

Abstract
A general catalytic method for asymmetric C-alkylation of nitroalkanes using nickel catalysis is described. This method enables the formation of highly enantioenriched β-nitroamides from readily available α-bromoamides using mild reaction conditions that are compatible with a wide range of functional groups. When combined with subsequent reactions, this method allows access to highly enantioenriched products with nitrogen-bearing fully substituted carbon centers.
Graphical Abstract
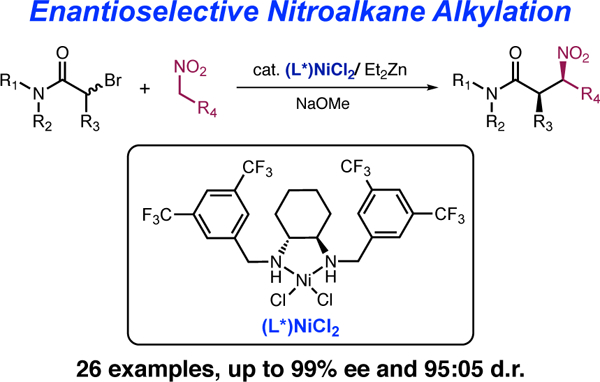
Nitroalkanes are broadly useful building blocks in organic synthesis.1 Not only can the nitro group be converted into a range of other functional groups, but nitroalkanes also participate in a variety of C–C bond-forming reactions, including Michael, Henry, nitro-Mannich, and palladium-catalyzed allylation and arylation reactions. However, despite this great synthetic versatility, for many years the simple C-alkylation of nitroalkanes – a potentially important reaction for converting simple nitroalkanes into more complex nitroalkanes –remained challenging due to the dominance of O-alkylation, which ultimately yields aldehydes instead of the desired nitroalkane products.2
Over the past several years, our group has begun to address this gap by developing transition metal-catalyzed alkylation reactions of nitroalkanes.3 By using a transition metal catalyst, we were able to change from the inherent two-electron chemistry of nitroalkanes to single-electron manifolds, thus changing the preference for C- vs. Oalkylation. With these new protocols, alkylation with a variety of alkyl halides, including aliphatic alkyl halides, is now possible.
Despite these advances in the ability to control the site-selectivity of the alkylation reactions, control of stereoselectivity has remained elusive. This is particularly noteworthy, because asymmetric variants of many other C–C bond-forming reactions of nitroalkanes have been described, and those reactions now constitute important ways to install nitrogencontaining stereocenters.4 The seeming inability to render nitroalkane alkylation asymmetric stems not only from that fact that the previously identified optimal ligands are not easily rendered chiral, but more significantly from the fact that all prior mechanistic data suggested that the reactions are proceeding via a radical pathway involving an outersphere C–C bond-forming step that does not directly involve the metal catalyst or ligand.3a, 3c, 3d Thus, prior data strongly suggested that asymmetric nitroalkane alkylation would not be possible using current catalytic methods, and the asymmetric alkylation of nitroalkanes has remained an open challenge.
Recently, while exploring copper-catalyzed reactions, we observed modest, ligand-dependent changes in diastereoselection in nitroalkane alkylation.5 Those experiments suggested a role for the ligand in C–C bond formation, prompting us to reevaluate an outer-sphere pathway and opening the possibility of asymmetric induction. Herein we report the nickel-catalyzed asymmetric alkylation of nitroalkanes using abromoamides (Scheme 1),6 which is the first example of an asymmetric nitroalkane alkylation using an alkyl halide electrophile. We show that alkylation of nitroalkanes with racemic α-bromoamides leads to highly enantioselective formation of α-substituted, β-nitroamides with good levels of diastereocontrol.7 We demonstrate that these products can be utilized to prepare asymmetric β-aminoamides with fully substituted bcarbons with outstanding levels of enantioselectivity. Moreover, these observations also cast new light onto transition metal-catalyzed nitroalkane alkylations, and suggest a more complex mechanism than previously understood.
Scheme 1.
General Method for Asymmetric Nitroalkane Alkylation.
