressed |
| ST8SIA6‐AS1 | – | 2.48 | 3.21 | Overexpressed |
| AC109826.1 | – | 2.12 | 2.99 | Overexpressed |
| RAMP‐AS1 | −1.38 | −1.43 | – | Downregulated |
| ADAMTS9‐AS2 | −1.65 | −3.31 | – | Downregulated |
| RP11‐490M8.1 | −2.32 | −1.8 | – | Downregulated |
| RP11‐92A5.2 | −3.53 | −5.05 | – | Downregulated |
| EPB41L4A‐AS1 | −1.55 | −1.18 | −1.5 | Downregulated |
| WDFY3‐AS2 | −1.68 | −1.44 | −1.65 | Downregulated |
| RP11‐392O17.1 | −2.69 | −2.72 | −2.63 | Downregulated |
| RP11‐161M6.2 | −2.44 | −2.11 | – | Downregulated |
| CTB‐92J24.3 | −2.42 | −2.42 | – | Downregulated |
| RP11‐295M3.4 | – | −2.79 | −2.77 | Downregulated |
Figure 2.
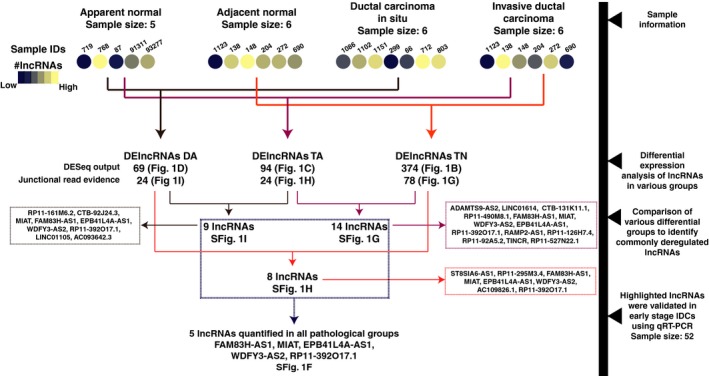
Schematic of lncRNA analysis and cross‐comparison of DElncRNAs in multiple comparison groups.
3.3. Validation of candidate lncRNA expression in breast tumor and paired normal
We selected 12 candidate lncRNAs (five upregulated lncRNAs: MIAT, FAM83H‐AS1, LINC01614, ST8SIA6‐AS1, and CTB‐131K11.1, and seven downregulated lncRNAs: ADAMTS9‐AS2, EPB41L4A‐AS1, WDFY3‐AS2, RP11‐161M6.2, RP11‐295M3.4, RP11‐490M8.1, and CTB‐92J24.3) for validation using TaqMan™ gene expression assays in n = 52 early‐stage IDC samples (Fig. 3A). We observed statistically significant dysregulation of seven out of 12 lncRNAs identified using RNA‐Seq. Among them, ADAMTS9‐AS2 (Fig. 3B) was observed to be the most commonly downregulated lncRNA in tumor tissues (13.59‐fold). We also confirmed significant downregulation of CTB92J24.3 (11.82‐fold), RP11‐295M3.4 (3.5‐fold), RP11‐490M8.1 (3.7‐fold), WDFY3‐AS2 (4.3‐fold), and EPB41L4A‐AS1 (2.09‐fold; Fig. 3C–G). FAM83H‐AS1 was the most significantly overexpressed lncRNA in tumors (8.9‐fold) compared to the paired normal tissues (Fig. 3H). Although, MIAT and LINC01614 were upregulated, they were statistically insignificants (Fig. 3I,J). Whereas, ST8SIA6‐AS1 and CTB‐131K11.1 were found to be down regulated contradicting our RNA sequencing results (Fig. 3K,L). Expression pattern of ST8SIA6‐AS1 and CTB‐131K11.1 in TCGA datasets were similar to the validation results (Fig S3). To evaluate the involvement of receptor status, expression levels of 12 DElncRNAs from validation cohort were correlated with receptors status (ER, PR, HER2; Fig. S4A–D). We observed that MIAT was overexpressed exclusively in samples that were ER+PR+Her2+ whereas RP11‐161 M6.2 was overexpressed in ER−PR−.
Figure 3.
