

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by the loss of upper and lower motor neurons. ALS exhibits high phenotypic variability including age and site of onset, and disease duration. To uncover epigenetic and transcriptomic factors that may modify an ALS phenotype, we used a cohort of Australian monozygotic twins (n = 3 pairs) and triplets (n = 1 set) that are discordant for ALS and represent sporadic ALS and the two most common types of familial ALS, linked to C9orf72 and SOD1. Illumina Infinium HumanMethylation450K BeadChip, EpiTYPER and RNA-Seq analyses in these ALS-discordant twins/triplets and control twins (n = 2 pairs), implicated genes with consistent longitudinal differential DNA methylation and/or gene expression. Two identified genes, RAD9B and C8orf46, showed significant differential methylation in an extended cohort of >1000 ALS cases and controls. Combined longitudinal methylation-transcription analysis within a single twin set implicated CCNF, DPP6, RAMP3, and CCS, which have been previously associated with ALS. Longitudinal transcriptome data showed an 8-fold enrichment of immune function genes and under-representation of transcription and protein modification genes in ALS. Examination of these changes in a large Australian sporadic ALS cohort suggest a broader role in ALS. Furthermore, we observe that increased methylation age is a signature of ALS in older patients.
Subject terms: DNA methylation, Gene expression, Methylation analysis, Gene expression analysis, Amyotrophic lateral sclerosis
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by the rapidly progressive loss of the upper and lower motor neurons. Disease onset commonly occurs in middle to late age1 and typically results in death within three to five years. Existing treatments are of limited effect, and despite intensive effort, the pathogenic mechanisms underlying disease are still poorly understood. A recognised family history (familial ALS, FALS) is seen in approximately 10% of cases while the remainder are considered sporadic (SALS)2. The familial and sporadic forms of the disease are clinically and pathologically indistinguishable3. To date, the only proven cause of ALS are gene mutations leading to motor neuron death. Pathogenic repeat expansions in the C9orf72 gene and missense mutations in the SOD1 gene are the most frequent known causes of ALS worldwide, yet no cause has been identified for the majority of patients (>80%2). Even in those individuals with a proven causal gene mutation, inter- and intra-familial phenotypic heterogeneity is commonly observed1,4. Age of disease onset may vary by more than 60 years and disease duration may be measured in months or in decades. Affected individuals, particularly those with a C9orf72 repeat expansion, may present with ALS or frontotemporal dementia (FTD), or a mixed phenotype. Causal mutations may show incomplete penetrance4 and indeed monozygotic twins are more commonly discordant for ALS than concordant5. Taken together, this phenotypic variability suggests a significant contribution from modifying factors in disease manifestation.
Epigenetic and transcriptional profiling have implicated differential DNA methylation and/or gene expression in ALS. C9orf72 has been shown to have increased methylation6,7 and decreased transcription8,9 in ALS/FTD patients with the pathogenic repeat expansion. Other major ALS genes, however, including SOD1, FUS and TARDBP, are generally unmethylated and show no differences between patients and controls10–12. Nevertheless, changes in expression of some ALS genes is apparent in sporadic disease13. Whole methylome and transcriptome studies in spinal cord and blood tissue have found global changes10,14,15 and implicated various genes, pathways and several overlapping themes including changes that affect immune response14,16,17 and cellular transport18,19.
Disease discordant monozygotic (MZ) twins hold great potential for studies that seek to identify epigenetic and transcriptomic factors that modify the phenotype of complex human diseases. Identical twin studies can account for confounding factors such as genetic variation and the early development environment. Such studies have informed understanding of phenotypic variation in Parkinson’s disease20, Alzheimer’s disease21, systemic lupus erythematosus22, and depression23, among others. Previous DNA methylation studies of known causal ALS genes in ALS-discordant MZ twins found no aberrant methylation between twins24,25, while twin-based methylome-wide studies suggested a different epigenetic age in affected twins25,26 and identified potentially altered GABA signalling25 and immune response27. Nevertheless, further studies are required because the differentially methylated sites implicated in initial screens have often failed to be validated in targeted studies using bisulphite pyrosequencing26. Similarly, candidate molecular pathways have shown limited overlap between twin sets25 and changes in methylation are yet to be linked to changes in transcription. It remains unclear which of the observed differences in either DNA methylation or gene expression reflect ALS discordance between co-twins. It is also unclear whether these differences in DNA methylation correlate with differential gene expression on a transcriptome-wide scale.
In this study, we undertook comprehensive methylome- and transcriptome-wide analysis of a longitudinal ALS-discordant cohort comprising MZ triplets and twins, representing the three most common types of ALS: C9orf72-linked ALS, SOD1-linked ALS and sporadic ALS. We analysed methylome- and transcriptome-wide data, independently and in combination, in an attempt to identify disease-relevant methylation changes and their downstream impact. Co-twin analyses indicated a significant interaction effect between age and disease status on DNA methylation age, with older twins showing a consistent difference between ALS-affected and unaffected co-twins in a longitudinal series. Furthermore, we identified several genes likely to contribute to ALS through integration of longitudinal twin genome-wide DNA methylation and transcription data, further assessed in a large sporadic ALS case-control cohort.
Results
ALS-discordant and control twin/triplet sets
Clinical and sample information for the three discordant MZ twin sets, one discordant MZ triplet set and two control twin sets are included in Table 1. Pedigrees and extended pedigrees are shown in Fig. 1. All individuals with ALS have been screened for causal mutations in known ALS genes. The FALS twin set has a pathogenic hexanucleotide repeat expansion in C9orf72. The FALS triplet set harbours a SOD1 p.I114T mutation.
Table 1.
Twin cohort details.
| MZ set | ALS | Disease Status | Sex | Mutation | Age of onset | Age at samplingA | Disease duration (months) | 450K samples (n)B | RNA-Seq samples (n) | EpiTYPER samples (n) |
|---|---|---|---|---|---|---|---|---|---|---|
| Female SALS twin set | SALS | ALS | F | 42.7 | 43.5–45.1 | Alive at 51 months | 7(+1) | — | — | |
| Unaffected | F | 43.9–45.1 | 7(+1) | — | — | |||||
| Male SALS twin set | SALS | ALS | M | 78.5 | 79.8–80.2 | 28.4 | 3(+1) | 3 | — | |
| Unaffected | M | 79.8–80.2 | 3(+1) | 3 | — | |||||
| C9orf72 twin set | FALS | ALS | M | C9orf72 HRE | 52 | 54.1 | 36 | 1 | — | 1 |
| Asymptomatic | M | C9orf72 HRE | 54.3–55 | 2 | — | 2 | ||||
| SOD1 triplet set | FALS | ALS | F | SOD1 p.I114T | 50 | 50.3 | Unknown | 1 | — | 1 |
| Asymptomatic | F | SOD1 p.I114T | 50.3 | 1 | — | 1 | ||||
| Asymptomatic | F | SOD1 p.I114T | 50.3 | 1 | — | 1 | ||||
| Control twin set 1 | NA | Control | F | NA | 46.1 | NA | 1 | — | ||
| Control | F | 46.1 | 1 | — | ||||||
| Control twin set 2 | NA | Control | M | NA | 36.8 | NA | — | — | 1 | |
| Control | M | 31.8–43.0C | — | — | 3C |
HRE: hexanucleotide repeat expansion; FALS: familial ALS; SALS: sporadic ALS; APresence of an age range indicates longitudinal samples were collected; BNumber of technical replicates during blood collection indicated in brackets; CMiddle sample matched to co-twin.
Figure 1.

ALS-discordant twin/triplet set pedigrees. Pedigrees for four sets of ALS-discordant twins/triplets, with gene mutations indicated. Circles represent females and squares represent males. Diagonal lines indicate deceased individuals. Filled shapes indicate ALS, open shapes with a dot indicate mutation carriers and open shapes are unaffected non-carriers. Horizontal lines between twins/triplets indicate confirmed monozygosity. HRE: hexanucleotide repeat expansion.
Targeted analysis of methylation in mutation-known MZ sets
To assess whether differential methylation of the C9orf72 or SOD1 CpG islands were associated with the disease discordance we observe in the C9orf72 twin set and SOD1 triplets, we investigated the status of CpG methylation of the C9orf72 and SOD1 CpG islands. To perform a high-density, targeted analysis, we used EpiTYPER, with additional support from a number of Infinium HumanMethylation450K CpG sites present in the same region.
SOD1 methylation in the SOD1 MZ triplet set shows a consistent methylation pattern
We used EpiTYPER to quantify methylation of the upstream SOD1 CpG island encompassing the SOD1 promoter region and exon 1 in the discordant MZ triplets carrying the SOD1 p.I114T mutation and a pair of control twins from another SOD1 p.I114T family that were negative for the SOD1 mutation. Additionally, five SOD1 CpG sites present in the Infinium HumanMethylation450K data set were located within the CpG island (Fig. 2A). Neither the 23 CpG units within the CpG island, nor the five 450K SOD1 CpG sites, showed any consistent methylation differences between ALS affected and ALS unaffected MZ triplets, nor control twins (Fig. 2A).
Figure 2.

Neither SOD1 nor C9orf72 CpG islands are differentially methylated between mutation-positive ALS-discordant twins/triplets. The relative location of targeted CpG islands (CGI) and exon 1 are indicated for SOD1 (A, top) and C9orf72 (B, top). (A) Methylation of the CpG island spanning the promoter region and exon 1 of SOD1 does not show differential methylation between an ALS-affected triplet and unaffected co-triplets, concordant for SOD1 p.I114T. Methylation status was determined using both EpiTYPER (bottom) and 450K (middle) assays. (B) Transcript variants (T1, T2, and T3) and the position of the repeat expansion (black diamond) relative to exon 1 are shown for C9orf72 (top). Methylation of the C9orf72 promoter region/expansion flanking CpG islands are not differentially methylated between ALS-discordant co-twins that carry the C9orf72 hexanucleotide repeat expansion in either EpiTYPER (bottom) or 450K data sets (middle).
No differences were observed in C9orf72 methylation in the C9orf72 MZ twin set
The quantitative methylation status of two CpG islands associated with C9orf72 was determined using EpiTYPER. The amplicons covered the entirety of both CpG islands, the promoter region and adjacent intronic/intergenic regions. The intronic pathogenic (GGGGCC)n repeat expansion (indicated with a black diamond in Fig. 2B) is flanked by the two CpG islands. In the disease discordant FALS twin set harbouring a C9orf72 expansion, methylation across the GpG island (CGI) measured by the EpiTYPER assay are highly concordant and generally unmethylated (Fig. 2B). Similarly, in the four 450K probes associated with C9orf72, none of the CpG sites show a clear difference in methylation between the co-twins (Fig. 2B).
