

Abstract
An early hallmark of Alzheimer’s disease is the accumulation of amyloid-β (Aβ), inspiring numerous therapeutic strategies targeting this peptide. An alternative approach is to destabilize the amyloid beta precursor protein (APP) from which Aβ is derived. We interrogated innate pathways governing APP stability using a siRNA screen for modifiers whose own reduction diminished APP in human cell lines and transgenic Drosophila. As proof of principle, we validated PKCβ—a known modifier identified by the screen—in an APP transgenic mouse model. PKCβ was genetically targeted using a novel adeno-associated virus shuttle vector to deliver microRNA-adapted shRNA via intracranial injection. In vivo reduction of PKCβ initially diminished APP and delayed plaque formation. Despite persistent PKCβ suppression, the effect on APP and amyloid diminished over time. Our study advances this approach for mining druggable modifiers of disease-associated proteins, while cautioning that prolonged in vivo validation may be needed to reveal emergent limitations on efficacy.
Introduction
Several major neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease and amyotrophic lateral sclerosis, are characterized by insoluble aggregates of normal cellular proteins. Where these aggregates are considered pathogenic, the most precise approach to intervention is to directly target the specific protein fragment accumulating in each disease. This approach can be challenging when the protein fragment acquires post-translational modifications that change over time or that have not been fully characterized. Both of these situations occur in AD, where the amyloid β peptide (Aβ) forms oligomeric structures that have not been structurally defined (1) and where deposited peptide can become truncated, phosphorylated and pyroglutaminated (2). In this setting, a complementary strategy is to reduce Aβ production before it can accumulate. Both β and γ secretases required to release Aβ from its precursor protein have been targeted pharmacologically, but clinical development has been hampered by unfavorable risk/benefit profiles (3).
Given these limitations, an alternative approach to intervention might target the full-length protein from which Aβ is derived. This strategy is particularly attractive for the amyloid β precursor protein (APP) because lifelong haploinsufficiency imparts no identifiable phenotype (4–6). Conversely, APP duplication causes early-onset AD, suggesting a relationship between APP levels and disease onset (7). In principle, lowering the synthesis or stability of APP should reduce production of Aβ peptide. Rather than screen libraries of chemical compounds to identify drug candidates influencing APP stability, we instead used a genetic screen to interrogate the innate cellular pathways controlling steady-state APP levels reasoning that these pathways might provide openings for pharmacologic intervention. We capitalized on the ease of siRNAs targeting to screen the druggable genome for APP modifiers, beginning with approximately 600 genes of the kinome (8,9). Our approach was based on the rationale that enzymes are easier to pharmacologically inhibit than to activate, and we therefore sought kinases whose own reduction via short interfering RNA (siRNA) lowered the steady-state level of APP. We initiated parallel genetic screens in both human cell lines and in transgenic Drosophila to provide cross-species validation of candidate modifiers (8–11).
Our screen identified multiple kinases capable of regulating full-length APP in these model systems, and we chose to advance one well-validated modifier, protein kinase C β (PRKCB, PKCβ), for proof of concept in a mouse model of Alzheimer’s amyloidosis. Translating our findings from the genetic screen into a preclinical model was hampered by the poor specificity of existing PKCβ inhibitors (12). To overcome this obstacle, we again took advantage of a genetic strategy to selectively target PKCβ in the mouse brain and here describe a novel adeno-associated virus (AAV) shuttle vector to deliver shRNA against PRKCB within a non-toxic micro-RNA backbone. Using this strategy, we demonstrate that neuronal reduction of PKCβ lowers steady-state levels of APP, decreases Aβ concentration and delays amyloid formation in the mouse brain, but does so only transiently. Taken together, our work outlines an approach for using the cell’s innate machinery to identify therapeutic opportunities for protein aggregation disorders and provides a modular viral vector for validating candidate drug targets in preclinical models of disease.
Results
Parallel cross-species genetic screens to identify evolutionarily conserved modifiers of APP stability
The first part of our screen to identify kinases controlling APP levels used a human medulloblastoma-derived Daoy cell line stably transfected with a bicistronic plasmid encoding wild-type APP695 fused to enhanced green fluorescent protein (eGFP) followed by IRES-DsRed (Fig. 1). The fluorescence signal of eGFP provided an indicator of APP levels, while the independently expressed DsRed signal provided a control for changes affecting global transcription or translation. The APP-expressing Daoy cell line was split into 96-well plates and each well transiently transfected with individual siRNAs from the Invitrogen kinase RNAi library targeting all 636 known human kinase and kinase-like genes (8). Cells with a selective change in APP stability were identified by fluorescence-activated cell sorting (FACS) based on the ratio of eGFP to DsRed fluorescence. This screen revealed a number of kinase targets that decreased the eGFP/DsRed ratio more than 1.5 standard deviations from the screen-wide mean (Fig. 2A and B). Candidate modifiers were then cross-examined in an independent Daoy cell line expressing DsRed-IRES-eGFP without APP to eliminate false positives. In total we identified 31 kinases that specifically decreased the ratio of APP-eGFP relative to DsRed (Table 1).
Figure 1.
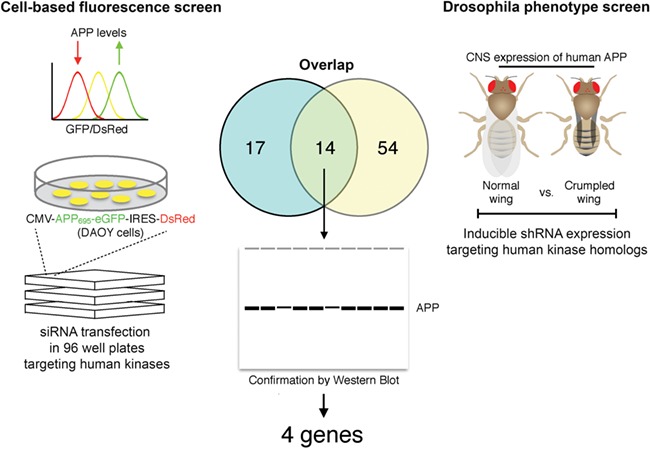
Schematic diagram of the cross-species kinome RNAi screen. Kinases capable of modifying APP stability were identified by fluorescence sorting in Daoy cells stably transfected with APP-eGFP-IRES-DsRed (left). A parallel screen was conducted in APP transgenic Drosophila using the crumpled wing phenotype as readout for neuronal APP levels (right). Candidate modifiers identified from each screen were compared to identify 14 shared genes (center). These 14 shared modifiers were tested individually in each system by Western blot to define 4 confirmed kinase modifiers of APP (center).
Figure 2.
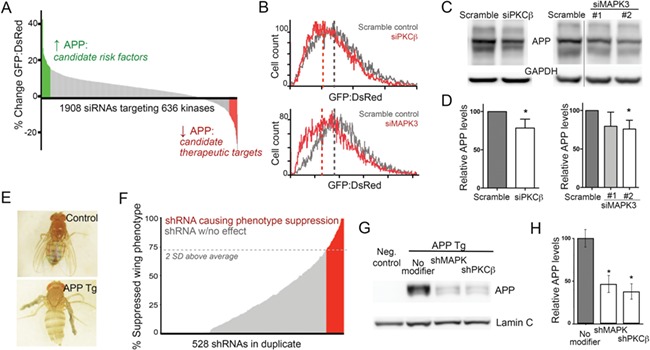
Integrative genetic screen identifies PKCβ as a modifier of APP stability. (A) The primary screen for APP modifiers was performed in Daoy cells stably transfected with a bicistronic plasmid to express APP-GFP and DsRed proteins. Graph shows the resulting change in green:red fluorescence ratio for each siRNA tested, arranged in rank order. Outcomes at the upper 10% of total are highlighted in green for modifiers increasing APP stability; the lower 10% are highlighted in red for those decreasing APP stability. (B) Histogram of APP-GFP:DsRed ratio in cells treated with siRNA (red) or scramble control (gray) against PKCβ (upper panel) or MAPK3 (lower panel). Note the leftward shift caused by siRNA repression of both PKCβ and MAPK3. (C) Western blot of wild-type Daoy cells confirmed the reduction of endogenous APP following siRNA knockdown of PKCβ (left) and MAPK3 (right). (D) Quantification of Daoy cell APP levels detected by Western blot following PKCβ (left) or MAPK3 (right) siRNA knockdown. APP levels were assessed relative to within-lane GAPDH signal. Values are normalized relative to the scramble control. (E) The crumpled wing phenotype observed in transgenic Drosophila overexpressing human wild-type APP was used as a surrogate readout for APP levels upon shRNA targeting of individual kinases. (F) The Drosophila screen was scored by measuring the percentage of total offspring from each shRNA line that displayed normal wing phenotype at eclosion. Red lines represent shRNAs considered to be positive modifiers that increased the percentage of wing extension by >2 SD above the control average. Gray lines represent shRNA considered to have no effect on the phenotype, changing the population outcome less than 2 SD from the control average. (G) Western blot for human APP in isolated fly heads was used to confirm the phenotypic screen for MAPK3 and PKCβ. (H) Quantification of Drosophila APP levels detected by Western blot following MAPK3 (left) or PKCβ (right) or shRNA knockdown. APP levels were assessed relative to within-lane lamin C signal. Values are normalized relative to the no modifier control group.
