

Abstract
UBE3A is a gene responsible for the pathogenesis of Angelman syndrome (AS), a neurodevelopmental disorder characterized by symptoms such as intellectual disability, delayed development and severe speech impairment. UBE3A encodes an E3 ubiquitin ligase, for which several targets have been identified, including synaptic molecules. Although proteolysis mainly occurs in the cytoplasm, UBE3A is localized to the cytoplasm and the nucleus. In fact, UBE3A is also known as a transcriptional regulator of the family of nuclear receptors. However, the function of UBE3A in the nucleus remains unclear. Therefore, we examined the involvement of UBE3A in transcription in the nuclei of neurons. Genome-wide transcriptome analysis revealed an enrichment of genes downstream of interferon regulatory factor (IRF) in a UBE3A-deficient AS mouse model. In vitro biochemical analyses further demonstrated that UBE3A interacted with IRF and, more importantly, that UBE3A enhanced IRF-dependent transcription. These results suggest a function for UBE3A as a transcriptional regulator of the immune system in the brain. These findings also provide informative molecular insights into the function of UBE3A in the brain and in AS pathogenesis.
Introduction
Genetic defects in the UBE3A gene are responsible for the pathogenesis of Angelman syndrome (AS; OMIM 105830), a human neurogenetic disorder characterized by intellectual disability, delayed development, severe speech impairment, epileptic seizures and problems with movement and balance. AS occurs in around 1 in 20 000 to 1 in 12 000 people (1,2). UBE3A is paternally imprinted in the brain, particularly in neurons (3–5), and loss of function of maternally-inherited UBE3A results in the development of AS (6). Most cases of AS are caused by deletion of the maternal copy of the UBE3A gene and to a lesser extent by mutations in UBE3A. Among the several AS mouse models that have been developed, one was generated by an insertional mutation of exon II of the Ube3a gene into the maternal germ line of Ube3a, which resulted in a null mutation that leads to loss of Ube3a expression (7). AS mouse models have been shown to recapitulate many of the phenotypic features of AS, including motor dysfunction, increased seizure susceptibility and hippocampal-dependent learning and memory deficits (7–9). Interestingly, studies using transgenic mice have shown that amplification of the Ube3a gene also contributes to phenotypes observed in 15q11-q13 duplication syndrome, which is associated with autism spectrum disorder (ASD) (10–12). While the dosage of UBE3A is critical for AS and ASD pathologies (10,13), an autism-linked mutation in UBE3A disrupts its protein kinase A-mediated phosphorylation and results in excess UBE3A activity and abnormal synaptic formation (14).
UBE3A protein was originally identified as a cellular protein that mediates the interaction between the human papillomavirus E6 oncoprotein and p53 and was accordingly named E6-associated protein (E6-AP) (15). Subsequently, UBE3A was categorized as a member of a class of functionally related E3 ubiquitin ligases characterized by the presence of a homologous to the E6-AP carboxyl terminus domain (16). A number of substrates of UBE3A ubiquitination other than p53 have been reported (1,2,17). In particular, three synaptic molecules, Arc, RhoGEF and ephexin5, have been identified as new targets of UBE3A (18,19). Among these, Arc stands out as a target of interest because its significance in synaptic regulation has been intensely studied. Studies have reported that UBE3A prevents the internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors in synaptic membranes by targeting Arc for degradation, suggesting that experience or activity-dependent synaptic regulation could be disrupted in AS. Additionally, γ-aminobutyric acid (GABA) transporter 1 and small-conductance potassium channels (SK2) are also reported as targets of UBE3A (20,21). More recently, ALDH1A2, the rate-limiting enzyme in retinoic acid (RA) synthesis, was also found to be a target of UBE3A. Excessive UBE3A dosage impairs RA-mediated neuronal homeostatic synaptic plasticity, and RA homeostasis regulates ASD-like phenotypes in mice with excessive UBE3A dosage (22).
In contrast, a report has shown that Arc is not a direct substrate of UBE3A but, instead, UBE3A controls Arc protein levels at the transcriptional level rather than at the posttranslational level (23). Given that UBE3A is known to function as a transcriptional coactivator of nuclear (N) hormone receptors (24–28), it is likely that UBE3A also regulates Arc at the transcriptional level. A recent report showed that increasing UBE3A in the nucleus leads to downregulation of the glutamatergic synapse organizer Cbln1 (29). Although UBE3A knockout (KO) mice show defects in reproductive function and tissue-specific steroid hormone resistance (24–26), there is little evidence to explain the phenotypic features of AS mouse models based on the function of UBE3A as a transcriptional regulator. To our knowledge, there is currently only one genome-wide transcriptome study of AS. This study conducted microarray analysis of mouse cerebelli and showed that gene expression implicated in three networks, cell signaling, nervous system development and cell death, were significantly changed in AS mice (30).
To determine whether the transcriptional regulatory function of UBE3A is associated with defects in the AS brain, we compared the transcriptome of the hippocampus between wild-type (WT) and AS mouse. We found that genes downstream of the interferon regulatory factor (IRF) transcription factor was significantly changed in AS mice, implying transcriptional regulation by UBE3A. As expected, UBE3A interacted with IRF and functioned as a coactivator of IRF. These findings suggest a role for Ube3a as a transcriptional regulator of immune-related transcription factors.
Results
UBE3A is localized to the nucleus of neurons
To confirm the subcellular localization of UBE3A in neurons, immunohistochemistry was performed on mouse brain tissue (Fig. 1A–I). UBE3A staining was broadly distributed in the cortices and dentate gyrus of WT mice (Fig. 1B, D and F) but was mostly absent in AS mice (Fig. 1A, C and E). UBE3A was mainly localized to the somata of neurons rather than their dendrites or axons as reported previously (5,10,31–34). On closer inspection, UBE3A was uniformly localized to the somata of most neurons (Fig. 1G and H), although, in some neurons of the dentate gyrus, expression was localized mainly to the cytoplasm and to a smaller extent to the nucleoplasm (Fig. 1I). A subcellular fractionation experiment also demonstrated that UBE3A was localized to both the cytoplasm and nucleoplasm in the brain (Fig. 1J). We therefore concluded that a large portion of UBE3A molecules was localized in the nucleus of neurons, which is consistent with the results of a recent study (35).
Figure 1.
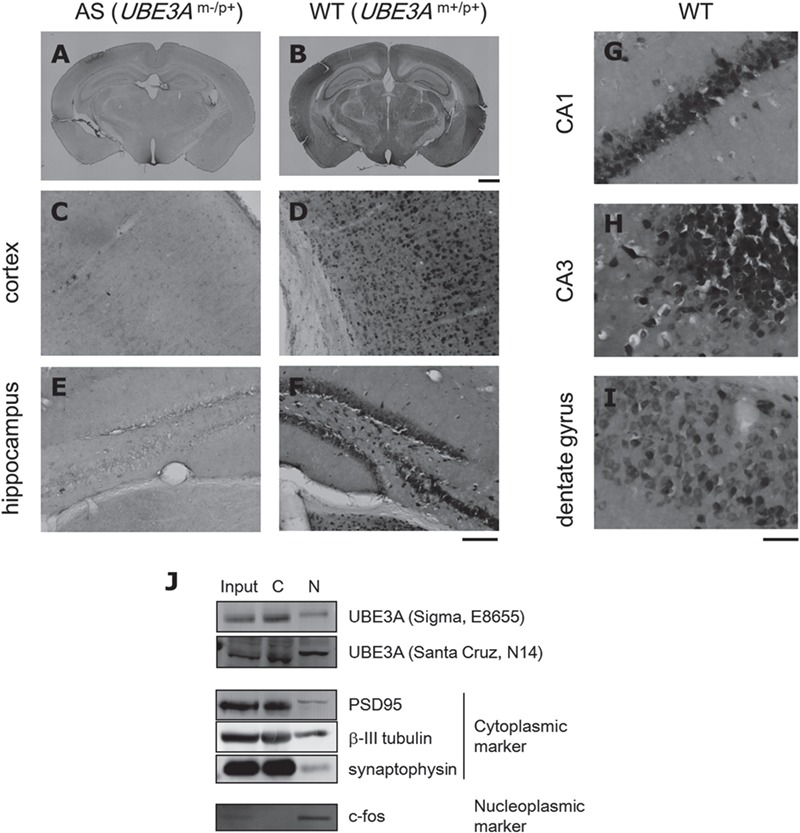
UBE3A is distributed in neuronal somata. (A–I) DAB immunostaining for UBE3A in coronal brain sections from WT (B, D, F and G–H) and AS (A, C and E) mice (3 weeks old). Both low and intermediate magnifications revealed broadly distributed UBE3A staining in the brain, which was mostly absent in AS mice. High magnification provided better resolution of UBE3A staining patterns in CA1 (G), CA3 (H) and the dentate gyrus (I). Scale bar beneath (B), 1 mm for (A) and (B); beneath (F), 100 μm for (C–F); beneath (I), 40 μm for (G–I). (J) Subcellular fractionation of mouse brain homogenates. UBE3A was localized to both the C and N fractions. PSD95, β-III tubulin and synaptophysin were used as cytoplasmic markers and c-fos as a nucleoplasmic marker.