Whole methylome analysis of disease discordant MZ twins/triplets
Co-twin/triplet differences in DNA methylation age (DNAm) reflects an age-dependant effect
Horvath’s DNA methylation age algorithm28 used methylation levels of 353 CpG sites to predict the epigenetic age from each twin/triplet sample in Table 1. DNA methylation age has a high correlation with chronological age across multiple tissue types, thus enabling calculation of the biological age of an individual. We tested the association of methylation age with disease status and chronological age in a mixed model while controlling for sex. The effect of disease status on methylation age was found to be highly dependent upon chronological age (p = 1.3E-5, Fig. 3A). Briefly, with increasing age, asymptomatic co-twins were estimated to have a younger epigenetic age than their ALS-affected twin. This result was most evident in the approximately 20-year difference in methylation age between twins in the oldest disease discordant twin set of this cohort (Fig. 3A).
Figure 3.

DNA methylation age (DNAm), but neither global mean methylation nor cell composition, varies between ALS-discordant twins/triplets. (A) DNA methylation age was more discrepant between ALS discordant twins with increasing chronological age (p = 1.3E-5), with greater DNAm aging in affected twins/triplets than their unaffected co-twin/-triplets, when controlling for age and sex. (B) Global mean methylation across 386183 CpG sites found no significant difference in global methylation between ALS-affected and unaffected co-twins/-triplets when controlling for age and sex (p = 0.08). (C) Proportions of six white blood cell types over time were estimated for ALS-affected and unaffected twins/triplets. Proportions for each cell type were not significantly associated with disease status of twin/triplet samples when controlling for age and sex (CD4+ T cells, p = 0.77; CD8+ T cells, p = 0.24; Monocytes, p = 0.60; B cells, p = 0.21; Natural killer cells, p = 0.52; granulocytes, p = 0.63). For collection times with technical replicate samples per person, points represent the mean at that time, with the standard deviation indicated with a line.
Global methylation and cell type proportions do not show any effect of disease status
Global methylation was calculated as the mean methylation across all Infinium HumanMethylation450K CpG sites passing data processing (n = 386183). No significant effect of disease on global methylation was found when controlling for sex and age at sample collection (p = 0.08, Fig. 3B). To better reflect influence on transcription, CpG sites were classified according to CpG density: high density CpG islands, intermediate density in islands, island shores, and low CpG density. Mean methylation within each of these four levels of CpG density does not show any effect of disease status (HC, p = 0.93; IC, p = 0.99; ICshore, p = 0.82; LC, p = 0.093, Supplementary Fig. S2). Full blood count data were unavailable from twin/triplet sets at time of sample collection. Therefore, blood cell proportions for each twin/triplet sample were estimated from methylation data using Houseman et al.’s algorithm29 and the six cell types were assessed for association with disease status. Disease status did not have a significant effect on any of the cell types when controlling for age at sample collection and sex (all p > 0.2, Fig. 3C).
Differentially methylated probes were identified across discordant MZ twins/triplets
The established DNA methylation twin-study method of statistical significance and the magnitude of pairwise methylation differences were combined to detect differentially methylated probes in discordant MZ twin studies. 59 probes were identified as differentially methylated across twin/triplet sets (full list in Supplementary Table S5, 9 top-ranked probes shown in Fig. 4A). All 59 probes were used for hierarchical clustering and principal components analysis (PCA) of the longitudinal MZ cohort to investigate the presence of a disease signature. Both hierarchical clustering and PCA did not indicate that samples cluster by disease status, but rather approximately by twin set and individual, where longitudinal samples were available (Fig. 4B,C). The 59 probes were subsequently investigated in our large case-control 450K methylation extended data set (n = 646 SALS cases and 533 unrelated controls). After FDR correction, 2 of the 59 probes showed significantly differential methylation between cases and controls when controlling for age and sex (RAD9B, cg00278366, p = 2.5E-5; C8orf46, cg15444185, p = 0.049, Fig. 4D; full results for all 59 CpGs in Supplementary Table S6). As observed in the MZ cohort, hierarchical clustering and PCA of this probe list in the case-control cohort does not indicate any power to discriminate between ALS and control samples (Fig. 4E,F), nor sex or age group.
Figure 4.

Top identified DMPs do not cluster by disease in MZ and case-control cohorts. (A) Of 59 DMPs found across all discordant twin sets, these 9 were top ranked for the combination of statistical significantly differences between affected and unaffected co-twins/-triplets and the magnitude of differences across twin/triplet sets. Per twin/triplet set differences are shown, with the ALS-affected sibling as the reference for direction of methylation. Gene annotation and CpG name are indicated as gene::cpg. Bar colour indicates hypomethylation of the ALS-affected twin (orange) or hypermethylation of the affected twin/triplet (blue) relative to their unaffected co-twin/-triplets. Grey bars represent the magnitude of difference in methylation in the control twin set and thus have no implicit directionality. They are shown with the same directionality as the discordant twins to facilitate comparison of the magnitude of the difference in methylation. (B) The 59 DMPs identified across discordant twin/triplet sets were used to cluster the samples in the MZ cohort (n = 34). Overall, methylation was similar across samples for most DMPs, and samples did not cluster by disease, nor perfectly by twin/triplet set. (C) Principal Components Analysis (PCA) across discordant twin/triplet sets and the control twin set using the same 59 DMPs also showed that samples did not cluster by disease, but approximately by individual for those where longitudinal samples were available. (D) Of the 59 DMPs identified across discordant twin/triplet sets, two were significantly different between cases (SALS) and controls in a large extended cohort (n SALS = 646, n controls = 533). Both cg15444185, annotated to C8orf46, and cg00278366, annotated to RAD9B, were hypomethylated in SALS samples (cg15444185, β = −0.06, adjusted p = 0.049; cg00278366, β = −0.0771, adjusted p = 2.5E-5) when controlling for age and sex. (E) The top 59 DMPs identified across all discordant twin set do not cluster by disease status in a sporadic case control cohort. (F) PCA also demonstrates that these top 59 twin DMPs do not cluster by disease status in a sporadic case control cohort.
Differentially methylated probes (DMPs) identified within discordant MZ twin/triplet sets implicates new genes and existing ALS genes
Given the clinical heterogeneity in our twin/triplet cohort, within-twin-set differential methylation was also investigated. Using a threshold of a difference in β-methylation ≥0.25 between co-twins or the affected triplet and the mean of the unaffected triplets, we identified 0 DMPs in female SALS twins, 6 in C9orf72 twins, 58 in SOD1 triplets, 2689 in male SALS, and 29 in control twins (Supplementary Fig. S3A–E). Up to 11 probes were annotated per gene in the male SALS twin list of DMPs, for a total of 1829 genes identified. The 506 genes to which multiple male SALS twin probes annotate are given in Supplementary Table S7, which includes two genes previously associated with ALS, DPP6 (Dipeptidyl Peptidase Like 6) and RAMP3 (Receptor Activity Modifying Protein 3) (Fig. 5A). No other discordant twin/triplet set had multiple probes annotated to the same gene. Across all discordant twin/triplet sets, 2 probes (Fig. 5B) and 13 genes (BDKRB2, CHRD, DYSF, HOXD11, IRX4, ISL1, JOSD1, mir_44, NKX2-5, NXN, OTX1, POU4F2, RFX4, Fig. 5C) were identified in multiple twin/triplet sets. None of these probes or genes were also identified in the control twin set. Each of the male SALS twins’ DMPs, C9orf72 twins’ DMPs and SOD1 triplets’ DMPs showed minimal overlap with the control twins DMPs (5, 1, and 1 DMPs, respectively, Fig. 5B, Supplementary Table S8). Similarly, minimal overlapping genes-annotated-to-DMPs were identified between the control and discordant twins/triplets, with 9, 1, and 1 genes respectively (Fig. 5C, Supplementary Table S8).
Figure 5.

Most differentially methylated probes (DMPs) per twin set were unique to one twin set including known ALS genes. DMPs within a twin/triplet set were those with a difference in β-methylation ≥0.25. (A) Within the male SALS twin set, two probes were identified which annotated to DPP6, and two additional probes annotated to RAMP3. Multiple data points per person at each probe indicate longitudinal sampling. For collection times with technical replicate samples per person, points represent the mean at that time, with the standard deviation indicated with a line. (B,C) Generally, DMPs were unique to a twin set, while no differences in methylation (>0.25) were detected in the female SALS twins. (B) The number of DMPs within a twin set varied from <10 in C9orf72 twins to >2500 male SALS twins (Supplementary Fig. S3B,D). Only two of these DMPs were found in multiple discordant twin sets. Each of the male SALS twins, SOD1 triplets and C9orf72 twins showed overlap with the control twins. (C) Within each of the three discordant twin sets and the control twin set DMP lists, multiple probes annotated to the same gene. When comparing these genes rather than individual probes, more shared genes were identified between discordant sets, with 13 genes containing a probe considered differentially methylated in multiple discordant twin/triplet sets.
Transcriptome-wide analysis of disease discordant MZ siblings
Differentially expressed genes within male SALS twins implicates immune function and cell signalling functional pathways in sporadic ALS
Using limma voom to detect genes differentially expressed between male SALS twins while controlling for repeated sampling, we identified 4179 genes as significant following FDR correction (p < 0.05). Of these, 750 genes also had a fold change of 1.5 or greater (Fig. 6A, top genes shown in Fig. 6C, full list in Supplementary Table S9). Notably, CCNF and CCS, both known ALS genes, were identified as significantly downregulated in the ALS twin compared to their unaffected co-twin (CCNF: logFC = 0.70, t = 3.99, FDR = 0.027; CCS: logFC = 0.70, t = 6.42, FDR = 0.008, Fig. 6B). Gene Ontology (GO) analysis of these 750 genes identified 74 terms significantly enriched in this list. Over-representation of genes was seen in 25 terms associated with immune function and cell signalling, while there was an under-representation of genes associated with 45 terms, largely related to transcription and protein modification (Fig. 7, Supplementary Table S10).
Figure 6.