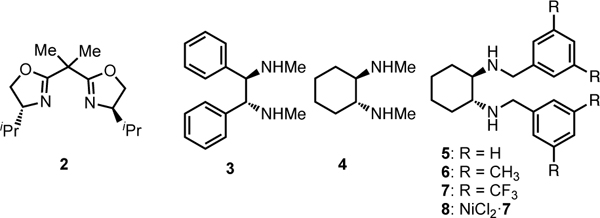
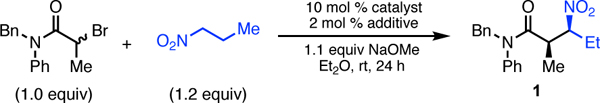
Our investigation began with reaction of commercially available racemic N-benzyl-2-bromo-N-phenylpropionamide with 1-nitropropane to make β-nitroamide 1 (Table 1). Using conditions similar to our prior non-stereocontrolled nickel-catalyzed alkylation reactions (10 mol% Ni(COD)2, slight excess of NaOMe),3d we began to systematically investigate a variety of chiral ligands. Although many classes of ligands provided either no enantioinduction or yield of product, we were pleased to find that use of commercially available bis(oxazoline) ligand 2 provided desired product 1 with a measurable 12% ee, albeit in 30% yield and no measurable diastereoselectivity (entry 1). Eventually we found that the C2 symmetric chiral 1,2-diamine 3 provided considerably higher levels of enantioselectivity (72% ee) and good diastereoselectivity (92:8, favoring the syn-isomer),8 but did not improve the yield of the reaction (entry 2). N,N’-Dimethylcyclohexane1,2-diamine (4) gave slightly lower dr and yield, but provided better enantioselectivity (78% ee, entry 3). However, increasing the size of the amino substituents provided much higher yield, good diastereoselectivity, and retained enantioselectivity (entry 4). Although increasing the size of the aromatic groups with the use of meta-methyl groups (ligand 6) did not provide substantially different results (entry 5), placing electron-withdrawing CF3 groups at the same position (ligand 7) provided a substantial increase in enantioselectivity and higher diastereoselectivity (entry 6). Several rounds of additional optimization led us to find that the optimal combination of enantioselectivity, diastereoselectivity, and reactivity was achieved by using the NiCl2 complex of this optimal ligand (complex 8), Et2Zn as an in situ reductant, and 0 °C as the reaction temperature (entry 7).5 These conditions resulted in high enantioselectivity, good diastereoselectivity, and outstanding yields.
Table 1.
Discovery of the Catalytic System.
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive | Yield of 1a | d.r syn/anti | %eeb syn |
| 1 | Ni(COD)2/2 | - | 30 | 50:50 | 12 |
| 2 | Ni(COD)2/3 | - | 27 | 92:08 | 72 |
| 3 | Ni(COD)2/4 | - | 18 | 78:22 | 78 |
| 4 | Ni(COD)2/5 | - | 80 | 82:18 | 79 |
| 5 | Ni(COD)2/6 | - | 80 | 84:16 | 82 |
| 6 | Ni(COD)2/7 | - | 82 | 85:15 | 88 |
| 7 | 8c | Et2Zn | 97 | 79:21 | 90 |
Determined via 1H NMR against internal standard
Determined using chiral HPLC analysis
0 °C.
With optimized conditions in hand, we investigated the scope of the nitroalkane (Scheme 2). A variety of primary nitroalkanes were subjected to the reaction using (±)-N-benzyl-2-bromo-Nphenylpropionamide as the alkylating reagent. High ee was observed for 1-nitropropane (1), as well as with a β-branched nitroalkane (9). A variety of functionalized nitroalkanes including those with alkene, aryl, aryl ether, acetate, free alcohol, ester, unprotected and protected ketone groups, were all alkylated with good to excellent ee (10–17). In all the above cases, good to excellent levels of dr were also observed. Nitromethane can also be alkylated albeit with low yield and ee (18).
Scheme 2.
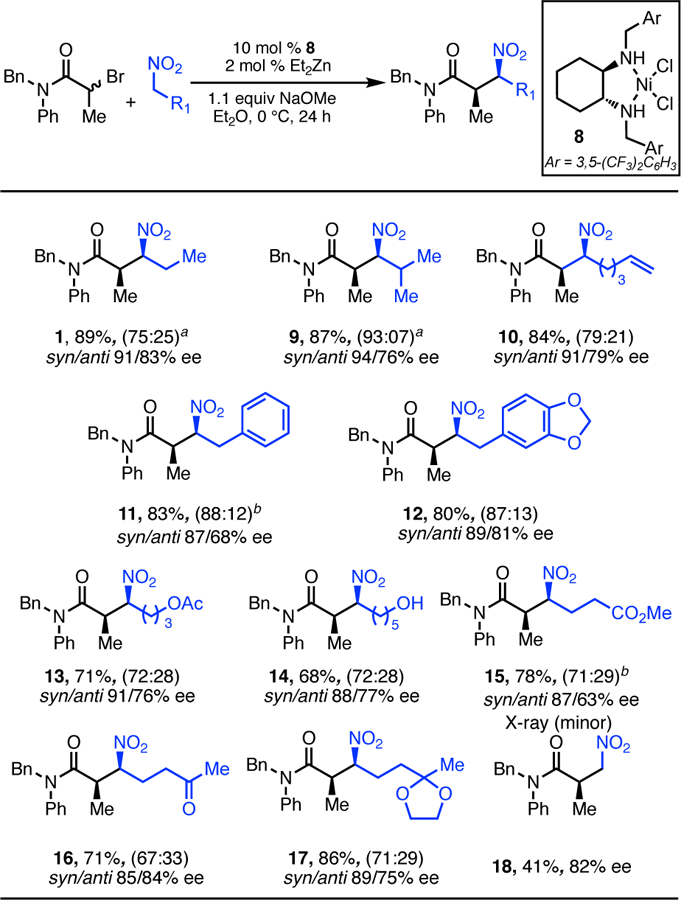
Scope of Nitroalkanes.
