

Abstract
Mutations in OCRL encoding the inositol polyphosphate 5-phosphatase OCRL (Lowe oculocerebrorenal syndrome protein) disrupt phosphoinositide homeostasis along the endolysosomal pathway causing dysfunction of the cells lining the kidney proximal tubule (PT). The dysfunction can be isolated (Dent disease 2) or associated with congenital cataracts, central hypotonia and intellectual disability (Lowe syndrome). The mechanistic understanding of Dent disease 2/Lowe syndrome remains scarce due to limitations of animal models of OCRL deficiency. Here, we investigate the role of OCRL in Dent disease 2/Lowe syndrome by using OcrlY/− mice, where the lethal deletion of the paralogue Inpp5b was rescued by human INPP5B insertion, and primary culture of proximal tubule cells (mPTCs) derived from OcrlY/− kidneys. The OcrlY/− mice show muscular defects with dysfunctional locomotricity and present massive urinary losses of low-molecular-weight proteins and albumin, caused by selective impairment of receptor-mediated endocytosis in PT cells. The latter was due to accumulation of phosphatidylinositol 4,5–bisphosphate PI(4,5)P2 in endolysosomes, driving local hyper-polymerization of F-actin and impairing trafficking of the endocytic LRP2 receptor, as evidenced in OcrlY/− mPTCs. The OCRL deficiency was also associated with a disruption of the lysosomal dynamic and proteolytic activity. Partial convergence of disease-pathways and renal phenotypes observed in OcrlY/− and Clcn5Y/− mice suggest shared mechanisms in Dent diseases 1 and 2.
These studies substantiate the first mouse model of Lowe syndrome and give insights into the role of OCRL in cellular trafficking of multiligand receptors. These insights open new avenues for therapeutic interventions in Lowe syndrome and Dent disease.
Introduction
The maintenance of body fluid and electrolyte homeostasis critically depends on the appropriate handling of solutes and water by the epithelial cells lining the proximal tubule (PT) of the kidney (1,2). These cells are characterized by an efficient endolysosomal pathway involving the apical multiligand receptors megalin (LRP2) and cubilin (3). By processing internalized cargos and recycling of receptors and transporters at the apical surface, the endolysosomal system of PT cells recover essential substances that are filtered through the glomerulus, including a large variety of low-molecular-weight (LMW) proteins that would otherwise be lost in the urine (2). Congenital and acquired disorders of the endolysosomal pathway cause PT dysfunction (renal Fanconi syndrome) with massive urinary loss of solutes, dehydration, electrolyte imbalance, rickets, growth retardation and the development of chronic kidney disease (CKD). Such PT dysfunctions are typically encountered in Dent disease, a rare, X-linked disorder characterized by LMW proteinuria, renal Fanconi syndrome, kidney stones, nephrocalcinosis and progressive renal failure (4,5).
Dent disease is genetically heterogeneous. The majority of cases (~60%) are due to mutations in the CLCN5 gene that encodes the electrogenic Cl−/H+ exchanger ClC-5 (Dent disease 1, MIM #300009)(6). In a subset of patients (~15–20%), the disease is caused by mutations in OCRL, the gene encoding the type II phosphatidylinositol (PI) bisphosphate 5-phosphatase OCRL (Dent disease 2, MIM #300555). Mutations in OCRL are also associated with the oculocerebrorenal syndrome of Lowe (MIM #309000), which includes systemic manifestations such as congenital cataracts, cognitive disability and hypotonia (4,7). OCRL is a protein of 110 kDa, which, in addition to the 5-phosphatase catalytic domain, comprises the following: (i) a pleckstrin homology (PH) domain containing a clathrin-binding site (8); (ii) an ASPM, SPD-2, Hydin (ASH) domain, characteristic of proteins that localize to centrosome and primary cilia (9); and (iii) a RhoGAP-like domain, which mediates interactions with proteins involved in cytoskeleton dynamics and progression of cell cycle (10). Almost all the mutations associated with Lowe syndrome occur in exons 8–23 that include the 5-phosphatase catalytic domain, the ASH domain and the RhoGAP-like domain. Conversely, the majority of mutations that cause Dent disease 2 are positioned in exons 1–7, which comprise the PH domain (11). Although mutations in the catalytic domain of OCRL have been described in both clinical profiles, the mutations in Dent disease 2 are always missense—compatible with a residual biological activity that could explain a less severe cellular phenotype (7).
The convergence of clinical phenotypes among patients with mutations in OCRL and CLCN5 raises the questions whether these gene products are part of the same pathways and how they cause dysfunction of endolysosomes in PT cells. ClC-5 is predominantly expressed in the early endosomes of PT cells of the kidney, potentially involved in their maturation toward the degradative compartments or the recycling route (6). Studies in mouse models showed that the functional loss of ClC-5 generates a trafficking defect involving megalin and cubilin, reflected by defective endocytosis and manifestations of PT dysfunction (6,12). OCRL is located at different stations of the endolysosomal pathway where it maintains the cellular metabolism of phosphatidylinositol 4,5-bisphosphate PI(4,5)P2, an essential regulator of membrane trafficking. Increased PI(4,5)P2 levels and marked vesicular trafficking defects affecting the endocytic network have been observed in cells lacking OCRL, resulting in defective receptor-mediated endocytosis (13).
The direct impact of OCRL on transport events leading to renal Fanconi syndrome cannot be reliably assessed on dedifferentiated cell systems, including non-kidney or non-epithelial cell types, or clonal cells isolated from urine (14). Renal biopsy material, usually obtained at an advanced disease stage, is of limited value. Furthermore, limitations of animal models of OCRL deficiency impede the development of translational studies. The first Ocrl KO mouse showed no kidney, eye or brain defects (15) due to a compensation by INPP5B, the closest paralogue of OCRL in mice and humans (16). Accordingly, a generalized endocytic defect (affecting both receptor-mediated and fluid-phase endocytosis) was detected in a conditional tubular deletion of Ocrl and Inpp5b mouse model (17). However, studies performed in this strain do not allow to discriminate the individual role of OCRL and INPP5B in the pathophysiology of Dent disease.
Recently a mouse model expressing human INPP5B in OcrlY/−; Inpp5b−/− background was generated (18). The replacement of mouse Inpp5b with human INPP5B in the whole body of OcrlY/− mouse provides a humanized background, which therefore allows to investigate the consequences related to the specific loss of OCRL activity. Preliminary studies revealed PT dysfunction in this line (18), with no investigation of the time-course, multi-systemic aspects and cellular basis of this defect. Here, we analyzed in detail the multi-systemic phenotype of the OcrlY/−; Inpp5b−/−; BAC-INPP5B mouse model, compared the kidney dysfunction to that of Clcn5 KO mice and investigated the mechanisms underpinning epithelial transport defects in Lowe syndrome.
Results
Ocrl Y/− mice show early manifestations of PT dysfunction
A humanized mouse model for Lowe syndrome/Dent disease 2 was generated by targeted disruption of both Ocrl and Inpp5b and oocyte injection of a bacterial artificial chromosome (BAC) containing the human gene INPP5B to avoid embryonic lethality (18). The mouse littermates lacking Inpp5b while harboring BAC-INPP5B expression with Ocrl are referred to as OcrlY/+ (OcrlY/+;Inpp5b−/−; BAC-INPP5B), whereas those lacking Ocrl are OcrlY/− (OcrlY/−; Inpp5b−/−; BAC-INPP5B) mice, respectively. All mice were born at Mendelian ratio and were viable and fertile. The genotype of Ocrl mice was demonstrated by genomic DNA analyses of Ocrl, Inpp5b and BAC-INPP5B (Supplementary Material, Fig. 1A) and immunoblotting for OCRL (Supplementary Material, Fig. 1B) in kidney samples derived from OcrlY/− and OcrlY/+ mice.
We first used the Ocrl mice to characterize the kidney phenotype over time. Growth retardation was observed in OcrlY/− mice starting from 16 weeks of age onwards (Fig. 1A), whereas manifestations of PT dysfunction appeared in OcrlY/− mice from 8 weeks of age, with albuminuria (Fig. 1B and C) and LMW proteinuria (Fig. 1D and E). The inappropriate loss of the LMW Clara cell secretory protein 16 (CC16) in the urine of OcrlY/− mice was observed at all time points analyzed (Fig. 1D). Western blotting of the urine confirmed the major loss of transferrin (TFR) and vitamin D binding protein (VDBP) along CC16 in the urine of OcrlY/− mice (Fig. 1E). The OcrlY/− mice did not show manifestations of renal failure and no polyuria, calciuria, glycosuria and phosphaturia during the timeframe of investigation, suggesting a partial renal Fanconi syndrome (Table 1). The expression levels of BAC-INPP5B were not associated with variable levels of PT dysfunction in OcrlY/− mice (Supplementary Material, Fig. 1C–F). Since mutations in CLCN5 and OCRL produce similar kidney defects in human patients, we compared the severity of PT dysfunction in the Clcn5Y/− and OcrlY/− mouse models. The OcrlY/− mice display a milder LMW proteinuria (CC16) than that observed in Clcn5Y/− mice (Fig. 1F). This difference is most likely reflecting specific roles of ClC-5 and OCRL along the endolysosomal pathway.
Figure 1.
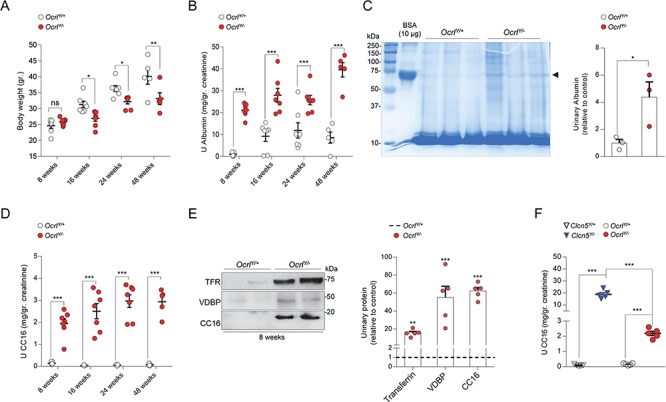
Growth retardation and renal phenotype in a whole body OcrlY/− mouse model. (A) Dot plot representing the measurement of body weight, (B) albumin and (D) CC16 urinary excretion in Ocrl male mice at 8, 16, 24 and 48 weeks after birth (8 weeks, n = 7 mice per group; 16 weeks, n = 7 mice per group; 24 weeks, n = 7 mice per group; 48 weeks, n = 5 mice per group). (C) Coomassie blue-stained SDS-PAGE analysis of urine derived from 8-week-old Ocrl mice and densitometry quantification of albumin (n = 3 mice per group). A total of 10 μg of BSA were loaded as positive control [first lane; molecular weight, ~66.5 kDa; arrowhead]. (E) Representative western blotting and densitometry quantification of TFR, VDBP and CC16 in urine derived from 8-week-old Ocrl mice (TFR, VDBP and CC16, n = 5 mice per group). (F) Quantification of CC16 urinary excretion in 8-weeks-old Clcn5 and Ocrl mice (n = 5 mice per group). All the urine parameters were normalized to urinary creatinine concentration. Plotted data represent mean ± SEM. Each dot of the graphs represents one mouse. Two-tailed unpaired Student’s t-test, *P < 0.05, **P < 0.01 and ***P < 0.001 relative to OcrlY/+ or Clcn5Y/+ mice. ns: not significant.