Transcriptome analysis of an AS mouse model
To examine whether UBE3A has an impact on neuronal transcription, we performed gene expression profiling of hippocampi from four WT and four AS mice using microarray analysis. The difference in global gene expression between WT and AS is shown in a volcano plot in Figure 2A. We set criteria (P < 0.05 by moderated t-test; fold change, >1.5) to extract differentially expressed genes (DEGs) in AS (Ube3a KO) mice. DEGs are listed in Supplementary Material, Table S1. Among 128 DEGs, 52 were upregulated while 76 were downregulated in AS mice. UBE3A itself was not included among the DEGs because abnormal transcripts due to the insertional mutation in the UBE3A gene could be detected in the microarray. We confirmed that there was no expression of mature (spliced) mRNA of UBE3A in the brain of AS mice (Supplementary Material, Fig. S1).
Figure 2.
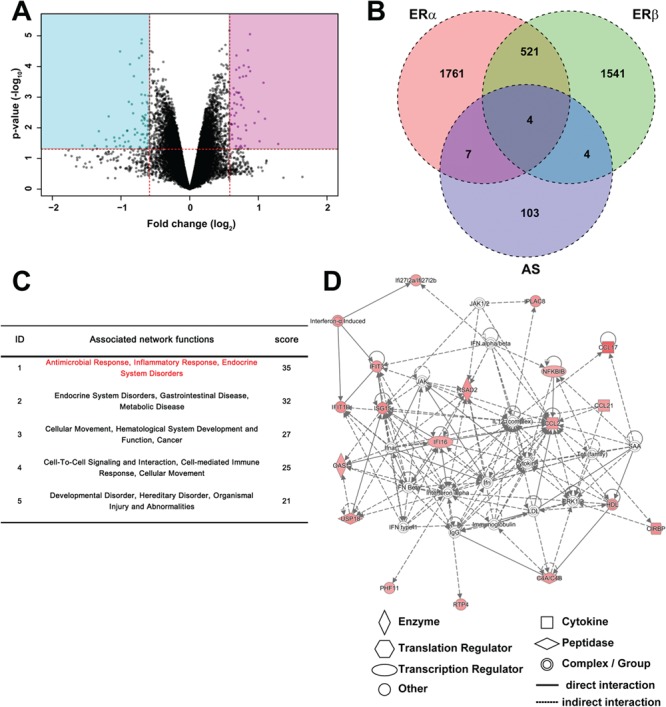
Gene expression profile of AS mouse hippocampus. (A) Volcano plot of the microarray analysis from WT and AS hippocampi. Vertical and horizontal red lines indicate 1.5-fold change and P = 0.05, respectively. Genes in the pink (up) and blue (down) rectangular areas were defined as DEGs in AS. (B) Comparison of DEGs in AS with those in ERα and ERβ KO mice. The values represent the number of genes. Pink, green and blue circles indicate DEGs compared with WT mice in ERα KO, ERβ KO and Ube3a KO mice, respectively. Gene expression profile data in ER KO mice were obtained from the GEO database (GEO accession no. GSE17869). (C) Network analysis of DEGs in AS mice using IPA. The core networks were algorithmically ranked by score based on their right-tailed Fisher’s exact test P-value, which reflects the likelihood that the genes were grouped in a network by chance. (D) The top-ranked network of upregulated DEGs in AS mice. This network was associated with ‘Antimicrobial Response, Inflammatory Response, Infectious Diseases’. Genes in red were upregulated in AS mice and the color intensity indicates the fold change in differential expression.
Given that UBE3A was previously shown to be a transcriptional coregulator for the estrogen receptor (ER) family (24,28,36), we examined whether the DEGs in AS were also affected in ER KO mice (whole brain) using microarray data retrieved from NCBI Gene Expression Omnibus (GEO) (Fig. 2B). The overall transcription of DEGs was not affected in either ER-α or ER-β KO mice, suggesting that the transcriptomic differences between WT and AS mice are not due to a deficiency in the ER pathway.
DEGs were then analyzed using pathway analysis according to gene ontology [GO; Database for Annotation, Visualization and Integrated Discovery (DAVID); Supplementary Material, Table S2] and Ingenuity Pathway Analysis (IPA; Fig. 2C and D). The significantly enriched relevant biological pathways obtained from GO analysis are listed in Supplementary Material, Table S2. We found that immune-related pathways such as ‘response to virus’, ‘defense response to virus’, ‘cellular response to interferon-alpha’, ‘inflammatory response’ and ‘innate immune response’ were prominent (Supplementary Material, Table S2). Causal network analysis by IPA also showed that many immunological factors were upstream regulators of DEGs in AS (Supplementary Material, Table S3). In particular, IRF family members repeatedly appeared as relevant upstream regulators among DEGs that were upregulated in Ube3a KO mice (Supplementary Material, Tables S4–6). IRFs are transcription factors that regulate the transcription of many antivirus factors including interferons and have diverse roles in regulating gene expression networks within the immune system. We subsequently focused on the IRF family as relevant factors of the N function of UBE3A. Additionally, a protein–protein interaction network comprising the DEG products was obtained by analysis using the biological database, Search Tool for Retrieval of Interacting Genes/Proteins (STRING; Supplementary Material, Fig. S2). This network showed a prominent cluster of interactions consisting of 16 proteins, many of which were immunological factors against virus infection (Supplementary Material, Table S7).
UBE3A interacts with IRFs and promotes IRF-dependent transcription
We first determined whether UBE3A can interact with IRF using immunoprecipitation (IP) and western blotting (Fig. 3). HEK293 cells overexpressing both UBE3A and IRF were first immunoprecipitated with anti-IRF antibodies and subsequently blotted using an anti-UBE3A antibody. Both WT and a catalytic mutant (C833A) of UBE3A were clearly detected in the IP of IRF1, 2 and 3 (Fig. 3), suggesting these IRF members can interact with UBE3A regardless of its E3 ubiquitin ligase activity. We also examined the opposite interaction, that is, whether IRF members could be detected in the IP of UBE3A. Weak signals for IRF1 and 2 but not 3 were detected in the IP of UBE3A (Supplementary Material, Fig. S3). IRF2 was only detected in the IP of WT UBE3A but not that of mutant UBE3A. These results suggest that IRF1 and 2 can interact with UBE3A at least to some extent in vitro. Endogenous expression of IRF members were difficult to detect even in total cell lysates using the antibodies at our disposal; we were therefore unable to demonstrate their endogenous interactions.
Figure 3.