Genes that showed consistent longitudinal differential expression in SALS twins included known ALS genes. (A) Seven hundred and fifty genes were identified as differentially expressed with a minimum fold change of 1.5 (vertical lines) and significant FDR-corrected p-value (horizontal line) in the male SALS twins. (B) Expression of two previously reported ALS genes, CCNF and CCS, identified as differentially expressed in male SALS twins. Gene expression is shown for all three collections in each twin. (C) Expression of the top 8 genes (as ranked by limma) are shown for all three longitudinal collections of the male SALS twins.
Figure 7.

Significantly enriched Gene Ontology (GO) terms implicate enrichment of immune function in the ALS co-twin. GO analysis of the 750 longitudinally differentially expressed genes from the male SALS twins identified 74 significantly enriched biological processes or pathways, shown on the y-axis of the graph. Adjusted p-value (using FDR method) is indicated by the height of the columns on the graph (x-axis). Log2 fold enrichment (logFoldEnrichment) of a GO biological process is indicated by depth of colour, and direction of gene representation (red = over-representation in affected co-twin, blue = under-representation in affected co-twin. Results demonstrate over-representation of genes associated with immune function and cell signalling, and under-representation of genes largely related to transcription and protein modification.
Validation of twin differentially expressed genes in a case-control cohort
Within the validation RNAseq data set of SALS and controls, 379 of the 750 genes identified in the male SALS twins were present. When analysed with limma while controlling for sex, 213 of the 379 genes were differentially expressed between cases and controls, yet none also showed a minimum fold change of 1.5 (top 8 of 213 genes shown in Fig. 8A, 213 genes in Supplementary Table S11). CCNF was not present in the case-control data set, while CCS was not validated (log FC = 0.13, t = 1.99, FDR = 0.075). Hierarchical clustering and PCA of the 379 genes did not identify clusters representing disease status (Fig. 8B,C).
Figure 8.
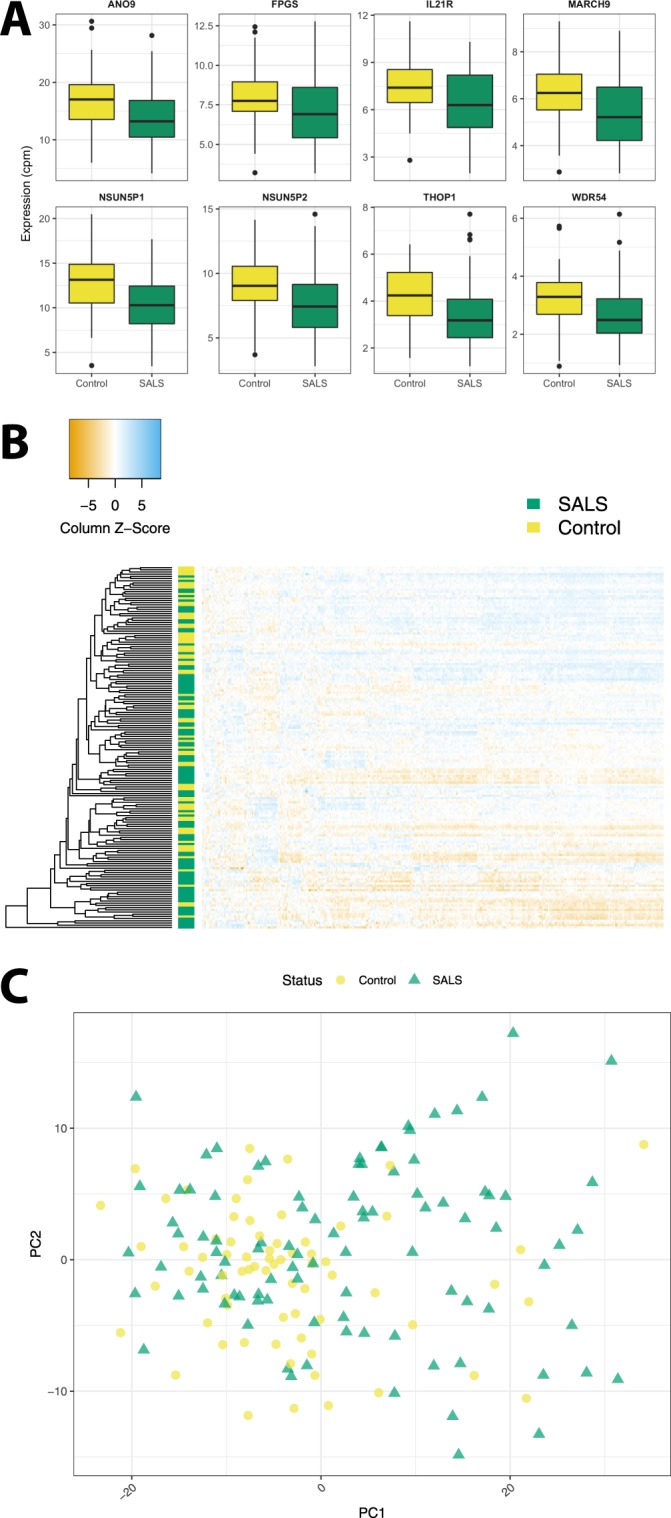
DEGs identified in male SALS twin are validated in a case-control cohort. Of the 750 DEGs identified in the male SALS twins, only 379 genes were present in the sporadic case-control cohort. (A) Two hundred and thirteen of these were validated as differentially expressed between SALS (n = 96) and controls (n = 69) when controlling for sex (Supplementary Table S7) and the top 8 are shown here. (B) Hierarchical clustering of the sporadic ALS and control cohort by these 379 genes did not identify disease-based clusters. (C) Similarly, principal Components Analysis (PCA) of the sporadic ALS and control cohort by these 379 genes did not identify disease-based clusters.
Integration of genome-wide methylation and transcriptome data sets identifies 12 genes both differentially methylated and differentially expressed
To increase the likelihood of detecting biologically meaningful disease-related alterations, RNA-Seq and Infinium HumanMethylation450K data sets were combined for the male SALS twins. Of 506 genes having at least one differentially methylated CpG probe annotated to them in the male SALS twins, 123 are also identified in the entire post-processing RNA-Seq data set of 13718 genes. Conversely, of the 750 genes present in our top DEG list, 642 also have at least one CpG probe mapped to the same gene in the full post-processing 450K set of 24073 genes. When comparing the gene lists of the 123 differentially methylated genes and the 642 differentially expressed genes from the same twin set, 12 genes (C11orf49, CD8A, COL7A1, EOMES, GATA6, GZMM, HOXA4, KANK3, OLIG2, QPRT, SMPD3, SNED1) were present in both gene lists (Fig. 9A–C).
Figure 9.

Twelve overlapping genes were identified in the male SALS twins DMPs and DEGs. (A) While 506 genes were identified as having multiple probes with a difference in β-methylation (≥0.25) between the ALS-discordant male SALS twins, only 123 of these genes were present in the matching RNA-Seq data. 642 of 750 genes identified as differentially expressed were present in the matching DNA methylation data. Twelve of these genes were both differentially expressed and differentially methylated (C11orf49, CD8A, COL7A1, EOMES, GATA6, GZMM, HOXA4, KANK3, OLIG2, QPRT, SMPD3, SNED1). (B) On the HumanMethylation 450K beadchip, multiple CpG probes are annotated to each gene. Methylation of all probes annotated to each of the twelve differentially expressed and differentially methylated genes shows strong consistency within each co-twin. Distance from the transcription start site in base pairs is shown on the x axis. Multiple data points per person at each probe indicate longitudinal sampling. Duplicate collections within a time point are shown as the mean with the standard deviation indicated by a line. (C) Longitudinal expression of the 12 shared genes, (C11orf49, CD8A, COL7A1, EOMES, GATA6, GZMM, HOXA4, KANK3, OLIG2, QPRT, SMPD3, SNED1), is consistently different between ALS discordant male SALS co-twins over time.
Discussion
Using a longitudinal cohort of MZ twins and triplets that are discordant for ALS, we have conducted both a targeted and genome-wide DNA methylation study in conjunction with a sample-matched transcriptomic study. Our cohort is representative of the clinical heterogeneity (age of disease onset, disease duration) frequently observed in ALS cohorts. We have shown that DNA methylation age is the most consistently altered epigenetic signature in ALS. In addition, we observed a higher frequency of unique peripheral blood methylation changes within twin/triplet sets compared to shared methylation changes across twin/triplet sets. However, combined analysis of peripheral blood methylation and transcription detected ALS-relevant changes. These data suggest that the epigenetic and transcriptomic landscape of ALS may be highly complex with numerous small perturbations and various pathways, only some of which are common, contributing to disease.
Epigenetic age was significantly associated with disease in an age-dependent manner, such that affected twins/triplets have an older DNA methylation age than their unaffected co-twins/-triplets while no such effect was observed in young discordant twins. A clear and consistent difference was apparent between the oldest twins in the study, and to a lesser extent, within both middle-aged twin/triplet sets. This pattern of increased methylation age in ALS affected twins is consistent with previous studies of SOD1, C9orf72 and SALS disease-discordant twins25,26,30. Increased DNA methylation age has been linked to increased mortality31 and age has been shown to be a major risk and prognostic factor for ALS32. Our results also reflect a contribution of ageing to disease risk. Methylation age has also been previously linked to age of onset in ALS patients with a C9orf72 repeat expansion30, while we observed a similar phenomenon in our sporadic ALS twin sets, with a much greater between-co-twin difference in DNA methylation age in our late onset twin set compared to our early onset twin set. Further investigation in extended ALS cohorts, specifically mutation-known FALS and SALS would be worthwhile to confirm the contribution of increased DNA methylation age to ALS.
When assessing genome-wide DNA methylation using a magnitude and statistical ranking method, we identified 59 probes differentially methylated in all ALS twins/triplets compared to their unaffected co-twin/-triplets. These 59 probes were selected from high CpG density regions of the genome, therefore considered biologically relevant as they are more likely to affect gene expression. Annotation of the probes to the closest gene transcription site and subsequent gene ontology analysis implicated developmental processes. However, clustering of these 59 DMPs were unable to discriminate between affected and healthy twins, or sporadic cases and controls. Yet, two of these probes were confirmed as significantly differentially methylated in the case-control analysis. C8orf46’s Xenopus homolog gene vexin is involved in neurogenesis and highly expressed in the brain33, while RAD9B responds to DNA damage by moving to the nucleus and contributes to control of the cell cycle34. There is a growing body of evidence that DNA damage response is a significant factor in ALS35.