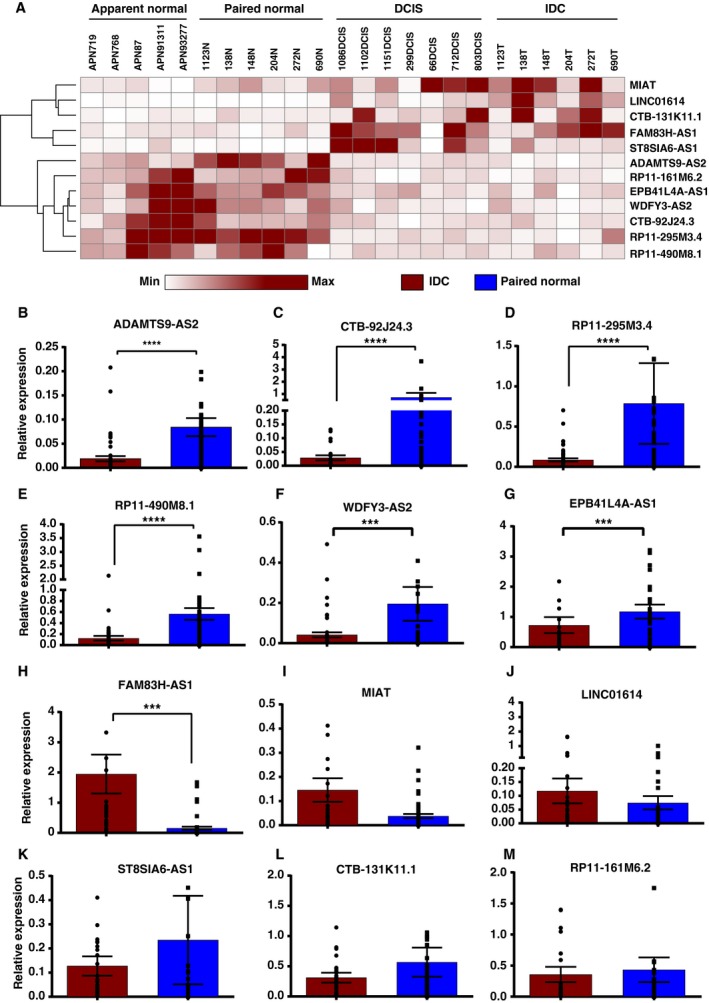
Expression validation of DElncRNAs using qRT‐PCR in cohort of 52 early‐stage breast cancer samples (A) Heatmap of differentially regulated lncRNAs showing expression trend in discovery set of samples. (B) Relative expression of ADAMTS9‐AS2. (C) Relative expression of CTB‐92J24.3. (D) Relative expression of RP11‐295M3.4. (E) Relative expression of RP11‐490M8.1. (F) Relative expression of WDFY3‐AS2. (G) Relative expression of EPB41L4A‐AS1. (H) Relative expression of FAM83H‐AS1. (I) Relative expression of MIAT. (J) Relative expression of LINC01614. (K) Relative expression of ST8SIA6‐AS1. (L) Relative expression of CTB‐131K11.1. (M) Relative expression of RP11‐161M6.2. (B–M are relative expression levels of lncRNA evaluated in validation set of samples); (Wilcoxon sign rank test P‐value < 0.0001 = ****, P < 0.001 = *** and not indicated for nonsignificant candidates).
3.4. ADAMTS9‐AS2 promoter is hypermethylated in breast tumors
Yao et al. (2014) reported the downregulation of ADAMTS9‐AS2 by promoter methylation in gliomas. Hence, methylation levels of the promoter region of ADAMTS9‐AS2 in our validation set of tumor and paired normal samples (n = 52) were done using pyrosequencing. We observed a nearly two‐fold (1.9) increase in methylation levels (P < 0.0001) in the promoter region (+879 to +929 bp from TSS) of tumor samples compared to paired normal samples (Fig. 4A).
Figure 4.
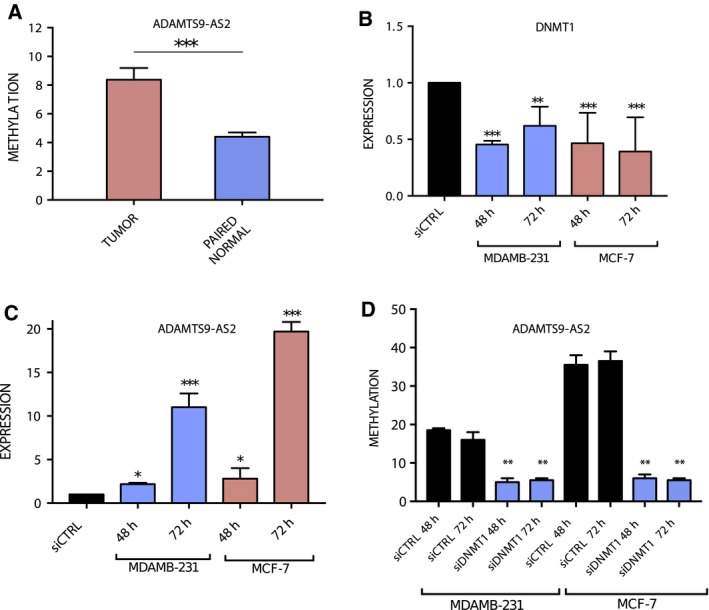
(A) Relative methylation levels of ADAMTS9‐AS2 promoter in tumor vs. paired normal tissue (N = 52). (B) Expression levels of DNMT1 with siRNA treatment in MDAMB‐231 and MCF7 cells. (C) Expression of ADAMTS9‐AS2 in MDAMB‐231 and MCF7 cells on DNMT1 knock‐down. (D) Relative methylation levels of ADAMTS9‐AS2 promoter in MDAMB‐231 and MCF7 cells with DNMT1 knock‐down (error bars represent SEM, Student's t‐test with two biological independent replicates were used to determine statistical significance; ***P < 0.001, **P < 0.01 and *P < 0.05).