The scope with respect to the α-bromoamide is also broad (Scheme 3). Good dr and high ee were observed for amides possessing electron-rich, electron-poor and sterically encumbered groups (19–21). aBromoamides possessing α-alkyl substituents larger than methyl were tolerated well, albeit with lower dr and ee (22, 23). Significantly, several amide backbones, including indoline, morpholine, aryl-alkyl, and Weinreb amides, were tolerated and all resulted in products with high dr and ee. These reactions were most effective when the nitroalkane starting material was β-branched (24–27), but substrates without bbranching also proceeded smoothly (28–31). Amides bearing tertiary bromides (32) and secondary nitroalkanes (33) could also be utilized in the alkylation reaction. In both cases, lower yields and ee were observed. However, these highly congested products would be challenging to prepare by other methods.
Scheme 3.
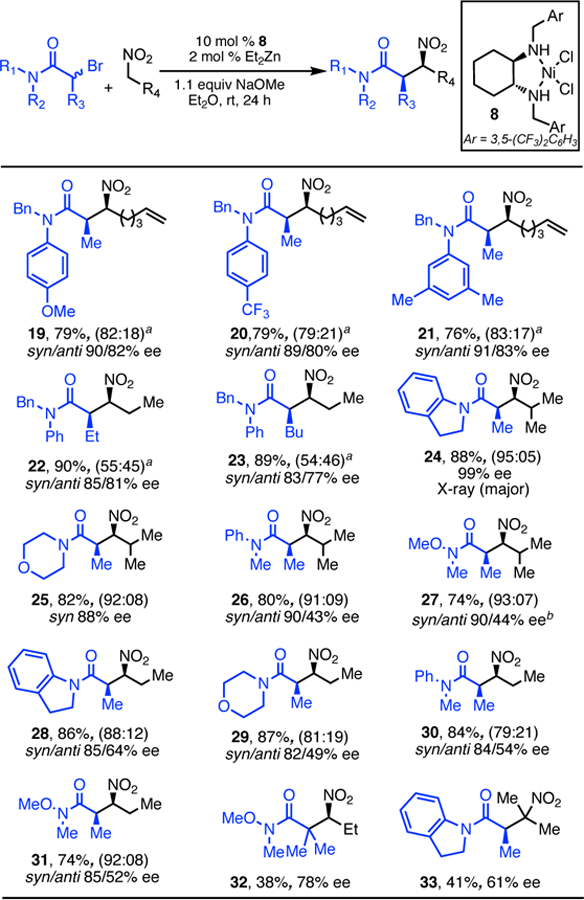
Scope of α-BromoamidesScheme.
As shown in Schemes 2 and 3, in most cases the asymmetric nitroalkane alkylation exhibits good to excellent levels of diastereoselectivity. In all cases, the major diastereomer was formed with higher enantioselectivity, but good enantioselection was also observed for the minor isomer. In many cases, the diastereomers can be easily separated by standard flash column chromatography. In two cases (15 and 24), we were able to determine the relative and absolute stereochemistry of one of the diastereomers using X-ray crystallography. In both cases, the (1R,2S)syn-isomer proved to be the major isomer using the R,R-catalyst.5 Diagnostic 1H NMR signals supported this relative configuration for the other entries as well.9
The ability to prepare enantioenriched β-nitroamides using this method has distinct advantages over other methods for construction of amides bearing β-nitrogen atoms (such as the Mannich reaction).4h, 10 Specifically, unlike Mannich products, the acidity of the proton α to the nitrogen atom allows β-nitroamides to be used in further synthetic transformations.11 These transformations lead to highly substituted products. For example, use of the alkylation products as nucleophiles in C–C bond-forming reactions leads to β-nitroamides with fully substituted β-carbons (Scheme 4). In these reactions, the stereocenter α to the carbonyl controls facial selection, leading to highly diastereoselective conjugate addition (top),11 trifluoromethylation (middle),12 and Tsuji-Trost allylation (bottom) reactions,13 all without erosion of ee. Consistent with our earlier studies,11–12 the syndiastereomer is observed in all cases, and the nitro groups of the products are readily reduced to the corresponding amines (37–39).