We conducted within-twin/triplet-set comparisons to show the SOD1 triplet set, C9orf72 twin set and the male SALS twin set each have a moderate number of probes with large differences in methylation (6, 58, and 2689 probes respectively with |Δβ| ≥ 0.25). In contrast, the female SALS twin set showed highly consistent methylation across all >386,000 probes (max |Δβ| = 0.11). Following annotation of these probes to genes, limited overlap of differentially methylated genes was observed between disease discordant twin sets, consistent with other genome-wide methylation studies in ALS-discordant SALS twin sets25,27. This may be reflective of the heterogenous nature of sporadic ALS, or the different methodologies and strategies used to determine differential methylation. A meta-analysis incorporating these studies would be worthwhile to give a broader picture of the methylation signature of sporadic ALS.
It is noteworthy that the four twin sets used in this study represent two distinct genetic forms of disease (SOD1 and C9orf72), along with two cases at extreme age-of-disease-onset ends of the clinical spectrum of sporadic ALS, suggesting again that there may be various epigenetic pathways impacting the phenotype. Some proportion of the observed differences unique to a twin set may result from epigenetic drift36, especially as the greatest number of unique differentially methylated probes was identified in the oldest twin set and the least in the youngest twin set. It is therefore likely that disease, as well as age, is contributing to the differential methylation observed. The metabolism and nutritional status of an individual with ALS, compared to their unaffected co-twin, may also influence methylation status37,38, and therefore potentially contribute to the differential methylation observed. However, detailed dietary data is not available for the individuals included in this study and therefore could not be assessed. Nevertheless, we identified multiple differentially methylated probes annotated to two genes previously associated with ALS, DPP639 and RAMP340 in our oldest twin set, the male SALS twins. DPP6 was the first gene to be associated with sporadic ALS41. It has roles regulating dendritic excitability, with membrane hyperexcitability observed in ALS42,43. It has also been associated with multiple sclerosis44 and spinal muscular atrophy45, and as such is worthy of further investigation in broader ALS.
Analysis of transcriptome-wide gene expression in a subset of our disease discordant MZ cohort, the male SALS twins, found 750 differentially expressed genes. 379 of these genes were assessed in our validation sporadic case-control cohort, and 213 were confirmed to be significantly differentially expressed in sporadic ALS. Gene Ontology analysis implicated primarily upregulation of the immune system, which has been previously identified as dysregulated in ALS14,16,17,27. Interestingly, CCNF and CCS were downregulated in the ALS-affected twin. CCNF has been identified as a causal ALS and FTD gene in several international cohorts46. While transient overexpression has been shown to have deleterious effects in CCNF zebrafish models47, this is the first report of altered CCNF mRNA expression in ALS. CCS has been previously linked to SOD1 in its implication in ALS48. Little is known about the effects of altered expression of CCS in ALS, but its overexpression in the G93A-SOD1 ALS mouse model has been linked to accelerated neurological deficits and worsened mitochondrial pathology49. It is interesting that we observed lower expression in the ALS twin than their unaffected co-twin, given that overexpression has been linked to disease in both genes. It was an unfortunate limitation of this study that neither gene featured in our post-processing HumanMethylation450K dataset, and that so few of the genes identified had data available in our case-control data set. As such, it would be worthwhile to further investigate disease-dependent expression of the remaining 371 genes.
Comparison of transcriptional and DNA methylation changes in ALS-discordant twin/triplet set(s) indicated that despite many genes being present in only one data set, there was overlap between the two datasets. Of the 750 differentially expressed genes identified in the male SALS twins, 642 had methylation data available, while of the 506 genes to which multiple of the 1366 differentially methylated probes annotated, only 123 were also represented in our gene expression data. When we compared these 642 expression-derived genes and 123 methylation-derived genes, we identified twelve genes: C11orf49, CD8A, COL7A1, EOMES, GATA6, GZMM, HOXA4, KANK3, OLIG2, QPRT, SMPD3, SNED1. Notably, the ALS genes identified from DMPs in the male SALS twin set, RAMP3 and DPP6, were not present in the post-processing male SALS twins RNA-Seq data set. C8orf46 and RAD9B were identified across all twin sets to have a single probe differentially methylated, which was confirmed in our sporadic case-control cohort, however, neither gene was present in our RNA-Seq data set. While CCNF and CCS were differentially expressed in the male SALS twins, neither gene was present in the methylation dataset. While none of the twelve genes have previously been directly linked to ALS, some indirect links exist. COL7A1, as part of the collagen gene family, is related to COL6A1, which has been linked to neurodegeneration through impaired autophagy and induction of apoptosis50. Additionally, collagen has also been identified as a significant gene ontology term in analysis of DNA methylation in sporadic ALS11. GZMM, granzyme M, is 1 of 4 gene products from the granzyme family. Granzymes A and B are elevated in ALS serum, with granzyme B correlated to ALS severity51. Granzyme B has been further implicated in inducing apoptosis in human ALS motor neurons52. SMPD3, neutral Sphingomyelinase II, is associated with apoptosis and cell cycle regulation, which have been previously linked to ALS53,54. KANK3 has been suggested as a possible gene contributing to an ALS-linked region on chromosome 1755. QPRT is involved in the kynurenine pathway, which has been implicated in ALS56. These twelve genes, identified when combining DNA methylation and gene expression data, may thus contribute to disease, and warrant further investigation.
Assessment of global methylation and blood cell composition showed no difference between ALS and healthy co-twins. Although a lack of global changes in methylation is consistent with five other sets of ALS-discordant twins25, not all studies agree10,14,15. It is also interesting that blood cell composition, as determined from whole blood methylation, was not found to vary between affected and unaffected twins, given that upregulation of the immune system and changes in white blood cell populations have previously been demonstrated in ALS57,58. This lack of effect in white blood cell estimates may be partly attributable to shared genetic background59,60, although a prior study reported differing methylation-derived cell proportion estimates in one ALS-discordant twin pair27.
High-density quantitative targeted analysis of the C9orf72 and SOD1 gene-associated CpG islands and gene promotors did not identify any differences in methylation status between ALS-discordant MZ twins/triplets carrying mutations in these genes. The general consistency observed in SOD1 methylation between carriers of SOD1 mutations suggests DNA methylation of the SOD1 promoter itself is not likely to be a major mechanism contributing to differences in penetrance in SOD1-linked ALS, in line with previous reports10. Methylation of C9orf72 was low in a twin set carrying the C9orf72 repeat expansion. Methylation of the C9orf72 promoter and/or the repeat expansion has been reported in the brain and blood of repeat expansion carriers6,9,61–65, in some cases with similar low levels of methylation as that observed here. Interestingly, neither of the two prior C9orf72 twin studies, one ALS concordant and one discordant, detected methylation of C9orf7224,66, suggesting that C9orf72 methylation is just one part of the epigenetic story in ALS.
In conclusion, our disease-discordant twin study, utilising longitudinal samples throughout disease progression, demonstrated significant association of DNA methylation age with disease in an age dependent manner. We have also identified an important set of DMPs and DEGs, and associated functional pathways, that may be involved in either ALS pathogenesis or protection from disease. These genes and pathways offer potential targets for future therapeutic treatment for ALS patients.
Methods
Participants
The cohort of 1806 total participants used in this study is summarised below. This study was approved by the human research ethics committees of Macquarie University (5201600387) and Sydney South West Area Health Service. Samples from ALS patients, family members, and unrelated controls were obtained from the Macquarie University Neurodegenerative Diseases Biobank, Molecular Medicine Laboratory at Concord Hospital, and the Australian MND DNA bank. Written informed consent was obtained from all study subjects and all methods were performed in accordance with the relevant guidelines and regulations. Most participants were of European descent and patients were clinically diagnosed with definite or probable ALS based on El Escorial criteria67. Genomic DNA was extracted from peripheral blood using standard protocols. RNA was extracted from peripheral blood with the QIAsymphony PAXgene blood RNA kit (Qiagen, Hilden, Germany).
Twin/triplet cohort
Three ALS discordant monozygotic twin pairs, one ALS discordant MZ triplet set and two control MZ twin pairs were included in this study (Fig. 1 and Table 1). Monozygosity for each twin/triplet set was confirmed using STR fragment analysis and/or SNP microarrays. Longitudinal samples were available from two twin sets (male and female sporadic ALS (SALS) twin sets in Fig. 1). The four discordant twin/triplet sets had previously undergone mutation analysis for known ALS genes and whole genome analysis for novel and/or rare de novo variants.
Data processing and extended cohorts
Additional samples were used in this study for data processing (Illumina HumanMethylation 450K, EpiTYPER methylation assays, and RNA-Seq) and for examination of significant findings (Illumina HumanMethylation 450K assay and RNA-Seq). Demographic characteristics between cases and controls in extended cohorts were assessed with t-tests for age and χ2 tests for sex.
For quality control and processing of the EpiTYPER data, 279 samples with C9orf72 EpiTYPER data (158 familial ALS/FTD samples, 56 asymptomatic samples (individuals harbouring a causal gene mutation but currently unaffected), and 65 control samples), and 261 samples with SOD1 EpiTYPER data (123 familial ALS, 65 asymptomatic, and 73 control samples) were used.
For the Infinium HumanMethylation 450K BeadChip, 1658 samples were used in data processing and normalisation. This comprised 889 individuals with sporadic or familial ALS, 92 asymptomatic and 668 controls. The familial ALS and asymptomatic cases largely overlap with the EpiTYPER cohort. The extended cohort subset comprised 650 sporadic ALS individuals and 539 unrelated controls.
One hundred and ninety samples were used for data processing and normalisation of the RNA-Seq data, comprising 114 individuals with ALS (99 sporadic ALS, 15 familial ALS) and 76 unrelated controls. The validation subset comprised of 96 sporadic cases and 69 controls. The majority of the 96 validation sporadic ALS cases were also present in the HumanMethylation 450K BeadChip SALS/control cohort.
Methylation assays and data processing
All quality control and data processing steps were carried out in R v 3.4.468.