Table 1.
Body weight, urine and blood parameters in Ocrl and Clcn5 mice
| 8 weeks | 8 weeks | 16 weeks | 24 weeks | 48 weeks | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ocrl Y/+ (n = 7) | Ocrl Y/− (n = 7) | Clcn5 Y/+ (n = 4) | Clcn5 Y/− (n = 4) | Ocrl Y/+ (n = 7) | Ocrl Y/− (n = 7) | Ocrl Y/+ (n = 7) | Ocrl Y/− (n = 7) | Ocrl Y/+ (n = 5) | Ocrl Y/− (n = 5) | |
| Body weight (gr) |
24.6 ± 0.8 | 25.8 ± 0.4 | 31.03 ± 1.3 | 27.2 ± 1.2 | 31.3 ± 0.9 | 26.6 ± 0.9a | 36.3 ± 0.9 | 32.2 ± 0.7a | 40.1 ± 2.5 | 33.1 ± 1.8b |
| U volume (μl/12 h) |
2267 ± 337 | 3122 ± 328 | 1400 ± 82 | 3025 ± 111c | 2414 ± 298 | 3571 ± 561 | 2388 ± 650 | 2462 ± 544 | 1655 ± 315 | 2138 ± 555 |
| Diuresis (μl/gr BW/min) |
0.12 ± 0.02 | 0.16 ± 0.01 | 0.06 ± 0.004 | 0.15 ± 0.004b | 0.1 ± 0.01 | 0.18 ± 0.03 | 0.09 ± 0.02 | 0.11 ± 0.02 | 0.06 ± 0.01 | 0.091 ± 0.02 |
| U Albumin (mg/gr creatinine) |
0.97 ± 0.3 | 21 ± 1.1c | 3.3 ± 0.2 | 69 ± 13b | 9.2 ± 2.3 | 27.9 ± 3.1c | 11.8 ± 3.4 | 25.7 ± 2.1c | 8.6 ± 2.5 | 39.5 ± 3.2c |
| U CC16 (mg/gr creatinine) |
0.14 ± 0.02 | 1.9 ± 0.2c | 0.09 ± 0.02 | 19.2 ± 1.9c | 0.03 ± 0.00 | 2.5 ± 0.3c | 0.05 ± 0.01 | 2.9 ± 0.3c | 0.06 ± 0.02 | 2.9 ± 0.2c |
| U calcium (mg/gr creatinine) |
94.9 ± 6.4 | 73.9 ± 14.5 | 58.2 ± 16.4 | 144 ± 11.6b | 135 ± 11.2 | 148 ± 14.3 | 70.4 ± 20.3 | 93.8 ± 8 | 148 ± 23 | 109 ± 11.5 |
| U glucose (mg/gr creatinine) |
1561 ± 136 | 1451 ± 95 | 1122 ± 76 | 1982 ± 341a | 1440 ± 171 | 1418 ± 163 | 1081 ± 179 | 1672.7 ± 493 | 3040 ± 993 | 1534 ± 93 |
| U phosphate (mg/gr creatinine) |
4051 ± 419 | 4316 ± 160 | 2684 ± 314 | 4294 ± 448a | 4436 ± 403 | 4017 ± 372 | 2584 ± 422 | 2761 ± 423 | 3072. ± 434 | 3101 ± 106 |
| U creatinine (mg/dl) |
26 ± 1.8 | 24 ± 1.5 | 31 ± 1.9 | 30 ± 2.5 | 23 ± 2.8 | 20 ± 1.9 | 35 ± 6.4 | 30 ± 3.8 | 25 ± 5 | 27 ± 1.9 |
| BUN (mg/dl) |
- | - | - | - | 22 ± 1.5 | 22 ± 1.3 | - | - | 24 ± 2.7 | 22 ± 1.2 |
| Plasma creatinine (mg/dl) |
- | - | - | - | 0.09 ± 0.02 | 0.08 ± 0.006 | - | - | 0.05 ± 0.007 | 0.06 ± 0.01 |
U, Urine; BUN, Blood urea nitrogen; BW, Body weight. All the measurements were performed on OcrlY/+ and OcrlY/− male mice matched per age.
Plotted data represent mean ± SEM. Two-tailed unpaired Student’s t-test was applied between genotypes for the indicated time point.
a P < 0.05 versus Ocrl Y/+ or Clcn5Y/+;
b P < 0.01 versus Ocrl Y/+or Clcn5Y/+;
c P < 0.001 versus Ocrl Y/+ or Clcn5Y/+.
Ocrl Y/− mice show a specific defect in receptor-mediated endocytosis
To determine the mechanism of LMW proteinuria, we followed the in vivo uptake of the LMW protein Cy5-labeled β-lactoglobulin in the kidneys of OcrlY/− mice compared to their control littermates. Fifteen minutes after injection, a substantial accumulation of fluorescent vesicles was observed in the brush border of PT cells of the OcrlY/+ mice, contrasting with a considerable reduction of the signal in PT cells from OcrlY/− mice (Fig. 2A). A similar defective uptake in Cy5-labeled β-lactoglobulin was observed in Clcn5Y/− tubules (Fig. 2B). By contrast, the internalization of Alexa 647-dextran, a marker of fluid phase endocytosis, was unaltered in OcrlY/− mice (Fig. 2C), suggesting that loss of OCRL function impacts specifically on receptor-mediated endocytosis. To substantiate this observation, we investigated the LRP2 receptor and found its expression dramatically decreased in PT cells of the OcrlY/− kidneys, whereas its mRNA level was unchanged (Fig. 2D–F). In line with absent phosphaturia and glycosuria, no modifications in the sodium-phosphate cotransporter IIa (NaPi-IIa), the sodium-glucose cotransporter 2 (SGLT2) or other PT components were observed in OcrlY/− kidneys (Fig. 2E and F).
Figure 2.
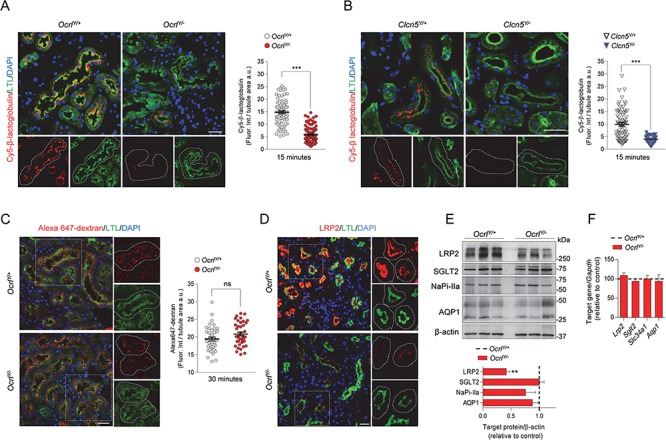
Defective receptor-mediated endocytosis in Dent disease mice. (A,B) Representative confocal micrographs showing Cy5-labeled β-lactoglobulin (red, 1 mg/kg B.W.) or (C) Alexa 647-labeled dextran uptake (red, 6 mg/kg B.W.) after 15 and 30 min from tail vein injections, respectively, and quantifications of the corresponding fluorescent signal in LTL+ (Lotus tetragonolobus Lectin, green) PTs from Ocrl and Clcn5 mouse kidneys (n = 90 Ocrl and n = 70 Clcn5 PTs for Cy5-labeled β-lactoglobulin uptake; n = 40 Ocrl PTs for Alexa 647 dextran uptake) n = 2 per group. Each dot represents fluorescence intensity in one PT; fluorescence intensity was normalized on tubule area; two-tailed unpaired Student’s t-test, ***P < 0.001 relative to OcrlY/+or Clcn5Y/+ kidneys. ns: not significant. Insets: high magnification of Cy5-labeled β-lactoglobulin+ or Alexa 647 dextran+ structures in LTL+ PTs. (D) Representative confocal micrographs showing LRP2 (red) expression in LTL+ (green) PTs of Ocrl mouse kidneys. Insets: high magnification of LRP2+ structures in LTL+ PTs. (E) Western blotting and densitometry analyses of LRP2, SGLT2, NaPi-IIa and AQP1 protein levels in whole-kidney lysates from Ocrl mice. β-actin was used as loading control. Protein levels normalized on β-actin and relative to OcrlY/+ mice (black dotted line; LRP2, n = 6 mice per group; SGLT2, NaPiIIa and AQP1, n = 3 mice per group; Mann–Whitney U test, **P < 0.01 relative to OcrlY/+ kidneys). (F) The mRNA kidney levels of Lrp2, Sglt2, Slc34a1 and Aqp1 were analyzed by real-time qPCR. Gene target expression normalized to Gapdh and relative to OcrlY/+ mice (black dotted line; n = 3 mice per group). Nuclei counterstained with DAPI (blue) in (A–D). Scale bars: 25 μm. Plotted data represent mean ± SEM.
These data reveal that a defective receptor-mediated endocytosis, caused by a decreased protein level of LRP2, characterizes OcrlY/− mice, similar to Clcn5Y/− mice. The renal Fanconi syndrome is restricted to LMW proteinuria in OcrlY/− mice, contrasting with a more severe PT dysfunction in Clcn5Y/− mice. At the age of 8 weeks, the latter showed a decreased expression of SGLT2 and NaPi-IIa (Supplementary Material, Fig. 2A), explaining glycosuria and phosphaturia along polyuria and calciuria (Table 1; Supplementary Material, Fig. 2C and D), and a defective uptake of fluid-phase endocytosis (dextran) markers (Supplementary Material, Fig. 2B).
Loss of OCRL increases PI(4,5)P2 and disrupts receptor-mediated endocytosis in vitro
In order to further analyze the endocytic defect observed in vivo, we developed primary cultures of PT cells (mPTC) from micro-dissected PT segments of OcrlY/− mice aged 8 weeks. This cell culture system preserves the differentiation and polarized transport processes and therefore represents a relevant in vitro model to investigate mechanisms underlying PT disorders (19). To validate the Ocrl cellular system, we evaluated whether the loss of OCRL in mPTCs reproduced the typical endosomal accumulation of PI(4,5)P2 observed in Lowe syndrome/Dent disease 2 (20). Compared to control, OcrlY/− mPTCs exhibited a remarkable increase of PI(4,5)P2 in early endosomal structures, as shown by colocalization with the early endosome marker EEA1 (Fig. 3A and B). The unchanged PI3P levels, the PI hallmark of the endosomal system, demonstrated that OCRL disruption in mPTCs exclusively targets the homeostasis of PI(4,5)P2 (Supplementary Material, Fig. 3A). These results, which are consistent with PI(4,5)P2 accumulation observed in a transgenic zebrafish model of Lowe syndrome or in OCRL-depleted immortalized cells, substantiate the reliability of Ocrl mPTC system (13,21).
Figure 3.