Table 1.
Genes found to selectively reduce the ratio of APP-eGFP:DsRed in Daoy cell lines
| APP modifiers identified in transfected Daoy cells | ||
|---|---|---|
| ACVRL1 | EIF2AK4 | PKN2 |
| ADK | GSK3B | PRKCB |
| BRSK1 | HK2 | PRKCQ |
| CDC42BPB | LATS1 | PSKH1 |
| CIT | MAP3K4 | RPS6KB1 |
| CRKL | MAPK3 | RPS6KC1 |
| CSF1R | MAPK14 | TAOK3 |
| CSNK1D | NEK3 | TRIM28 |
| CSNK2A2 | NPR2 | YES1 |
| DYRK1A | PDPK1 | |
| EGFR | PFKFB4 | |
In parallel to the Daoy cell screen, we conducted an RNAi screen in transgenic Drosophila expressing wild-type human APP695 under control of the pan-neuronal driver elav-GAL4. Rather than using fluorescence to measure APP levels, we took advantage of the wing phenotype caused by neuronal APP overexpression to screen for modifiers that lowered APP levels. Fruit flies extend their wings shortly after eclosion by pumping fluid into the veins. Transgenic APP expression in Drosophila neurons impairs wing extension and results in adult flies with crumpled wings (Fig. 2E). This phenotype can be easily quantified and used as a proxy for APP levels (Fig. 2E). The transgenic APP695 line was crossed with 1056 individual shRNA lines targeting the Drosophila homologs of 606 human kinases. Offspring expressing both APP and each shRNA were examined for the percentage of animals with crumpled wings. From this screen we identified 68 Drosophila kinases that lowered the APP-induced wing phenotype by more than 2 standard deviations from the screen average (Fig. 2F; Table 2). These 68 Drosophila genes corresponded to 153 human homologs (Table 2).
Table 2.
Genes found to reduce penetrance of the APP-induced wing phenotype in Drosophila and their human homologs
| APP modifiers identified in transgenic Drosophila and their human homologs | |||||||
|---|---|---|---|---|---|---|---|
| Drosophila gene | Human homolog | Drosophila gene | Human homolog | Drosophila gene | Human homolog | Drosophila gene | Human homolog |
| Ack-like | ACK1 | CG3008 | RIOK3 | InR | IGF1R, INSR, INSRR | Pi3K92E | PIK3CA, PIK3CB, PIK3CD, PIK3CG |
| Akt1 | AKT1, AKT2, AKT3, SGK494 | CG32944 | STK32A, STK32B, STK32C | IP3K2 | ITPKA, ITPKB, ITPKC | Pitslre | CDC2L2, CDK10 |
| Alk | ALK | CG33156 | NADK | KP78a | MARK1, MARK2, MARK3, MARK4 | plum | AGER |
| bon | TRIM28 | CG34357 | GUCY2D, GUYCY2F | l(2)k01209 | UCKL1 | Pvr | FLT4, KDR |
| Btk29A | BMX, BTK, ITK, LMTK2, RIPK1, TEC, TEX14, TXK | CG3544 | XYLB | Lerp | IGR2R | rdgA | DGKI, DGKZ |
| CASK | CASK, MPP1, MPP2 | CG4839 | PRKG1, PRKG2 | LIMK1 | LIMK1, LIMK2 | RIOK2 | RIOK2 |
| Cdc7 | CDC7 | CG6800 | CDK20 | Madm | MRBP1, NRBP2 | rl | MAPK3 |
| Cdk5alpha | CDK5R1 | CG7335 | KHK | mri | BTBD10 | Rok | ROCK1, ROCK2 |
| Cdk8 | CDK8 | CG9886 | GLYCTK | Mvd | MVD | S6k | RPS6KB1 |
| Cdk9 | CDK9 | dlg1 | DLG1, DLG2 DLG3, DLG4 | niki | NEK1, NEK3, NEK4, NEK5, NEK7, NEK8, NEK9, NEK11 | sax | ACVRL1 |
| CG10268 | PMVK | Drak | STK17A, STK17B | nmdyn-D7 | NME7 | sdt | MMP1, MMP2, MMP3, MMP4, MMP5, MMP6, MMP7 |
| CG10738 | NPR2 | Egfr | EGFR | Nrk | AATK, MUSK, PTK7 | Sik2 | SIK1, SIK2, SIK3, PIM1 |
| CG11255 | ADK | Eip63E | CDK14, CDK15, CDK16, CDK17, CDK18 | Pak | PAK1, PAK2, PAK3 | Slik | SLK, PDIK1L, STK10 |
| CG13369 | RBKS | fh | FXN | Pask | PASK, PIM3 | sqa | DAPK1, DAPK3 |
| CG14721 | TPK1 | gskt | GSK3B | Pdk | PDK1, PDK2, PDK3, PDK4, BCKDK | Tao | TAOK1, TAOK2, TAOK3 |
| CG15547 | NME5 | Hex-A | GCK, HK1, HK2, HK3 | Pdk1 | PDPK1 | Tlk | TLK1 |
| CG2577 | CSNK1A1, CSNK1A1L, CSNK1D, CSNK1E | inaC | PRKCA, PRKCB, PRKCG | Pi3K68D | PIK3C2A, PIK3C2B, PIK3C2G | trc | STK38, STK38L |
We next compared the candidate modifiers identified from the cell-based and Drosophila screens to derive 14 kinases that were shared between them (Table 3). Because our initial screening strategies used indirect measures of APP expression, our secondary screen of these 14 candidates directly measured APP levels by Western blot. We targeted each of these kinases in unmodified Daoy cells and in APP transgenic Drosophila using an independent set of si/shRNAs and quantified the expression of human full-length APP in cell extracts and Drosophila tissue (Fig. 2C, D, G and H). From the shared set of 14 candidate modifiers, 4 genes were confirmed to effectively decrease steady-state APP protein levels when targeted in both Drosophila and human neuroblastoma cells (ADK, PDPK1, MAPK3 and PRKCB).
Table 3.
Candidate APP modifiers common to both Daoy and Drosophila screens
| Shared modifiers of APP stability in Daoy cells and Drosophila | ||
|---|---|---|
| ACVRL1 | HK2 | PRKCB |
| ADK | MAPK3 | RPS6KB1 |
| CSNK1D | NEK3 | TAOK3 |
| EGFR | NPR2 | TRIM28 |
| GSK3B | PDPK1 | |
Genes confirmed by APP Western blot in both systems are indicated in boldface.
Genetic reduction of PKCβ but not MAPK3 lowers transgenic APP levels in a mouse model of AD
We advanced two of our confirmed APP modifiers for testing in the mammalian brain. Mouse cell lines were used to identify the two most effective shRNA sequences against MAPK3 and PRCKB for in vivo testing. We built an AAV shuttle vector to deliver each gene-specific shRNA into the mouse brain along with a fluorescent protein that would label transduced cells (Fig. 3A). Because viral delivery of naked shRNA can be neurotoxic in vivo (13–15), we took advantage of a commercialized strategy to embed the shRNA within artificial human micro RNA 30 (miR30) arms (Dharmacon, GIPZ collection) (16,17). We expressed the miR-adapted shRNA under control of the U6 polymerase III promoter followed by the fluorescent protein tdTomato under control of an independent CAG polymerase II promoter. The resulting AAV vector allows for modular exchange of one shRNA for another using traditional restriction-digest cloning from the Dharmacon GIPZ shRNA plasmid library. Packaging in AAV has the advantage of allowing widespread transduction of the mouse brain by viral injection of neonatal pups (Fig. 3B) (18). AAV8 in particular biases for neuronal transduction and can result in viral expression lasting a year or more (19,20). We cloned two independent shRNAs against either murine MAPK3 or PRKCB, as well as a non-silencing control sequence, into our AAV shuttle vector, packaged each into AAV8 and injected each into the brains of neonatal mice overexpressing a pathogenic variant of APP. Animals were harvested 4 weeks after injection to measure the efficiency of kinase knock-down in the brain and the effect on APP protein levels.
Figure 3.