EpiTYPER assay
Custom EpiTYPER assays (Sequenom, San Diego, USA) were used to quantify CpG methylation of 56 and 39 CpG units respectively of the two gene-associated CpG islands for C9orf72 and the gene-associated CpG island upstream of SOD1. EpiTYPER uses base-specific cleavage of bisulphite-converted DNA and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MADL-TOF MS) to quantify DNA methylation69. Primers for overlapping amplicons were designed with Sequenom’s EpiDesigner software to target the CpG island regions, and therefore the promoter regions, as shown in Supplementary Fig. S1. Primer and assay details are available in Supplementary Table S1. Samples were assayed in one or two batches, and either in duplicate or as singletons (Supplementary Table S1). Sample processing was performed by Agena Bioscience (Brisbane, Queensland, Australia). As each gene assay was run across several plates of samples, the highly methylated DNA control was used to calculate the between-plate coefficient of variation, determined to be 4.9% and 2.3% for SOD1 and C9orf72 plates, respectively. CpG methylation was quantified as the percentage of methylated cytosines for each CpG unit, where CpG units consist of one or more CpG sites. For units with multiple CpG sites, methylation percentages were normalised by averaging across the number of sites.
EpiTYPER data processing
EpiTYPER data processing was adapted from a previously established method70. Twin samples were processed together with the full familial cohorts to leverage the increased sample size. In brief, CpG units that failed to meet assay reliability standards were discarded and samples in duplicate were averaged for the remaining CpG units. Given the relatively small number of CpG units remaining after removal of those determined to be unreliable and the relatively high failure rate of samples and units, a two-step sample/unit filtering process was used. First, failed samples, with ≥90% of CpG unit readings missing, were removed, followed by CpG units which were missing data for ≥90% of samples. Second, samples with a low detection success (missing data for ≥15% of units) were removed, and the same threshold applied to remove CpG units with low detection success (units missing data for ≥15% of samples). Finally, any remaining missing values were imputed with the mean for that unit. Following data processing and filtering, 28 of 56 and 23 of 39 CpG units (for C9orf72 and SOD1, respectively) remained for analysis.
Infinium Human Methylation 450K v1.2 BeadChip array
Genome-wide methylation was investigated using the Infinium HumanMethylation 450K v1.2 BeadChip (Illumina, San Diego, USA). This microarray provides qualitative methylation values for approximately 480,000 CpG sites distributed throughout the genome. Bisulphite-converted DNA was hybridised to the Infinium HumanMethylation 450K BeadChip. Fluorescence imaging of the BeadChip using an Illumina HiScan SQ scanner successfully generated raw Intensity Data files (.idat) for all samples.
450K data processing
Data processing of the .idat files was adapted from the method presented by71. Twin samples were processed together with the full cohort to leverage larger sample sizes. All default settings were used except where otherwise specified. In brief, samples with less than 99% of CpGs detected were removed. shinyMethyl (v. 1.12.072) was used to visually identify possible outliers, with confirmation of sex queries using RnBeads (v. 1.0.0.73). Samples with any possibility of incorrect identification were removed. Data were normalised with the dasen function from wateRmelon (v. 1.20.374). Probes that had failed to be detected (threshold p > 0.05) with the minfi (v. 1.22.175) function detectionP were removed (n = 10270 probes). Normalised data were submitted to Horvath’s online DNAm age calculator28. Samples that did not strongly correlate (r < 0.85) with the DNAm age results gold standard were removed. Leveraging technical replicate/duplicate samples (n = 30), both 1) in the form of multiple blood collections at the same time and resulting independent DNA extractions (technical replicates) and 2) multiple aliquots of single DNA extractions (duplicates), a custom filtering step was included to identify and remove highly variable probes. Any probe identified to have multiple pairs of technical replicate or duplicate samples with differences greater than three standard deviations from the probe’s mean difference was discarded (n = 38697). Of the remaining probes, any known to cross hybridise, be located on sex chromosomes, or bind to SNPs, were removed (n = 50362)76.
Following raw data processing, quantitative CpG methylation values for 1215 samples (including 34 twin samples from Table 1 and 1179 case/control extended cohort samples outlined in Supplementary Table S2) and 386183 probes remained for analysis and examination. Comparison of the case-control extended cohort (Supplementary Table S2) showed that sex ( = 33.8, p < 0.01) and age (t1174 = 4.20, p < 0.01) were significantly different between ALS cases and controls.
Analysis of methylation data
All statistical analyses were carried out in R v. 3.4.468.
Gene-specific targeted methylation analysis of SOD1 and C9orf72 in the FALS twin/triplet sets
Methylation of SOD1 or C9orf72, as quantified by both EpiTYPER and 450K assays, was visualised in the relevant monozygotic disease discordant twin/triplets. Four and five 450K CpGs were available in the post processing data set in the targeted region of C9orf72 (cg05990720, cg11613875, cg14363787, cg23074747) and SOD1 (cg16086310, cg17253939, cg18126791, cg19948014, cg26893544), respectively. Since only one twin set and one triplet set are available in our cohort for each respective variant, results are descriptive only.
Observed differences in DNA methylation age, blood cell composition and global methylation within a twin/triplet set
DNA methylation age was determined from 450K methylation data using the method of Horvath28. Blood cell proportions in whole blood derived methylation was estimated from 450K methylation data with the minfi implementation of Houseman et al.’s algorithm29. Global methylation levels were determined as the mean methylation estimate across all post-processing 450K CpG sites per sample. CpG sites were also divided into one of four categories based on HIL CpG classes (high-density CpG island (HC), intermediate-density CpG island (IC) and non-island (LC); ICshore, intermediate-density CpG island shore that borders HCs)77,78 and the mean methylation for each was calculated.
Methylome-wide analysis in MZ sets to identify differentially methylated probes
The list of differentially methylated probes (DMP) across all MZ sets was identified using an established ranked magnitude-significance method79. In brief, statistical significance per CpG site was determined using a paired t-test on methylation M-values, using the per-patient mean of longitudinal samples and unaffected triplets. The magnitude of the difference in methylation was calculated as the mean difference in β-methylation between co-twins. Both methods were used to rank all CpGs, and a final ranked list was determined from the mean of these two ranking methods. Top DMPs were the subset of all CpG probes that met the following two criteria, 1) they were in high CpG density regions of the genome and 2) the ranked list of high-density probes was truncated immediately prior to the first probe to show a difference in the direction of change across the four discordant MZ sets. The ability of these probes to discriminate between ALS and healthy individuals was assessed by hierarchical clustering and principal components analysis of all twin/triplet sets.
Within-twin/triplet set DMPs were also identified. A CpG probe was considered to be differentially methylated within a twin/triplet set where there was an absolute difference in β-methylation ≥0.25 between the affected twin and their unaffected co-twin/triplet.
Examination of identified twin DMPs in a sporadic ALS cohort
Twin/triplet DMPs were examined in the larger sporadic case-control cohort. Differences between cases and controls for each of the identified probes were analysed, along with the ability of the DMP list to cluster cases and controls separately.
Gene expression
RNA sequencing
Raw sequencing reads in fastq format were generated for male SALS twins (based on longitudinal sample availability) and the sporadic case-control validation cohort as outlined in Supplementary Table S3.
RNA-Seq data processing
The quality of raw sequencing reads was evaluated using fastQC (v 0.11.780) for both datasets. Trimming and alignment was performed as outlined in Table 1 using either Trimmomatic (v. 0.3681) or Cutadapt (version 1.8.182) and HISAT2 (v2.0.583). All subsequent data processing and analysis was completed in R (v. 3.4.4), using BioConductor packages edgeR (v. 3.18.184) and limma (v. 3.32.1085). A standard edgeR TMM normalisation and filtering pipeline was used in data processing, with only those genes where expression was greater than 0.3 counts per million in a minimum of 3 samples (male SALS twins) or 2 counts per million in a minimum of 75 samples (case-control cohort) retained for analysis, which is equivalent to approximately 12–15 raw counts in the smallest library size for each dataset. For the male SALS twins RNA-Seq data, of the 27685 human genes present in the per-gene read counts generated by HTSeq86, 13718 genes remained following raw data processing using edgeR84. Whereas in the case-control cohort, of the 23368 human genes present in the per-gene read counts generated by HTSeq86, 7354 genes remained following raw data processing using edgeR84. MDS (multi-dimensional scaling) indicated the presence of three outliers in the case-control cohort, 1 control and 2 SALS samples. All three were removed and final clinical details for the cohort can be found in Supplementary Table S4. Comparison of the RNA-Seq case-control validation cohort (Supplementary Table S4) showed that there were no significant differences in age (t157.9 = 1.74, p = 0.08) between the ALS cases and controls, but a difference was observed in sex between cases and controls ( = 6.5, p = 0.01).
Differentially expressed genes in MZ twins
To identify differentially expressed genes (DEGs) using the paired longitudinal RNA-Seq samples from the ALS-discordant male SALS twins (Table 1), read count data was analysed using limma85, including model terms for longitudinal sample collection and disease status. Voom87 transformation was applied prior to modelling. Multiple testing correction using the BH-FDR method88 was applied to the full list of post-processing genes.
Validation of twin DEGs in a sporadic cohort
Genes identified in twin analyses were investigated for an effect of disease in the full case-control cohort with limma, including sex as a covariate. Data were voom transformed, given the highly variable library sizes. Multiple testing corrections using the BH-FDR method was applied only on the subset of genes identified as differentially expressed in twins. Hierarchical clustering and principal components analysis of the expression of these DEGs in the case-control cohort was assessed.
Combined methylation and expression analysis
Intersect of top CpGs and genes
To identify genes most likely to be altered in disease, results from independent analysis of genome-wide methylation and expression data sets were integrated. Longitudinal RNA-Seq data is only available for one male SALS twin set, therefore we first identified the overlap between top DEGs and the genes annotated to the most differentially methylated probes within that twin set. We extended this analysis by overlapping the same list of DEGs with the genes annotated to the top differentially methylated probes across all combined twin sets.
Gene Ontology analysis
Gene Ontology enrichment analysis89,90 for biological processes was applied to the genes identified as differentially expressed in male SALS twins. The gene list was analysed with PANTHER overrepresentation tests (GO Ontology database release 2018-08-0991). Enrichment was tested relative to all genes detected in the appropriate post-processing data set. Fisher’s exact test with FDR correction was used.