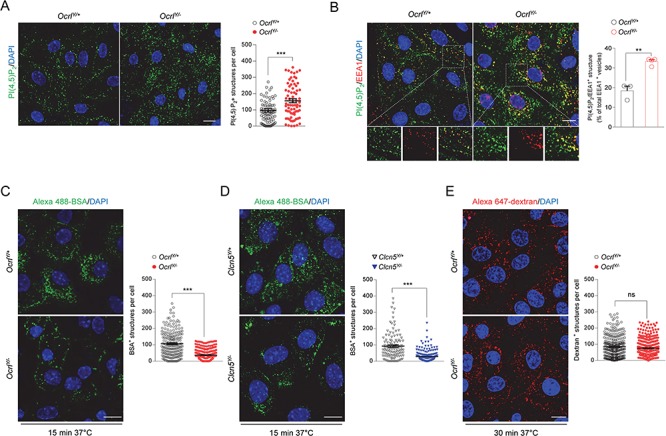
Altered PI(4,5)P2 subcellular distribution and receptor-mediated endocytosis in mPTCs derived from Dent disease mouse models. (A) Representative confocal micrographs and quantification of PI(4,5)P2+ structures (green) in Ocrl mPTCs (n ≈ 80 cells pooled from 3 mouse kidneys per condition; each dot representing the number of PI (4,5)P2+ structures in a cell). (B) Representative confocal micrographs of Ocrl mPTCs immunostained with anti-PI(4,5)P2 (green) and anti-EEA1 (red, early endosomes) and quantification (adjacent panel) of the number of PI(4,5)P2/EEA1+ structures by confocal microscopy (percentage of total EEA1+ vesicles: n = 3 OcrlY/+ and n = 4 OcrlY/− randomly selected fields per condition, each containing approximately 15–20 cells). Insets: high magnification of PI(4,5)P2/EEA1+ structures. (C,D) Ocrl and Clcn5 mPTCs were loaded with Alexa 488-BSA (green, 100 μg ml−1 for 15 min at 37°C), fixed and analyzed by confocal microscopy. Quantification of the number of Alexa 488-BSA+ structures (n ≈ 150–250 cells pooled from 3 mouse kidneys per condition; each dot representing the number of BSA+ structures in a cell). (E) Ocrl mPTCs were loaded with Alexa 647-dextran 10 kDa (red, 250 μg ml−1 for 30 min at 37°C), fixed and analyzed by confocal microscopy. Quantification of the number of Alexa 647-dextran+ structures (n ≈ 200–250 cells pooled from 3 mouse kidneys per condition; each dot representing the number of dextran+ structures in a cell). Nuclei counterstained with DAPI (blue). Scale bars: 15 μm in (A) and (B) and 10 μm in (C–E). Plotted data represent mean ± SEM. Two-tailed unpaired Student’s t-test, **P < 0.01, ***P < 0.001 relative to OcrlY/+or Clcn5Y/+ mPTCs. ns: not significant.
Given that the tight regulation of PI(4,5)P2 homeostasis is fundamental for a proper endocytic trafficking, we asked whether its increase at the early endosomes might impair the endocytic function of mPTCs. Functional studies demonstrated that albumin uptake was markedly reduced in OcrlY/− (−84%) when compared to control mPTCs (Fig. 3C). A similar disruption of the endocytic capacity was observed in Clc5Y/− mPTCs (Fig. 3D). Confirming the observations made in vivo, fluid phase endocytosis was unaltered in OcrlY/− mPTCs (Fig. 3E), highlighting the specificity of the defect in receptor-mediated endocytosis.
OCRL deficiency perturbs expression of LRP2 through aberrant F-actin polymerization
We next examined whether the defective uptake capacity of OcrlY/− mPTCs was reflected by alterations of the endogenous LRP2 expression. LRP2 protein level was reduced in the total lysates of mPTCs lacking OCRL (Fig. 4A), similarly to Clcn5Y/− mPTCs (Supplementary Material, Fig. 4A). This reduction was confirmed by the analysis of the confocal Z-stack images, evidencing a decreased LRP2 fluorescent signal in each focal plane throughout the entire volume of OcrlY/− mPTCs (Fig. 4B; Supplementary Material, Fig. 4B). The latter analysis showed a shift of LRP2 signal from the apical plasma membrane toward a more intracellular compartment in OcrlY/− mPTCs, whose polarization is defined by the typical apical [wheat germ agglutinin (WGA)] and basolateral (Na+, K+-ATPase) markers (Fig. 4C; Supplementary Material, Fig. 4C). This result was corroborated by a striking decrease of LRP2 level in the plasma membrane fraction of OcrlY/− mPTCs (Fig. 4D). These changes were not associated with LRP2 transcriptional modifications or alterations of other PT receptors or transporters (Supplementary Material, Fig. 3B).
Figure 4.
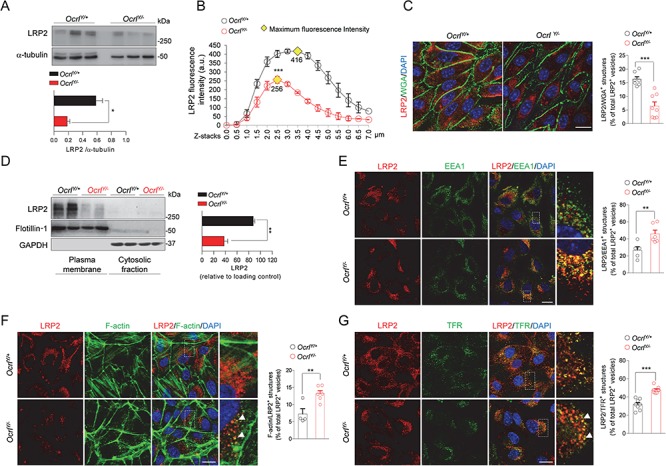
OcrlY/− mPTCs exhibit decreased expression of LRP2 and defective endocytic recycling. (A) Representative western blotting and quantification of LRP2 in whole Ocrl mPTCs lysates. α-tubulin was used as loading control (n = 4 mice per group). (B) Dot plot representing distribution and average fluorescence intensity of LRP2 along Z-stacks projections in Ocrl mPTCs. Yellow rhombus represents the maximum peak of fluorescence intensity. Quantifications of Z-stacks were obtained from 3 randomly selected fields per condition, with each containing approximately 15–20 cells. (C) Representative confocal micrographs and quantification of LRP2/WGA+ structures by confocal microscopy (percentage of total LRP2+ vesicles: n = 7 OcrlY/+ and n = 8 OcrlY/− randomly selected fields per condition, each containing approximately 10–15 cells). (D) Representative western blotting and quantification of LRP2 protein levels in plasma membrane fractions derived from Ocrl mPTCs. Flotillin-1 and GAPDH were used as purity marker and loading control of the plasma membrane and cytosolic fractions respectively (n = 3 independent experiment). (E) Representative confocal micrographs of Ocrl mPTCs immunostained with anti-LRP2 (red) and anti-EEA1 (green, early endosomes) and quantification (adjacent panel) of the number of LRP2/EEA1+ structures by confocal microscopy (percentage of total LRP2+ vesicles: n = 5 OcrlY/+ and n = 6 OcrlY/− randomly selected fields per condition, each containing approximately 15–20 cells). Insets: high magnification of LRP2/EEA1+ structures. (F) Representative confocal micrographs of Ocrl mPTCs immunostained with anti-LRP2 (red) and Alexa-Fluor-488-phalloidin (green, F-actin). Quantification (adjacent panel) of the number of F-Actin/LRP2+ structures (percentage of total LRP2+ vesicles: n = 4 OcrlY/+ and n = 6 OcrlY/− randomly selected fields per condition, each containing approximately 10–15 cells). Insets: high magnification of F-actin/LRP2+ structures. (G) Representative confocal micrographs of Ocrl mPTCs stained with anti-TFR (green) and anti-LRP2 (red) and quantification of the number of TFR/LRP2+ structures by confocal microscopy (percentage of total LRP2+ vesicles: n = 8 randomly selected fields per condition, each containing approximately 20–25 cells). Insets: high magnification of TFR/LRP2+ structures. Nuclei counterstained with DAPI (blue) in (C) and (E–G). Scale bars: 15 μm in (E–G) and 20 μm in (C). Plotted data represent mean ± SEM. Two-tailed unpaired Student’s t-test, *P < 0.05, **P < 0.01 and ***P < 0.001 relative to OcrlY/+ mPTCs.
The intracellular localization of LRP2 in OcrlY/− mPTCs prompted us to examine its organelles compartmentalization. OCRL is known to modulate the trafficking of endocytic receptors by orchestrating the recycling of endosomal vesicles to the surface (13). Thus, lack of OCRL function might disrupt the return of the endocytic receptors to the apical membrane, leading to their accumulation in endosomal structures. Imaging studies confirmed LRP2 increase within EEA1+ early endosomes in OcrlY/− mPTCs, suggesting that loss of OCRL function impacts on both expression and distribution of this endocytic scavenger (Fig. 4E).
As PI(4,5)P2 promote the recruitment of the actin nucleating machinery, we asked whether their endosomal accumulation, observed in OcrlY/− mPTCs, might locally induce an aberrant actin polymerization and, in turn, affect the trafficking of endocytic receptors (22). We observed that, in OcrlY/− mPTCs, the levels of actin stress fibers decreased and foci of filamentous actin (F-actin) accumulated on internal membranes ruffles (Supplementary Material, Fig. 4D).
Compared to controls, an increase of F-actin structures, colocalizing with EEA1, was observed in OcrlY/− mPTCs (Supplementary Material, Fig. 4E). This uncontrolled actin dynamics might impede the entrance of the endosomes in the recycling route and coerce LRP2 to stick inside them. Supporting this hypothesis, a considerable amount of LRP2 vesicles were coalescent with F-actin membranes (Fig. 4F). Furthermore, by high-magnification confocal microscopy, it was possible to visualize the formation of F-actin basket-like structure surrounding LRP2 (Supplementary Material, Fig. 4F). These data indicate that the increased association of F-actin with early endosomes may affect the endocytic trafficking and prevent the recycling of LRP2 to the apical surface. In order to substantiate the role of OCRL in maintaining the recycling activity, we investigated the localization of the canonical recycling marker transferrin receptor (TFR), which crosses the same endocytic stations traveled by LRP2 (plasma membrane—early endosomes/recycling endosomes)(23). Our data showing the redistribution of TFR from the plasma membrane to enlarged cytoplasmic structures, which also trap LRP2, provide another evidence of defective endocytic recycling in OcrlY/− mPTCs (Supplementary Material, Fig. 5A; Fig. 4G).
We also tested whether the lack of OCRL might affect the trafficking of the cation-independent mannose 6-phosphate receptor (CI-MPR), which is required for the transport of lysosomal enzymes and travels through intracellular itineraries diverging from those exploited by LRP2 (trans Golgi network–late endosomes–plasma membrane) (24). An increase in the CI-MPR associated with peripheral structures and a decrease in the perinuclear pool was observed in the kidneys and mPTCs from OcrlY/− mice compared to controls (Supplementary Material, Fig. 5B and C), indicating a defective retrograde transport of CI-MPR from the endosomes to the Golgi. All together, these data suggest that the actin-trapping mechanism impairing the recycling of LRP2 extends to other receptors traveling through the endosomal structures of OcrlY/− mice, and it is therefore highly relevant for the molecular basis of Lowe syndrome.