Scheme 4.
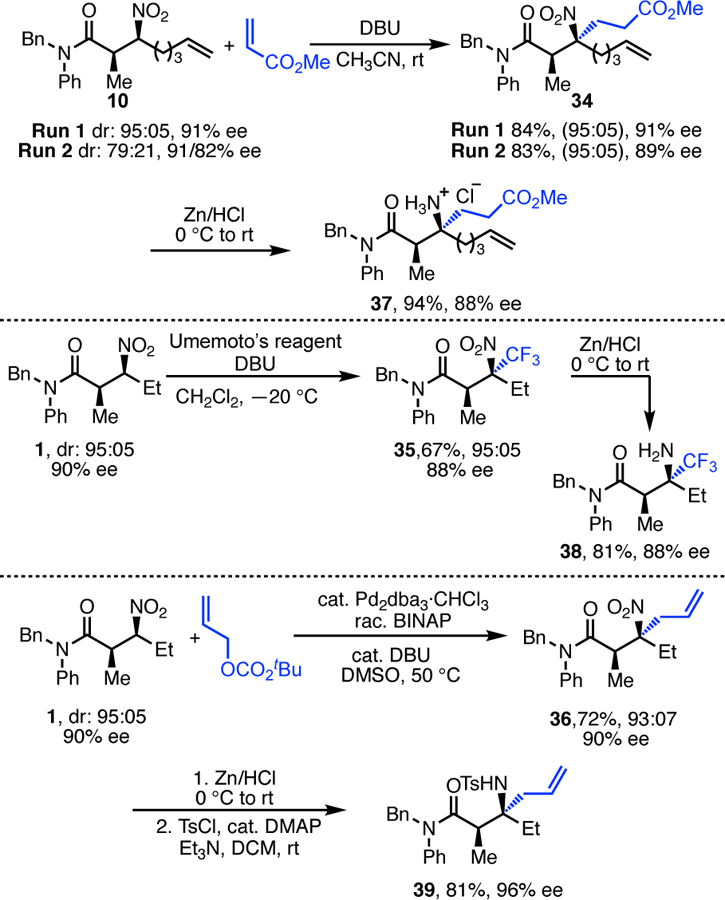
Downstream Functionalization of Alkylated Products.
Significantly, isolation of a single diastereomer of the alkylation product is not required for use in these downsteam reactions. As shown at the top of Scheme 4, the conjugate addition reaction can be conducted either with a single isolated diastereomer or with the mixture of diastereomers obtained from the nickel-catalyzed reaction. In both cases, identical diastereoselectivity and nearly identical enantiopurity of product are obtained. These results indicate that the diastereomers observed in the alkylation reaction are epimeric at the β-center, and that the diastereomers converge upon deprotonation of the nitroalkane in the subsequent reactions.14 From a practical standpoint, this is highly advantageous when utilizing the alkylation products in this way.
Several mechanistic experiments were carried out to probe the nature of the transformation. Consistent with our earlier non-stereoselective nitroalkane alkylation reactions, these studies indicate a mechanism involving radical intermediates. First, when the reaction was run in the presence of 1 equiv TEMPO, a known radical scavenger,15 no alkylation product 1 was formed (Scheme 5, top). Second, cyclopropylcarbinyl rearrangement is observed with substrate 40, resulting exclusively in ring-opened product 41 (Scheme 5, middle).16 Finally, the enantiopurity of the starting α-bromoamide does not affect the stereoselectivity of the reaction; both enantiomers of 42 lead to identical dr and ee of products, albeit with slightly different yields. Additionally, when starting material was re-isolated from reactions stopped at partial conversion, no erosion of ee of the bromoamide was observed (Scheme 5, bottom).5 This result indicates that activation of the C–Br is irreversible.
Scheme 5.