Statistics
All analyses were carried out in R (v. 3.4.4)68. Linear mixed effects models were used to analyse DNAm age, blood cell type proportions and global mean M-methylation. Modelling was carried out using the lmer function in the package lme4 (v. 1.1.1492) for DNAm age and mean methylation, while a mixed effects beta regression for cell type proportions was applied with the glmmTMB function from the glmmTMB package (v. 0.2.2.093). Blood cell type proportions were increased by 0.001 to all estimates to avoid taking the log of zero. All mixed models assessed the effect of disease status while controlling for age at sample collection and sex. When analysing DNAm age, the interaction of disease and age at collection was also tested. Random effects were introduced for repeated sampling within co-twins, and a random intercept per twin/triplet set. When modelling cell types, due to convergence issues, the random slope for repeated sampling was dropped, leaving random intercepts for each co-twin and twin set. Likelihood ratio tests were used to determine significance of model terms. Linear models were used for case-control examination of probes identified in the MZ cohort, with the same fixed effect terms of age at sample collection and sex as described for mixed models. Hierarchical clustering utilised the package cluster (v. 2.0.6), with Manhattan distance and ward clustering methods for 450K data94, and Spearman correlation distance and average linkage clustering for log-transformed RNA-Seq count data95. Where appropriate, technical replicates are shown as means with error bars indicating standard deviation (unless otherwise stated).
Supplementary information
Supplementary Tables S1-S4 and Figures S1-S3
Acknowledgements
We thank Carolyn Cecere and Lorel Adams for their assistance in compiling family information, Elisa Cachia and Dr Sarah Furlong for providing patient materials, clinical and technical assistance, and Janette Edson for technical assistance. This work was funded by the Motor Neurone Disease Research Institute of Australia (grant to K.L.W.), National Health and Medical Research Council of Australia (grant 1083187 to I.P.B., grant 1079583 to B.B., fellowship 1084417 to B.B. and fellowship 1092023 to K.L.W.) and Macquarie University (grant to K.L.W.).
Author Contributions
I.S.T. and K.L.W. conceived and designed the study with input from S.J.C. and I.P.B. I.S.T., E.M., B.B., T.J.P., N.A.T., K.Y.Z., Q.Z., Z.-H.Z., D.B. and K.L.W. performed the experiments/data analysis and interpretation. D.B.R. and G.A.N. collected clinical information and samples. I.S.T. and K.L.W. wrote the manuscript with input from I.P.B. All authors read and approved the final manuscript.
Data Availability
The datasets generated and/or analysed during the current study are not publicly available since our ethics permission does not cover sharing of data to third parties but are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-44765-4.
References
- 1.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10:661–670. doi: 10.1038/nrneurol.2014.184. [DOI] [PubMed] [Google Scholar]
- 2.Renton Alan E, Chiò Adriano, Traynor Bryan J. State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience. 2013;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iguchi Yohei, Katsuno Masahisa, Ikenaka Kensuke, Ishigaki Shinsuke, Sobue Gen. Amyotrophic lateral sclerosis: an update on recent genetic insights. Journal of Neurology. 2013;260(11):2917–2927. doi: 10.1007/s00415-013-7112-y. [DOI] [PubMed] [Google Scholar]
- 4.McCann E.P., Williams K.L., Fifita J.A., Tarr I.S., O'Connor J., Rowe D.B., Nicholson G.A., Blair I.P. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clinical Genetics. 2017;92(3):259–266. doi: 10.1111/cge.12973. [DOI] [PubMed] [Google Scholar]
- 5.Al-Chalabi A., Fang F., Hanby M. F., Leigh P. N., Shaw C. E., Ye W., Rijsdijk F. An estimate of amyotrophic lateral sclerosis heritability using twin data. Journal of Neurology, Neurosurgery & Psychiatry. 2010;81(12):1324–1326. doi: 10.1136/jnnp.2010.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belzil Veronique V., Bauer Peter O., Gendron Tania F., Murray Melissa E., Dickson Dennis, Petrucelli Leonard. Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Research. 2014;1584:15–21. doi: 10.1016/j.brainres.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xi Zhengrui, Rainero Innocenzo, Rubino Elisa, Pinessi Lorenzo, Bruni Amalia C, Maletta Raffaele G, Nacmias Benedetta, Sorbi Sandro, Galimberti Daniela, Surace Ezequiel I, Zheng Yonglan, Moreno Danielle, Sato Christine, Liang Yan, Zhou Ye, Robertson Janice, Zinman Lorne, Tartaglia Maria Carmela, St. George-Hyslop Peter, Rogaeva Ekaterina. Hypermethylation of the CpG-island near the C9orf72 G4C2-repeat expansion in FTLD patients. Human Molecular Genetics. 2014;23(21):5630–5637. doi: 10.1093/hmg/ddu279. [DOI] [PubMed] [Google Scholar]
- 8.Belzil Veronique V., Bauer Peter O., Prudencio Mercedes, Gendron Tania F., Stetler Caroline T., Yan Irene K., Pregent Luc, Daughrity Lillian, Baker Matthew C., Rademakers Rosa, Boylan Kevin, Patel Tushar C., Dickson Dennis W., Petrucelli Leonard. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathologica. 2013;126(6):895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xi Zhengrui, Zinman Lorne, Moreno Danielle, Schymick Jennifer, Liang Yan, Sato Christine, Zheng Yonglan, Ghani Mahdi, Dib Samar, Keith Julia, Robertson Janice, Rogaeva Ekaterina. Hypermethylation of the CpG Island Near the G4C2 Repeat in ALS with a C9orf72 Expansion. The American Journal of Human Genetics. 2013;92(6):981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coppedè Fabio, Stoccoro Andrea, Mosca Lorena, Gallo Roberta, Tarlarini Claudia, Lunetta Christian, Marocchi Alessandro, Migliore Lucia, Penco Silvana. Increase in DNA methylation in patients with amyotrophic lateral sclerosis carriers of not fully penetrant SOD1 mutations. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2017;19(1-2):93–101. doi: 10.1080/21678421.2017.1367401. [DOI] [PubMed] [Google Scholar]
- 11.Morahan Julia M., Yu Bing, Trent Ronald J., Pamphlett Roger. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2009;10(5-6):418–429. doi: 10.3109/17482960802635397. [DOI] [PubMed] [Google Scholar]
- 12.Oates Nathan, Pamphlett Roger. An epigenetic analysis of SOD1 and VEGF in ALS. Amyotrophic Lateral Sclerosis. 2007;8(2):83–86. doi: 10.1080/17482960601149160. [DOI] [PubMed] [Google Scholar]
- 13.Morello, G., Spampinato, A. G. & Cavallaro, S. Molecular taxonomy of sporadic amyotrophic lateral sclerosis using disease-associated genes. Front. Neurol., 10.3389/fneur.2017.00152 (2017). [DOI] [PMC free article] [PubMed]
- 14.Figueroa-Romero Claudia, Hur Junguk, Bender Diane E., Delaney Colin E., Cataldo Michael D., Smith Andrea L., Yung Raymond, Ruden Douglas M., Callaghan Brian C., Feldman Eva L. Identification of Epigenetically Altered Genes in Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE. 2012;7(12):e52672. doi: 10.1371/journal.pone.0052672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tremolizzo Lucio, Messina Paolo, Conti Elisa, Sala Gessica, Cecchi Matteo, Airoldi Luisa, Pastorelli Roberta, Pupillo Elisabetta, Bandettini Di Poggio Monica, Filosto Massimiliano, Lunetta Christian, Agliardi Cristina, Guerini Franca, Mandrioli Jessica, Calvo Andrea, Beghi Ettore, Ferrarese Carlo, Cotelli M.S., Corbo M., Maestri E., Georgoulopoulou E., Marzorati L., Susani E., Arosio A., Chiò A., Fuda G., Pisa F. Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2013;15(1-2):98–105. doi: 10.3109/21678421.2013.851247. [DOI] [PubMed] [Google Scholar]
- 16.Andrés-Benito Pol, Moreno Jesús, Aso Ester, Povedano Mónica, Ferrer Isidro. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: implications in frontotemporal lobar degeneration. Aging. 2017;9(3):823–851. doi: 10.18632/aging.101195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Weihua, Beers David R., Hooten Kristopher G., Sieglaff Douglas H., Zhang Aijun, Kalyana-Sundaram Shanker, Traini Christopher M., Halsey Wendy S., Hughes Ashley M., Sathe Ganesh M., Livi George P., Fan Guo-Huang, Appel Stanley H. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurology. 2017;74(6):677. doi: 10.1001/jamaneurol.2017.0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebbert Mark T. W., Ross Christian A., Pregent Luc J., Lank Rebecca J., Zhang Cheng, Katzman Rebecca B., Jansen-West Karen, Song Yuping, da Rocha Edroaldo Lummertz, Palmucci Carla, Desaro Pamela, Robertson Amelia E., Caputo Ana M., Dickson Dennis W., Boylan Kevin B., Rademakers Rosa, Ordog Tamas, Li Hu, Belzil Veronique V. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72-associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathologica. 2017;134(5):715–728. doi: 10.1007/s00401-017-1760-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Rheenen Wouter, Diekstra Frank P., Harschnitz Oliver, Westeneng Henk-Jan, van Eijk Kristel R., Saris Christiaan G. J., Groen Ewout J. N., van Es Michael A., Blauw Hylke M., van Vught Paul W. J., Veldink Jan H., van den Berg Leonard H. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLOS ONE. 2018;13(6):e0198874. doi: 10.1371/journal.pone.0198874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaut Oliver, Schmitt Ina, Tost Jörg, Busato Florence, Liu Yi, Hofmann Per, Witt Stephanie H., Rietschel Marcella, Fröhlich Holger, Wüllner Ullrich. Epigenome-wide DNA methylation analysis in siblings and monozygotic twins discordant for sporadic Parkinson’s disease revealed different epigenetic patterns in peripheral blood mononuclear cells. neurogenetics. 2016;18(1):7–22. doi: 10.1007/s10048-016-0497-x. [DOI] [PubMed] [Google Scholar]