OCRL depletion alters lysosomal dynamics and function
It has been recently shown that, under lysosomal overload conditions, OCRL translocates on the lysosomal membranes, where it ensures adequate levels of PI(4,5)P2 necessary for fusion and subsequent degradation of cargo vesicles (25). We thus explored whether loss of OCRL function might generate a rearrangement of PI(4,5)P2 at the lysosomal membranes and, consequently, affect lysosomal morphology. Confocal microscopy analysis showed that Ocrl deletion increased the number of PI(4,5)P2-positive structures colocalized with LAMP1-labeled lysosomes (Fig. 5A). These changes were associated with dramatic modifications in the dynamics of lysosomes as evidenced by their abnormal increase in number and size (Fig. 5B). As changes in lysosomal dynamics could affect their proteolytic activity, we examined whether OCRL disruption impairs the lysosomal cargo degradation in OcrlY/− mPTCs. To this end we used dequenched bovine serum albumin (BSA-DQ) reagent, which is readily incorporated by cells via fluid-phase endocytosis. Upon fusion with endo-lysosomes, BSA-DQ is digested into smaller fragments, thereby relieving its self-quenching properties and causing a fluorescent signal that reflects lysosomal degradative capacity (26). OcrlY/− mPTCs showed a remarkable decrease in BSA-DQ fluorescent puncta colocalized with LAMP1 (Fig. 5C), indicating a specific impairment of lysosomal proteolysis as fluid phase endocytosis was not affected by the loss of OCRL. To substantiate the defective lysosomal activity, we tested the processing of the epidermal growth factor receptor (EGFR), an endogenous protein that, upon EGF-induced internalization, is normally sorted to endolysosomal proteolytic pathway for degradation (27). Control mPTCs, 90 min after EGF stimulation, showed an efficient degradation of EGFR. On the contrary, at the same time frame, the levels of EGFR remained high in OcrlY/− mPTCs, suggesting a delayed lysosomal processing of the receptor (Fig. 5D).
Figure 5.
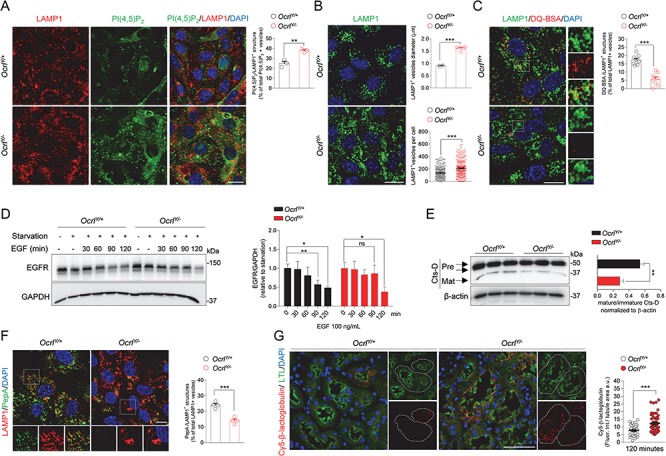
Altered lysosomal dynamics and degradative capacity in OcrlY/− mPTCs. (A) Ocrl mPTCs were immunostained with anti-PI(4,5)P2 (green) and anti-LAMP1 (red, lysosomes) and the number of PI(4,5)P2/LAMP1+ structures were quantified by confocal microscopy (percentage of total PI(4,5)P2+ vesicles: n = 3 randomly selected fields per condition, each containing approximately 40–50 cells). (B) Representative confocal micrographs of Ocrl mPTCs immunostained with anti-LAMP1 (green). Quantification of the average LAMP1+ vesicles diameter (top, n = 4 OcrlY/+ and n = 6 OcrlY/− randomly selected fields per condition, each containing approximately 50–60 cells) and number of structures (bottom, n ≈ 200–220 cells pooled from 3 Ocrl kidneys per group, each point representing the number of LAMP1+ structure in a cell). (C) Ocrl mPTCs were loaded with DQ Red BSA (red, 10 μg ml−1 for 1 h at 37°C), immunostained with anti-LAMP1 (green, lysosomes) fixed and analyzed by confocal microscopy. Quantification of number of DQ Red BSA/LAMP1+ structures (percentage of total LAMP1+ structures: n = 8 randomly selected fields per condition, with each containing approximately 10–15 cells). Insets: high magnification of DQ Red BSA/LAMP1+ vesicles. (D) Ocrl mPTCs were serum starved for 24 h and then stimulated with EGF (100 ng ml-1) for the indicated times. EGFR protein levels were evaluated by western blotting and quantified relative to time 0 (starved cells; n = 3 mice per group; two-tailed unpaired Student’s t-test, *P < 0.05, **P < 0.01 relative to OcrlY/+ or OcrlY/− starved mPTCs. ns: not significant). (E) Western blotting and densitometry analyses of Cathepsin D (Cts-D) protein levels in Ocrl mPTCs (n = 4 mice per group). (F) Ocrl mPTCs were loaded with Bodipy-FL-PepA (1 μm, for 1 h at 37°C, green), immunostained with anti-LAMP1 antibody (red) and analyzed by confocal microscopy. Quantification of numbers of PepA/LAMP1+ structures (percentage of total LAMP1+ structures: n = 4 randomly selected fields per condition, with each containing approximately 20–25 cells). (G) Representative confocal micrographs showing Cy5-labeled β-lactoglobulin (red) after 120 min from tail vein injections and quantifications of the corresponding fluorescent signal in LTL+ PTs from Ocrl mouse kidneys (n = 50 Ocrl PTs; each dot representing fluorescence intensity in one PT; fluorescence intensity was normalized on tubule area). Nuclei counterstained with DAPI (blue) in (A–C), (F) and (G). Scale bars: 15 μm in (A–C), 10 μm in (F) and 50 μm in (G). Plotted data represent mean ± SEM. Two-tailed unpaired Student’s t-test. **P < 0.01, ***P < 0.001 relative to OcrlY/+ mPTCs or kidneys.
One mechanism by which cargo clearance might be impeded is a defective maturation of lysosomal cathepsins. Western blot analyses of Cathepsin D (Cts-D) showed a decreased generation of the 32 kDa mature Cts-D in OcrlY/− mPTCs compared to control (Fig. 5E). We next tested the lysosomal Cts-D activity by incubating the cells with Bodipy-FL-PepstatinA (PepA), a fluorescence-tagged PepA that binds to the active site of Cts-D in acidic lysosomes. Although the majority of lysosomes were co-stained with PepA in control mPTCs, the number of PepA-labeled vesicles colocalizing with LAMP1 were substantially lower in OcrlY/− mPTCs (Fig. 5F). Similarly, the lysosome-based processing of the LMW Cy5-β-lactoglobulin, which is normally internalized and degraded by endolysosomes, was dramatically reduced in OcrlY/− PTs compared to wild-type (Fig. 5G).
To investigate whether missorting of lysosomal hydrolases in the extracellular space might contribute to the aberrant lysosomal proteolysis observed in OcrlY/− mice, we tested for and detected significantly increased levels of the lysosomal protease Cts-D (immature form) in the plasma of these mice (Supplementary Material, Fig. 6A). This result was in line with the mistrafficking of CI-MPR at the cell periphery and with previous studies performed in patients with Lowe syndrome (13). Collectively these data indicate that OCRL is important to maintain lysosome homeostasis, itself crucial for PT function. Of note, impaired lysosomal proteolysis and insufficient degradation of Cy5-β-lactoglobulin was also observed in Clcn5Y/− PTs (Supplementary Material, Fig. 6B–D) indicating that OCRL and ClC-5 are part of the same cellular pathways.
The OcrlY/− mice show dysfunctional locomotricity associated with muscular defects
In order to test whether the deletion of OCRL in this model was reflected by extra-renal manifestations, we performed comprehensive behavioral studies on Ocrl mice to examine basic motor activity, learning and memory skills, social behavior and vision.
To identify disturbances in locomotor activity, we evaluated the general mobility of the mice by challenging them with the open field large arena test, which has been extensively used for analyzing the locomotor defects exhibited by Parkinson and Huntington mouse models (28,29). While velocity was comparable between genotypes (data not shown), OcrlY/− mice exhibited an overall decreased locomotor activity, as scored by the measurement of their trajectories (Fig. 6A and B). The OcrlY/− mice presented more resting episodes than controls, as well as fewer lingering and walking events during the test (Fig. 6C). Of note, center-field avoidance was robust and equal in both genotypes (data not shown), suggesting that the impaired motor activity in OcrlY/− mice was more likely driven by a muscular defect rather than an abnormal anxiety-related response.
Figure 6.
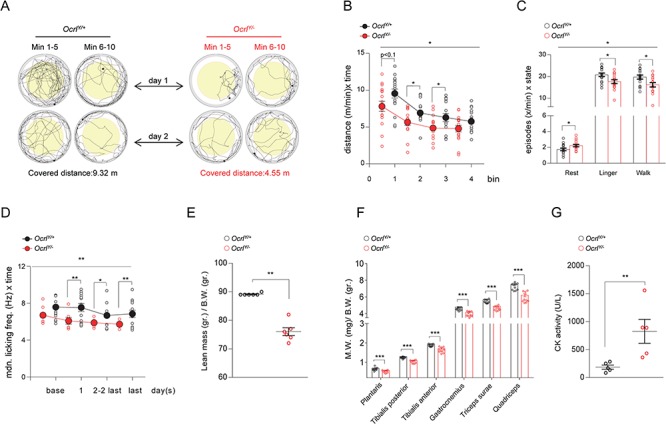
Impaired locomotor activity and muscular defects in OcrlY/− mice. (A–C) Open-field large-arena test was performed during 10 min of two subsequent days in a circular arena with a diameter of 150 cm on a cohort of 16 OcrlY/+and 16 OcrlY/− age-matched male mice. (A) Illustrative pictures representing the trajectories traveled by Ocrl mice during the open field large arena test. (B) Dot plot representing the distance covered by Ocrl mice during the open-field large-arena test. The distance traveled by each mouse is normalized to 1 min observation time and plotted as function of time for each bin (bin = 5 min; split ANOVA effects followed by post-hoc tests between genotype for each bin: Bin1, *P < 0.1; Bin2, *P < 0.05; Bin3, *P < 0.05). (C) Dotted bar graph representing the frequency of resting, lingering and walking episodes exhibited by Ocrl mice during the open field large arena test (split ANOVA effects followed by post-hoc tests between genotype for each state: rest, *P < 0.05; linger, *P < 0.05; walk, *P < 0.05). (D) IntelliCage experiments were performed on a cohort of age matched male Ocrl mice (13 OcrlY/+and 13 OcrlY/−). Dot plot representing the median licking frequency during drinking restriction protocol. Base indicates the licking frequency during the last 24 h of nose-poke adaptation phase (split ANOVA effects followed by post-hoc tests between genotype for each time point: day 1, **P < 0.01; day 2 to last day, *P < 0.05; last day, **P < 0.001). (E) Lean mass/body weight ratio determined by whole body composition analysis in Ocrl mice (n = 6 mice per group). (F) Muscle mass, relative to body weight, of selected hind limb muscles obtained by bilateral dissection of 5 pairs of Ocrl mice (n = 10 values for symmetrical leg muscles). (G) Plasma CK activity in the corresponding five pairs of Ocrl mice (as in F). Plotted data represent mean ± SEM. In (B–D), one-way ANOVA between subject factor genotype: *P < 0.05, **P < 0.01 (on the top of each graph); in (E–G), Mann Withney test, **P < 0.01, ***P < 0.001 relative to OcrlY/+ mice.