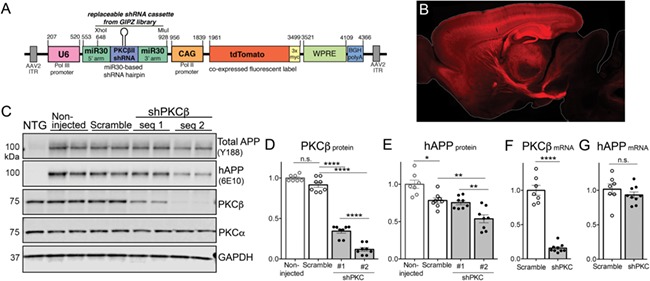
AAV-mediated PKCβ knock-down in APP transgenic mouse brain. (A) Diagram of the pAAV vector used to deliver microRNA-adapted shRNA targeting PKCβ (or MAPK3) and tdTomato fluorescent label. The microRNA was placed under control of the U6 RNA polymerase III promoter, while tdTomato was controlled by the CAG RNA polymerase II promoter. Note that unique MluI and XhoI restriction sites within the microRNA cassette allow for exchange of PKCβ shRNA with other miR30-embedded shRNA sequences from the pGIPZ library. (B) Sagittal brain section from a mouse harvested 1 month after stereotaxic P0 injection with the AAV construct illustrated in (A). tdTomato fluorescence serves as a marker of viral transduction prominent throughout the forebrain. (C) Western blot showing the relative expression of total APP (endogenous + transgenic; detected with antibody Y188), transgenic APP (detected with 6E10; hAPP), PKCβII, PKCα and GAPDH in cortical homogenates from mice harvested 1 month after P0 AAV injection. (D) Quantification of PKCβII protein in cortex of APP/TTA mice harvested 1 month after P0 injection with AAV-shPKCβ or scramble shRNA relative to expression in non-injected APP/TTA control mice. 1-way ANOVA, F(3, 13) = 301.5, P < 0.0001 (E) Quantification of transgenic APP (hAPP) protein in cortical homogenates of mice virally injected at P0. 1-way ANOVA, F(3, 13) = 18.15, P < 0.0001. (F) Quantitation of PKCβ mRNA in cortical extracts of mice injected at P0 with either scramble or shPKCβ #2 AAV. Student’s t-test, P < 0.0001. (G) Quantitation of transgenic APP mRNA in cortical extracts of mice injected at P0 with either scramble or shPKCβ #2 AAV. Student’s t-test, P = 0.33.
MicroRNA-adapted shRNA targeting reduced the expression of both kinases in vivo. PKCβ levels decreased by 62.4 ± 2.8% and 87.1 ± 2.6% (shRNA#1 and #2, respectively) compared to the expression in animals transduced by non-silencing shRNA control virus (Fig. 3C and D). MAPK3 levels decreased by 40.0 ± 2.2% and 58.2 ± 10.4% (shRNA#1 and #2, respectively) compared to expression in animals transduced by non-silencing shRNA control virus (data not shown). Despite effective reduction of each kinase with at least one shRNA, only knockdown of PKCβ impacted APP protein levels in vivo. Mice treated with MAPK3 shRNA showed no significant change in transgenic APP expression compared to animals injected with non-silencing shRNA control virus (+1.4 ± 6.9% and 6.5 ± 6.4% for shRNA#1 and #2, respectively; data not shown). In contrast, both transgenic and total APP levels were markedly diminished by targeting PKCβ. Transgenic APP levels decreased by 3.7 ± 3.6% and 31.3 ± 6.8% (shRNA#1 and #2, respectively; Fig. 3C and E), while total APP levels decreased by 4.1 ± 3.2% and 15.3 ± 3.2% (Fig. 3C) in the PKCβ-targeted animals compared to non-silencing controls. Importantly, targeting of PKCβ had no effect on the levels of related kinases PKCα (Fig. 3C) or PKCδ (data not shown). Based on these findings, we pursued further study of PKCβ using the more effective shRNA #2 sequence for all future experiments.
Based on the initial screens in stably transfected Daoy cells and transgenic Drosophila, we predicted that PKCβ reduction in our APP transgenic mice affected APP expression by changing protein stability rather than transcription. To ensure that this was the case and that PKCβ levels did not affect activity of the tetracycline transactivator used to control transgenic APP expression in our model, we performed quantitative Reverse-transcription polymerase chain reaction (RT-PCR) measurements on mRNA from cortical tissue harvested 1 month after P0 injection with scramble or PKCβ shRNA virus. As expected, PKCβ mRNA was lowered by 84.4 ± 2.1% similar to that of PKCβ protein (Fig. 3F). In contrast, there was no change in transgenic APP mRNA expression between scramble control and PKCβ shRNA-treated groups (Fig. 3G). This qPCR data confirms that PKCβ knockdown affected APP expression at the level of protein stability rather than transcription.
PKCβ directly phosphorylates APP-CTF in vitro
Having identified PKCβ as our top candidate for further in vivo testing, we wanted to determine whether this kinase acted directly on APP or indirectly through another signaling pathway. PKCβ is expressed in two different splice variants, PKCβI and PKCβII, which are identical throughout their regulatory and catalytic domains and predicted to share target specificity. For these experiments, we therefore chose βII as representative of the two PKCβ kinases.
The APP intracellular domain is identical between mouse and human and contains multiple phosphorylation sites concentrated within two internalization motifs (Fig. 4A). Early studies on APP cleavage and trafficking indicated that PKC family members could phosphorylate APP directly upon phorbol ester treatment, but investigators were not able to identify which isoforms were responsible (21–24). Consistent with our genetic targeting in the mouse brain, application of the PCKβ inhibitor enzastaurin to Neuro 2a cells stably transfected with human APPswe produced a dose-dependent decrease in APP levels (Fig. 4B and C) (12,25). We next tested for a direct interaction between recombinant human PKCβII and purified APP C-terminal fragment (CTF) using a non-radioactive ADP-Glo assay. Indeed, in vitro incubation of PKCβII with APP-CTF produced a dose-dependent increase in ATP conversion that could be suppressed by enzastaurin (Fig. 4D). We then turned to a radioactive phosphorylation reaction to prove that APP-CTF was in fact being phosphorylated by PKCβII. The known PKCβII substrate histone H1 served as a positive control for PKC activity (26), while addition of enzastaurin served as a negative control (Fig. 4E). Finally, we used liquid-chromatography-mass spectrometry (LC–MS/MS) to identify the specific APP residue phosphorylated by PKCβII. Tryspin digest analysis defined a single residue, serine 655, as the PKCβII phosphorylation site on APP-CTF (Fig. 4F). This site is identical to that defined by Greengard and colleagues 30 years ago, but now can be specifically attributed to the PKCβ isoform.
Figure 4.
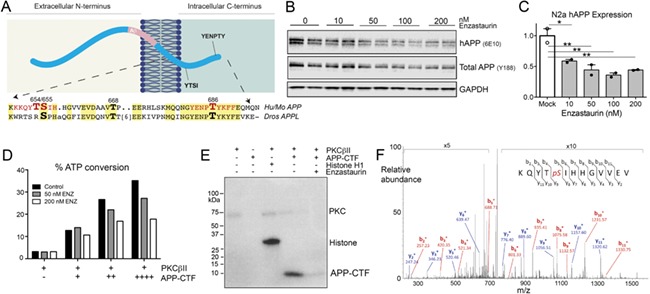
APP is directly phosphorylated by PKCβ. (A) Diagram of APP illustrating the YTSI and YENPTY internalization motifs that harbor key phosphorylation sites. Sequence alignment of the intracellular domain between mouse/human APP and Drosophila APPL highlights the conservation of these sites. (B) Western blot showing dose-dependent reduction of APP levels in stably transfected N2a cells upon treatment with PKCβ inhibitor enzastaurin. Transgenic APP was detected with antibody 6E10 (hAPP), total APP detected with antibody Y188. (C) Quantification of transgenic APP (6E10) from enzastaurin-treated N2a cells. All western blot measurements were assessed relative to within-lane GAPDH signal. Values are normalized to the average of mock-transfected controls. 1-way ANOVA, F(4, 5) = 14.82, P = 0.0056. (D) ADP-Glo assay was used to measure the percent of ATP conversion by recombinant PKCβII upon addition of purified APP C-terminal fragment (APP-CTF). ATP conversion increased with the amount of APP-CTF substrate and dose-dependently decreased upon addition of enzastaurin. (E) Radiolabeled in vitro phosphorylation assay demonstrates direct interaction between recombinant PKCβII and purified APP-CTF. Histone H1 served as a positive control, enzastaurin as a negative control. (F) Mass spectrometry confirmed that S655 was the sole PKCβII phosphorylation site within APP-CTF.