3.5. Knock‐down of DNA methyltransferase 1 increases ADAMTS9‐AS2 expression
In order to investigate promoter methylation‐mediated regulation of ADAMTS9‐AS2 expression, DNMT1 was knocked down in MDAMB‐231 and MCF7 using short interfering RNA. The downregulation of DNMT1 led to subsequent overexpression of ADAMTS9‐AS2 by 1.93‐fold (P < 0.001) and 2.32‐fold (P < 0.001) in MDAMB‐231 and MCF7, respectively (Fig. 4B,C). Loss of promoter methylation was observed using pyrosequencing in DNMT1 siRNA‐transfected MDAMB‐231 (2.6‐fold; P = 0.001) and MCF‐7 cells (6.7‐fold; P = 0.007; Fig. 4D). These results show that ADAMTS9‐AS2 is overexpressed in both MDAMB‐231 and MCF7 cells following DNMT1 silencing indicating methylation‐mediated suppression of ADAMTS9‐AS2 in breast cancer cells.
3.6. Prognostic lncRNAs in early‐stage breast cancer
Survival analysis was done to investigate the prognostic potential of candidate lncRNA using TCGA datasets. We observed FAM83H‐AS1 was significantly overexpressed by ~ 4‐fold in TN, TA, as well as DA pairs and its overexpression is associated with overall poor survival in luminal A, ER‐positive tumors, stage 3 datasets, and overall breast tumor datasets irrespective of subtypes (Fig. 5A–D). Overexpression of WDFY3‐AS2 in luminal A, ER‐positive tumors, and breast tumor datasets irrespective of subtypes (Fig. 5E,F,H) is significantly associated with adverse outcomes, whereas downregulation of RP11‐161M6.2 in breast cancer and CTB‐92J24.3 in stage 3 was observed significantly associated with poor overall survival (Fig. 5K). We observed significant association with overexpression of WDFY3‐AS2 (Fig. 5G) and downregulation of RP11‐161M6.2 in stage 2 of breast cancer based on TANRIC analysis indicating them as potential early prognostic markers (Fig. 5G,J).
Figure 5.
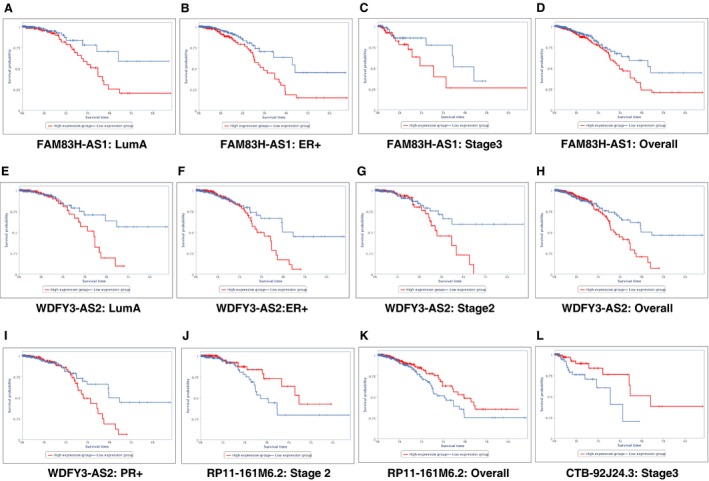
Kaplan–Meier plots derived from TANRIC depicting significant overall poor survival of patients associated with DElncRNAs (A) FAM83H‐AS1 in luminal A molecular subtype. (B) FAM83H‐AS1 in ER+ molecular subtype. (C) FAM83H‐AS1 in stage 3 dataset. (D) FAM83H‐AS1 in overall breast cancer dataset. (E) WDFY3‐AS2 in luminal A molecular subtype. (F) WDFY3‐AS2 in ER+ molecular subtype. (G) WDFY3‐AS2 in stage 2 dataset. (H) WDFY3‐AS2 in overall breast cancer dataset. (I) WDFY3‐AS2 in PR+ molecular subtype. (J) RP11‐161M6.2 in stage 2 dataset. (K) RP11‐161M6.2 in overall breast cancer dataset. (L) CTB‐92J24.3 in stage 3 dataset.
3.7. Co‐expression and pathway analysis
Guilt‐by‐association method was employed to speculate the putative functions of lncRNAs. This approach investigates the association of mRNA expression patterns with lncRNAs using Pearson's correlation analysis. A correlation analysis between DElncRNA‐DEmRNA pairs was done, and only those with PCC ≥ |0.9| were considered significantly co‐expressed. The co‐expressed pairs were filtered for lncRNA with typical junctional read evidence which led to the identification of 2398 pairs consisting of 78 lncRNA and 1097 mRNA between IDC and paired normal samples and 385 pairs consisting of 24 lncRNA and 245 mRNA between IDC and apparent normal samples.
Similarly, 26 pairs were co‐expressed in DCIS vs. apparent normal samples consisting of 11 lncRNA and 26 mRNA and 10 co‐expressed lncRNA‐mRNA pairs in IDC compared to DCIS representing three lncRNA and 10 mRNA (Tables [Link], [Link], [Link], [Link]). Among 2398 co‐expressed lncRNA‐mRNA pairs in IDC vs. paired normal samples, 2225 (92.83%) harbor on different chromosomes (trans‐acting) whereas remaining pairs are cis‐acting. Similarly, 351 (91.64%) out of 383 in IDC vs. apparent normal samples and 23 (85.17%) out of 27 in DCIS vs. apparent normal samples are located on different chromosomes.