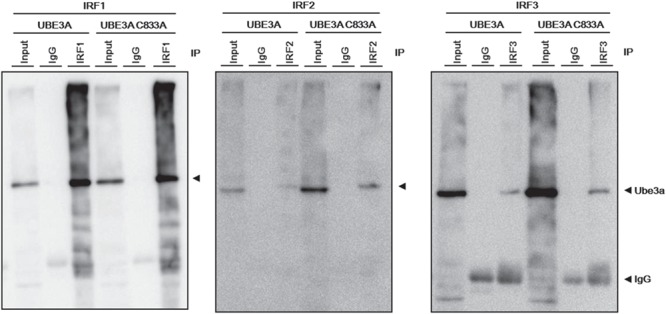
Molecular interactions between UBE3A and IRF in vitro IP following western blotting of samples from HEK293 cells that overexpressed each IRF, with UBE3A (WT or C833A mutant). UBE3A was successfully co-immunoprecipitated using antibodies against IRF1, 2 and 3 (from left to right). UBE3A C833A is the ligase-dead mutant. Input: total cell lysate; IgG: normal IgG negative control.
We next investigated whether UBE3A had an impact on the transcriptional activity of IRF by the luciferase reporter assay using a reporter construct containing the consensus binding site for IRF, interferon-stimulated responsive element (ISRE), upstream of a luciferase gene. Transcriptional activity of endogenous IRF in Neuro2a cells was augmented by WT but not mutant UBE3A (Fig. 4A). When ectopically expressed, IRF1 and 3 showed marked transcriptional activity, whereas the activity of IRF2 and 7 was much weaker (Fig. 4B). No IRF9 activity was detected in this assay. Addition of UBE3A to these assays again upregulated the transcriptional activity of all members (IRF1, 2, 3 and 7; Fig. 4C). The UBE3A mutant partially upregulated the activity of IRF1 but had no effect on the other members. In contrast, WT and mutant UBE3A had no effect on the transcriptional activity of p53 or NF-κB (Fig. 4D). These results indicate that UBE3A can specifically potentiate IRF-mediated transcription at least partially through its E3 ubiquitin ligase activity.
Figure 4.
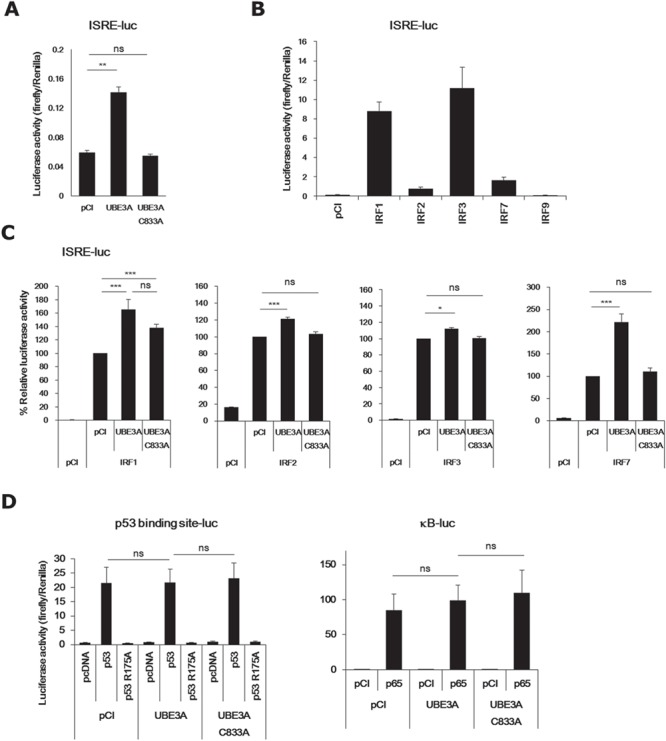
Transcriptional activities of IRF family members are augmented by UBE3A. (A) UBE3A upregulated endogenous IRF activity in Neuro2a cells. (B) Transcriptional activities of IRF1, 2, 3, 7 and 9. (C) UBE3A upregulated the transcriptional activities of IRF1, 2, 3 and 7 (from left to right). (D) UBE3A had no effect on p53 (left) or NF-κB (right). p53 R175A is a mutant p53 without transcriptional activity. p65 is a major component of the NF-κB transcription factor. Bars represent mean ±S.E.M. of 3–9 samples (*P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant). pCI and pcDNA are vehicle vectors.
IRF2 suppressive complex is a major target of UBE3A
Although in vitro reporter assays clearly indicated that UBE3A was a coactivator for IRF-mediated transcription, according to our microarray analysis, most of the DEGs downstream of IRF were upregulated in AS mice (see ‘Activation state’ column in Supplementary Material, Table S5). This suggests that UBE3A may have inhibitory actions on IRF in the mouse brain. To explain this apparent discrepancy between in vitro and in vivo studies, we focused on one of the IRF members, IRF2. IRF2 has been described as a transcriptional repressor and is thought to function by competing with IRF1 (37). At the same time, reports indicate that IRF2 functions as a transcriptional activator for several genes (38–40). We first sought to determine the degree to which IRF2 can suppress the transcriptional activity of IRF1. ISRE reporter assays showed that IRF2 markedly suppressed IRF1 activity (>94%; Fig. 5A, left). This suppression was more pronounced when IRF2 was expressed with IRF2 binding partner (IRF2BP)1, IRF2BP2 or both. Intriguingly, the IRF2 complex suppressed IRF3 to a similar extent (Fig. 5A, right). According to our microarray results, one of the two IRF2 binding partners, IRF2BP2, was most highly expressed among the IRF family members in the brain (Fig. 5B). IRF2BP1 and IRF2BP2 have been identified as components of the IRF2 corepressor complex (41). They both have a zinc finger domain in the N-terminus and a RING finger domain in the C-terminus, with the latter being sufficient for interaction with IRF2. Of note, IRF2BP1 and IRF2BP2 themselves have been demonstrated to function as transcriptional repressors, even when not bound to IRF2 (42–45).
Figure 5.
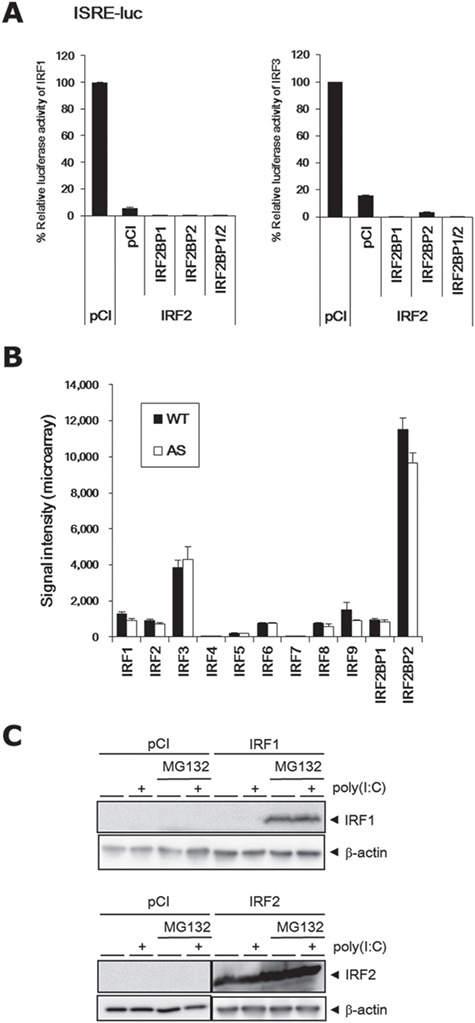
IRF2 complex is a potent suppressor of IRF. (A) Marked suppression of the transcriptional activities of IRF1 (left) and IRF3 (right) by IRF2 and IRF2BP1/2 complex. (B) Comparison of mRNA expression levels between IRF family members, IRF2BP1 and IRF2BP2, in microarray analysis. (C) Overexpressed IRF1 was only detected in the presence of MG132 (upper), whereas overexpressed IRF2 was detected even in the absence of MG132 (lower) in HEK293 cells. MG132 and poly (I:C) were used as a proteasome inhibitor and an IRF stimulant, respectively. pCI is a vehicle vector.