To test the latter hypothesis, we pursued mice analyses in IntelliCage, an automated system, which allowed the evaluation of the spontaneous behavior of the animals (30). During free adaptation phase, OcrlY/− mice made fewer corner visits compared to controls, confirming the reduced locomotor and exploratory activity shown in the test above (data not shown). OcrlY/− mice showed an incompetent licking pattern, which became more evident during the drinking restriction protocol and was associated with slower progress of water intake during drinking sessions (Fig. 6D). We excluded an influence of the kidney defect on the licking pattern as similar water intake was detected between genotypes (Supplementary Material, Fig. 7A). Thus, the impaired licking of OcrlY/− mice was most likely caused by a global muscular dysfunction, which affected also the oro-lingual motor apparatus.
To explore whether the defective locomotricity evidenced in OcrlY/− mice was driven by muscular defects, we performed whole body composition analysis (EchoMRI) which evidenced a striking decrease in lean mass/body weight ratio in OcrlY/− mice compared to controls (Fig. 6E), with a global reduction of the mass of several hind limb (gastrocnemius, triceps surae, plantaris, tibialis posterior/anterior and quadriceps) muscles relative to body weight (Fig. 6F) demonstrating skeletal muscle atrophy. These changes were paralleled by a strong increase in plasma creatine kinase (CK) activity, suggesting membrane fragility of striated muscles. (Fig. 6G). These events, which are in line with the muscle hypotonia observed in Lowe Syndrome patients (7), support the motor defect detected in OcrlY/− mice.
To assess the cognitive function of Ocrl mice we applied IntelliCage standard protocols, which investigated the hippocampus-dependent spatial learning ability of the animals. No sign of dysfunctional learning was observed in OCRL deficient mice, even during the most challenging chaining task (data not shown). Aggressive behavior reported in patients with Lowe syndrome was also not reflected in OcrlY/− mice, which showed intact sociability (Supplementary Material, Fig. 7B). Given that congenital cataract and glaucoma are hallmarks of Lowe syndrome, we also examined the visuospatial and visuoperceptual ability of Ocrl mice. During water maze cue navigation, OcrlY/− mice exhibited a robust learning without evidences for an inadequate vision (Supplementary Material, Fig. 7C). In addition no noticeable differences were observed in lens epithelium and in retinal morphology between control and OcrlY/− mice. Irrespective of the Ocrl genotype, an impairment of retinal photoreceptor layer was observed in mice harboring rd8 (retinal degeneration 8) mutation in Crb1 gene, a spontaneous mutation associated with C57BL/6N genetic background, leading to photoreceptor rosette-like structures when expressed homozygously (rd8/rd8). (Supplementary Material, Fig. 7D and E) (31).
Discussion
In these studies we present the first OCRL-deficient mouse model that associates renal and extra-renal manifestations encountered in patients with Lowe syndrome. The absence of OCRL in kidney triggers endolysosomal defects and PT epithelial dysfunction, with a consistent LMW proteinuria reflecting defective receptor-mediated endocytosis due to decreased expression of LRP2 endocytic receptor, in absence of renal failure. We also evidence a partial convergence of disease-pathways and kidney tubular phenotype between mouse models deficient in ClC-5 (Dent disease 1) and OCRL (Lowe syndrome/Dent disease 2). These results provide insights into the mechanisms of endocytosis and the pathophysiology of Dent disease/Lowe syndrome.
In contrast with a previous kidney tubular conditional Ocrl and Inpp5b KO mouse model, where the phenotype reflects the combined loss of OCRL and INPP5B (17), the genetic architecture of the mouse line investigated here allows to directly address dysfunctions related to the single loss of OCRL activity (18). The reinsertion of human INPP5B in the Ocrl−/−;Inpp5b−/− background, irrespective of its level of expression, was fundamental for survival but it was not compensating for the renal defect. This observation is in line with studies on cells derived from patients with Lowe syndrome, which exhibit INPP5B-independent phenotypic variability (32). Although the renal defect was detected in all the OcrlY/− mice analyzed, a noticeable spread was observed comparing individual animals. This individual variability was not due to different levels of INPP5B expression. The possibility that compensatory changes in kinases or phosphatases crossing the same metabolic pathway than OCRL may contribute to such variability should be further investigated.
The differences between the conditional double Ocrl and Inpp5b KO and the humanized transgenic OcrlY/− mice studied here are evident when comparing the defective tubular endocytic phenotypes. The conditional deletion of OCRL and INPP5B resulted in an unspecific impairment of the clathrin-dependent and clathrin-independent endocytosis. By contrast, the single lack of OCRL in OcrlY/− transgenic mice resulted in a selective dysfunction of the clathrin receptor-mediated endocytosis, as evidenced by the defective uptake of β-lactoglobulin and the unaffected internalization of the fluid-phase marker dextran. These differences are in line with the function of OCRL in regulating the membrane trafficking of clathrin coated vesicles (33).
Defective receptor-mediated endocytosis was reflected in OcrlY/− mice by a consistent LMW proteinuria, occurring early, in absence of renal failure. In contrast, the OcrlY/− mice did not show glycosuria, phosphaturia and calciuria, even at old age, mimicking the partial renal Fanconi syndrome typically present in the majority of patients carrying mutations in OCRL (7,34). The Clcn5Y/− mice showed a similar defect in receptor-mediated endocytosis, causing a severe LMW proteinuria. The Clcn5Y/− mice also showed a defective uptake of fluid phase endocytosis markers, in line with previous data from mouse models and patient-derived cells (35–37) but not with others (12). The presence of a more complete form of renal Fanconi syndrome in the Clcn5Y/− mouse model was evidenced by the hypercalciuria, phosphaturia and glucosuria detected at 8 weeks, paralleled by a decreased expression of SGLT2 and NaPi-IIa in addition to LRP2.
Filtered LMW proteins are reabsorbed through megalin/LRP2 and cubilin endocytic receptors expressed at the apical membrane of PT cells. This reabsorptive function is maintained by the degradative and recycling activity of the endolysosomal system, as evidenced by severe defects in tubular homeostasis associated with endolysosomal disorders (1,2). Our investigations characterize the involvement of LRP2 in the pathogenesis of Lowe syndrome and demonstrate analogies with the endocytic defect associated with the loss of ClC-5 in Dent disease 1/Clcn5Y/− mouse model (12). Previous studies, relying on OCRL-deficient immortalized cells expressing LRP2 mini-receptor, showed a redistribution of LRP2 from the apical membrane to the intracellular compartments rather than a defective expression (13). Here, analyses on kidney tissues reveal a remarkable decrease of LRP2 protein in the PT from OcrlY/− mice, similar to that observed in Clcn5Y/− mice. These findings are in line with observations made in zebrafish pronephros, demonstrating the evolutionary conservation of the role of OCRL in endocytosis (21). The identification of converging kidney phenotypes in mouse models for Lowe syndrome and Dent disease supports the view that the endosomal exchanger ClC-5 and the 5-phosphatase OCRL impact on common pathways operating in kidney tubular cells. In absence of transcriptional changes, the mechanism by which the loss of ClC-5 or OCRL might affect LRP2 protein levels remains to be clarified. First, the decrease of apical LRP2 may reflect an increased shedding of the receptor in the urine in response to the endolysosomal engorgement observed in OcrlY/− mice. Previous studies showed that LRP2 is a substrate for metalloproteases and is constitutively subjected to ectodomain shedding (38). This event produces LRP2 membrane-associated fragments, which in turn form the substrate for γ-secretase, the main player of regulated intramembrane proteolysis. Preliminary data showing a higher abundance of LRP2 fragments in OcrlY/− urine, together with a transcriptional increase of all subunits composing the γ-secretase (39) are in line with this hypothesis (data not shown). Alternatively, the loss of LRP2 could be explained by an increased urinary excretion of (full length) LRP2-containing exosomes. A third mechanism might be an aberrant degradation of LRP2, mediated by the proteasome in response to altered lysosomal proteolysis (see below). Further studies of post-translational modifications potentially triggering LRP2 to the proteasome are necessary to test this hypothesis (40).
By establishing differentiated and polarized proximal tubular cell cultures (mPTCs) directly derived from OcrlY/− mouse kidneys (19), we were able to reconstitute critical aspects of the disease in vitro. The OcrlY/− mPTCs recapitulate the selective impairment of receptor-mediated endocytosis and the ectopic accumulation of PI(4,5)P2 in early endosomes (20). The latter is due to the loss of 5-phospatase activity of OCRL that regulates the transition from high (plasma membrane) to low (early endosomes) levels of PI(4,5)P2 in clathrin coated vesicles, enabling progression of the cargo/receptor along the endocytic pathway (33,41). The PI(4,5)P2 dynamic is critical for actin assembly at the plasma membrane ruffles and early endosomes, hence, for the regulation of membrane trafficking (42,43). Accordingly, the endosomal accumulation of PI(4,5)P2 in OcrlY/− mPTCs triggers an aberrant F-actin polymerization, which impairs the recycling of residual LRP2, thus impeding the endocytic uptake. The actin-trapping mechanism impairing LRP2 recycling might be extended to other receptors, as evidenced by the redistribution of the canonical recycling marker TfR in OcrlY/− mPTCs. These defects in receptor recycling represent an appealing druggable target for rescuing the epithelial dysfunction associated with the disease.
It has been shown recently that OCRL can translocate to the lysosome under cargo overload conditions, where its fine regulation of PI(4,5)P2 balance is instrumental for maintaining lysosomal function (25). Here, we demonstrate that lack of OCRL induces an abnormal lysosomal accumulation of PI(4,5)P2 leading to altered lysosomal dynamics and defective lysosomal function, as scored by decreased expression and activity of lysosomal cathepsin D and impaired processing of lysosomal substrates in OcrlY/− kidney and mPTCs. These results, which are consistent with previous observations on biopsies derived from Lowe patients (25), raise the issue of the mechanisms responsible for lysosomal dysfunction associated with loss of OCRL. The peripheral mislocalization of the CI-MPR, a protein regulating the transport of lysosomal hydrolases from the Golgi to lysosomes, along with increased levels of lysosomal protease Cathepsin D (immature form) in the plasma, indicates that misrouting of lysosomal enzymes could be involved. These findings are in line with previous studies performed in Lowe syndrome patients (13). Alternatively, the lysosomal engulfment of PI(4,5)P2 driven by the loss of OCRL may somehow alter the vacuolar-type H+-ATPase (V-ATPase) complex, thereby impairing lysosome acidification and activation of cathepsins. It should be noted that similar defects in receptor-mediated endocytosis and lysosomal dynamic and activity are observed in mouse and cellular models deficient for the endosomal ClC-5 or lysosomal cystinosin transporters, highlighting the role of the endolysosome network as crucial signaling hub to ensure epithelial homeostasis (44).