Reduction of PKCβ in APP transgenic mice has a potent but transient impact on Aβ pathology
After demonstrating that PKCβ could directly phosphorylate APP, and that PKCβ inhibition decreased steady-state APP levels in vitro and in vivo, we predicted that PKCβ manipulation should also decrease the production of Aβ peptide and delay amyloid onset in APP transgenic mice. To test this prediction, we measured the impact of preventive PKCβ reduction on APP levels, Aβ concentration, and plaque load at three time points during disease progression. The APP/TTA model used here develops amyloid pathology within 2 months of transgenic APP expression, accumulating plaque levels equivalent to CERAD stage B human deposits by 6 months (27,28). Animals were intracranially injected with AAV8 encoding microRNA-adapted PKCβ shRNA #2 or non-silencing shRNA control immediately after birth and harvested for study after 1, 3 or 6 months of APP expression to examine time points before, during and after amyloid onset.
As described above, shRNA targeting lowered PKCβ levels by 87 ± 2.6% at 1 month, and this effect largely persisted at 3 months (74 ± 6.8%) and 6 months (71 ± 6.1%; Fig. 5A and B). Although knockdown of PCKβ remained strong, the PCKβ-mediated reduction of transgenic APP lost potency over time. Compared to the 31 ± 6.8% reduction at 1 month, transgenic APP levels remained lower than controls at 3 months (18 ± 8.3%) and 6 months (14 ± 6.7%); however, the differences were not statistically significant (Fig. 5A and C). As a result, the strength of correlation between PKCβ and APP diminished with prolonged shRNA suppression, suggesting the possibility of compensatory mechanisms emerging over time (Fig. 5D).
Figure 5.
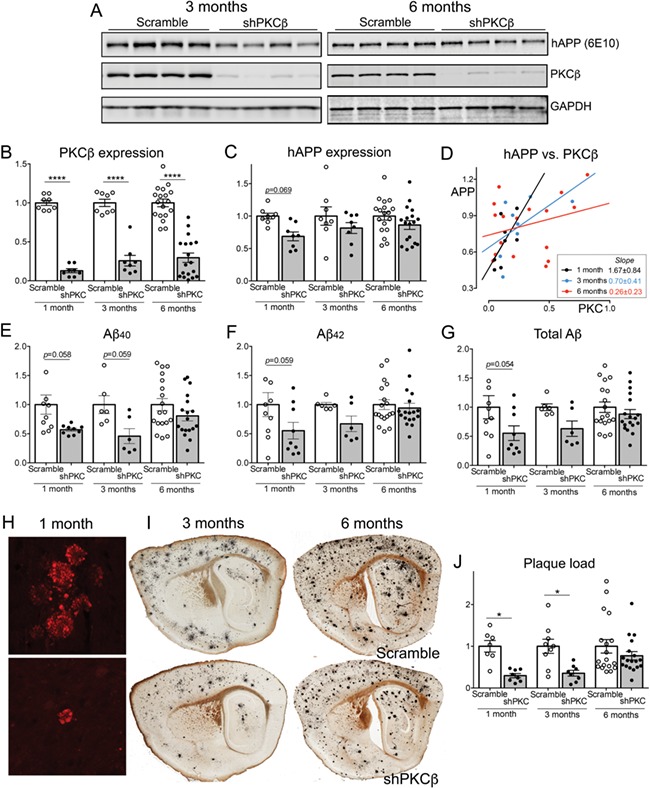
Chronic shRNA knockdown of PKCβ transiently lowers transgenic APP and Aβ. Animals were injected at birth with AAV-shRNA #2 against PKCβII and compared against control littermates receiving scramble shRNA. Protein and plaque levels were analyzed 1, 3 or 6 months later. All graphs values are normalized to the average of non-silencing shRNA controls for each age. (A) Western blots showing expression of transgenic APP (6E10), PKCβII and GAPDH in cortical homogenates from mice harvested 3 (left) and 6 months (right) after P0 AAV injection. (B) Quantification of PKCβII relative to GAPDH in cortex of APP/TTA mice harvested 1, 3 or 6 months after P0 injection with AAV-shPKCβ #2 or scramble shRNA. Values for all scatter plots are normalized to age-matched scramble control group. Note that the APP and PKCβ protein expression data for animals harvested 1 month after viral injection are taken from Figure 3D and E. 2-way ANOVA, main effect of treatment F(1, 62) = 231.5, P < 0.0001; treatment x age interaction F(2, 62) = 1.018, P = 0.367. (C) Quantification of transgenic APP (6E10) relative to GAPDH in mice virally injected at P0. 2-way ANOVA, main effect of treatment F(1, 62) = 9.140, P = 0.0036; treatment x age interaction F(2, 62) = 0.588, P = 0.559. (D) Transgenic APP (6E10) as a function of PCKβII at 1 month (black; R2 = 0.397, P = 0.094), 3 months (blue; R2 = 0.323, P = 0.141) and 6 months (red; R2 = 0.078, P = 0.278) after viral shRNA delivery. (E) Aβ40 concentration measured by MSD assay from guanidine extracts of cortical tissue in APP/TTA mice injected with AAV-shPKCβ or scramble at P0. 2-way ANOVA, main effect of treatment F(1, 61) = 13.58, P = 0.0005; treatment x age interaction F(2, 61) = 1.141, P = 0.3361. (F) Aβ42 concentration measured by MSD from cortical extracts. 2-way ANOVA, main effect of treatment F(1, 61) = 6.472, P = 0.0135; treatment x age interaction F(2, 61) = 1.557, P = 0.2190. (G) Total Aβ concentration (Aβ40 + 42) measured by MSD assay from cortical extracts. 2-way ANOVA, main effect of treatment F(1, 61) = 8.362, P = 0.0053; treatment x age interaction F(2, 61) = 1.194, P = 0.310. (H) Dystrophic neurites labeled by cytoplasmic accumulation of virally-delivered tdTomato mark the location of nascent Aβ deposits in APP/TTA mice harvested 1 month after P0 viral injection. (I) Sagittal sections stained using the Campell–Switzer silver method illustrate plaque burden in APP/TTA mice harvested at 3 (left) or 6 months (right) after P0 viral injection. (J) Quantitation of plaque burden measured by number of tdTomato-labeled neuritic foci (1 month) or silver area (3 and 6 months). 2-way ANOVA, main effect of treatment F(1, 62) = 17.45, P < 0.0001; treatment x age interaction F(2, 62) = 1.831, P = 0.169.
Even a transient reduction in APP might impact the onset and progression of amyloid pathology and so we further explored both the concentration of Aβ peptides and the area of plaque deposits in the virally-transduced APP/TTA mice. We measured the concentration of transgene-derived human Aβ40 and Aβ42 peptides in guanidine extracts from cortical tissue by MesoScale Discovery assay. This analysis revealed a strong reduction of both peptides in animals transduced with PKCβ shRNA compared to non-silencing shRNA at 1 month of age (−43% and −45%, respectively; Fig. 5E and F). The effect diminished over time for both peptides, albeit to different extents. The difference in Aβ40 between PKCβ-targeted and non-silencing controls was 54% at 3 months and 19% at 6 months (Fig. 5E). Aβ42 was reduced by 33% at 3 months but differed by just 6% at 6 months (Fig. 5F). Taken together, the difference in total Aβ concentration approached significance between groups only at the earliest time point (Fig. 5G), closely paralleling the pattern observed for transgenic APP levels over time.
The extent of plaque pathology for a given transgenic model is usually well-aligned with the Aβ concentration; however, differences in the 40:42 ratio can influence this relationship between biochemistry and histology. We therefore examined how shRNA-mediated reduction of PKCβ affected the onset and progression of plaque pathology in the APP/TTA mice. As expected from a past study with this model, no plaques were detected by silver stain in mice harvested at 1 month of age. To our surprise, however, the viral fluorescent label used to identify transduced cells revealed dystrophic neurites in plaque-like clusters even before true plaques could be detected (Fig. 5H). We used these intensely fluorescent neurites as a surrogate marker for counting nascent deposits. The number of neuritic clusters was 70% lower in mice treated with PKCβ shRNA compared to animals treated with non-silencing control shRNA (Fig. 5J). At later ages, Aβ deposits could be detected by standard histological methods (Fig. 5I). At 3 months of age, mice treated with PKCβ shRNA still had 65% less cortical surface covered by plaque than animals treated with control shRNA. By 6 months, treated mice still had 23% less plaque burden, although the difference was no longer statistically significant (Fig. 5J).
The possibility that other APP kinases may have compensated for the chronic loss of PKCβ lead us to examine the other main serine/threonine phosphorylation site controlling APP trafficking and thereby affecting protein stability. Threonine 668 in the APP intracellular domain is the target for phosphorylation by c-Jun N-terminal kinases (JNK1/2/3), cyclin-dependent protein kinases (Cdk1 and Cdk5) and GSK3β (29). Of these, GSK3β is especially well studied and increased activity of this kinase is associated with greater Aβ production in transgenic mice (30–32). GSKβ activity can be negatively regulated by phosphorylation at serine 9, and so we examined both total and serine 9-phosphorylated GSK3β, as well as total and threonine 668-phosphorylated APP in cortical homogenates from mice treated with scramble or PKCβ shRNA (Fig. 6A and B). We focused this experiment on the 6 month treatment group, where PKCβ expression had been reduced for the greatest length of time and where we knew APP protein had recovered to control levels. Despite the importance of threonine 668 phosphorylation in APP regulation and GSK3β as a major kinase for this site, we found no difference in total or serine 9-phosphorylated GSK3β levels between mice treated with PKCβ or scramble shRNA (Fig. 6C and D), nor did we find any change in total or threonine 668-phosphorylated APP (Fig. 6E and F). In this case, compensation for loss of one kinase was not simply a case of increasing another, at least for GSK3β and APP regulation via threonine 668 more broadly.