Co‐expressed mRNAs were further analyzed using StringDB for network analysis. To augment guilt‐by‐association concept, we further focused on mRNA network that is reported to co‐express irrespective of lncRNA. We observe that partial sets of mRNAs from 22 DElncRNAs in IDC compared to paired normal samples were co‐expressed according to StringDB analysis. After removing disconnected nodes and filtering high confidence nodes from the network, genes co‐expressed with RP11‐142C4.6 (Fig. S5A) were found enriched for extracellular regions (red nodes) and overrepresented for extracellular matrix organization (green nodes) and disassembly (blue nodes) whereas genes co‐expressed with RAMP2‐AS1 were enriched on the cell membrane (red nodes; Fig. S5A,B). Genes co‐expressed with RP11‐701H24.4 were enriched for integral component of membrane (green nodes) and activation of cellular processes (blue nodes; Fig. S5C). In case of PSMB8‐AS1, we observed overrepresentation of immune response and (red nodes) involved in type I interferon signaling pathway (blue nodes; Fig. S5D). We observed enrichment of biological process such as cell division (yellow nodes), cell cycle process (pink nodes), and microtubule cytoskeleton (red nodes) in genes positively co‐expressed with TINCR and negatively co‐expressed with LINC01359 (Figs [Link], [Link]). Interestingly, most genes co‐expressed with PSMB8‐AS1, TINCR, and LINC01359 are also known to co‐express with each other according to StringDB. Using Cytoscape, we were able to segregate the subnetwork of 76 genes potentially governed jointly by TINCR (65 genes) and LINC01359 (55 genes), which resulted in submodules of genes with core histone protein domains (green nodes) and involved in pathways in cancer (blue nodes).
4. Discussion
Aberrant expression of lncRNAs is documented in various cancers (Huarte, 2015; Prensner and Chinnaiyan, 2011). In recent years, lncRNAs have gained importance in early detection and better prognosis of tumors (Chandra Gupta and Nandan Tripathi, 2017). Although several lncRNAs associated with breast cancer have been reported previously, studying aberrantly expressed lncRNAs specific to early‐stage breast cancer will provide insight into molecular mechanisms associated with breast cancer development. It will also result in identification of putative markers that might be useful in diagnosis or prognosis of breast cancer. Previous studies have associated altered expression of lncRNAs with specific breast cancer subtypes. For example, HOTAIR is a lncRNA that is highly expressed in HER2+ breast cancers whereas HOTAIRM1 is highly expressed in basal‐like subgroup of breast cancers (Su et al., 2014). Luminal A types showed overexpression of LINC00160, and abundance of DSCAM‐AS1 was reported in luminal B subtypes of breast cancer (Jonsson et al., 2015; Vu et al., 2016). MALAT, lncRNA‐ATB, BC200, XIST, and H19 are some of other lncRNAs frequently associated with breast tumorigenesis and progression (Hansji et al., 2014). Functionally important lncRNAs in early‐stage breast cancers are less reported. Our study evaluated the landscape of lncRNA expression in early‐stage breast cancer [IDC (stage I–IIA) and DCIS breast tissues] to identify aberrantly expressed lncRNAs.
The DESeq analysis resulted in identification of 375 DElncRNAs in IDC compared to paired normal samples and 94 DElncRNAs in IDC compared to apparent normal samples. The analysis also identified 69 DElncRNAs in DCIS compared to apparent normal samples. We identified several antisense lncRNAs including ADAMTS9‐AS2, EPB41L4A‐AS1, WDFY3‐AS2, FAM83H‐AS1, ST8SIA6‐AS1, CTB‐92J24.3, and CTB‐131K11.1 that were aberrantly expressed. Twelve candidate lncRNAs that showed significant differential expression were further validated in 52 paired tumor and normal breast samples. We observed significant downregulation of ADAMTS9‐AS2, WDFY3‐AS2, RP11‐295M3.4, RP11‐490M8.1, and CTB‐92J24.3 and significant overexpression of FAM83H‐AS1 in breast cancer. We found ADAMTS9‐AS2 to be significantly downregulated in tumor compared to paired normal breast tissues. ADAMTS9‐AS2 is an antisense transcript originating from the opposite stand coding for ADAMTS9 which is a known inhibitor of angiogenesis and is implicated to have a tumor‐suppressive role. Functional importance of ADAMTS9 in nasopharyngeal and esophageal cancers has been described (Lo et al., 2010). ADAMTS9‐AS2 like ADAMTS9 is downregulated in glioblastoma (Yao et al., 2014), colorectal cancer (Li et al., 2016), bladder cancer, lung adenocarcinoma, and ER+ breast cancers (Li et al., 2017). Yao et al. have shown that promoter methylation regulates ADAMTS9‐AS2 expression by knocking down DNMT1 in glioma cells. We found that methylation of ADAMTS9‐AS2 controls its expression through correlative DNMT1 knock‐down in MDAMB‐231 and MCF7 cells. Similar results were observed when methylation levels at ADAMTS9‐AS2 promoter were compared between tumors and paired normal tissues using pyrosequencing. We observed DNA methylation‐mediated loss of ADAMTS9‐AS expression in stage I breast cancer. Among other downregulated lncRNAs, WDFY3‐AS2 has recently been reported with TGF‐B‐induced EMT of breast cancer cells through hnRNP‐R modulated positive regulation of STAT3 and WDFY3 (Richards et al., 2016). Downregulation of WDFY‐AS2 was found in diffuse glioma and strongly associated with poor prognosis (Wu et al., 2018). EPB41L4A‐AS1 (also known as TIGA1) has been shown to be transcribed during growth arrest but has not been extensively studied in cancer to elucidate its role (Yabuta et al., 2006). RP11‐161M6.2 was found to be overexpressed in ER/PR−negative and HER2− positive breast cancers in our samples. The finding indicates an association of RP11‐161M6.2 and estrogen receptor and is possibly downregulated in estrogen‐mediated signaling. Similarly, MIAT was dominantly expressed in ER/PR/HER2+ breast cancers samples.