To evaluate the protein stability of IRF1 and 2, these proteins were ectopically expressed in HEK293 cells in the presence or absence of the proteasome inhibitor, MG132 (Fig. 5C). Endogenous IRF1 and 2 were undetectable even in the presence of MG132. Surprisingly, overexpressed IRF1 was only detectable in the presence of MG132, indicating rapid turnover of IRF1 by the ubiquitin-proteasome system. In contrast, overexpressed IRF2 was readily detectable both in the absence and presence of MG132. These results suggest that IRF2 is much more stable than IRF1.
Based on combined evidence that the IRF2 complex was an extremely potent suppressor of IRF1 and 3 and that the expression of IRF2BP2 was markedly higher than that of other IRF members, we hypothesized that the IRF2 suppressor complex predominantly functions under unstimulated conditions. Moreover, given that UBE3A likely activates IRF2 at least in normal conditions, IRF2 function is likely weakened in UBE3A-deficient AS mice. We speculate that this may explain the overall upregulation of the transcriptional activity of IRF in AS mice despite UBE3A likely being a coactivator of IRF in vitro. Although still hypothetical, this may explain the discrepancy between the in vivo and in vitro findings described above.
IRF dysfunction in AS model mice
IRF family members are critical transcription factors in the immune system, particularly for genes involved in protection against virus infection. If the effect of UBE3A on IRF is biologically significant, we would expect to see some sign of immunologic disease in AS patients. However, there are no such case reports. Another possibility is that another function of IRF, other than immunoreaction, is lost in AS patients. In the brain, the IRF family of proteins has also been implicated as transcription factors for genes involved in neuronal survival (46,47), neuroinflammation (48) and microglia activation (49–53). We compared the levels of brain apoptosis, neuroinflammation and the morphology of microglia and astrocytes between WT and AS mouse but found no clear differences (Supplementary Material, Fig. S3, data not shown). We next attempted to find changes in the number of microglia. Fluorescence activated cell sorting (FACS) analysis using anti-CD11b revealed that the number of microglia was unchanged in the AS mouse brain (Supplementary Material, Fig. S5). Collectively, in our hand, we were not able to find obvious morphological defects related to compromised immunity in the AS brain.
Discussion
As mentioned above, many groups have shown that endogenous UBE3A is localized to neuronal somata (5,10,31–34) and that UBE3A functions as a coregulator of N hormone receptors (24–28,32,36). Using immunohistochemistry, we confirmed that UBE3A was indeed localized mainly to the somata of neurons rather than dendrites or axons (Fig. 1). Therefore, while there is some evidence suggesting that UBE3A may be involved in the turnover of AMPA receptors (18) and GABA transporters (20) in the synapse, this is likely controlled by a limited population of UBE3A molecules. In contrast, the function of UBE3A in the nucleus, if any, is expected to be its primary function in the brain, as reported recently (29).
Microarray analysis of the hippocampus revealed that 128 genes were significantly affected at the mRNA level in AS mice. We found that a large number of immune-related genes were enriched among the upregulated genes in AS. In particular, genes downstream of a family of antivirus transcription factor genes, IRF, were affected in the AS brain, suggesting that UBE3A regulates IRF-dependent transcription. Our results differed from those of a previous microarray study of the cerebellum from the same AS mouse model (30), probably due to differences in tissue specificity. Moreover, given that IRF is a general transcription factor of immune system genes that protect against virus infections, our findings are likely not hippocampus-specific and require further extensive work.
Given that our in vitro experiments indicated that UBE3A can bind IRF and enhance IRF-dependent transcription (Figs 3 and 4; Supplementary Material, Fig. S2), we propose that UBE3A may function as a coactivator for IRF transcription in the neuronal nucleus. It remains unclear how UBE3A works as a coactivator for IRF-dependent transcription, although, according to the results of our luciferase reporter assay using a catalytic-dead mutant of UBE3A, ubiquitination may be involved in its coactivator function. Among the IRF family members, the IRF2 repressive complex was potent, abundant and relatively stable (Fig. 5). Therefore, we hypothesize that the IRF2 repressive complex is the primary IRF family member that restricts transcription of IRF target genes until the proper stimulation becomes available. This hypothesis is consistent with findings from previous studies on IRF2 (54,55). If this is the case, IRF2 should be the main target of UBE3A as a coactivator at least under normal conditions. Given that IRF2 and p53 share the same E3 ubiquitin ligase, MDM2 (56), it is possible that UBE3A also targets both p53 and IRF2 as E3 ligase targets. If this is the case, the IRF2 repressive complex may not exhibit proper function in the UBE3A-deficient AS mouse model, providing an explanation for the upregulation of genes downstream of IRF in the hippocampus of AS mice (Supplementary Material, Table S5).
That UBE3A was originally identified as a binding partner for the human papillomavirus E6 oncoprotein and was called E6-AP is consistent with its involvement in antiviral machinery. However, there are no case reports of immunologic disease pathology in AS patients other than obesity, an immunity-relevant disease, which was previously reported as an (20–80%) associated clinical feature of AS (57). IRF has been implicated in functions other than its typical role in antiviral pathways. Several studies have demonstrated that IRF1 acts as a coregulator of p53 in the apoptosis pathway (58–62). In the brain, IRF1 and 4 are involved in the immunoreactivity and survival of neurons after ischemic stroke (46–48). Moreover, IRF5 and 8 act as vital transcription factors for microglial activation (49–53). IRF2 has also been shown to be a key factor for the maintenance and/or differentiation of hematopoietic stem cells, progenitor cells of microglia (63–65). However, we found little difference in neuroinflammation, brain apoptosis and the microglia population between the WT and AS mouse brain. Recently, however, a prospective trial on the safety and tolerability of minocycline treatment and its effect on cognitive function and adaptive behavior among AS children provided strikingly supportive evidence for our findings (66). This study showed that minocycline was well tolerated and caused significant improvements in the adaptive behaviors in AS children. Minocycline is a tetracycline antibiotic medication that readily crosses the blood brain barrier and has been shown to be effective in Fragile X syndrome patients (67) and several animal models of degenerative neuropathology such as Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (68,69), probably by recovering synaptic dysfunction. Importantly, studies have demonstrated that minocycline is an inhibitor of microglial activity (70–72). We speculate that an abnormality in microglia due to IRF hyperactivation in AS mice might explain the effectiveness of minocycline in AS patients.
Mouse Ube3a has three alternative spliced transcripts (10). Isoform 1 is reported to be expressed as a non-coding RNA and regulates dendritic complexity or spine morphogenesis by regulating a miRNA pathway (73). Isoform 2 corresponds to the open reading frame of Ube3a, called ‘canonical form’. On the other hand, isoform 3 lacks 21 amino acids from its N-terminal. Although the differences including their function, localization or expression pattern between isoform 2 and 3 remain unclear, Miao et al. reported that isoform 2 is distributed mainly in the cytoplasm, whereas isoform 3 is localized largely in the nucleus (74). Therefore, the data in this study may reflect the role of UBE3A isoform 3.
UBE3A is located on the human chromosome 15q11-q13 region and copy number variations in this gene have been implicated in two neurological disorders other than AS: Prader–Willi syndrome (PWS) and ASD (75). Paternally- or maternally-inherited deletions of 15q11-q13 occur quite frequently and manifest as PWS or AS, respectively. In contrast, duplication of the same region is the only recurrent cytogenetic aberration associated with autism, occurring in up to 5% of autism cases. Therefore, AS is considered to be pathophysiologically associated with ASD. Immune dysfunction has been proposed as a potential mechanism for the etiology of autism. For example, high levels of inflammation have been observed in post-mortem samples of brains from individuals with autism (76). Epidemiological studies have also shown that children with autism tend to have a family history of autoimmune diseases (77), and the rate of autism is higher in the offspring of mothers who contract influenza during pregnancy (78–80). A number of other studies have shown that cytokine levels in the blood, brain and cerebrospinal fluid of autistic subjects are elevated compared to those of healthy individuals (81,82).