- 21.Mastroeni Diego, McKee Ann, Grover Andrew, Rogers Joseph, Coleman Paul D. Epigenetic Differences in Cortical Neurons from a Pair of Monozygotic Twins Discordant for Alzheimer's Disease. PLoS ONE. 2009;4(8):e6617. doi: 10.1371/journal.pone.0006617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Javierre B. M., Fernandez A. F., Richter J., Al-Shahrour F., Martin-Subero J. I., Rodriguez-Ubreva J., Berdasco M., Fraga M. F., O'Hanlon T. P., Rider L. G., Jacinto F. V., Lopez-Longo F. J., Dopazo J., Forn M., Peinado M. A., Carreno L., Sawalha A. H., Harley J. B., Siebert R., Esteller M., Miller F. W., Ballestar E. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Research. 2009;20(2):170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byrne E M, Carrillo-Roa T, Henders A K, Bowdler L, McRae A F, Heath A C, Martin N G, Montgomery G W, Krause L, Wray N R. Monozygotic twins affected with major depressive disorder have greater variance in methylation than their unaffected co-twin. Translational Psychiatry. 2013;3(6):e269–e269. doi: 10.1038/tp.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xi Z., Yunusova Y., van Blitterswijk M., Dib S., Ghani M., Moreno D., Sato C., Liang Y., Singleton A., Robertson J., Rademakers R., Zinman L., Rogaeva E. Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology. 2014;83(16):1476–1478. doi: 10.1212/WNL.0000000000000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young Paul E., Kum Jew Stephen, Buckland Michael E., Pamphlett Roger, Suter Catherine M. Epigenetic differences between monozygotic twins discordant for amyotrophic lateral sclerosis (ALS) provide clues to disease pathogenesis. PLOS ONE. 2017;12(8):e0182638. doi: 10.1371/journal.pone.0182638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Ming, Xi Zhengrui, Ghani Mahdi, Jia Peixin, Pal Mrinal, Werynska Karolina, Moreno Danielle, Sato Christine, Liang Yan, Robertson Janice, Petronis Arturas, Zinman Lorne, Rogaeva Ekaterina. Genetic and epigenetic study of ALS-discordant identical twins with double mutations inSOD1andARHGEF28. Journal of Neurology, Neurosurgery & Psychiatry. 2016;87(11):1268–1270. doi: 10.1136/jnnp-2016-313592. [DOI] [PubMed] [Google Scholar]
- 27.Lam Larry, Chin Lydia, Halder Ramesh C., Sagong Bien, Famenini Sam, Sayre James, Montoya Dennis, Rubbi Liudmilla, Pellegrini Matteo, Fiala Milan. Epigenetic changes in T-cell and monocyte signatures and production of neurotoxic cytokines in ALS patients. The FASEB Journal. 2016;30(10):3461–3473. doi: 10.1096/fj.201600259RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horvath Steve. DNA methylation age of human tissues and cell types. Genome Biology. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma., 10.1186/1471-2105-13-86 (2012). [DOI] [PMC free article] [PubMed]
- 30.Zhang Ming, Tartaglia Maria Carmela, Moreno Danielle, Sato Christine, McKeever Paul, Weichert Anna, Keith Julia, Robertson Janice, Zinman Lorne, Rogaeva Ekaterina. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathologica. 2017;134(2):271–279. doi: 10.1007/s00401-017-1713-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christiansen Lene, Lenart Adam, Tan Qihua, Vaupel James W., Aviv Abraham, McGue Matt, Christensen Kaare. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2015;15(1):149–154. doi: 10.1111/acel.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiò, A. et al. Prognostic factors in ALS: A critical review, 10.3109/17482960802566824 (2009). [DOI] [PMC free article] [PubMed]
- 33.Moore Kathryn B., Logan Mary A., Aldiri Issam, Roberts Jacqueline M., Steele Michael, Vetter Monica L. C8orf46 homolog encodes a novel protein Vexin that is required for neurogenesis in Xenopus laevis. Developmental Biology. 2018;437(1):27–40. doi: 10.1016/j.ydbio.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Castro A. J., Freire R. Rad9B responds to nucleolar stress through ATR and JNK signalling, and delays the G1-S transition. Journal of Cell Science. 2012;125(5):1152–1164. doi: 10.1242/jcs.091124. [DOI] [PubMed] [Google Scholar]
- 35.Coppedè Fabio, Migliore Lucia. DNA damage in neurodegenerative diseases. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2015;776:84–97. doi: 10.1016/j.mrfmmm.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Fraga M. F., Ballestar E., Paz M. F., Ropero S., Setien F., Ballestar M. L., Heine-Suner D., Cigudosa J. C., Urioste M., Benitez J., Boix-Chornet M., Sanchez-Aguilera A., Ling C., Carlsson E., Poulsen P., Vaag A., Stephan Z., Spector T. D., Wu Y.-Z., Plass C., Esteller M. From The Cover: Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulrey CL, Liu L, Andrews LG, Tollefsbol TO. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005;14:R139–R147. doi: 10.1093/hmg/ddi100. [DOI] [PubMed] [Google Scholar]
- 38.Waterland RA. Assessing the Effects of High Methionine Intake on DNA Methylation. The J. Nutr. 2006;136:1706S–1710S. doi: 10.1093/jn/136.6.1706S. [DOI] [PubMed] [Google Scholar]
- 39.van Es Michael A, van Vught Paul WJ, Blauw Hylke M, Franke Lude, Saris Christiaan GJ, Van Den Bosch Ludo, de Jong Sonja W, de Jong Vianney, Baas Frank, van't Slot Ruben, Lemmens Robin, Schelhaas Helenius J, Birve Anna, Sleegers Kristel, Van Broeckhoven Christine, Schymick Jennifer C, Traynor Bryan J, Wokke John HJ, Wijmenga Cisca, Robberecht Wim, Andersen Peter M, Veldink Jan H, Ophoff Roel A, van den Berg Leonard H. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nature Genetics. 2007;40(1):29–31. doi: 10.1038/ng.2007.52. [DOI] [PubMed] [Google Scholar]
- 40.Daoud, H. et al. Resequencing of 29 candidate genes in patients with familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol., 10.1001/archneurol.2010.351 (2011). [DOI] [PubMed]
- 41.Kim Jinhyun, Nadal Marcela S., Clemens Ann M., Baron Matthew, Jung Sung-Cherl, Misumi Yoshio, Rudy Bernardo, Hoffman Dax A. Kv4 Accessory Protein DPPX (DPP6) is a Critical Regulator of Membrane Excitability in Hippocampal CA1 Pyramidal Neurons. Journal of Neurophysiology. 2008;100(4):1835–1847. doi: 10.1152/jn.90261.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park Susanna B., Kiernan Matthew C., Vucic Steve. Axonal Excitability in Amyotrophic Lateral Sclerosis. Neurotherapeutics. 2016;14(1):78–90. doi: 10.1007/s13311-016-0492-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wainger Brian J., Kiskinis Evangelos, Mellin Cassidy, Wiskow Ole, Han Steve S.W., Sandoe Jackson, Perez Numa P., Williams Luis A., Lee Seungkyu, Boulting Gabriella, Berry James D., Brown Robert H., Cudkowicz Merit E., Bean Bruce P., Eggan Kevin, Woolf Clifford J. Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cell Reports. 2014;7(1):1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brambilla Paola, Esposito Federica, Lindstrom Eva, Sorosina Melissa, Giacalone Giacomo, Clarelli Ferdinando, Rodegher Mariaemma, Colombo Bruno, Moiola Lucia, Ghezzi Angelo, Capra Ruggero, Collimedaglia Laura, Coniglio Gabriella, Celius Elisabeth G., Galimberti Daniela, Sørensen Per Soelberg, Martinelli Vittorio, Oturai Annette B., Harbo Hanne F., Hillert Jan, Comi Giancarlo, Martinelli-Boneschi Filippo. Association between DPP6 polymorphism and the risk of progressive multiple sclerosis in Northern and Southern Europeans. Neuroscience Letters. 2012;530(2):155–160. doi: 10.1016/j.neulet.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 45.van Es MA, et al. DPP6 is associated with susceptibility to progressive spinal muscular atrophy. Neurol. 2009;72:1184–1185. doi: 10.1212/01.wnl.0000345368.01098.7e. [DOI] [PubMed] [Google Scholar]
- 46.Williams, K. L. et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun., 10.1038/ncomms11253, arXiv:1011.1669v3 (2016). [DOI] [PMC free article] [PubMed]
- 47.Hogan Alison L., Don Emily K., Rayner Stephanie L., Lee Albert, Laird Angela S., Watchon Maxinne, Winnick Claire, Tarr Ingrid S., Morsch Marco, Fifita Jennifer A., Gwee Serene S. L., Formella Isabel, Hortle Elinor, Yuan Kristy C., Molloy Mark P., Williams Kelly L., Nicholson Garth A., Chung Roger S., Blair Ian P., Cole Nicholas J. Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Human Molecular Genetics. 2017;26(14):2616–2626. doi: 10.1093/hmg/ddx136. [DOI] [PubMed] [Google Scholar]
- 48.Beckman Joseph S, Estévez Alvaro G, Crow John P, Barbeito Luis. Superoxide dismutase and the death of motoneurons in ALS. Trends in Neurosciences. 2001;24(11):S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- 49.Son M., Puttaparthi K., Kawamata H., Rajendran B., Boyer P. J., Manfredi G., Elliott J. L. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proceedings of the National Academy of Sciences. 2007;104(14):6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cescon Matilde, Gregorio Ilaria, Bonaldo Paolo. Lack of collagen VI promotes neurodegeneration by impairing autophagy and inducing apoptosis during aging. Aging. 2017;8(5):1083–1101. doi: 10.18632/aging.100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iłżecka Joanna. Granzymes A and B levels in serum of patients with amyotrophic lateral sclerosis. Clinical Biochemistry. 2011;44(8-9):650–653. doi: 10.1016/j.clinbiochem.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 52.Song, S. ALS Astrocytes Adopt Natural Killer Properties to Induce Motor Neuron Death. Ph.D. thesis (The Ohio State University, 2014).