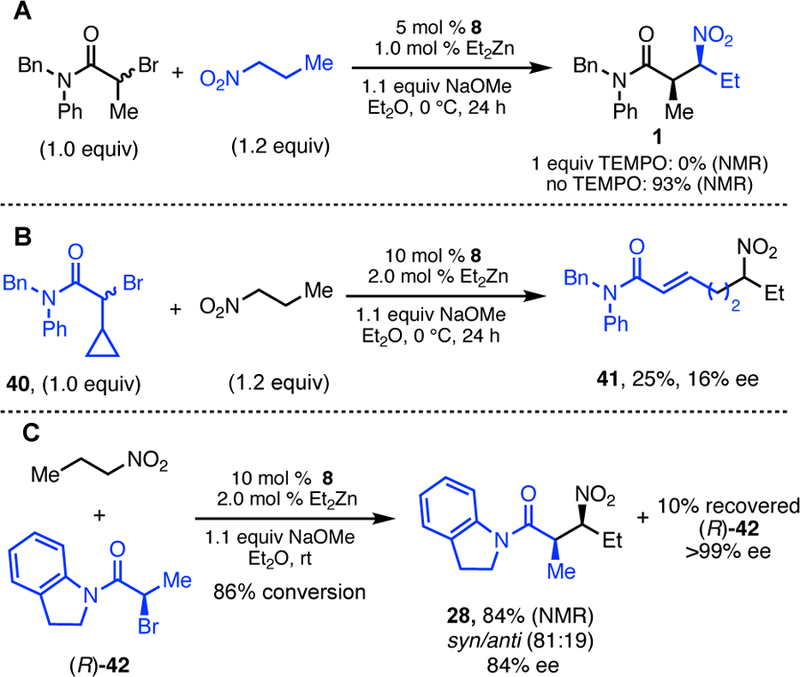
Mechanistic Probes.
Although further studies will be required to fully elucidate the mechanism, at present we favor the NiI/NiIII catalytic cycle shown in Scheme 6.17 Initial reduction of the Ni(II) precatalyst by Et2Zn results in formation of a Ni(0) complex. Comproportionation with excess Ni(II) complex then results in a Ni(I) catalyst.18 This pathway would explain the need for excess Ni(II) compared to Et2Zn. Simultaneously, exothermic deprotonation of the acidic nitroalkane by the alkoxide base results in an insoluble (or sparingly soluble) nitronate anion, which undergoes anion exchange with the Ni(I) complex resulting in a soluble Ni(I) nitronate. This electron-rich Ni(I) complex then reacts with the alkyl bromide via a stepwise oxidative addition to form a Ni(III) alkyl nitronate. Reductive elimination then provides the observed product and regenerates the catalyst.5
Scheme 6.
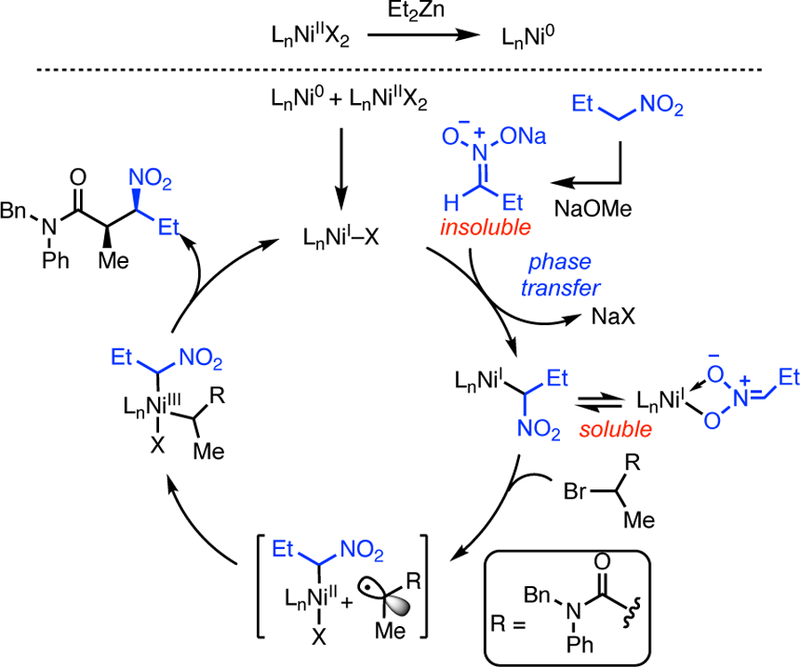
Possible Mechanistic Pathway.
In conclusion, the first Ni-catalyzed asymmetric C-alkylation of nitroalkanes using an alkyl halide has been developed. This method enables formation of highly enantioenriched β-nitroamide from readily available α-bromoamides using mild reaction conditions that are compatible with a wide range of functional groups. Significantly, due to both the acidity of the β-proton and the ability of the a stereocenter to control subsequent reactions, these products can be easily manipulated to access a range of highly substituted β-aminoamides, providing distinct advantages over competing technologies. This study also demonstrates that the mechanism of transition metal-catalyzed nitroalkane alkylation reactions are more complex than earlier believed, and indicate that nickel-catalyzed nitroalkane alkylation occurs via metal-mediated C–C bond formation. Current efforts are directed at further expanding the scope of asymmetric nitroalkane alkylation reactions and better defining the mechanisms by which they proceed.