In conclusion, we found that IRF activity was prominently affected in the UBE3A-deficient AS mouse brain. Our finding that UBE3A interacts with IRF and induces IRF-mediated transcription in vitro suggests a novel function for UBE3A as a transcriptional coactivator of IRF. We also found that a potent suppressor member, IRF2, likely predominantly functions with the aid of UBE3A under normal unstimulated conditions. Further studies are needed to determine the effect of UBE3A on compromised immunity in the AS brain.
Materials and Methods
Animals
AS model mice (B6.129S7-Ube3a<tm1Alb>/J, stock#: 016590) (7) were purchased from Charles River Laboratories Japan (Yokohama, Japan). Tissues were immediately frozen in liquid nitrogen and stored at −80°C until required for processing for RNA. All experimental protocols using animals in the present study were approved by the RIKEN Animal Research Committee.
Immunohistochemistry
Mice were anesthetized prior to transcardial perfusion with phosphate buffer and phosphate-buffered 4% paraformaldehyde. Perfused brains were removed and postfixed for 4 h at 4°C prior to cryoprotection by submerging for 48 h in 30% sucrose in phosphate-buffered saline. Cryoprotected brains were embedded in OCT compound (Sakura Finetek Japan, Tokyo, Japan), frozen on dry ice and cut into 40 μm thick sections using a cryostat (Hyrax C50, Thermo Fisher Scientific, Waltham, MA). For chromogenic staining, sections were incubated in 0.3% H2O2 to quench endogenous peroxidases and incubated in primary antibodies for 24 h at 4°C before rinsing several times and incubating with biotinylated anti-mouse/rabbit secondary antibodies (1:500, Vector Laboratory, Burlingame, CA) for 1 h at room temperature. Sections were rinsed again prior to amplification using reagents from the Vectastain ABC standard kit (Vector Laboratory). Finally, the immune complex was visualized using the 3′3′-diaminobenzidine (DAB) substrate kit for peroxidases (SK-4100, Vector Laboratory). For immunofluorescent staining, sections were incubated in primary antibodies for 24 h at 4°C and rinsed before incubation in secondary antibodies conjugated to Alexa Fluor 488 or 568 (1:1000; Thermo Fisher Scientific). Stained sections were mounted in VECTASHIELD (H1200, Vector Laboratory). Primary antibodies used were anti-Ube3a (1:1000; E8655, clone 330, Sigma-Aldrich, St. Louis, MO), anti-Iba1 (1:500; 019-19741, Wako, Osaka, Japan), anti-GFAP (1:200; MAB3402, clone GA5, Merck Millipore, Guyancourt, France) and anti-TNFA (1:200; SAB4502982, Sigma-Aldrich). Images of brain sections were acquired using a BZ-9000 microscope (Keyence, Osaka, Japan). Sections compared in the figures were stained at the same time under identical conditions.
Subcellular fractionation
Whole brains from mice (3 weeks old) were minced and suspended in low-salt HEPES buffer (10 mm KCl). Brain lysates were homogenized using a Potter-Elvehjem grinder. Total homogenates were spun down at 1000g to yield the cytosolic (C) fraction. The pellet was resuspended in high-salt HEPES buffer (400 mm NaCl) and briefly sonicated before spinning down at 20 000g to yield the N fraction.
Western blotting
Cell lysates or brain homogenates were fractionated using Sodium Dodecyl Sulfate-Poly-Acrylamide Gel Electrophoresis and proteins were transferred onto nitrocellulose membranes. The membranes were blocked in 3% Bovine Serum Albumin in Tris-buffered solution with Tween-20 at room temperature for 1 h. Blocked membranes were incubated with primary antibody at 4°C overnight. The following primary antibodies were used: anti-Ube3a clone 330 (E8655, Sigma-Aldrich), anti-Ube3a (sc-8926, Santa Cruz, CA), anti-PSD95 clone 7E3-1B8 (MA1-046, Thermo Fisher Scientific), anti-beta-III tubulin (MMS-435P, Covance Laboratories, Harrogate, UK), anti-synaptophysin (10706, Progen, Heidelberg, Germany), anti-c-fos (PC38T, Merck Millipore), anti-IRF1 (sc-497, Santa Cruz, CA), anti-IRF2 (sc-498, Santa Cruz, CA), anti-IRF3 (sc-FL-425, Santa Cruz, CA) and anti-beta-actin clone AC-15 (A1978, Sigma-Aldrich). After washing, the membranes were incubated with peroxidase-conjugated secondary antibody (mouse: NA9310, GE Healthcare, Waukesha, WI; rabbit: 111-035-003, Jackson ImmunoResearch, West Grove, PA). Signals on the membranes were developed using a chemiluminescence assay kit (Chemi-Lumi One L, Nacalai Tesque, Kyoto, Japan) prior to imaging with luminescent image analyzer LAS3000 (Fujifilm, Tokyo, Japan).
Microarray
Microarray was performed as described previously (83,84). Total RNA from the hippocampus was extracted using TRIzol (Life Technologies, Gaithersburg, MD), followed by column purification with DNaseI treatment (Promega, Madison, WI). The quantity and quality of RNA was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). Microarray analysis was performed according to the manufacturer’s instructions (mouse GE 4x44K v2 microarray kit, Agilent Technologies). Briefly, 200 ng of total RNA was processed for Cy3 labeling with T7 RNA polymerase amplification prior to hybridization onto a microarray chip. The raw expression data were acquired using Feature Extraction Software 9.5. Data processing was performed as described previously (83). Briefly, raw signal intensities were corrected using the ‘backgroundCorrect’ function in the limma package in R Bioconductor. Between-array normalization was performed using the quantile method. Linear model and Bayes moderated t-statistics were applied to extract DEGs. DEGs in AS mice were defined and ranked using moderated t-tests (P < 0.05 and fold change >1.5). Data are available from GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE119415.
Quantitative Reverse Transcription-Polymerase Chain Reaction
DNaseI-treated total RNA (1 μg) was reverse-transcribed using a random hexamer primer (Hokkaido System Science, Hokkaido, Japan) and Superscript reverse transcriptase (Invitrogen, Carlsbad, CA). cDNA equivalent to 30 ng total RNA was amplified by quantitative Polymerase Chain Reaction using Power SYBR Green master mix in a 7900HT sequence detection system or StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA). Gapdh genes were used as internal controls. Forward and reverse primers were as follows: Gapdh sense, 5′-ACGGGAAGCTCACTGGCATGGCCTT-3′; Gapdh antisense, 5′-CATGAGGTCCACCACCCTGTTGCTG-3′; UBE3A sense, 5′-TGAACAAGAAGGAAGGAAAAGA-3′; UBE3A antisense, 5′-GGGGAATAATCCTCACTCTCTC-3′.
Network/pathway analysis
DEGs were analyzed using DAVID, STRING or IPA to identify the relevant biological networks/pathways or interactions. DAVID is a web-based tool for GO used to identify enriched biological GO terms (http://david.abcc.ncifcrf.gov/home.jsp). STRING is a database of known and predicted protein interactions (http://string-db.org/). IPA is pathway analysis software (Ingenuity, Qiagen, Hilden, Germany). Briefly, a list of DEGs along with their P-values and fold changes were uploaded to the IPA server and causal network analysis was performed to generate plausible regulatory networks that may explain the gene expression changes in the data set.