- 53.Barbosa Livea F., Cerqueira Fernanda M., Macedo Antero F.A., Garcia Camila C.M., Angeli José Pedro F., Schumacher Robert I., Sogayar Mari Cleide, Augusto Ohara, Carrì Maria Teresa, Di Mascio Paolo, Medeiros Marisa H.G. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2010;1802(5):462–471. doi: 10.1016/j.bbadis.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Marcuzzo Stefania, Bonanno Silvia, Kapetis Dimos, Barzago Claudia, Cavalcante Paola, D’Alessandro Sara, Mantegazza Renato, Bernasconi Pia. Up-regulation of neural and cell cycle-related microRNAs in brain of amyotrophic lateral sclerosis mice at late disease stage. Molecular Brain. 2015;8(1):5. doi: 10.1186/s13041-015-0095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sher Roger B., Heiman-Patterson Terry D., Blankenhorn Elizabeth A., Jiang Juliann, Alexander Guillermo, Deitch Jeffrey S., Cox Gregory A. A major QTL on mouse chromosome 17 resulting in lifespan variability in SOD1-G93A transgenic mouse models of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2014;15(7-8):588–600. doi: 10.3109/21678421.2014.932381. [DOI] [PubMed] [Google Scholar]
- 56.Chen Yiquan, Brew Bruce J., Guillemin Gilles J. Characterization of the kynurenine pathway in NSC-34 cell line: implications for amyotrophic lateral sclerosis. Journal of Neurochemistry. 2011;118(5):816–825. doi: 10.1111/j.1471-4159.2010.07159.x. [DOI] [PubMed] [Google Scholar]
- 57.Mantovani Stefania, Garbelli Silvia, Pasini Alessandra, Alimonti Dario, Perotti Cesare, Melazzini Mario, Bendotti Caterina, Mora Gabriele. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. Journal of Neuroimmunology. 2009;210(1-2):73–79. doi: 10.1016/j.jneuroim.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 58.Rentzos M., Evangelopoulos E., Sereti E., Zouvelou V., Marmara S., Alexakis T., Evdokimidis I. Alterations of T cell subsets in ALS: a systemic immune activation? Acta Neurologica Scandinavica. 2011;125(4):260–264. doi: 10.1111/j.1600-0404.2011.01528.x. [DOI] [PubMed] [Google Scholar]
- 59.Evans David M, Frazer Ian H, Martin Nicholas G. Genetic and environmental causes of variation in basal levels of blood cells. Twin Research. 1999;2(04):250–257. doi: 10.1375/136905299320565735. [DOI] [PubMed] [Google Scholar]
- 60.Thein, S. L. et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood (2000). [PubMed]
- 61.Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Engelborghs S, De Bleecker J, Ivanoiu A, Deryck O, Edbauer D, Zhang M, Heeman B, Bäumer V, Van den Broeck M, Mattheijssens M, Peeters K, Rogaeva E, De Jonghe P, Cras P, Martin J-J, de Deyn P P, Cruts M, Van Broeckhoven C. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Molecular Psychiatry. 2015;21(8):1112–1124. doi: 10.1038/mp.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Elaine Y., Russ Jenny, Wu Kathryn, Neal Donald, Suh Eunran, McNally Anna G., Irwin David J., Van Deerlin Vivianna M., Lee Edward B. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathologica. 2014;128(4):525–541. doi: 10.1007/s00401-014-1286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McMillan C. T., Russ J., Wood E. M., Irwin D. J., Grossman M., McCluskey L., Elman L., Van Deerlin V., Lee E. B. C9orf72 promoter hypermethylation is neuroprotective: Neuroimaging and neuropathologic evidence. Neurology. 2015;84(16):1622–1630. doi: 10.1212/WNL.0000000000001495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Russ Jenny, Liu Elaine Y., Wu Kathryn, Neal Donald, Suh EunRan, Irwin David J., McMillan Corey T., Harms Matthew B., Cairns Nigel J., Wood Elisabeth M., Xie Sharon X., Elman Lauren, McCluskey Leo, Grossman Murray, Van Deerlin Vivianna M., Lee Edward B. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathologica. 2014;129(1):39–52. doi: 10.1007/s00401-014-1365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xi Zhengrui, Zhang Ming, Bruni Amalia C., Maletta Raffaele G., Colao Rosanna, Fratta Pietro, Polke James M., Sweeney Mary G., Mudanohwo Ese, Nacmias Benedetta, Sorbi Sandro, Tartaglia Maria Carmela, Rainero Innocenzo, Rubino Elisa, Pinessi Lorenzo, Galimberti Daniela, Surace Ezequiel I., McGoldrick Philip, McKeever Paul, Moreno Danielle, Sato Christine, Liang Yan, Keith Julia, Zinman Lorne, Robertson Janice, Rogaeva Ekaterina. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathologica. 2015;129(5):715–727. doi: 10.1007/s00401-015-1401-8. [DOI] [PubMed] [Google Scholar]
- 66.Conforti Francesca Luisa, Tortelli Rosanna, Morello Giovanna, Capozzo Rosa, Barulli Maria Rosaria, Cavallaro Sebastiano, Logroscino Giancarlo. Clinical features and genetic characterization of two dizygotic twins with C9orf72 expansion. Neurobiology of Aging. 2018;69:293.e1-293.e8. doi: 10.1016/j.neurobiolaging.2018.05.002. [DOI] [PubMed] [Google Scholar]
- 67.Brooks Benjamin Rix, Miller Robert G, Swash Michael, Munsat Theodore L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. 2000;1(5):293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 68.R Core Team. R: A Language and Environment for Statistical Computing, https://www.r-project.org/ (R Foundation for Statistical Computing, Vienna, Austria, 2018).
- 69.Ehrich M., Nelson M. R., Stanssens P., Zabeau M., Liloglou T., Xinarianos G., Cantor C. R., Field J. K., van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proceedings of the National Academy of Sciences. 2005;102(44):15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho Vikki, Ashbury Janet E., Taylor Sherryl, Vanner Stephen, King Will D. Quantification of gene-specific methylation of DNMT3B and MTHFR using sequenom EpiTYPER®. Data in Brief. 2016;6:39–46. doi: 10.1016/j.dib.2015.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maksimovic Jovana, Phipson Belinda, Oshlack Alicia. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research. 2017;5:1281. doi: 10.12688/f1000research.8839.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fortin Jean-Philippe, Fertig Elana, Hansen Kasper. shinyMethyl: interactive quality control of Illumina 450k DNA methylation arrays in R. F1000Research. 2014;3:175. doi: 10.12688/f1000research.4680.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Assenov Yassen, Müller Fabian, Lutsik Pavlo, Walter Jörn, Lengauer Thomas, Bock Christoph. Comprehensive analysis of DNA methylation data with RnBeads. Nature Methods. 2014;11(11):1138–1140. doi: 10.1038/nmeth.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pidsley Ruth, Y Wong Chloe C, Volta Manuela, Lunnon Katie, Mill Jonathan, Schalkwyk Leonard C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14(1):293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aryee Martin J., Jaffe Andrew E., Corrada-Bravo Hector, Ladd-Acosta Christine, Feinberg Andrew P., Hansen Kasper D., Irizarry Rafael A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Yi-an, Lemire Mathieu, Choufani Sanaa, Butcher Darci T., Grafodatskaya Daria, Zanke Brent W., Gallinger Steven, Hudson Thomas J., Weksberg Rosanna. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Price Magda E, Cotton Allison M, Lam Lucia L, Farré Pau, Emberly Eldon, Brown Carolyn J, Robinson Wendy P, Kobor Michael S. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics & Chromatin. 2013;6(1):4. doi: 10.1186/1756-8935-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Triche, J. T. FDb.InfiniumMethylation.hg19: Annotation package for Illumina Infinium DNA methylation probes. (2014).
- 79.Dempster Emma L., Pidsley Ruth, Schalkwyk Leonard C., Owens Sheena, Georgiades Anna, Kane Fergus, Kalidindi Sridevi, Picchioni Marco, Kravariti Eugenia, Toulopoulou Timothea, Murray Robin M., Mill Jonathan. Disease-associated epigenetic changes in monozygotic twins discordant for schizophrenia and bipolar disorder. Human Molecular Genetics. 2011;20(24):4786–4796. doi: 10.1093/hmg/ddr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andrews, S. & Babraham Bioinformatics. FastQC: A quality control tool for high throughput sequence data, citeulike-article-id:11583827, arXiv:0906.2747v1 (2010).
- 81.Bolger Anthony M., Lohse Marc, Usadel Bjoern. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin Marcel. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17(1):10. [Google Scholar]
- 83.Kim Daehwan, Langmead Ben, Salzberg Steven L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Robinson M. D., McCarthy D. J., Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ritchie Matthew E., Phipson Belinda, Wu Di, Hu Yifang, Law Charity W., Shi Wei, Smyth Gordon K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7):e47–e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anders S., Pyl P. T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2014;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Law Charity W, Chen Yunshun, Shi Wei, Smyth Gordon K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology. 2014;15(2):R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hochberg, B. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J. Royal Stat. Soc., 10.2307/2346101, 95/57289 (1995).
- 89.Carbon, S. et al. Expansion of the gene ontology knowledgebase and resources: The gene ontology consortium. Nucleic Acids Res., 10.1093/nar/gkw1108 (2017). [DOI] [PMC free article] [PubMed]
- 90.Ashburner Michael, Ball Catherine A., Blake Judith A., Botstein David, Butler Heather, Cherry J. Michael, Davis Allan P., Dolinski Kara, Dwight Selina S., Eppig Janan T., Harris Midori A., Hill David P., Issel-Tarver Laurie, Kasarskis Andrew, Lewis Suzanna, Matese John C., Richardson Joel E., Ringwald Martin, Rubin Gerald M., Sherlock Gavin. Gene Ontology: tool for the unification of biology. Nature Genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mi Huaiyu, Huang Xiaosong, Muruganujan Anushya, Tang Haiming, Mills Caitlin, Kang Diane, Thomas Paul D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Research. 2016;45(D1):D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nelson Heather H., Marsit Carmen J., Kelsey Karl T. Global Methylation in Exposure Biology and Translational Medical Science. Environmental Health Perspectives. 2011;119(11):1528–1533. doi: 10.1289/ehp.1103423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brooks, M. E. et al. glmmTMB balances speed and flexibility among packages for zero-inflated generalized linear mixed modeling. The R J., 10.3929/ETHZ-B-000240890 (2017).
- 94.Clifford, H., Wessely, F., Pendurthi, S. & Emes, R. D. Comparison of clustering methods for investigation of genome-wide methylation array data. Front. Genet., 10.3389/fgene.2011.00088 (2011). [DOI] [PMC free article] [PubMed]
- 95.Jaskowiak Pablo Andretta, Costa Ivan G., Campello Ricardo J.G.B. Clustering of RNA-Seq samples: Comparison study on cancer data. Methods. 2018;132:42–49. doi: 10.1016/j.ymeth.2017.07.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables S1-S4 and Figures S1-S3
Data Availability Statement
The datasets generated and/or analysed during the current study are not publicly available since our ethics permission does not cover sharing of data to third parties but are available from the corresponding author on reasonable request.