Studies on the locomotor, neurological and vision abnormalities detected in patients with Lowe syndrome have been hindered by the limitations of available mouse models. Deep phenotyping analyses unveiled a defective locomotricity in OcrlY/−mice, which reflects a global impairment of the muscular apparatus, as indicated by the increased plasma activity of protein CK and atrophy of several hind limb muscles. This phenotype was reminiscent of the muscle hypotonia associated with Lowe syndrome and therefore highly relevant (45). Whether this defective muscular/motor pattern originates from a primary myopathy or is rather a consequence of neurological alterations and how OCRL contributes in maintaining muscular tone remains to be further investigated. Behavior, learning-memory function and vision were not altered in OcrlY/−mice. The discrepant clinical manifestations in mice and humans could be partly explained by differences in tissue-specific expression of enzymes with overlapping functions or by specific roles of OCRL in the tissues affected by the disease (46,47).
Collectively, these studies validate the first mouse model of Lowe syndrome and give insights into the role of OCRL in cellular trafficking of multiligand receptors. Partial convergence of disease-pathways and renal phenotypes observed in OcrlY/− and Clcn5Y/− mice suggest shared mechanisms in Dent diseases 1 and 2. These insights open new avenues for therapeutic interventions in Lowe syndrome and Dent disease.
Materials and Methods
Antibodies and reagents
The following antibodies were used: rabbit anti-human transferrin (A0061, Dako); rabbit anti-human Gc-globulin (also known as VDBP, A0021, Dako); rabbit anti-uteroglobin (also known as CC16, ab40873, Abcam); rabbit anti-SLC1A5 (also known as SGLT2, ab84903, Abcam); rabbit NaPi-IIa (gift from C.A.Wagner, University of Zurich, Zurich, Switzerland); rabbit anti-human AQP1 (ab2219, Millipore); mouse anti-β-actin (A2228, Sigma-Aldrich); mouse conjugated to Fluorescein (FITC) anti-PI(4,5)P2 (Z-G045, Echelon Biosciences Inc.); mouse anti-EEA1 (610456, BD Bioscience); rabbit anti-RFP (600–401-379, ROCKLAND); sheep anti-LRP2 (gift from P. Verroust and R. Kozyraki, INSERM, Paris, France); mouse anti Flotillin-1(610821, BD Bioscience); mouse anti-α-tubulin (T5168, Sigma-Aldrich); rabbit anti-GAPDH (2118, Cell Signaling Technology); rat anti-LAMP1 (sc-19992, Santa Cruz Biotechnology); goat anti-Cathepsin-D (Cts-D; sc-6486, Santa Cruz Biotechnology); rabbit anti-EGFR (1005 sc-03, Santa Cruz Biotechnology); Alexa-488 Phalloidin (F-actin, A12379, Thermofisher Scientific); mouse anti-Transferrin Receptor Antibody (H68,4, ThermoFisher Scientific); WGA FITC Conjugate (L 4895, Sigma-Aldrich); mouse anti-Na+/K+-ATPase subunit α1 (C464.6 EMD Millipore); rabbit anti-MPR and rabbit anti-OCRL (gift from A. De Matteis, Telethon Institute of Genetics and Medicine (TIGEM), Pozzuoli, Italy).
Mouse models
Experiments were conducted on age- and gender-matched OcrlY/+;Inpp5b−/− and OcrlY/−;Inpp5b−/− mouse littermates harboring BAC-INPP5B expression in equal copies (BAC1) (129S/SvEv * 129S6/SvEvTac * FVB/N * C57BL/6 background), (18) and Clcn5Y/+and Clcn5Y/− mouse littermates (C57BL/6 background) (48). All the Ocrl mice used in this study were expressing similar levels of INPP5B as analyzed by RT-qPCR (see below). Mice were maintained under temperature-and humidity-controlled conditions with 12 h light/12 h dark cycles with free access to appropriate standard diet in accordance with the institutional guidelines of National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Renal function parameters
Mice were placed overnight in metabolic cages with ad libitum access to food and drinking water. Urines were collected over ice and body weight, water intake and diuresis were measured at the indicated time point (49). Blood (from sublingual vein) was obtained after anesthesia with ketamine/xylazine or isoflurane. Urine and blood parameters were measured using a UniCel DxC 800 pro Synchron (Beckman Coulter, Fullerton, CA, USA), whereas urinary Clara cell protein (CC16) concentration was measured in duplicate by enzyme-linked immunosorbent assay (ELISA; BIOMATIK EKU03200) (44). Albuminuria was measured by Coomassie Blue staining by using ProtoBlue Safe (EC-722, national diagnostics) according to manufacturer instructions.
Kidney isolation and primary cultures of proximal tubule cells
The kidneys were harvested from OcrlY/+ and OcrlY/− mice and Clcn5Y/+and Clcn5Y/− mice as previously described (44). Briefly, one half of the kidney was fixed in 4% PFA and processed for immunostaining while the other half was used for protein isolation or RT-qPCR analysis. The contralateral kidney was taken to generate mPTCs according to a previously established protocol (14). Confluent monolayers of mPTCs, characterized by a high endocytic uptake capacity, were expanded from tubular segments after 6–7 days.
Genotyping
Genomic DNA was isolated from ear biopsies from OcrlY/+;Inpp5b−/−;BAC-INPP5B and OcrlY/−;Inpp5b−/−;BAC-INPP5B mice by using E.Z.N.A Forensic DNA Kit (OMEGA bio-tek) according to the manufacturer instruction. PCR was performed by using the following primers. Ocrl wild-type: the forward primer 5′-CCC TTT TCATCTGTTAGGAGAAATC-3′ is located at the junction of intron 18 and the 5′ end of exon 19; the reverse primer 5′-GCATGG TTAAACGCACTATGTGG-3′ is located in intron 19, which is deleted in the OcrlY/− line. Ocrl knock-out: the forward primer 5′-GCCCTTTGATTCTAATCCCTTTTC ATC-3′ is located in the intron positioned just before the exon 19; the reverse primer 5′-TCT GAGCCCAGAAAGCGAAG-3′ is located in the PGK promoter, which is part of the neo-cassette gene targeting vector. Inpp5b knock-out: the forward primer 5′-TAAAGTCTGAAA ATCCAAGGC-3′ is located in exon 25; the reverse primer 5′-CTCATTTCTCCTTGATTC CAAT-3′ is located in exon 34. BAC-INPP5B: the forward primer 5′-CCACCCCACGATTGACTC-3′ is localized in exon 1; the reverse primer 5′-GGTGTCCCAGCCCTCAG-3′ is localized also in exon 1. PCR conditions were 32 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 1 min.
Quantitative real-time PCR
Total RNA was extracted from mouse kidney tissues using Aurum Total RNA Fatty and Fibrous Tissue Kit according to manufacturer’s protocol (Bio-Rad, Hercules, CA). DNAse I treatment was performed to eliminate genomic DNA contamination. Total RNA was extracted from primary cell cultures with RNAqueous kit (Applied Biosystems, Life Technologies). 1 μg of RNA was used to perform the reverse transcriptase reaction with iScript™ cDNA Synthesis Kit (Bio-Rad). Changes in target gene mRNA levels were determined by relative RT-qPCR with a CFX96™ Real-Time PCR Detection System (BioRad) by using iQ™ SYBR Green Supermix (Bio-Rad). RT-qPCR analyses were performed in duplicate. Specific primers were designed by using Primer3 (Supplementary Material, Table 1). PCR conditions were 95°C for 3 min followed by 40 cycles of 15 s at 95°C, 30 s at 60°C. The PCR products were sequenced with the BigDye terminator kit (Perkin Elmer Applied Biosystems, Thermo Fischer Scientific). The efficiency of each set of primers was determined by dilution curves (Supplementary Material, Table 1). The relative changes in targeted genes over Gapdh, Actb, Hprt1, Ppiase, 18S, 36B4 mRNAs were calculated using the 2−ΔΔCt formula (50).
Immunofluorescence and confocal microscopy
Mouse kidneys were fixed by perfusion with 50–60 ml of 4% PFA in PBS before being snap-frozen in cryogenic Tissue-Tek OCT compound (Electron Microscopy Sciences, Hatfield, USA). The embedded tissues were sectioned at 5 μm and processed for immunofluorescent staining as previously described (44). The slides were acquired on Leica SP5 confocal laser scanning microscope (Center for Microscopy and Image Analysis, University of Zurich) equipped with a Leica APO 63x NA 1.4 oil immersion objective at a definition of 1024 x 1024 pixels, adjusting the pinhole diameter to 1 Airy unit for each emission channel. Quantitative image analysis was performed by selecting random visual fields containing at least 3–5 PTs (LTL-positive) each one and acquired with the same setting parameters. For quantification of β-lactoglobulin and dextran internalization, the integrated density signal detected within each LTL-positive tubule was calculated. Internalization was then expressed as a ratio between the internal fluorescence and tubule area. Tubules at the border of the picture were excluded. ImageJ software was used for the analysis (17).
The mPTCs were fixed in 4% PFA and processed for immunofluorescent staining as previously described (44). Briefly, after incubation with blocking/permeabilization solution (0.1% Saponin,0.5% BSA and 50 mM NH4Cl in PBS), mPTCs were stained overnight with the appropriate primary antibodies and 45 min with the suitable fluorophore-conjugated Alexa secondary antibodies (Invitrogen) or with Alexa-488 Phalloidin (F-actin) or 10 min with WGA FITC Conjugate. Immunostained mPTCs were analyzed by a Leica SP5 confocal laser scanning microscope using the settings described above. The quantitative cell image analyses were performed by using ImageJ software and the open-source cell image analysis software CellProfiler™ (51). More details regarding the quantifications are described in Supplementary Materials.
Endocytosis assays
PTs endocytic capacity of Ocrl and Clcn5 mice was examined by measuring β-lactoglobulin and dextran uptake. β-lactoglobulin was tagged with Cy5 using TM2 Ab labeling kit (Amersham) in accordance with the manufacturer’s instructions. Fifteen minutes after tail-vein injection of Cy5-β-lactoglobulin (1 mg/kg B.W., L3908, Sigma) or 30 min after injection of 10 kDa Alexa 647-dextran (6 mg/kg B.W.; D22914, Thermo Fisher Scientific), mice were anesthetized and their kidneys were harvested and processed for confocal microscopy. The endocytic capacity of Ocrl and Clcn5 mPTCs was examined by measuring albumin and dextran uptake as described previously (44). Briefly mPTCs were incubated at 37°C with 100 μg ml−1 Alexa488-BSA (A13100, Thermo Fisher Scientific) or 250 μg ml−1 Alexa 647-dextran diluted in medium without FBS supplementation, for 15 and 30 min, respectively. After washing, the cells were fixed in 4% PFA and processed for confocal microscopy.