Cell culture
HEK293 and Neuro2a cells were cultured in Dulbecco’s modified Eagle’s medium (Nacalai Tesque) supplemented with 10% heat-inactivated fetal bovine serum (ICN Biomedicals, Aurora, OH) and antibiotics penicillin/streptomycin. Cells were treated with 10 μm MG132 (Enzo Life Sciences, Farmingdale, NY) for 12 h to inhibit proteasome activity. Poly (I:C) (Invivogen, San Diego, CA), a synthetic analog of dsRNA, was used to stimulate IRF (20 μg/ml; 12 h).
Immunoprecipitation
HEK293 cells transfected with the appropriate expression plasmids using Lipofectamine 3000 (Invitrogen) were cultured for 2 days and treated with proteasome inhibitor (10 μm MG132) for 12 h. Cells were lysed in Tris NaCl EDTA buffer using a protease inhibitor cocktail (Nacalai Tesque) and precleared with protein A/G+agarose (Roche Penzberg, Germany) and immunoprecipitated with antibodies against anti-Ube3a (E8655, Sigma-Aldrich), IRF1 (sc-497, Santa Cruz), IRF2 (sc-498, Santa Cruz) or IRF3 (sc-9082, Santa Cruz). After washing three times, the precipitates were resuspended in the 2× SDS-PAGE sample buffer, boiled for 5 min, and separated on a 10% SDS-PAGE gel before performing western blot analysis.
Luciferase assay
Firefly luciferase vectors used as experimental reporters were pGL4.45 [luc2P/ISRE/Hygro] (Promega) for IRF transcriptional activity, 3× κB luciferase reporter construct for NF-κB activity and a reporter construct containing p53 DNA binding consensus sites for p53 activity. Renilla luciferase vector used as a reference reporter for the normalization of the transfection efficiencies was phRL-TK (Promega) expressing Renilla luciferase driven by a herpes simplex virus thymidine kinase promoter. Neuro2a cells were transfected with the reporter plasmids together with expression plasmids for Ube3a and/or IRFs using Lipofectamine LTX reagents (Invitrogen). Cells were harvested 2 days after transfection, and luciferase assays were performed according to the manufacturer’s protocol (Dual Luciferase Reporter Assay System, Promega). Promoter activity was determined from the ratio of firefly luciferase activity to Renilla luciferase activity.
FACS analysis
P0 mice were anesthetized on ice and perfused intracardially using phosphate-buffered saline. Brains were dissected and digested in papain solution (Sumitomo Bakelite, Tokyo, Japan). The cell pellet was suspended in medium and layered over 30% Percoll. Samples were centrifuged at 700g for 10 min at room temperature, and the pellet was further cleared through 70 μm cell strainers (Corning Inc., Corning, NY). Isolated cells were washed in FACS buffer and suspended in anti-CD11b antibody (M1/70, Biolegend, San Diego, CA) for 20 min. Cells were washed and analyzed in Cell Sorter SH800 (Sony, Tokyo, Japan). Microglia populations were quantified by calculating the percentage of microglia obtained from flow cytometry analysis compared to live cell counts.
Supplementary Material
Acknowledgements
We thank the RIKEN Research Resources Center, the Support Unit for Animal Resources Development for breeding the mice and the Support Unit for Bio-Material Analysis for conducting FACS. We also thank A. Homma, H. Okubo, A. Furuya and other technical staff of the Takumi Laboratory for their technical assistance.
Conflict of Interest statement. None declared.
Funding
KAKENHI (Grant-in-Aid for Scientific Research) from the Japan Society of Promotion of Science and Ministry of Education, Culture, Sports, Science and Technology (15K08305, 16H06316, 16H06463); CREST (Core Research for Evolutional Science and Technology) from Japan Science and Technology Agency; Intramural Research Grant for Neurological and Psychiatric Disorders from NCNP (National Institute of Mental Health); Takeda Science Foundation.
References
- 1. Buiting K., Williams C. and Horsthemke B. (2016) Angelman syndrome—insights into a rare neurogenetic disorder. Nat. Rev. Neurol., 12, 584–593. [DOI] [PubMed] [Google Scholar]
- 2. Mabb A.M., Judson M.C., Zylka M.J. and Philpot B.D. (2011) Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci., 34, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vu T.H. and Hoffman A.R. (1997) Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet., 17, 12–13. [DOI] [PubMed] [Google Scholar]
- 4. Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J. and Kishino T. (2003) Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum. Mol. Genet., 12, 837–847. [DOI] [PubMed] [Google Scholar]
- 5. Gustin R.M., Bichell T.J., Bubser M., Daily J., Filonova I., Mrelashvili D., Deutch A.Y., Colbran R.J., Weeber E.J. and Haas K.F. (2010) Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol. Dis., 39, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kishino T., Lalande M. and Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet., 15, 70–73. [DOI] [PubMed] [Google Scholar]
- 7. Jiang Y.H., Armstrong D., Albrecht U., Atkins C.M., Noebels J.L., Eichele G., Sweatt J.D. and Beaudet A.L. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron, 21, 799–811. [DOI] [PubMed] [Google Scholar]
- 8. Miura K., Kishino T., Li E., Webber H., Dikkes P., Holmes G.L. and Wagstaff J. (2002) Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol. Dis., 9, 149–159. [DOI] [PubMed] [Google Scholar]
- 9. Woerden G.M., Harris K.D., Hojjati M.R., Gustin R.M., Qiu S., Avila Freire R., Jiang Y.H., Elgersma Y. and Weeber E.J. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat. Neurosci., 10, 280–282. [DOI] [PubMed] [Google Scholar]
- 10. Smith S.E., Zhou Y.D., Zhang G., Jin Z., Stoppel D.C. and Anderson M.P. (2011) Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med., 3, 103ra197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Copping N.A., Christian S.G.B., Ritter D.J., Islam M.S., Buscher N., Zolkowska D., Pride M.C., Berg E.L., LaSalle J.M., Ellegood J. et al. (2017) Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q11.2-q13.3 duplication syndrome. Hum. Mol. Genet., 26, 3995–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takumi T. and Tamada K. (2018) CNV biology in neurodevelopmental disorders. Curr. Opin. Neurobiol., 48, 183–192. [DOI] [PubMed] [Google Scholar]
- 13. Silva-Santos S., Woerden G.M., Bruinsma C.F., Mientjes E., Jolfaei M.A., Distel B., Kushner S.A. and Elgersma Y. (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest., 125, 2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yi J.J., Berrios J., Newbern J.M., Snider W.D., Philpot B.D., Hahn K.M. and Zylka M.J. (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell, 162, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huibregtse J.M., Scheffner M. and Howley P.M. (1991) A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J., 10, 4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huibregtse J.M., Scheffner M., Beaudenon S. and Howley P.M. (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U. S. A., 92, 2563–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martínez-Noël G., Luck K., Kühnle S., Desbuleux A., Szajner P., Galligan J.T., Rodriguez D., Zheng L., Boyland K., Leclere F. et al. (2018) Network analysis of UBE3A/E6AP-associated proteins provides connections to several distinct cellular processes. J. Mol. Biol., 430, 1024–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greer P.L., Hanayama R., Bloodgood B.L., Mardinly A.R., Lipton D.M., Flavell S.W., Kim T.K., Griffith E.C., Waldon Z., Maehr R. et al. (2010) The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell, 140, 704–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Margolis S.S., Salogiannis J., Lipton D.M., Mandel-Brehm C., Wills Z.P., Mardinly A.R., Hu L., Greer P.L., Bikoff J.B., Ho H.Y. et al. (2010) EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell, 143, 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Egawa K., Kitagawa K., Inoue K., Takayama M., Takayama C., Saitoh S., Kishino T., Kitagawa M. and Fukuda A. (2012) Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci. Transl. Med., 4, 163ra157. [DOI] [PubMed] [Google Scholar]