FAM83H‐AS1 was consistently overexpressed in breast tumor samples and overall survival, analysis of TCGA datasets showed poor prognosis of the upregulated group which are in agreement with other studies in breast, colorectal, and lung cancers (Lu et al., 2018; Yang et al., 2016a,2016c; Zhang et al., 2017). Functional studies have demonstrated proliferative potential of FAM83H‐AS1 through MET/EGFR signaling in lung adenocarcinoma and NOTCH1 signaling pathway in colorectal cancer. Overexpression of FAM83H‐AS1 in luminal‐type breast cancer was associated with good prognosis in patients (Yang et al., 2016a). Detection of FAM83H‐AS1 expression levels in plasma could be a potential diagnostic and prognostic biomarker for breast cancer.
5. Conclusion
In summary, this study has shed light on novel lncRNA and substantiated several previous findings on lncRNA involved in early‐stage breast cancers. We report 375 and 94 lncRNA differentially expressed in tumor samples compared to paired and apparent normal samples, respectively, and 69 DElncRNAs in DCIS compared to apparent normal samples. Seven downregulated and five upregulated lncRNA were further validated to discover significant lncRNA candidate with potential role in breast carcinogenesis. ADAMTS9‐AS2 was one of the lncRNAs consistently downregulated in patient samples, and experimental evidence proved promoter methylation as major cause of ADAMTS9‐AS2 downregulation in breast cancer. Moreover, LINC01614, RP11‐490M8.1, and CTB‐92J24.3 are novel lncRNA reported in our study that has not been associated with breast cancer earlier. Our study also contributes to the existing evidence on MIAT and FAM83H‐AS1 as crucial lncRNA expressed at preliminary stages of breast cancer.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AKDMR, KP, HG, and SM planned the experiments. AKDMR, KP, SKJ, and BM carried out the experiments. VS and SS contributed to surgical excision of tissue samples and histopathological confirmation. AKDMR, KP, AC, HG, and SM contributed to the interpretation of the results. KP and HG contributed to visualizations. AKDMR and SM validated the results. AKDMR and KP wrote the original manuscript. AC, HG, and SM reviewed, edited, and finalized the manuscript. AC, HG, AP, TR, and SM designed the study. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Supporting information
Fig. S1. Expression pattern of lncRNAs and protein coding genes in various pathological subtype and comparison of DElncRNAs in different groups. (A) Comparative histogram represents relatively lower expression of lncRNAs (blue bars) compared to protein coding genes (grey bars) based on raw read count profile of apparent normal samples (n = 5) (B) Comparative histogram represents relatively lower expression of lncRNAs (green bars) compared to protein coding genes (grey bars) based on raw read count profile of paired normal samples (n = 6) (C) Comparative histogram represents relatively lower expression of lncRNAs (yellow bars) compared to protein coding genes (grey bars) based on raw read count profile of DCIS samples (n = 7) (D) Comparative histogram represents relatively lower expression of lncRNAs (red bars) compared to protein coding genes (grey bars) based on raw read count profile of IDC samples (n = 6) (E) PCA using normalized read counts of protein coding genes. Color legend. Apparent normal samples: Yellow, DCIS samples: Purple, Paired normal samples: Green, IDC samples: Red (F) Venn diagram depicting comparison of differential expression analysis group IDC vs. paired normal, IDC vs. apparent normal and DCIS vs. apparent normal samples (G) Venn diagram depicting comparison of differential expression analysis group IDC vs. paired normal and IDC vs. apparent normal samples (H) Venn diagram depicting comparison of differential expression analysis group DCIS vs. apparent normal and IDC vs. paired normal samples (I) Venn diagram depicting comparison of differential expression analysis group IDC vs. apparent normal and DCIS vs. apparent normal samples.
Fig. S2. Summary of lncRNA expression profile in IDC vs. DCIS. (A) Volcano plot representing expression pattern in IDC vs. DCIS (B) Heatmap depicting expression trend of differentially expressed gene in IDC vs. DCIS.
Fig. S3. LncRNA expression profile in various molecular subtype of breast cancer obtained from TCGA dataset using TANRIC platform (A) RP11‐161M6.2 (B) ADAMTS9‐AS2 (C) CTB‐92J24.3 (D) CTB‐131K11.1 (E) EPB41L4A‐AS1 (F) FAM83H‐AS1 (G) LINC01614 (H) MIAT (I) RP11‐295M3.4 (J) RP11‐490M8.1 (K) ST8SIA6‐AS1 (L) WDFY3‐AS2.
Fig. S4. Expression levels of deregulated lncRNAs in various combination of receptors (ER, PR, HER2) positivity in TCGA dataset (A) ER+ or PR+ or Her2+ (B) ER+ or PR+ along with Her2− (C) ER+ and PR− and Her2+ (D) ER− or PR− along with Her2+ (E) Molecular subtype stratification of validation cohort; Red background: Upregulated lncRNAs and Blue background: Downregulated lncRNAs.