Figure 6.
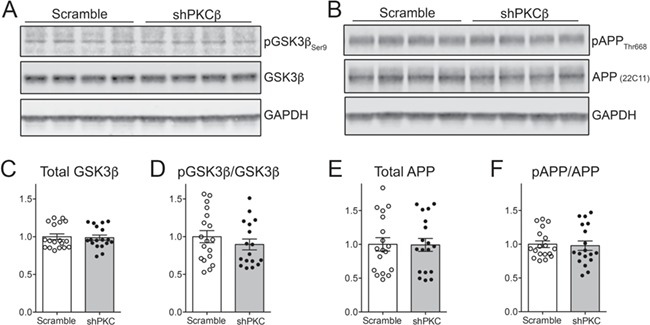
Chronic knockdown of PKCβ does not cause compensatory changes in GSK3β or in APP phosphorylation at Thr668. (A,B) Western blots for total GSK3β and GSK3β phosphorylated at serine 9 (A) and for total APP and APP phosphorylated at threonine 668 (B) in cortical homogenates from mice harvested 6 months after P0 AAV injection with scramble or shPKCβ. Note that the same samples were probed for APP and PKCβ in Figure 6A. (C) Quantification of total GSK3β relative to GAPDH. Values for all graphs were normalized to the respective scramble control groups. Student’s t-test, P = 0.82. (D) Quantification of serine 9-phosphorylated GSK3β relative to total GSK3β. Student’s t-test, P = 0.35. (E) Quantification of total APP (detected with antibody 22C11) relative to GAPDH. Student’s t-test, P = 0.96. (F) Quantification of threonine 668-phosphorylated APP relative to total APP. Student’s t-test, P = 0.80.
Discussion
We set out to test whether the power of forward genetics might reveal new therapeutic opportunities for Aβ reduction in AD by interrogating evolutionarily conserved pathways controlling APP stability. Our approach was based on parallel unbiased RNAi screens of the kinome originally pioneered to identify modifiers of ataxin 1 protein in models of spinocerebellar ataxia 1 (8). Here we modified this strategy to target another disease-associated protein, APP, on the hypothesis that even modest reduction of aggregating proteins—or their full-length precursor—may be sufficient to delay disease onset or slow progression. In addition to ataxin 1 and APP, cross-species unbiased RNAi screens have now been applied to several disease-causing proteins, including survival of motor neuron protein (33), α-synuclein (10,11) and microtubule-associated tau protein (9). The current study of APP modifiers combines two advances from recent work using similar approaches (11,34). First, we introduce a high-throughput screen using a scorable phenotype caused by neuron-specific expression of human APP in the Drosophila CNS. By screening on neurological impairment rather than cytotoxicity, we predict this approach may be sensitive to small functional benefits that would not be apparent from ommatidia morphology. Second, we developed a modular viral vector for delivery of micro RNA-adapted shRNA to rapidly move candidate modifiers identified by the screen into mouse models of AD. The use of shRNA for target reduction eliminated any issues with drug penetrance into the brain and provided greater specificity for the β isoform than available drug candidates (12). Moreover, we designed the AAV transfer plasmid to allow easy exchange of micro RNA-embedded shRNA for any gene of interest using standard restriction enzyme-based cloning from the commercially available GIPZ plasmid collection. This vector system and AAV delivery method should simplify validation of candidate modifiers in mouse models of neurological disease.
The first-line RNAi screens in Drosophila and human cell lines yielded 14 shared candidate modifiers of APP stability. Of these, MAPK3 and PRKCB were confirmed by Western blotting to lower APP protein expression using independent RNAi reduction of each kinase. While MAPK3 (ERK1) had previously been shown to impact APP levels via transcriptional regulation in vitro (35), it is not known to phosphorylate APP directly. Instead, the effect on APP stability observed here was likely indirect as ERK1/2 signaling has been shown to affect expression of all three APP secretases (36–39). Although we achieved >50% knockdown of MAPK3 in the mouse brain, we suspect the lack of effect on APP was due to our choice of transgenic mouse model. The APP/TTA model expresses an artificial APP variant containing familial AD mutations at both the β and γ cleavage sites, which may have precluded any further influence on APP processing by MAPK knockdown. In contrast to MAPK3, PKC is a well-established APP modifier, shown to directly phosphorylate APP and increase α-secretase cleavage (21–24). The mechanism by which PKC modulates APP processing and secretion may have both direct and indirect components. The most striking evidence in support of an indirect mechanism comes from early studies showing that the entire APP cytoplasmic tail could be deleted without dampening α-secretase processing following PKC activation (40). Subsequent work has shown that PKC activation can enhance non-amyloidogenic processing by promoting APP trafficking while sequestering BACE1 (41–43). Complementary studies also support the possibility that PKC phosphorylation directly affects APP processing. The PKC phosphorylation site, S655, resides in the 653YTSI656 region of the APP cytoplasmic domain, which forms a conserved motif for clathrin-mediated internalization (44). Phospho-mimetic substitution of glutamic acid at S655 increased plasma membrane trafficking and subsequent internalization into endocytic vesicles resulting in prolonged APP half-life in vitro, while dephospho-mimetic S655A delayed membrane delivery and diminished APP stability (45,46). Based on this evidence, we predict that both direct and indirect mechanisms contributed to the effects of PKCβ knockdown in our APP transgenic mice.
Since the original studies showing that PKC could directly phosphorylate APP, at least 10 mammalian isoforms of PKC have been identified and cloned (47). The PKC isoform(s) studied by Greengard and colleagues in their pioneering work on APP phosphorylation were activated by phorbol esters, which includes both conventional (α, βI, βII, and γ) and novel PKC isoforms (δ, ɛ, θ, and η). Experiments to pinpoint the contribution of specific PKC isoforms in APP processing have been limited, but have generally converged on PKC α, δ and ɛ. Early in vitro studies demonstrated that stable transfection of PKCα or ɛ enhanced the phorbol ester-mediated rise in soluble APP secretion (48). Subsequent studies showed that neuronal overexpression of PKCɛ significantly reduced plaque accumulation in APP transgenic mice; however, the proposed mechanism was through increased Aβ degradation by endothelin converting enzyme rather than diminished Aβ production (49). Together these studies suggest that PKCɛ may have different actions depending on context. Whereas transgenic overexpression of PKCɛ diminished Aβ via degradation, PKCδ knockdown in mice lowered Aβ via BACE1. Reduction of PKCδ in primary neurons from APP transgenic mice decreased BACE1 expression, β-site APP cleavage and Aβ production, while chronic treatment with rotterlin to inhibit PKCδin vivo reiterated these effects and delayed plaque onset (50). These two experiments hint that PKC isoforms may act in opposition to control Aβ levels, yet few isoforms have been selectively manipulated in vivo to isolate their effect. Here we genetically perturb PKCβ both in vivo and in vitro and show that a specific PKC isoform is sufficient to mediate direct phosphorylation of APP. Our work suggests a direct connection between PKCβ and Aβ via control of steady-state APP levels.
While having PKCβ emerge from our unbiased cross-species screen served to validate our approach, it is also worth examining which additional kinases known to modify APP trafficking and stability were not identified by our strategy. APP has at least eight serine and threonine residues that undergo phosphorylation. In particular, threonine 668 within the APP 667VTPEER672 motif can be phosphorylated by c-Jun N-terminal kinases (JNK1/2/3), cyclin-dependent protein kinases (Cdk1 and Cdk5) and GSK3β to govern interaction with intracellular binding partners, and through them, control APP trafficking within the neuron (29). In addition, tyrosine 682 within the APP 682YENPTY687 motif can be phosphorylated by TrkA/NGFR and Src kinase to regulate clathrin-mediated internalization and processing of APP (29,51). Of these known APP kinases, only GSK3β was identified in addition to PKCβ from either Daoy cells or Drosophila, but was not confirmed by secondary validation. Genetic screens such as ours are subject to false negative findings, and the very features that made each screen feasible may have also limited their sensitivity. The Daoy cell screen relied on a C-terminal GFP fusion for detection, which may have blocked access to APP phosphorylation sites or interaction domains required for trafficking or processing. The Drosophila screen relied on a wing phenotype controlled by an unmapped neural circuit, which may not express known APP kinases (52). The strength of cross-species screening is that false-positive findings are minimized to focus validation efforts on high-value targets. Our study substantiated this principle in a well-studied class of proteins, nominating a small number of kinases for in vivo testing and proving one capable of delaying pathology in a mouse model of AD.