- 21. Sun J., Zhu G., Liu Y., Standley S., Ji A., Tunuguntla R., Wang Y., Claus C., Luo Y., Baudry M. and Bi X. (2015) UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Rep., 12, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu X., Li C., Gao X., Xia K., Guo H., Li Y., Hao Z., Zhang L., Gao D., Xu C. et al. (2018) Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res., 28, 48–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kühnle S., Mothes B., Matentzoglu K. and Scheffner M. (2013) Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. U. S. A., 110, 8888–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nawaz Z., Lonard D.M., Smith C.L., Lev-Lehman E., Tsai S.Y., Tsai M.J. and O’Malley B.W. (1999) The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol., 19, 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith C.L., DeVera D.G., Lamb D.J., Nawaz Z., Jiang Y.H., Beaudet A.L. and O’Malley B.W. (2002) Genetic ablation of the steroid receptor coactivator-ubiquitin ligase, E6-AP, results in tissue-selective steroid hormone resistance and defects in reproduction. Mol. Cell. Biol., 22, 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khan O.Y., Fu G., Ismail A., Srinivasan S., Cao X., Tu Y., Lu S. and Nawaz Z. (2006) Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol. Endocrinol., 20, 544–559. [DOI] [PubMed] [Google Scholar]
- 27. Li L., Li Z., Howley P.M. and Sacks D.B. (2006) E6AP and calmodulin reciprocally regulate estrogen receptor stability. J. Biol. Chem., 281, 1978–1985. [DOI] [PubMed] [Google Scholar]
- 28. Catoe H.W. and Nawaz Z. (2011) E6-AP facilitates efficient transcription at estrogen responsive promoters through recruitment of chromatin modifiers. Steroids, 76, 897–902. [DOI] [PubMed] [Google Scholar]
- 29. Krishnan V., Stoppel D.C., Nong Y., Johnson M.A., Nadler M.J., Ozkaynak E., Teng B.L., Nagakura I., Mohammad F., Silva M.A. et al. (2017) Autism gene Ube3a and seizures impair sociability by repressing VTA Cbln1. Nature, 543, 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Low D. and Chen K.S. (2010) Genome-wide gene expression profiling of the Angelman syndrome mice with Ube3a mutation. Eur. J. Hum. Genet., 18, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dindot S.V., Antalffy B.A., Bhattacharjee M.B. and Beaudet A.L. (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet., 17, 111–118. [DOI] [PubMed] [Google Scholar]
- 32. Godavarthi S.K., Dey P., Maheshwari M. and Jana N.R. (2012) Defective glucocorticoid hormone receptor signaling leads to increased stress and anxiety in a mouse model of Angelman syndrome. Hum. Mol. Genet., 21, 1824–1834. [DOI] [PubMed] [Google Scholar]
- 33. Maheshwari M., Samanta A., Godavarthi S.K., Mukherjee R. and Jana N.R. (2012) Dysfunction of the ubiquitin ligase Ube3a may be associated with synaptic pathophysiology in a mouse model of Huntington disease. J. Biol. Chem., 287, 29949–29957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Judson M.C., Sosa-Pagan J.O., Del Cid W.A., Han J.E. and Philpot B.D. (2014) Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol., 522, 1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burette A.C., Judson M.C., Burette S., Phend K.D., Philpot B.D. and Weinberg R.J. (2017) Subcellular organization of UBE3A in neurons. J. Comp. Neurol., 525, 233–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun J., Zhou W., Kaliappan K., Nawaz Z. and Slingerland J.M. (2012) ERα phosphorylation at Y537 by Src triggers E6-AP-ERα binding, ERα ubiquitylation, promoter occupancy and target gene expression. Mol. Endocrinol., 26, 1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harada H., Fujita T., Miyamoto M., Kimura Y., Maruyama M., Furia A., Miyata T. and Taniguchi T. (1989) Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell, 58, 729–739. [DOI] [PubMed] [Google Scholar]
- 38. Birnbaum M.J., Zundert B., Vaughan P.S., Whitmarsh A.J., Wijnen A.J., Davis R.J., Stein G.S. and Stein J.L. (1997) Phosphorylation of the oncogenic transcription factor interferon regulatory factor 2 (IRF2) in vitro and in vivo. J. Cell. Biochem., 66, 175–183. [PubMed] [Google Scholar]
- 39. Jesse T.L., LaChance R., Iademarco M.F. and Dean D.C. (1998) Interferon regulatory factor-2 is a transcriptional activator in muscle where it regulates expression of vascular cell adhesion molecule-1. J. Cell Biol., 140, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oshima S., Nakamura T., Namiki S., Okada E., Tsuchiya K., Okamoto R., Yamazaki M., Yokota T., Aida M., Yamaguchi Y. et al. (2004) Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol. Cell. Biol., 24, 6298–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Childs K.S. and Goodbourn S. (2003) Identification of novel co-repressor molecules for interferon regulatory factor-2. Nucleic Acids Res., 31, 3016–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kimura M. (2008) IRF2-binding protein-1 is a JDP2 ubiquitin ligase and an inhibitor of ATF2-dependent transcription. FEBS Lett., 582, 2833–2837. [DOI] [PubMed] [Google Scholar]
- 43. Koeppel M., Heeringen S.J., Smeenk L., Navis A.C., Janssen-Megens E.M. and Lohrum M. (2009) The novel p53 target gene IRF2BP2 participates in cell survival during the p53 stress response. Nucleic Acids Res., 37, 322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carneiro F.R., Ramalho-Oliveira R., Mognol G.P. and Viola J.P. (2011) Interferon regulatory factor 2 binding protein 2 is a new NFAT1 partner and represses its transcriptional activity. Mol. Cell. Biol., 31, 2889–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yeung K.T., Das S., Zhang J., Lomniczi A., Ojeda S.R., Xu C.F., Neubert T.A. and Samuels H.H. (2011) A novel transcription complex that selectively modulates apoptosis of breast cancer cells through regulation of FASTKD2. Mol. Cell. Biol., 31, 2287–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Paschen W., Gissel C., Althausen S. and Doutheil J. (1998) Changes in interferon-regulatory factor-1 mRNA levels after transient ischemia in rat brain. Neuroreport, 9, 3147–3151. [DOI] [PubMed] [Google Scholar]
- 47. Guo S., Li Z.Z., Jiang D.S., Lu Y.Y., Liu Y., Gao L., Zhang S.M., Lei H., Zhu L.H., Zhang X.D., Liu D.P. and Li H. (2014) IRF4 is a novel mediator for neuronal survival in ischaemic stroke. Cell. Death. Differ., 21, 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alexander M., Forster C., Sugimoto K., Clark H.B., Vogel S., Ross M.E. and Iadecola C. (2003) Interferon regulatory factor-1 immunoreactivity in neurons and inflammatory cells following ischemic stroke in rodents and humans. Acta Neuropathol., 105, 420–424. [DOI] [PubMed] [Google Scholar]
- 49. Masuda T., Tsuda M., Yoshinaga R., Tozaki-Saitoh H., Ozato K., Tamura T. and Inoue K. (2012) IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep., 1, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minten C., Terry R., Deffrasnes C., King N.J. and Campbell I.L. (2012) IFN regulatory factor 8 is a key constitutive determinant of the morphological and molecular properties of microglia in the CNS. PLoS One, 7, e49851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kierdorf K., Erny D., Goldmann T., Sander V., Schulz C., Perdiguero E.G., Wieghofer P., Heinrich A., Riemke P., Hölscher C. et al. (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci., 16, 273–280. [DOI] [PubMed] [Google Scholar]