Fig. S5. High confidence interaction network (score: 0.7) representing differentially expressed mRNA that are known to co‐express with each other as per String analysis and with lncRNA with Pearson correlation coefficient ≥ 0.9 (A) RP11‐142C4.6 (B) RAMP2‐AS2 (C) RP11‐701H24.4 (D) PSMB8‐AS1.
Fig. S6. High confidence interaction network (score: 0.7) representing differentially expressed mRNA that are known to co‐express with each other as per String analysis and with lncRNA with Pearson correlation coefficient ≥ 0.9 with TINCR.
Fig. S7. High confidence interaction network (score: 0.7) representing differentially expressed mRNA that are known to co‐express with each other as per String analysis and with lncRNA with Pearson correlation coefficient ≤ −0.9 with LINC01359.
Table S1. List of clinicopathological features of patients’ tissue samples used in discovery and validation cohort in the study.
Table S2. Read alignment statistics and number of genes identified in different samples.
Table S3. Complete list of DElncRNAs identified to be differentially expressed in IDC (T) vs. paired normal (N) samples with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S4. Complete list of DElncRNAs identified to be differentially expressed in IDC (T) vs. apparent normal (APN) with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S5. Complete list of DElncRNAs identified to be differentially expressed in DCIS vs. apparent normal (APN) with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S6. Complete list of DElncRNAs identified to be differentially expressed in IDC (T) vs. DCIS with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S7. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in IDC vs. paired normal with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S8. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in IDC vs. apparent normal with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S9. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in DCIS vs. apparent normal with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S10. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in IDC vs. DCIS with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S11. List of gene expression assays
Acknowledgements
We thank Dr. Uma Devi K.R. and Dr. S. Sivakumar, National Institute for Research in Tuberculosis, ICMR, Chennai for providing pyrosequencing facility. Krishna Patel is recipient of Senior Research Fellowship from Council of Scientific and Industrial Research (CSIR). This research study was fully funded by Department of Biotechnology, Govt. of India (BT/PR8152/AGR/36/739/2013). We acknowledge DST Research and Development for infrastructural facility at Department of Molecular Oncology, Cancer Institute (WIA).
Arunagiri Kuha Deva Magendhra Rao and Krishna Patel contributed equally to this manuscript.
Contributor Information
Harsha Gowda, Email: harsha@ibioinformatics.org.
Samson Mani, Email: samsonn.m@gmail.com.
Data availability
Raw sequencing data are available in Sequence Read Archive hosted by National Center for Biotechnology Information (NCBI) search database with accession number PRJNA484546.
References
- Adriaenssens E, Dumont L, Lottin S, Bolle D, Lepretre A, Delobelle A, Bouali F, Dugimont T, Coll J and Curgy JJ (1998) H19 overexpression in breast adenocarcinoma stromal cells is associated with tumor values and steroid receptor status but independent of p53 and Ki‐67 expression. Am J Pathol 153, 1597–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S and Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT and Huber W (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA and Grant GR (2017) Simulation‐based comprehensive benchmarking of RNA‐seq aligners. Nat Methods 14, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra Gupta S and Nandan Tripathi Y (2017) Potential of long non‐coding RNAs in cancer patients: from biomarkers to therapeutic targets. Int J Cancer 140, 1955–1967. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F (2013) GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. International Agency for Research on Cancer, Lyon, France: Available from: http://globocan.iarc.fr, accessed on 06/08/2018. [Google Scholar]
- Hansji H, Leung EY, Baguley BC, Finlay GJ and Askarian‐Amiri ME (2014) Keeping abreast with long non‐coding RNAs in mammary gland development and breast cancer. Front Genet 5, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M (2015) The emerging role of lncRNAs in cancer. Nat Med 21, 1253–1261. [DOI] [PubMed] [Google Scholar]
- Iacoangeli A, Lin Y, Morley EJ, Muslimov IA, Bianchi R, Reilly J, Weedon J, Diallo R, Bocker W and Tiedge H (2004) BC200 RNA in invasive and preinvasive breast cancer. Carcinogenesis 25, 2125–2133. [DOI] [PubMed] [Google Scholar]
- Jonsson P, Coarfa C, Mesmar F, Raz T, Rajapakshe K, Thompson JF, Gunaratne PH and Williams C (2015) Single‐molecule sequencing reveals estrogen‐regulated clinically relevant lncRNAs in breast cancer. Mol Endocrinol 29, 1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Dai Y, Wang F and Hou S (2016) Differentially expressed long non‐coding RNAs and the prognostic potential in colorectal cancer. Neoplasma 63, 977–983. [DOI] [PubMed] [Google Scholar]