One unexpected finding of our study was the transient reduction of APP despite persistent PCKβ knockdown. Past studies of PKC modulators in APP transgenic mice have generally tested just a single time point of shorter duration than examined here, and none have suggested a mechanism based on APP stability (49,50,53–56). We found that transgenic APP levels were significantly reduced following 1 month of PKCβ knockdown, but this effect lost potency as the animals aged. The result was a temporary reduction in amyloid load that ultimately lost statistical significance after 6 months of treatment. The discrepancy between short-term and longer treatments suggests the possibility of homeostatic regulation over APP itself or over an intermediary pathway linking PKCβ to APP. While we know of no precedent for this slow loss of APP suppression in models of AD treatment, cancer therapeutics have grappled with a related phenomenon of acquired drug resistance. For example, tyrosine kinase inhibitors such as erlotinib and gefitinib used to treat non-small cell lung cancer can be initially successful, but lose efficacy as the activation of non-EGFR signaling pathways bypass EGFR inhibition (57). In our AD model, although we rule out the possibility of compensation through one alternative phosphorylation site, chronic knockdown of a single PKC isoform may have simply been offset by upregulation of another. Both examples make good cases for multi-drug therapy—already accepted practice in cancer treatment—targeting multiple pathways in combination or in series to attain long term effect. Genetic screens such as ours are ideally suited to this effort with the potential to identify conserved pathways converging on the regulation of disease-associated proteins.
Materials and Methods
Generation of APP-expressing stable cell line
The Daoy human neuroblastoma cell line expressing a bicistronic CMV-APP695-eGFP-IRES-DsRed transgene was generated as previously described (8). Briefly, the APP-GFP:DsRed construct was cloned into a pHAGE vector and packaged into lentiviruses. Daoy cells were infected at low multiplicity (0.3) to promote single copy integration. Transduced cells were selected with puromycin and then processed by fluorescence activated cell sorting (Aria II cell sorter, BD Biosciences, San Jose, CA) to isolate individual red-green double fluorescent cells. Clones were expanded and one was selected for screening based on the following traits: low transgene expression, low variation, and homogenous population. Following similar procedures, we also generated a DsRed-IRES-eGFP cell line to serve as a control.
Cell-based kinase screen
The APP-GFP:DsRed Daoy cell clone was plated into 96-well round bottom plates sufficient for testing three independent siRNAs for each kinase (1908 siRNAs, 636 genes; tests performed in triplicate). The following day, each well was transfected at 20 nM with a single siRNA from the Stealth RNAi Human Kinase Collection (Invitrogen, Carlsbad, CA; #12938200) as previously reported (8). Cells were grown for 3 days before being trypsinized and suspended in PBS + 5% FBS for analysis. The green:red fluorescence ratio for each well was analyzed by flow cytometry using a LSRII Fortessa coupled with an HTS module (BD Biosciences). A Z-score for each siRNA was calculated by comparing the average fluorescence ratio for the triplicate sample against the plate mean. siRNA with a Z-score of ±1.5 were considered positive hits. Next, individual t-tests were performed to compare the fluorescence ratios of each hit against those obtained for scrambled siRNA sequences containing low, medium, and high GC content. Hits with a P value of <0.05 were kept for further analysis. Kinases that met both the Z-score and P value thresholds were confirmed using a distinct set of siRNA for each gene, but this time including a DsRed-IRES-eGFP cell line as a negative control. Candidates that significantly altered the fluorescence ratio in this control cell line in the same direction as the APP-eGFP:DsRed clone were excluded.
The effect of PRKCB as a candidate modifier of endogenous human APP levels was confirmed using wild-type (non-transfected) Daoy cells. Individual siRNAs against PRKCB (ThermoFisher Scientific, Pittsburgh, PA, #HSS108495) were transfected using DharmaFECT (Dharmacon, Lafayette, CO) and cells harvested 3 days later for Western blotting against APP (mouse anti-APP clone 22C11).
Drosophila kinase screen
APP transgenic Drosophila lines carrying human wild-type APP695 (P5) or an APP695 -N-myc fusion construct (P(UAS-APP695-N-myc)) as well as the pan-neuronal driver line elav-Gal4C155 (P(GawB)elavC155) were obtained from the Bloomington Drosophila Stock Center at the University of Indiana (http://flystocks.bio.indiana.edu). Hairpin RNA lines for the kinome screen were obtained from the Vienna Drosophila RNAi Center (http://stockcenter.vdrc.at/control/main). For the wing phenotype primary screen, we used the UAS-APP695-N-myc controlled by the elav-Gal4C155 driver line. Animals were raised at 25°C and non-balancer females were scored for the ratio of animals with normal wings compared to the total number of animals. Hairpin RNAs that improved the APP wing phenotype by 2 standard deviations above the average of the experiment were considered primary hits; these hits were selected for protein quantification by Western blotting in Drosophila tissue. Drosophila heads were homogenized in NuPAGE LDS sample buffer (ThermoFisher, #NP0007) and electrophoresed on 4–12% NuPAGE Bis-Tris gels (ThermoFisher) followed by standard transfer to nitrocellulose membrane. Transgene-derived APP was detected with mouse antibody 6E10 (Biolegend, San Diego, CA, #803001) diluted 1:1000 in TBS containing 0.1% Triton X-100 (TBST) and 3% BSA. Lamin C was used as an internal control (clone LC26.28, Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, IA) diluted 1:1000 in TBST containing 5% non-fat dry milk. Blots were then incubated with HRP-conjugated secondary antibody followed by ECL detection and imaged using an ImageQuant LAS 4000 (GE Healthcare Life Sciences, Pittsburgh, PA). Quantification was carried out using ImageJ.
In vitro PKCβ inhibition
Neuro 2a cells stably expressing human APP695 encoding the Swedish mutation (25) were cultured for 24 hr in DMEM (Lonza, Basel, Switzerland, #12-604F) containing 10% fetal bovine serum (ThermoFisher, #MT35010CV), 100 IU/ml Penicillin and 100 μg/ml of streptomycin (ThermoFisher, #15140122). The following day, media was changed to add varying doses of enzastaurin (LY317615; ApexBio Technology, Houston, TX, #A1670), and cells were allowed to grow for an additional 48 h. Cells were trypsinized, collected and lysed by sonication in one volume of pre-chilled PEPI buffer (1x PBS plus 5 mM EDTA, 1x protease inhibitor (Roche, Basel, Switzerland, #05892970001) and 1x PhosSTOP (Roche, #04906845001), mixed with an equal volume of modified RIPA buffer (1x PBS, 5 mM EDTA, 1% NP40, 1% deoxycholate, 2% SDS), vortexed and centrifuged at 20 000g for 10 min at room temperature to prepare sample for APP immunoblotting as described below.
APP-CTF peptide purification
DNA encoding the cytoplasmic domain of wild-type mouse APP (APP-CTF, amino acids 649–695 using the APP695 numbering convention; identical to human APP-CTF) was amplified by PCR from an adult mouse brain cDNA library (forward primer: GCGCGCATATGAAGAAGAAACAGTACACATCCATC; reverse primer: GCGCGAAGCTTTTAGTTCTGCATTTGCTCAAAGAA). The purified PCR fragment was digested with NdeI and HindIII and ligated into the NdeI/HindIII sites of pKW32 (58). This construct was used to express a fusion protein of APP-CTF and His-tagged Vitreoscilla hemoglobin (VHB) in BL21 E. coli. Following growth in lysogeny broth (LB) media at 37°C and IPTG induction, the culture was collected, resuspended in binding buffer (20 mM Tris pH 8.0, 0.5 M NaCl and 5 mM imidazole) and lysed by 30 min incubation with lysozyme. Following a 30 min centrifugation at 15 000g, the supernatant was loaded onto a Ni–NTA column (Qiagen, Germantown, MD, #30600) at a flow rate of 0.5 ml/min. The column was washed (20 mM Tris pH 8.0, 0.5 M NaCl and 60 mM imidazole) and then eluted (20 mM Tris pH 8.0, 0.5 M NaCl and 250 mM imidazole). After imidazole was removed by dialysis (Novagen, Madison, WI, #71508), the VHB-APPCTF fusion protein was cleaved with thrombin for 6 h at 30°C (Sigma-Aldrich, St. Louis, MO, #RECOMT). After cleavage, the reaction was loaded onto a new Ni-NTA column and the eluent containing APPCTF was collected. Fractions from each step of the purification were analyzed by SDS-PAGE on a 4–20% Tris-HCl gel (Bio-Rad, Hercules, CA, #3450032).