Supplementary Material
ACKNOWLEDGMENT
Dr. Glenn Yap (UD) is acknowledged for assistant with X-ray crystallography.
Funding Sources
The University of Delaware (UD), the University of Delaware Research Foundation, the Research Corp. Cottrell Scholars Program, and the NIH NIGMS (R01 GM102358) are gratefully acknowledged for support. Data was acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF CHE0421224, CHE0840401, CHE1229234, CHE1048367; NIH S10 OD016267, S10 RR026962, P20 GM104316, P30 GM110758).
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental Procedures (PDF)
Crystallographic data (CIF)
REFERENCES
- (1).(a) Ono N, The Nitro Group In Organic Synthesis John Wiley And Sons: New York, 2001; [Google Scholar]; (b) Ballini R; Palmieri A Synthetic Procedures for the Preparation of Nitroalkanes. Adv. Synth. Catal 2018, 360, 2240, doi: 10.1002/adsc.201800163 [DOI] [Google Scholar]
- (2).(a) Weisler L; Helmkamp RW The Action of Some Benzyl Halides on Salts of Phenylnitromethane and Phenylnitro-acetonitrile. J. Am. Chem. Soc 1945, 67, 1167, doi: 10.1021/ja01223a040; [DOI] [Google Scholar]; (b) Hass HB; Bender ML The Reaction of Benzyl Halides with the Sodium Salt of 2-Nitropropane. A General Synthesis of Substituted Benzaldehydes. J. Am. Chem. Soc 1949, 71, 1767, doi: 10.1021/ja01173a066; [DOI] [Google Scholar]; (c) Kornblum N Substitution Reactions Which Proceed via Radical Anion Intermediates. Angew. Chem., Int. Ed. Engl 1975, 14, 734, doi: 10.1002/anie.197507341; [DOI] [Google Scholar]; (d) Seebach D; Henning R; Lehr F; Gonnermann J Carbon Alkylations of α.α- and α.βdoubly Deprotonated Nitroalkanes. Tetrahedron Lett 1977, 18, 1161, doi: 10.1016/S0040-4039(01)92859-X; [DOI] [Google Scholar]; (e) Seebach D; Henning R; Lehr F Double Deprotonation of 3-Nitropropene and 4-Nitro-1-butene: Derivatives of Their Dianions as Novel Reagents for CC-Linkage. Angew. Chem., Int. Ed. Engl 1978, 17, 458, doi: 10.1002/anie.197804581; [DOI] [Google Scholar]; (f) Katritzky AR; de Ville G; Patel RC Carbon-Alkylation of Simple Nitronate Anions. J. Chem. Soc., Chem. Commun 1979, 602, doi: 10.1039/C3979000602A; [DOI] [Google Scholar]; (g) Katritzky AR; Kashmiri MA; De Ville GZ; Patel RC Kinetics and Mechanism of the C-Alkylation of Nitroalkane Anions by 1-Alkyl-2,4,6-triphenylpyridiniums: A Nonchain Reaction with Radicaloid Characteristics. J. Am. Chem. Soc 1983, 105, 90, doi: 10.1021/ja00339a016; [DOI] [Google Scholar]; (h) Russell GA; Khanna RK The Reaction of Carbanions with tert-Butyl Radicals. Tetrahedron 1985, 41, 4133, doi: 10.1016/S0040-4020(01)97189-3; [DOI] [Google Scholar]; (i) Branchaud PB; Yu G-X Cobaloxime-Mediated Radical Alkyl-Nitroalkylanion Cross Coupling. Tetrahedron Lett 1988, 29, 6545, doi: 10.1016/S0040-4039(00)82393-X [DOI] [Google Scholar]
- (3).(a) Gildner PG; Gietter AAS; Cui D; Watson DA Benzylation of Nitroalkanes Using Copper-Catalyzed Thermal Redox Catalysis: Toward the Facile C-Alkylation of Nitroalkanes. J. Am. Chem. Soc 2012, 134, 9942, doi: 10.1021/ja304561c; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gietter AAS; Gildner PG; Cinderella AP; Watson DA General Route for Preparing βNitrocarbonyl Compounds Using Copper Thermal Redox Catalysis. Org. Lett 2014, 16, 3166, doi: 10.1021/ol5014153; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shimkin KW; Gildner PG; Watson DA Copper-Catalyzed Alkylation of Nitroalkanes with αBromonitriles: Synthesis of β-Cyanonitroalkanes. Org. Lett 2016, 18, 988, doi: 10.1021/acs.orglett.6b00093; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rezazadeh S; Devannah V; Watson DA Nickel-Catalyzed C-Alkylation of Nitroalkanes with Unactivated Alkyl Iodides. J. Am. Chem. Soc 2017, 139, 8110, doi: 