- Li J, Han L, Roebuck P, Diao L, Liu L, Yuan Y, Weinstein JN and Liang H (2015) TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res 75, 3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li B, Zheng Y, Li M, Shi L and Pu X (2017) Exploring functions of long noncoding RNAs across multiple cancers through co‐expression network. Sci Rep 7, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Li C, Xing Z, Hu Q, Liang K, Han L, Wang C, Hawke DH, Wang S, Zhang Y et al (2016) The LINK‐A lncRNA activates normoxic HIF1alpha signalling in triple‐negative breast cancer. Nat Cell Biol 18, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X, Lin L, Yao H, Su F, Li D et al (2015) A cytoplasmic NF‐kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell 27, 370–381. [DOI] [PubMed] [Google Scholar]
- Lo PH, Lung HL, Cheung AK, Apte SS, Chan KW, Kwong FM, Ko JM, Cheng Y, Law S, Srivastava G et al (2010) Extracellular protease ADAMTS9 suppresses esophageal and nasopharyngeal carcinoma tumor formation by inhibiting angiogenesis. Cancer Res 70, 5567–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Dong W, Zhao P and Liu Z (2018) lncRNA FAM83H‐AS1 is associated with the prognosis of colorectal carcinoma and promotes cell proliferation by targeting the Notch signaling pathway. Oncol Lett 15, 1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niknafs YS, Han S, Ma T, Speers C, Zhang C, Wilder‐Romans K, Iyer MK, Pitchiaya S, Malik R, Hosono Y et al (2016) The lncRNA landscape of breast cancer reveals a role for DSCAM‐AS1 in breast cancer progression. Nat Commun 7, 12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Zhang L, Yuan C, Zhou L, Xu S, Lin Y, Zhang J, Yin W and Lu J (2017) Expression profile analysis of long noncoding RNA in ER‐positive subtype breast cancer using microarray technique and bioinformatics. Cancer Manag Res 9, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA et al (2000) Molecular portraits of human breast tumours. Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Pertea G (2015) Fqtrim: v0. 9.4 release (2015) 10.5281/zenodo.20552. Available online at: https://zenodo.org/record/20552
- Prensner JR and Chinnaiyan AM (2011) The emergence of lncRNAs in cancer biology. Cancer Discov 1, 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Rajkumar T and Mani S (2017) Perspectives of long non‐coding RNAs in cancer. Mol Biol Rep 44, 203–218. [DOI] [PubMed] [Google Scholar]
- Richards E, Challa S, Li Y, Permuth‐Wey J, Bui M, Coppola D, Sellers T and Cheng J (2016) LncRNA WDFY3‐AS2 contributes to the EMT and metastasis in breast cancer. AACR. [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Xie B, Ma Z, Yu W, Wang W, Xu D, Yan X, Chen B, Yu L, Li J et al (2015) Identification of novel long non‐coding RNAs in triple‐negative breast cancer. Oncotarget 6, 21730–21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98, 10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Malouf GG, Chen Y, Zhang J, Yao H, Valero V, Weinstein JN, Spano JP, Meric‐Bernstam F, Khayat D et al (2014) Comprehensive analysis of long non‐coding RNAs in human breast cancer clinical subtypes. Oncotarget 5, 9864–9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al (2015) STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Grembergen O, Bizet M, de Bony EJ, Calonne E, Putmans P, Brohee S, Olsen C, Guo M, Bontempi G, Sotiriou C et al (2016) Portraying breast cancers with long noncoding RNAs. Sci Adv 2, e1600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TN, Pramana S, Calza S, Suo C, Lee D and Pawitan Y (2016) Comprehensive landscape of subtype‐specific coding and non‐coding RNA transcripts in breast cancer. Oncotarget 7, 68851–68863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ye C, Xiong H, Shen Y, Lu Y, Zhou J and Wang L (2017) Dysregulation of long non‐coding RNA in breast cancer: an overview of mechanism and clinical implication. Oncotarget 8, 5508–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, Wang L, Sarver A, Koller A, Zhi J et al (2013) BRCA1 is a negative modulator of the PRC2 complex. EMBO J 32, 1584–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zhao Z, Chai R, Liu Y, Wang K, Wang Z, Li G, Huang R, Jiang H and Zhang K (2018) Expression profile analysis of antisense long non‐coding RNA identifies WDFY3‐AS2 as a prognostic biomarker in diffuse glioma. Cancer Cell Int 18, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Kong D, Chen Q, Ping Y and Pang D (2017) Oncogenic long noncoding RNA landscape in breast cancer. Mol Cancer 16, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuta N, Onda H, Watanabe M, Yoshioka N, Nagamori I, Funatsu T, Toji S, Tamai K and Nojima H (2006) Isolation and characterization of the TIGA genes, whose transcripts are induced by growth arrest. Nucleic Acids Res 34, 4878–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lv SX, Lv L, Liu YH, Dong SY, Yao ZH, Dai XX, Zhang XH and Wang OC (2016a) Identification of lncRNA FAM83H‐AS1 as a novel prognostic marker in luminal subtype breast cancer. Onco Targets Ther 9, 7039–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lyu S, Dong S, Liu Y, Zhang X and Wang O (2016b) Expression profile analysis of long noncoding RNA in HER‐2‐enriched subtype breast cancer by next‐generation sequencing and bioinformatics. Onco Targets Ther 9, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xu L, Wang Q, Wang M and An G (2016c) Dysregulation of long non‐coding RNA profiles in human colorectal cancer and its association with overall survival. Oncol Lett 12, 4068–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhou B, Zhang J, Geng P, Liu K, Zhu Y and Zhu W (2014) A new tumor suppressor LncRNA ADAMTS9‐AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumour Biol 35, 7935–7944. [DOI] [PubMed] [Google Scholar]