Non-radioactive kinase assay
The conversion of ATP to ADP by PKCβII in the presence of purified APP-CTF was assessed using the ADP-Glo Kinase Assay (Promega, Madison, WI, #V9101) according to manufacturer’s instructions. Briefly, the reaction was carried out with 0.5 μg of recombinant purified human PKCβII (Life Technologies, Carlsbad, CA, #P2251), 1 μg purified APP-CTF and 1 μM ATP, with or without 200 nM of enzastaurin (ApexBio, #A1670). Following 30 min incubation at 30°C, ADP-Glo reagent was added and the reaction continued for another 40 min at room temperature. Finally, detection buffer was added and incubated for another 60 min at room temperature before measuring luminescence on a Synergy 2 Multi-Mode Reader (BioTek, Winooski, VT).
Radioactive in vitro kinase assay
A total of 1 μg of purified APP-CTF was incubated with 0.5 μg of recombinant human PKCβII in reaction buffer (100 mM HEPES pH 7.4, 6.5 mM CaCl2, 5 mM DTT, 50 mM MgCl2, 5 mM ATP and 1 mg/ml phosphatidylserine) spiked with 1.5 μCi [γ32P] ATP (Perkin Elmer, Waltham, MA, #BLU502A250UC) for 1 h at 30°C. Histone H1 (Calbiochem, San Diego, CA, #382150) was used as a positive control PKCβII substrate (26); the PKCβ inhibitor enzastaurin was used as a negative control. The reaction was stopped by adding Laemmli sample buffer and heating to 95°C for 10 min. Radiolabeled proteins were separated by SDS-PAGE on a 4–20% Tris–HCl gel (BioRad, #3450032) and detected by autoradiography.
Mass spectrometry
Purified APP-CTF was phosphorylated in vitro by incubation with recombinant PKCβII in reaction buffer as above. The reaction was stopped by addition of NuPAGE LDS sample buffer (Invitrogen, #NP0008) and subjected to SDS-PAGE (Invitrogen, NuPAGE 10% Bis-Tris gel, #NP0315BOX). Following electrophoresis, the gel was stained with Coomassie Brilliant blue R250 and the APP-CTF fragment identified and excised based on molecular weight. The gel piece was de-stained and subject to in-gel trypsin digestion (GenDEPOT Corp., Barker, TX, #T9600) in 50 mM ammonium bicarbonate buffer for 4 h at 37°C, followed by digestion with Asp-N endoprotease (Promega, #V1621) for 4 h at 37°C. The digested peptides were extracted from the gel using 100% acetonitrile and dried in a Speedvac. Peptides were suspended in 10 μl of loading solution (5% methanol containing 0.1% formic acid) and subjected to nanoflow LC-MS/MS analysis with an Ultimate 3000 HPLC system (ThermoFisher Scientific, San Jose, CA) coupled to an Orbitrap Fusion Tribrid mass spectrometer (ThermoFisher). Peptides were loaded onto a 100 μm x 2 cm pre-column packed in-house with 1.9 μm Reprosil-Pur Basic C18 beads (Dr Maisch GmbH, Germany, #R119.b9.3). The pre-column was switched in-line with a 150 μm x 50 mm analytical column packed in-house with 1.9 μm Reprosil-Pur Basic C18 beads equilibrated in 0.1% formic acid/water. The peptides were eluted using a 35 min discontinuous gradient of 4–26% acetonitrile/0.1% formic acid at a flow rate of 800 nl/min. The eluted peptides were directly electro-sprayed into an Orbitrap Fusion mass spectrometer operated in the data-dependent acquisition mode, acquiring fragmentation spectra of the top 35 strongest ions under control of Xcalibur software 4.0 (ThermoFisher). Raw files from the Orbitrap Fusion were searched against NCBI’s human RefSeq protein database (updated June 10, 2015) using the MASCOT search engine (Matrix Science, Boston, MA, Mascot 2.4) and validated with the Percolator-based q-value in Proteome Discoverer software (ThermoFisher, PD1.4). Dynamic modification was allowed for phosphorylation (serine, threonine and tyrosine), oxidation (methionine), protein N-terminal acetylation and de-streak (cysteine). Maximum tolerance for precursor ions was set to 20 ppm, fragment mass tolerance set to 0.5 dalton and a maximum of two missed cleavages was allowed. Phosphorylation assignments were manually validated.
Screening PRKCB and MAPK3 shRNAs for efficacy in mouse cell lines
Mouse J774.1 macrophage cells (gift of Dr Susan F. Venable) were used to identify optimal murine shRNA sequences for PRKCB knock-down before constructing AAV vectors for use in vivo. Mouse NIH 3T3 cells were used to identify optimal murine shRNA sequences for MAPK3 knockdown. Lentivirus was packaged according to protocols from the RNAi Consortium at the Broad Institute (https://portals.broadinstitute.org/gpp/public/resources/protocols). Cells were transduced with lentivirus carrying each shRNA, media was changed the following day and cells were harvested for analysis 3 days later. The extent of gene knockdown for each sequence was measured relative to non-silencing control shRNA by Western blotting of cell extracts using an anti-PKCβ antibody (ThermoFisher #PA5–13741, 1:250) or anti-MAPK/ERK1/2 antibody (Cell Signaling, Danvers, MA, #4695, 1:5,000).
PRKCB 5′ stem shRNA sequences from Dharmacon:
V3LMM_433130 CGGAGCAAACACAAGTTTA (shRNA #1)
V3LMM_433131 TGGCTGGTTCAAGTTACTA (shRNA #2)
MAPK3 5′ stem shRNA sequences from Dharmacon:
V2LMM_24968 GTGCCTGTATCTAATATAT (shRNA #1)
V2LMM_37333 GACCTTAATTGCATCATTA (shRNA #2)
Non-silencing control 5′ stem shRNA sequence from Dharmacon:
RHS4346 TCTCGCTTGGGCGAGAGTAAG
Viral constructs
A pAAV expression vector was generated to deliver gene-specific shRNAs embedded within an artificial human micro RNA 30 (miR 30) transcript under control of the mouse U6 RNA polymerase III promoter (13,17). The 19 nucleotide loop and approximately 125 nucleotides of flanking sequence on either side of the gene-specific dsRNA hairpin stem were derived from miR30 to provide increased shRNA processing and potency (Dharmacon, GIPZ Lentiviral shRNA Manual). The AAV transfer plasmid contained a second domain encoding the tandem dimer Tomato fluorescent protein (tdTomato) under control of the CAG RNA polymerase II promoter. TdTomato was followed by the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) and a bovine growth hormone (BGH) polyadenylation signal, and the entire expression cassette flanked by AAV2 inverted terminal repeats (ITRs). The original pAAV backbone plasmid and the pAAV-CTR4-tdTomato-3x-Myc used to clone CAG-tdTomato-3xMyc were gifts from Dr Todd Golde. The U6 promoter was cloned from pCS/U6 purchased from Addgene (Cambridge, MA, #15173). The miR30-adapted shRNA was cloned in part from pGIPZ clone V3LMM_440513 targeting murine APP, and in part from clones V3LMM_433130 or V3LMM_433131 targeting murine PRKCB, or from clones V2LMM_24968 or V2LMM_37333 targeting MAPK3 (Dharmacon). A detailed protocol for the cloning is provided below.
Briefly, the vector was made in three steps. The miR-adapted shRNA for APP was inserted into the multiple cloning sites of the pCS/U6 plasmid. The U6-miR/shRNA cassette was isolated from this vector and ligated into pAAV immediately downstream of the 5′ ITR. Simultaneously, the CAG promoter and tdTomato coding sequence were isolated from pAAV-CTR4-tdTomato-3x-Myc and ligated into the pAAV-U6-miRshRNA plasmid to produce an AAV transfer plasmid in which miR-adapted shRNA against APP was under control of the U6 promoter and tdTomato followed by WPRE was under control of the CAG promoter. Finally, the shRNA sequence targeting APP was replaced by shRNAs targeting murine PRKCB or MAPK3.
Detailed viral expression vector cloning
pCS/U6 (Addgene #15173) was linearized with ApaI followed by Klenow reaction to blunt the ends and then digested with PstI. A linker oligonucleotide was designed to add MluI-PacI-BglII-AgeI-KpnI-PstI restriction sites to the multiple cloning site (MCS) of pCS/U6 (5ʹ-CGGACACGCGTTAATTAAGATCTACCGGTGGTACCTG-CAGCCCGG-3′). The MCS oligo and its reverse complement were annealed, digested with MluI and PstI and purified. In parallel, the pGIPZ clone #V3LMM_440513 targeting murine APP was digested with DraIII, followed by a Klenow reaction to blunt the ends and then digested with MluI to isolate the complete 407 bp miR-adapted shRNA cassette. The blunt end/MluI-digested miR/shRNAAPP fragment and the MluI/PstI-digested MCS oligonucleotide were combined in a three-way ligation with the blunt end/PstI-linearized pCS/U6 plasmid to generate pCS/U6-miR/shRNAAPP. This vector was then digested with KpnI to isolate an 880 bp U6-miR/shRNAAPP fragment, which was ligated into pAAV that had been linearized with KpnI, generating pAAV-U6-miR/shRNAAPP, which also carried the CAG promoter, WPRE and BGH polyA signal.