- 52. Masuda T., Iwamoto S., Yoshinaga R., Tozaki-Saitoh H., Nishiyama A., Mak T.W., Tamura T., Tsuda M. and Inoue K. (2014) Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain. Nat. Commun., 5, 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Masuda T., Nishimoto N., Tomiyama D., Matsuda T., Tozaki-Saitoh H., Tamura T., Kohsaka S., Tsuda M. and Inoue K. (2014) IRF8 is a transcriptional determinant for microglial motility. Purinergic Signal., 10, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Matsuyama T., Kimura T., Kitagawa M., Pfeffer K., Kawakami T., Watanabe N., Kündig T.M., Amakawa R., Kishihara K., Wakeham A. et al. (1993) Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell, 75, 83–97. [PubMed] [Google Scholar]
- 55. Senger K., Merika M., Agalioti T., Yie J., Escalante C.R., Chen G., Aggarwal A.K. and Thanos D. (2000) Gene repression by coactivator repulsion. Mol. Cell, 6, 931–937. [DOI] [PubMed] [Google Scholar]
- 56. Pettersson S., Kelleher M., Pion E., Wallace M. and Ball K.L. (2009) Role of Mdm2 acid domain interactions in recognition and ubiquitination of the transcription factor IRF-2. Biochem. J., 418, 575–585. [DOI] [PubMed] [Google Scholar]
- 57. Williams C.A., Beaudet A.L., Clayton-Smith J., Knoll J.H., Kyllerman M., Laan L.A., Magenis R.E., Moncla A., Schinzel A.A., Summers J.A. and Wagstaff J. (2006) Angelman syndrome 2005: updated consensus for diagnostic criteria. Am. J. Med. Genet. A, 140, 413–418. [DOI] [PubMed] [Google Scholar]
- 58. Tanaka N., Ishihara M., Lamphier M.S., Nozawa H., Matsuyama T., Mak T.W., Aizawa S., Tokino T., Oren M. and Taniguchi T. (1996) Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature, 382, 816–818. [DOI] [PubMed] [Google Scholar]
- 59. Chapman R.S., Duff E.K., Lourenco P.C., Tonner E., Flint D.J., Clarke A.R. and Watson C.J. (2000) A novel role for IRF-1 as a suppressor of apoptosis. Oncogene, 19, 6386–6391. [DOI] [PubMed] [Google Scholar]
- 60. Park S.Y., Seol J.W., Lee Y.J., Cho J.H., Kang H.S., Kim I.S., Park S.H., Kim T.H., Yim J.H., Kim M., Billiar T.R. and Seol D.W. (2004) IFN-gamma enhances TRAIL-induced apoptosis through IRF-1. Eur. J. Biochem., 271, 4222–4228. [DOI] [PubMed] [Google Scholar]
- 61. Kim P.K., Armstrong M., Liu Y., Yan P., Bucher B., Zuckerbraun B.S., Gambotto A., Billiar T.R. and Yim J.H. (2004) IRF-1 expression induces apoptosis and inhibits tumor growth in mouse mammary cancer cells in vitro and in vivo. Oncogene, 23, 1125–1135. [DOI] [PubMed] [Google Scholar]
- 62. Porta C., Hadj-Slimane R., Nejmeddine M., Pampin M., Tovey M.G., Espert L., Alvarez S. and Chelbi-Alix M.K. (2005) Interferons alpha and gamma induce p53-dependent and p53-independent apoptosis, respectively. Oncogene, 24, 605–615. [DOI] [PubMed] [Google Scholar]
- 63. Sato T., Onai N., Yoshihara H., Arai F., Suda T. and Ohteki T. (2009) Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med., 15, 696–700. [DOI] [PubMed] [Google Scholar]
- 64. Yamamizu K., Piao Y., Sharov A.A., Zsiros V., Yu H., Nakazawa K., Schlessinger D. and Ko M.S. (2013) Identification of transcription factors for lineage-specific ESC differentiation. Stem Cell Reports, 1, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li Y., Esain V., Teng L., Xu J., Kwan W., Frost I.M., Yzaguirre A.D., Cai X., Cortes M., Maijenburg M.W. et al. (2014) Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev., 28, 2597–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grieco J.C., Ciarlone S.L., Gieron-Korthals M., Schoenberg M.R., Smith A.G., Philpot R.M., Heussler H.S., Banko J.L. and Weeber E.J. (2014) An open-label pilot trial of minocycline in children as a treatment for Angelman syndrome. BMC Neurol., 14, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Paribello C., Tao L., Folino A., Berry-Kravis E., Tranfaglia M., Ethell I.M. and Ethell D.W. (2010) Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol., 10, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yong V.W., Wells J., Giuliani F., Casha S., Power C. and Metz L.M. (2004) The promise of minocycline in neurology. Lancet Neurol., 3, 744–751. [DOI] [PubMed] [Google Scholar]
- 69. Garrido-Mesa N., Zarzuelo A. and Gálvez J. (2013) Minocycline: far beyond an antibiotic. Br. J. Pharmacol., 169, 337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tikka T., Fiebich B.L., Goldsteins G., Keinanen R. and Koistinaho J. (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J. Neurosci., 21, 2580–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tikka T.M. and Koistinaho J.E. (2001) Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J. Immunol., 166, 7527–7533. [DOI] [PubMed] [Google Scholar]
- 72. Giuliani F., Hader W. and Yong V.W. (2005) Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell–microglia interaction. J. Leukoc. Biol., 78, 135–143. [DOI] [PubMed] [Google Scholar]
- 73. Valluy J., Bicker S., Aksoy-Aksel A., Lackinger M., Sumer S., Fiore R., Wüst T., Seffer D., Metge F., Dieterich C. et al. (2015) A coding-independent function of an alternative Ube3a transcript during neuronal development. Nat. Neurosci., 18, 666–673. [DOI] [PubMed] [Google Scholar]
- 74. Miao S., Chen R., Ye J., Tan G.H., Li S., Zhang J., Jiang Y.H. and Xiong Z.Q. (2013) The Angelman syndrome protein Ube3a is required for polarized dendrite morphogenesis in pyramidal neurons. J. Neurosci., 33, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Takumi T. (2011) The neurobiology of mouse models syntenic to human chromosome 15q. J. Neurodev. Disord., 3, 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vargas D.L., Nascimbene C., Krishnan C., Zimmerman A.W. and Pardo C.A. (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol., 57, 67–81. [DOI] [PubMed] [Google Scholar]
- 77. Atladóttir H.O., Pedersen M.G., Thorsen P., Mortensen P.B., Deleuran B., Eaton W.W. and Parner E.T. (2009) Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics, 124, 687–694. [DOI] [PubMed] [Google Scholar]
- 78. Patterson P.H. (2002) Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Curr. Opin. Neurobiol., 12, 115–118. [DOI] [PubMed] [Google Scholar]
- 79. Brown A.S., Begg M.D., Gravenstein S., Schaefer C.A., Wyatt R.J., Bresnahan M., Babulas V.P. and Susser E.S. (2004) Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry, 61, 774–780. [DOI] [PubMed] [Google Scholar]
- 80. Smith S.E., Li J., Garbett K., Mirnics K. and Patterson P.H. (2007) Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci., 27, 10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Xu N., Li X. and Zhong Y. (2015) Inflammatory cytokines: potential biomarkers of immunologic dysfunction in autism spectrum disorders. Mediators Inflamm., 2015, 531518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Estes M.L. and McAllister A.K. (2016) Maternal immune activation: implications for neuropsychiatric disorders. Science, 353, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu X., Tamada K., Kishimoto R., Okubo H., Ise S., Ohta H., Ruf S., Nakatani J., Kohno N., Spitz F. and Takumi T. (2015) Transcriptome profiling of white adipose tissue in a mouse model for 15q duplication syndrome. Genom. Data, 5, 394–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kishimoto R., Tamada K., Liu X., Okubo H., Ise S., Ohta H., Ruf S., Nakatani J., Kohno N., Spitz F. and Takumi T. (2015) Model mice for 15q11-13 duplication syndrome exhibit late-onset obesity and altered lipid metabolism. Hum. Mol. Genet., 24, 4559–4572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.