10.1021/jacs.7b04312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Trost BM; Surivet J-P Diastereo- and Enantioselective Allylation of Substituted Nitroalkanes. J. Am. Chem. Soc 2000, 122, 6291, doi: 10.1021/ja001062s; [DOI] [Google Scholar]; (b) Ooi T; Takada S; Doda K; Maruoka K Highly Diastereo- and Enantioselective Formal Conjugate Addition of Nitroalkanes to Nitroalkenes by Chiral Ammonium Bifluoride Catalysis. Angew. Chem., Int. Ed 2006, 45, 7606, doi: 10.1002/anie.200602787; [DOI] [PubMed] [Google Scholar]; (c) Palomo C; Oiarbide M; Laso A Recent Advances in the Catalytic Asymmetric Nitroaldol (Henry) Reaction. Eur. J. Org. Chem 2007, 2007, 2561, doi: 10.1002/ejoc.200700021; [DOI] [Google Scholar]; (d) Dobish MC; Johnston JN Chiral Brønsted Base-Promoted Nitroalkane Alkylation: Enantioselective Synthesis of sec-Alkyl-3-Substituted Indoles. Org. Lett 2010, 12, 5744, doi: 10.1021/ol1025712; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Roca-Lopez D; Sadaba D; Delso I; Herrera RP; Tejero T; Merino P Asymmetric organocatalytic synthesis of γnitrocarbonyl compounds through Michael and Domino reactions. Tetrahedron: Asymmetry 2010, 21, 2561, doi: 10.1016/j.tetasy.2010.11.001; [DOI] [Google Scholar]; (f) Blay G; Hernández-Olmos V; Pedro JR Development of New N,NLigands for the Enantioselective Copper(II)-Catalyzed Henry Reaction. Synlett 2011, 2011, 1195, doi: 10.1055/s-0030-1260558; [DOI] [Google Scholar]; (g) Ohmatsu K; Ito M; Kunieda T; Ooi T Ion-paired Chiral Ligands for Asymmetric Palladium Catalysis. Nature Chem 2012, 4, 473, doi: 10.1038/nchem.1311; [DOI] [PubMed] [Google Scholar]; (h) Noble A; Anderson JC Nitro-Mannich Reaction. Chem. Rev 2013, 113, 2887, doi: 10.1021/cr300272t; [DOI] [PubMed] [Google Scholar]; (i) Qian H; Yu X; Zhang J; Sun J Organocatalytic Enantioselective Synthesis of 2,3-Allenoates by Intermolecular Addition of Nitroalkanes to Activated Enynes. J. Am. Chem. Soc 2013, 135, 18020, doi: 10.1021/ja409080v; [DOI] [PubMed] [Google Scholar]; (j) Serdyuk OV; Heckel CM; Tsogoeva SB Bifunctional Primary Amine-Thioureas in Asymmetric Organocatalysis. Org. Biomol. Chem 2013, 11, 7051, doi: 10.1039/C3OB41403E; [DOI] [PubMed] [Google Scholar]; (k) Tsakos M; Kokotos CG Primary and Secondary Amine-(thio)ureas and Squaramides and Their Applications in Asymmetric Organocatalysis. Tetrahedron 2013, 69, 10199, doi: 10.1016/j.tet.2013.09.080; [DOI] [Google Scholar]; (l) Manna MS; Mukherjee S Organocatalytic Enantioselective Formal C(sp2)–H Alkylation. J. Am. Chem. Soc 2015, 137, 130, doi: 10.1021/ja5117556; [DOI] [PubMed] [Google Scholar]; (m) Vara BA; Johnston JN Enantioselective Synthesis of β-Fluoro Amines via β-Amino α-Fluoro Nitroalkanes and a Traceless Activating Group Strategy. J. Am. Chem. Soc 2016, 138, 13794, doi: 10.1021/jacs.6b07731; [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Kawada M; Nakashima K; Hirashima S.-i.; Yoshida A; Koseki Y; Miura T Asymmetric Conjugate Addition of Nitroalkanes to Enones Using a Sulfonamide–Thiourea Organocatalyst. J. Org. Chem 2017, 82, 6986, doi: 10.1021/acs.joc.7b00835; [DOI] [PubMed] [Google Scholar]; (o) Li Y; Huang Y; Gui Y; Sun J; Li J; Zha Z; Wang Z Copper-Catalyzed Enantioselective Henry Reaction of β,γ-Unsaturated α-Ketoesters with Nitromethane in Water. Org. Lett 2017, 19, 6416, doi: 10.1021/acs.orglett.7b03299; [DOI] [PubMed] [Google Scholar]; (p) Lu N; Bai F; Fang Y; Wei Z; Cao J; Liang D; Lin Y; Duan H Bifunctional Phase-Transfer Catalysts Catalyzed Diastereo- and Enantioselective Aza-Henry Reaction of β,γ-Unsaturated Nitroalkenes With Amidosulfones. Adv. Synth. Catal 2017, 359, 4111, doi: 10.1002/adsc.201700787 [DOI] [Google Scholar]
- (5).See Supporting Information for further detail.