PI(4,5)P2 and PI3P staining
mPTCs were grown on 35 mm dishes and PI(4,5)P2 or PI3P staining were performed according to previously established protocols (25). Briefly, mPTCs were fixed for 15 min in 2% PFA and permeabilized for 5 min with 20 μm digitonin in buffer A (20 mm PIPES pH 6.8, 137 mm NaCl, 2.7 mm KCl) for PI(4,5)P2 or buffer A1 (150 mm NaCl, 20 mm Hepes, pH 7.4, and 2 mm EDTA) for PI3P. Next, mPTCs were incubated for 45 min with buffer A containing 10% fetal bovine serum and anti-PI(4,5)P2 antibody or in buffer A1 supplemented with 5% goat serum and mCherry-2 × FYVE PI3P-binding domain (gift from J.Gallop, Gurdon Institute, University of Cambridge). Anti-RFP was used for amplifying m-cherry staining. Studies of colocalization with early endosomes or lysosomes were performed by 1 h incubation at room temperature with anti-EEA1 or anti-LAMP1 antibodies respectively. After washing and subsequent incubation with appropriate secondary antibodies, mPTCs were post-fixed for 5 min in 2% PFA and analyzed by confocal microscopy.
Lysosomal activity and degradation assays
The detection of lysosomal activity and degradative capacity in mPTCs was performed by using Bodipy-FL-PepstatinA (P12271, Thermo Fischer Scientific) and DQ-RED BSA (D12051, Thermo Fischer Scientific), respectively. Briefly, mPTCs were pulsed with 1 μm Bodipy-FL-Pepstatin A or with 10 μg ml−1 DQ-RED BSA in pre-warmed media at 37°C for 1 h. After washing, cells were fixed, immunostained with anti-LAMP1 and suitable secondary antibody and subsequently analyzed by confocal microscopy (14,26). Lysosomal processing in Ocrl and Clcn5 mice was measured by confocal analysis of kidney PTs after 120 min from tail-vein injection of Cy5-β-lactoglobulin.
Western blotting
Proteins were extracted from mouse kidney tissues or primary cultured cells and lysed as previously described (44). Samples were normalized for protein or urinary creatinine levels before loading (20 μg/lane or 4 μg/lane, respectively), diluted in Laemmli buffer and separated by SDS-PAGE in reducing conditions. Briefly, gels were blotted onto PVDF membranes, blocked in 5% non-fat milk and probed with appropriate primary and peroxidase-labeled secondary antibody. Protein bands were visualized by chemiluminescence (WBKLS0050, Millipore, Life technologies). Image intensity was measured by ImageJ.
Plasma membrane isolation
mPTCs were lysed in a fractionation buffer (250 mm sucrose, 20 mm HEPES, 10 mm KCL, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA) containing protease and phosphatase inhibitors and passed through a 25G needle. Cell lysates were kept on ice for 20 min and centrifuged at 720g (5 min at 4°C). The resultant postnuclear supernatants were centrifuged at 10 000g (5 min at 4°C) to eliminate mitochondria. The postmitochondria supernatant were centrifuged at 100 000g (1 h at 4°C) to separate cytosolic (supernatant) and membrane fractions (pellet). The membrane pellets were washed in fractionation buffer by centrifugation (100 000g for 45 min at 4°C) and suspended in lysis buffer (10% glycerol and 0.1% SDS supplemented with protease and phosphatase inhibitors). The cytosolic and membrane fractions were analyzed by SDS-PAGE and western blotting.
Behavioral tests
Open-field large-arena test, water maze cue navigation and three chambers sociability test were performed on a cohort of 32 male mice aged 32 weeks (16 OcrlY/+;Inpp5b−/−;BAC-INPP5B and 16 OcrlY/−;Inpp5b−/−;BAC-INPP5B) in order to investigate motor coordination, vision and spatial learning and the social attitude of the mice respectively. Behavior testing procedures will be described briefly here and in more detail in Supplementary Materials.
Open-field large-arena test
Each subject was released near the wall of the arena and observed for 10 min on 2 subsequent days. Movements were tracked using Noldus EthoVision.
Water-maze cue navigation
Animals were trained for 2 days to reach the goal platform hidden under milky water and marked by a salient cue. The localization of the platform in the pool changed for every trial. All subjects were trained on the same sequence of goal positions and released from the same start points (52).
Three-chamber sociability test
The test consisted of a 10-min trial where a test mouse was given the choice to spend time in the chamber with the novel object (the cylinder) plus the unfamiliar mouse (social stimulus) and the chamber containing only the empty cylinder. The time spent by the test mouse in the lateral chambers was noted. (53).
IntelliCages experiments
Intellicages experiments were performed on a cohort of 28 male mice aged 24 weeks (13 OcrlY/+;Inpp5b−/−;BAC-INPP5B and 15 OcrlY/−;Inpp5b−/−;BAC-INPP5B). More details regarding the description of IntelliCage apparatus and mice preparation for IntelliCage experiments are provided in Supplementary Materials.
Exploration and free adaptation
During the first 4–7 days in IntelliCage all doors were open providing free access to all 8 drinking bottles (free adaptation). Data of the first 24 h of free adaptation were analyzed separately to monitor exploration and habituation of the mice in the new environment (IntelliCage exploration).
Nose-poke adaptation
During 3–7 days, all doors were closed but can be opened at any time with a nose poke for 5 s once per visit.
Drinking session adaptation
During 3–7 days the mice were adapted to a fixed drinking schedule with doors opening in response to nose pokes only in the following time points: 04:00–05:00, 11:00–12:00, 16:00–17:00 and 23:00–00:00 (30).
Epon embedding and retinal morphology
Thirty-six-week-old mice were euthanized and perfused with PFA 4%. Eyes were enucleated and post-fixed in 4% PFA or 2.5% glutaraldehyde before being embedded in paraffin or epon plastic, respectively. For light microscopy, paraffin sections (including the lens, 5 μm) were stained with hematoxylin–eosin, while plastic-embedded semi-thin cross-sections (0.5 μm) were counterstained with toluidine blue as described previously (54,55). Retinal morphology was analyzed by light microscopy (Zeiss, Axioplan, Jena, Germany).
Muscle analyses
Muscles analyses were performed on OcrlY/+ versus OcrlY/− male mice aged 24 weeks. Lean mass/body weight ratio was determined in six pairs of live mice by using the whole body composition analyzer EchoMRI™ (Zinsser Analytic, Frankfurt, Germany) (56). Five pairs of mice were used for muscle and blood sampling. The mice were slightly sedated using isoflurane before being anesthetized with an i.p. injection of a mixture containing urethane (1.5 g/kg) and diazepam (5 mg/kg) and killed by exsanguination. Blood and muscle tissues were collected as previously described (57). Selected leg muscles (triceps surae, soleus, plantaris, gastrocnemius, tibialis anterior, tibialis posterior, quadriceps) were carefully dissected bilaterally and weighed. Muscle mass was expressed relative to body weight. CK activity in plasma was determined by spectrophotometry using a kit (C184-0B; Catachem, Nuuchem, Oxford, CT, USA) according to the manufacturer’s instructions (57).
Data analysis and Statistics
The quantitative data were expressed as means ± standard error of the mean (s.e.m.). Differences between experimental groups were evaluated using analysis of variance followed by post hoc test, when appropriate. When only two groups were compared, unpaired or paired two tailed Student’s t-tests were used as appropriate. No statistical methods were used to predetermine the sample size. The sample size (n of biological replicates derived from distinct mice) of each experimental group is described in figure legends. GraphPad Prism software was used for all statistical analyses. Statistical significance was set at a P < 0.05.
Supplementary Material
Acknowledgements
We acknowledge Jennifer Gallop, Jonathan R. Gadsby, Andrew Hall and Eric Olinger for fruitful discussions, Marcello Polesel, Claus-Dieter Schuh, Huguette Debaix and Nadine Nagele for providing technical assistance, Jonathan R. Gadsby, Renata Kozyraki, Pierre Verroust and Carsten A. Wagner for providing reagents. We thank the Center for Microscopy and Image Analysis of the University of Zurich (Zurich, Switzerland) for providing the equipment for imaging acquisition and the support for imaging analysis.
Conflict of Interest statement. None declared.
Funding
Cystinosis Research Foundation (Irvine, CA, USA); the Swiss National Science Foundation (project grant 31003A-169850); the clinical research priority program RADIZ (Rare Disease Initiative Zurich) of the UZH; the Swiss National Centre of Competence in Research Kidney Control of Homeostasis (Kidney.CH); the Fondation Suisse de Recherche sur les Maladies Musculaires.