- Zhang J, Feng S, Su W, Bai S, Xiao L, Wang L, Thomas DG, Lin J, Reddy RM, Carrott PW et al (2017) Overexpression of FAM83H‐AS1 indicates poor patient survival and knockdown impairs cell proliferation and invasion via MET/EGFR signaling in lung cancer. Sci Rep 7, 42819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Wang J, Ghoshal T, Wilkins D, Mo YY, Chen Y and Zhou Y (2018) lncRNA gene signatures for prediction of breast cancer intrinsic subtypes and 33 prognosis. Genes (Basel) 9, E65 10.3390/genes9020065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression pattern of lncRNAs and protein coding genes in various pathological subtype and comparison of DElncRNAs in different groups. (A) Comparative histogram represents relatively lower expression of lncRNAs (blue bars) compared to protein coding genes (grey bars) based on raw read count profile of apparent normal samples (n = 5) (B) Comparative histogram represents relatively lower expression of lncRNAs (green bars) compared to protein coding genes (grey bars) based on raw read count profile of paired normal samples (n = 6) (C) Comparative histogram represents relatively lower expression of lncRNAs (yellow bars) compared to protein coding genes (grey bars) based on raw read count profile of DCIS samples (n = 7) (D) Comparative histogram represents relatively lower expression of lncRNAs (red bars) compared to protein coding genes (grey bars) based on raw read count profile of IDC samples (n = 6) (E) PCA using normalized read counts of protein coding genes. Color legend. Apparent normal samples: Yellow, DCIS samples: Purple, Paired normal samples: Green, IDC samples: Red (F) Venn diagram depicting comparison of differential expression analysis group IDC vs. paired normal, IDC vs. apparent normal and DCIS vs. apparent normal samples (G) Venn diagram depicting comparison of differential expression analysis group IDC vs. paired normal and IDC vs. apparent normal samples (H) Venn diagram depicting comparison of differential expression analysis group DCIS vs. apparent normal and IDC vs. paired normal samples (I) Venn diagram depicting comparison of differential expression analysis group IDC vs. apparent normal and DCIS vs. apparent normal samples.
Fig. S2. Summary of lncRNA expression profile in IDC vs. DCIS. (A) Volcano plot representing expression pattern in IDC vs. DCIS (B) Heatmap depicting expression trend of differentially expressed gene in IDC vs. DCIS.
Fig. S3. LncRNA expression profile in various molecular subtype of breast cancer obtained from TCGA dataset using TANRIC platform (A) RP11‐161M6.2 (B) ADAMTS9‐AS2 (C) CTB‐92J24.3 (D) CTB‐131K11.1 (E) EPB41L4A‐AS1 (F) FAM83H‐AS1 (G) LINC01614 (H) MIAT (I) RP11‐295M3.4 (J) RP11‐490M8.1 (K) ST8SIA6‐AS1 (L) WDFY3‐AS2.
Fig. S4. Expression levels of deregulated lncRNAs in various combination of receptors (ER, PR, HER2) positivity in TCGA dataset (A) ER+ or PR+ or Her2+ (B) ER+ or PR+ along with Her2− (C) ER+ and PR− and Her2+ (D) ER− or PR− along with Her2+ (E) Molecular subtype stratification of validation cohort; Red background: Upregulated lncRNAs and Blue background: Downregulated lncRNAs.
Fig. S5. High confidence interaction network (score: 0.7) representing differentially expressed mRNA that are known to co‐express with each other as per String analysis and with lncRNA with Pearson correlation coefficient ≥ 0.9 (A) RP11‐142C4.6 (B) RAMP2‐AS2 (C) RP11‐701H24.4 (D) PSMB8‐AS1.
Fig. S6. High confidence interaction network (score: 0.7) representing differentially expressed mRNA that are known to co‐express with each other as per String analysis and with lncRNA with Pearson correlation coefficient ≥ 0.9 with TINCR.
Fig. S7. High confidence interaction network (score: 0.7) representing differentially expressed mRNA that are known to co‐express with each other as per String analysis and with lncRNA with Pearson correlation coefficient ≤ −0.9 with LINC01359.
Table S1. List of clinicopathological features of patients’ tissue samples used in discovery and validation cohort in the study.
Table S2. Read alignment statistics and number of genes identified in different samples.
Table S3. Complete list of DElncRNAs identified to be differentially expressed in IDC (T) vs. paired normal (N) samples with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S4. Complete list of DElncRNAs identified to be differentially expressed in IDC (T) vs. apparent normal (APN) with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S5. Complete list of DElncRNAs identified to be differentially expressed in DCIS vs. apparent normal (APN) with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S6. Complete list of DElncRNAs identified to be differentially expressed in IDC (T) vs. DCIS with adjusted P‐values < 0.1 in this study along with normalized read counts from individual samples.
Table S7. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in IDC vs. paired normal with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S8. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in IDC vs. apparent normal with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S9. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in DCIS vs. apparent normal with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S10. Complete list of dysregulated mRNA co‐expressed with dysregulated lncRNAs supported by split reads in IDC vs. DCIS with Pearson correlation coefficient (PCC) ≥ 0.9.
Table S11. List of gene expression assays
Data Availability Statement
Raw sequencing data are available in Sequence Read Archive hosted by National Center for Biotechnology Information (NCBI) search database with accession number PRJNA484546.