- (6).For recent reviews of asymmetric reactions involving alkyl halides, see: (a) Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents To Construct C–C Bonds. Chem. Rev 2015, 115, 9587, doi: 10.1021/acs.chemrev.5b00162; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi J; Fu GC Transition Metal–Catalyzed Alkyl-Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry. Science 2017, 356, doi: 10.1126/science.aaf7230; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bhat V; Welin ER; Guo X; Stoltz BM Advances in Stereoconvergent Catalysis from 2005 to 2015: TransitionMetal-Mediated Stereoablative Reactions, Dynamic Kinetic Resolutions, and Dynamic Kinetic Asymmetric Transformations. Chem. Rev 2017, 117, 4528, doi: 10.1021/acs.chemrev.6b00731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wilson JE; Casarez AD; MacMillan DWC Enantioselective Aldehyde α-Nitroalkylation via Oxidative Organocatalysis. J. Am. Chem. Soc 2009, 131, 11332, doi: 10.1021/ja904504j [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lundin PM; Fu GC Asymmetric Suzuki Cross-Couplings of Activated Secondary Alkyl Electrophiles: Arylations of Racemic αChloroamides. J. Am. Chem. Soc 2010, 132, 11027, doi: 10.1021/ja105148g [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).The absolute configuration of the other beta-nitroamide products were assigned by analogy.
- (10).Arend M; Westermann B; Risch N Modern Variants of the Mannich Reaction. Angew. Chem., Int. Ed 1998, 37, 1044, doi: [DOI] [PubMed] [Google Scholar]
- (11).Gietter-Burch AAS; Mitrut RE; Watson DA Highly Diastereoselective Michael Reactions Using β-Nitrocarbonyl Nucleophiles. Org. Lett 2015, 17, 5468, doi: 10.1021/acs.orglett.5b02832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gietter-Burch AAS; Devannah V; Watson DA Trifluoromethylation of Secondary Nitroalkanes. Org. Lett 2017, 19, 2957, doi: 10.1021/acs.orglett.7b01196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Maki K; Kanai M; Shibasaki M Pd-Catalyzed Allylic Alkylation of Secondary Nitroalkanes. Tetrahedron 2007, 63, 4250, doi: 10.1016/j.tet.2007.03.062 [DOI] [Google Scholar]
- (14).Interestingly, the use of weaker bases, such as DBU leads to inconsistent stereoselection in the alkylation reaction due to epimerization of both the alpha and beta stereocenters. We attribute the stability of the product under the tert-butoxide conditions to be due to the solubility of the base. DBU is soluble under the reaction condition; tert-butoxide quantitively deprotonates the nitronate substrate, which then precipitates from the reaction media. The fact that there is little to no free base in solution during the reaction protects the product against epimerization. See Supporting Information for more details.
- (15).Bowry VW; Ingold KU Kinetics of Nitroxide Radical Trapping. 2. Structural Effects. J. Am. Chem. Soc 1992, 114, 4992, doi: 10.1021/ja00039a006 [DOI] [Google Scholar]
- (16).Newcomb M Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49, 1151, doi: 10.1016/S0040-4020(01)85808-7 [DOI] [Google Scholar]
- (17).(a) Hu X Nickel-Catalyzed Cross Coupling of Non-Activated Alkyl Halides: A Mechanistic Perspective. Chemical Science 2011, 2, 1867, doi: 10.1039/C1SC00368B; [DOI] [Google Scholar]; (b) Schley ND; Fu GC Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc 2014, 136, 16588, doi: 10.1021/ja508718m [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jones GD; Martin JL; McFarland C; Allen OR; Hall RE; Haley AD; Brandon RJ; Konovalova T; Desrochers PJ; Pulay P; Vicic DA Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl−Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc 2006, 128, 13175, doi: 10.1021/ja063334i [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.