References
- 1. Eckardt K.U., Coresh J., Devuyst O., Johnson R.J., Kottgen A., Levey A.S. and Levin A. (2013) Evolving importance of kidney disease: from subspecialty to global health burden. Lancet, 382, 158–169. [DOI] [PubMed] [Google Scholar]
- 2. Devuyst O. and Luciani A. (2015) Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J. Physiol., 593, 4151–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Christensen E.I., Verroust P.J. and Nielsen R. (2009) Receptor-mediated endocytosis in renal proximal tubule. Pflugers Arch., 458, 1039–1048. [DOI] [PubMed] [Google Scholar]
- 4. Devuyst O. and Thakker R.V. (2010) Dent's disease. Orphanet J. Rare Dis., 5, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Devuyst O., Knoers N.V., Remuzzi G. and Schaefer F. (2014) Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet, 383, 1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Devuyst O., Christie P.T., Courtoy P.J., Beauwens R. and Thakker R.V. (1999) Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease. Hum. Mol. Genet., 8, 247–257. [DOI] [PubMed] [Google Scholar]
- 7. De Matteis M.A., Staiano L., Emma F. and Devuyst O. (2017) The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat. Rev. Nephrol., 13, 455–470. [DOI] [PubMed] [Google Scholar]
- 8. Mao Y., Balkin D.M., Zoncu R., Erdmann K.S., Tomasini L., Hu F., Jin M.M., Hodsdon M.E. and Camilli P. (2009) A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J., 28, 1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ponting C.P. (2006) A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics, 22, 1031–1035. [DOI] [PubMed] [Google Scholar]
- 10. Faucherre A., Desbois P., Satre V., Lunardi J., Dorseuil O. and Gacon G. (2003) Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. Hum. Mol. Genet., 12, 2449–2456. [DOI] [PubMed] [Google Scholar]
- 11. Hichri H., Rendu J., Monnier N., Coutton C., Dorseuil O., Poussou R.V., Baujat G., Blanchard A., Nobili F., Ranchin B. et al. (2011) From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. mutat., 32, 379–388. [DOI] [PubMed] [Google Scholar]
- 12. Christensen E.I., Devuyst O., Dom G., Nielsen R., Van der Smissen P., Verroust P., Leruth M., Guggino W.B. and Courtoy P.J. (2003) Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc. Natl. Acad. Sci. U. S. A., 100, 8472–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vicinanza M., Di Campli A., Polishchuk E., Santoro M., Di Tullio G., Godi A., Levtchenko E., De Leo M.G., Polishchuk R., Sandoval L. et al. (2011) OCRL controls trafficking through early endosomes via PtdIns4,5P(2)-dependent regulation of endosomal actin. EMBO J., 30, 4970–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luciani A., Sirac C., Terryn S., Javaugue V., Prange J.A., Bender S., Bonaud A., Cogne M., Aucouturier P., Ronco P. et al. (2016) Impaired lysosomal function underlies monoclonal light chain-associated renal fanconi syndrome. J. Am. Soc. Nephrol., 27, 2049–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janne P.A., Suchy S.F., Bernard D., MacDonald M., Crawley J., Grinberg A., Wynshaw-Boris A., Westphal H. and Nussbaum R.L. (1998) Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J. Clin. Invest., 101, 2042–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Norris F.A., Atkins R.C. and Majerus P.W. (1997) The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. Evidence for conserved alternative splicing in the 4-phosphatase family. J. Biol. Chem., 272, 23859–23864. [DOI] [PubMed] [Google Scholar]
- 17. Inoue K., Balkin D.M., Liu L., Nandez R., Wu Y., Tian X., Wang T., Nussbaum R., De Camilli P. and Ishibe S. (2017) Kidney tubular ablation of Ocrl/Inpp5b phenocopies Lowe syndrome tubulopathy. J. Am. Soc. Nephrol., 28, 1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bothwell S.P., Chan E., Bernardini I.M., Kuo Y.M., Gahl W.A. and Nussbaum R.L. (2011) Mouse model for Lowe syndrome/Dent disease 2 renal tubulopathy. J. Am. Soc. Nephrol., 22, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Terryn S., Jouret F., Vandenabeele F., Smolders I., Moreels M., Devuyst O., Steels P. and Van Kerkhove E. (2007) A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am. J. Physiol. Renal. Physiol., 293, F476–F485. [DOI] [PubMed] [Google Scholar]
- 20. Zhang X., Hartz P.A., Philip E., Racusen L.C. and Majerus P.W. (1998) Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem., 273, 1574–1582. [DOI] [PubMed] [Google Scholar]
- 21. Oltrabella F., Pietka G., Ramirez I.B., Mironov A., Starborg T., Drummond I.A., Hinchliffe K.A. and Lowe M. (2015) The Lowe syndrome protein OCRL1 is required for endocytosis in the zebrafish pronephric tubule. PLoS Genet., 11, e10050581–0.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Senju Y., Kalimeri M., Koskela E.V., Somerharju P., Zhao H., Vattulainen I. and Lappalainen P. (2017) Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc. Natl. Acad. Sci. U. S. A., 114, E8977–e8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perez Bay A.E., Schreiner R., Benedicto I., Paz Marzolo M., Banfelder J., Weinstein A.M. and Rodriguez-Boulan E.J. (2016) The fast-recycling receptor Megalin defines the apical recycling pathway of epithelial cells. Nat. Commun., 7, 11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pfeffer S.R. (2009) Multiple routes of protein transport from endosomes to the trans Golgi network. FEBS Lett., 583, 3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Leo M.G., Staiano L., Vicinanza M., Luciani A., Carissimo A., Mutarelli M., Di Campli A., Polishchuk E., Di Tullio G., Morra V. et al. (2016) Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell. Biol., 18, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perera R.M., Stoykova S., Nicolay B.N., Ross K.N., Fitamant J., Boukhali M., Lengrand J., Deshpande V., Selig M.K., Ferrone C.R. et al. (2015) Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature, 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miaczynska M. (2013) Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol., 5, a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taylor T.N., Greene J.G. and Miller G.W. (2010) Behavioral phenotyping of mouse models of Parkinson's disease. Behav. Brain. Res., 211, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fowler S.C. and Muma N.A. (2015) Use of a force-sensing automated open field apparatus in a longitudinal study of multiple behavioral deficits in CAG140 Huntington's disease model mice. Behav. Brain. Res., 294, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vannoni E., Voikar V., Colacicco G., Sanchez M.A., Lipp H.P. and Wolfer D.P. (2014) Spontaneous behavior in the social homecage discriminates strains, lesions and mutations in mice. J. Neurosci. Methods., 234, 26–37. [DOI] [PubMed] [Google Scholar]
- 31. Luhmann U.F., Carvalho L.S., Holthaus S.M., Cowing J.A., Greenaway S., Chu C.J., Herrmann P., Smith A.J., Munro P.M., Potter P. et al. (2015) The severity of retinal pathology in homozygous Crb1rd8/rd8 mice is dependent on additional genetic factors. Hum. Mol. Genet., 24, 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Montjean R., Aoidi R., Desbois P., Rucci J., Trichet M., Salomon R., Rendu J., Faure J., Lunardi J., Gacon G. et al. (2015) OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum. Mol. Genet., 24, 994–1006. [DOI] [PubMed] [Google Scholar]
- 33. Nandez R., Balkin D.M., Messa M., Liang L., Paradise S., Czapla H., Hein M.Y., Duncan J.S., Mann M. and Camilli P. (2014) A role of OCRL in clathrin-coated pit dynamics and uncoating revealed by studies of Lowe syndrome cells. eLife, 3, e02975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bockenhauer D., Bokenkamp A., van't Hoff W., Levtchenko E., Kist-van Holthe J.E., Tasic V. and Ludwig M. (2008) Renal phenotype in Lowe syndrome: a selective proximal tubular dysfunction. Clin. J. Am. Soc. Nephrol., 3, 1430–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Piwon N., Gunther W., Schwake M., Bosl M.R. and Jentsch T.J. (2000) ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature, 408, 369–373. [DOI] [PubMed] [Google Scholar]
- 36. Gorvin C.M., Wilmer M.J., Piret S.E., Harding B., Heuvel L.P., Wrong O., Jat P.S., Lippiat J.D., Levtchenko E.N. and Thakker R.V. (2013) Receptor-mediated endocytosis and endosomal acidification is impaired in proximal tubule epithelial cells of Dent disease patients. Proc. Natl. Acad. Sci. U. S. A., 110, 7014–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Novarino G., Weinert S., Rickheit G. and Jentsch T.J. (2010) Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science, 328, 1398–1401. [DOI] [PubMed] [Google Scholar]
- 38. Biemesderfer D. (2006) Regulated intramembrane proteolysis of megalin: linking urinary protein and gene regulation in proximal tubule? Kidney. Int., 69, 1717–1721. [DOI] [PubMed] [Google Scholar]
- 39. Zou Z., Chung B., Nguyen T., Mentone S., Thomson B. and Biemesderfer D. (2004) Linking receptor-mediated endocytosis and cell signaling: evidence for regulated intramembrane proteolysis of megalin in proximal tubule. J. Biol. Chem., 279, 34302–34310. [DOI] [PubMed] [Google Scholar]
- 40. Norden A.G., Lapsley M., Igarashi T., Kelleher C.L., Lee P.J., Matsuyama T., Scheinman S.J., Shiraga H., Sundin D.P., Thakker R.V. et al. (2002) Urinary megalin deficiency implicates abnormal tubular endocytic function in Fanconi syndrome. J. Am. Soc. Nephrol., 13, 125–133. [DOI] [PubMed] [Google Scholar]
- 41. Zoncu R., Perera R.M., Balkin D.M., Pirruccello M., Toomre D. and De Camilli P. (2009) A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell, 136, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Di Paolo G. and De Camilli P. (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature, 443, 651–657. [DOI] [PubMed] [Google Scholar]
- 43. Symons M.H. and Mitchison T.J. (1991) Control of actin polymerization in live and permeabilized fibroblasts. J. Cell. Biol., 114, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Festa B.P., Chen Z., Berquez M., Debaix H., Tokonami N., Prange J.A., Hoek G.V., Alessio C., Raimondi A., Nevo N. et al. (2018) Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun., 9, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park E., Choi H.J., Lee J.M., Ahn Y.H., Kang H.G., Choi Y.M., Park S.J., Cho H.Y., Park Y.H., Lee S.J. et al. (2014) Muscle involvement in Dent disease 2. Pediatr. Nephrol., 29, 2127–2132. [DOI] [PubMed] [Google Scholar]
- 46. Erdmann K.S., Mao Y., McCrea H.J., Zoncu R., Lee S., Paradise S., Modregger J., Biemesderfer D., Toomre D. and De Camilli P. (2007) A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev. Cell., 13, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bothwell S.P., Farber L.W., Hoagland A. and Nussbaum R.L. (2010) Species-specific difference in expression and splice-site choice in Inpp5b, an inositol polyphosphate 5-phosphatase paralogous to the enzyme deficient in Lowe syndrome. Mamm. Genome., 21, 458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang S.S., Devuyst O., Courtoy P.J., Wang X.T., Wang H., Wang Y., Thakker R.V., Guggino S. and Guggino W.B. (2000) Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum. Mol. Genet., 9, 2937–2945. [DOI] [PubMed] [Google Scholar]
- 49. Raggi C., Luciani A., Nevo N., Antignac C., Terryn S. and Devuyst O. (2014) Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum. Mol. Genet., 23, 2266–2278. [DOI] [PubMed] [Google Scholar]
- 50. Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A. and Speleman F. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol., 3, Research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J. et al. (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol., 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mohajeri M.H., Madani R., Saini K., Lipp H.P., Nitsch R.M. and Wolfer D.P. (2004) The impact of genetic background on neurodegeneration and behavior in seizured mice. Genes Brain Behav., 3, 228–239. [DOI] [PubMed] [Google Scholar]
- 53. Nadler J.J., Moy S.S., Dold G., Trang D., Simmons N., Perez A., Young N.B., Barbaro R.P., Piven J., Magnuson T.R. et al. (2004) Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav., 3, 303–314. [DOI] [PubMed] [Google Scholar]
- 54. Grimm C., Wenzel A., Williams T., Rol P., Hafezi F. and Reme C. (2001) Rhodopsin-mediated blue-light damage to the rat retina: effect of photoreversal of bleaching. Invest. Ophthalmol. Vis. Sci., 42, 497–505. [PubMed] [Google Scholar]
- 55. Heynen S.R., Tanimoto N., Joly S., Seeliger M.W., Samardzija M. and Grimm C. (2011) Retinal degeneration modulates intracellular localization of CDC42 in photoreceptors. Mol. Vis., 17, 2934–2946. [PMC free article] [PubMed] [Google Scholar]
- 56. Krizo J.A., Moreland L.E., Rastogi A., Mou X., Prosser R.A. and Mintz E.M. (2018) Regulation of Locomotor activity in fed, fasted, and food-restricted mice lacking tissue-type plasminogen activator. BMC Physiol., 18, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dorchies O.M., Reutenauer-Patte J., Dahmane E., Ismail H.M., Petermann O., Patthey-Vuadens O., Comyn S.A., Gayi E., Piacenza T., Handa R.J. et al. (2013) The anticancer drug tamoxifen counteracts the pathology in a mouse model of duchenne muscular dystrophy. Am. J. Pathol., 182, 485–504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.