In order to add the tdTomato coding sequence downstream of the CAG promoter, pAAV-U6-miR/shRNAAPP was linearized with HindIII, followed by a Klenow reaction to blunt the ends and then digested with NdeI, to remove most of the original pAAV CAG promoter. This sequence was replaced by a CAG-tdTomato fragment isolated from pAAV-CTR4-tdTomato-3x-Myc by digestion with EcoRI, followed by Klenow reaction and then digestion with NdeI to yield the 2435 bp CAG-tdTomato fragment. The blunt end/NdeI-digested CAG-tdTomato fragment was ligated into the blunt end/NdeI-digested pAAV-U6-miR/shRNAAPP to generate the final pAAV-U6-miR/shRNAAPP-CAG-tdT plasmid. MiR30-adapted shRNAs for PRKCB and MAPK3 (Dharmacon, PRKCB #1 V3LMM_433130, PRKCB #2 V3LMM_433131, MAPK3 #1 V2LMM_24968, and MAPK3 #2 V2LMM_37333) were removed from pGIPZ by digestion at XhoI and MluI and transferred to pAAV-U6-miR/shRNA-CAG-tdT, generating the final transfer plasmids used for AAV packaging.
Viral packaging
All AAVs were prepared by the Gene Vector Core at Baylor College of Medicine as described previously (20). HEK293T cells were grown in DMEM (GenDEPOT) supplemented with 10% FBS (Sigma) and 1x antibiotic/antimycotic (GenDEPOT). Serotype 8 AAV was prepared by co-transfection of three plasmids (expression vector, p5E18-VD2/8 Rep-Cap plasmid and pAdΔF6 helper plasmid) using iMFectin Poly DNA Transfection Reagent (GenDEPOT). AAV purification was performed using a modified protocol based on Ayuso et al. (59). Three days after transfection, cells were collected while the media was retained for subsequent polyethylene glycol (PEG) precipitation. The cell pellet was re-suspended in 1 ml per dish of 50 mM Tris pH 8.0 containing 5 mM MgCl2 and 0.15 M NaCl, lysed by adding 0.1 volume of 5% sodium deoxycholate at RT for 30 min and then incubated with 10 μg/ml of DNase I and RNase A for 1 h at 37°C. Cell lysates were clarified by centrifugation at 5000g for 10 min at 4°C. The culture media was incubated with 10 μg/ml of DNase I and RNase A for 1 h at 37°C and then incubated at 4°C overnight in 8% PEG (stock 40% PEG 8000 plus 2.5 M NaCl). AAV was collected from this mixture by centrifugation at 2500g for 30 min. The pellet containing AAV was resuspended in a minimal volume of HBS (50 mM HEPES, 0.15 M NaCl, 1% sarcosyl and 20 mM EDTA pH 8.0). Cell-associated and secreted AAV preparations were combined for iodixanol density centrifugation. The resulting AAV particles were dialyzed against Mg- and Ca-free PBS using an Amicon Ultra-15 centrifugal filter (100 000 kDa nominal limit, Millipore, Burlington, MA,) and the titer determined by real time PCR.
Mice
The tet-responsive APP transgenic lines 102 (tetO-APPswe/ind 102; MMRRC, Bar Harbor, ME, #034845-JAX, (60) and the CaMKIIα-tTA tetracycline transactivator line B (Jackson Laboratory, Bar Harbor, ME, #3010, (61)) were independently backcrossed to C57BL/6J for >30 generations before being intercrossed for these studies. The resulting double-transgenic male offspring were then mated with wild-type FVB females to produce experimental cohorts on a FVBB6 F1 background. All transgenic mice were thus heterozygous and animals of both genders were included in all cohorts. Mice for the 6 month cohort were reared on doxycycline (dox) for 6 weeks from P2-P42 to allow behavioral testing as adults (data not shown). At 6 weeks of age, animals were returned to unmedicated chow and allowed to express transgenic APP for 6 months prior to harvest. This treatment was previously shown to slightly delay the accumulation of Aβ compared to mice that had never been on dox; however, it does not affect the expression level of APP once dox is removed from the diet and does not diminish the severity of amyloid burden (13).
P0 intraventricular injections
Within 6 h after birth, neonates were collected from the cage and prepared for injection by cryoanesthesia. Following cessation of movement, animals were head-fixed into a neonatal stereotaxic device to identify two sites on each hemisphere used to target the lateral ventricles for viral delivery (X, Y, Z) = (± 0.8, ± 1.5, −1.5 mm) and (± 1.35, ± 2.0, −1.7 mm) (20,62). rAAV was diluted in sterile PBS containing 0.05% trypan blue to deliver 6.0 × 1010 AAV particles in a total volume of 4 μl. Viral preparations were injected using a 10 μl syringe (Hamilton Company, Reno, NV, #7653–01) fitted with a 32 gauge needle (Hamilton, #7803–04, RN 6PK PT4) to deliver 1 μl of viral solution per site. After the injections were complete, pups were placed on a warming pad until they regained normal color and resumed movement. All injected animals were then returned to their biological mother for care.
Tissue harvest
Mice were killed by sodium pentobarbital overdose and transcardially perfused with cold PBS. Brains were removed and dissected along the midline. The cortex and hippocampus were isolated from one hemisphere and frozen for biochemistry; the remaining hemisphere was immersion fixed in 4% paraformaldehyde and sectioned at 35 μm for histology.
Campbell–Switzer silver stain
A detailed protocol for this stain can be found at the NeuroScience Associates website: https://www.neuroscienceassociates.com/reference/papers/alzheimers-disease-pathology-silver-stain/
Quantification of plaque burden
For each animal, 5–7 sections spaced at 420 μm intervals were analyzed by native tdTomato fluorescence (1 month) or stained using the Campbell–Switzer silver method (3 and 6 months). Plaque number within the cortex of 1 month animals was sparse enough to count manually by fluorescence at 10× magnification. Tissue from the 3 and 6 month animals was stained in a single batch and imaged under the same exposure conditions. Tiled brightfield images of each section were captured on an AxioImager.Z1 using Zen Pro 2012 Blue Edition software (Carl Zeiss AG, Oberkochen, Germany) at 5× magnification. Each image was converted to grayscale using ImageJ software, and the region of interest (ROI) was manually defined to outline the cortex. Within each ROI, signal was separated from background using the Yen thresholding method. The area occupied by signal was then obtained using the ImageJ Analyze Particles tool.
RNA extraction, cDNA preparation and qPCR
Animals for qPCR analysis were harvested at 1 month of age and cortical tissue isolated from one hemisphere for analysis. RNA was extracted using PureLink RNA Mini Kit (ThermoFisher, #12183018A) according to manufacturer’s instructions. A total of 1 μg of RNA was used to prepare cDNA using qScript cDNA Supermix (ThermoFisher, #95048–025) according to manufacturer’s instructions. Samples were incubated 5 min at 25°C, 30 min at 42°C and 5 min at 85°C using a SimpliAmp Thermal Cycler (ThermoFisher #A24811). A total of 10 ng of cDNA was amplified to analyze expression of PKCβ, transgenic APP and GAPDH using SYBR Green PCR Master Mix (ThermoFisher, #4309155) according to the manufacturer’s instructions (PKCβ: For GTGTCAAGTCTGCTGCTTTGT, Rev GTAGGACTGGAGTACGTGTGG; hAPP: For CCAAGATGCAGCAGAACGGA, Rev AGACCACGAGAATGCGAAGG; and GAPDH: For TCAAGAAGGTGGTGAAGCAGG, Rev ACCAGGAAATGAGCTTGACAAA). Reactions were run on a StepOne Plus and analyzed using either StepOne or QuantStudio 6 Flex software (Applied Biosystems, Waltham, MA).
Tissue homogenization
Frozen cortical tissue was sonicated in 5 volumes of pre-chilled PBS containing 5 mM EDTA, 1x protease inhibitor (Roche, #05892970001) and 1x PhosSTOP (Roche, #04906845001).
One half of each sample would be used for immunoblotting and was diluted 1:1 with modified RIPA buffer (1x PBS, 5 mM EDTA, 1% NP40, 1% deoxycholate, 2% SDS) vortexed and centrifuged at 20 000g for 10 min at RT.
The other half of each sample would be used for Aβ extraction, which began by centrifugation at 100 000g for 30 min at 4°C. Supernatant was saved as the TBS soluble fraction. The pellet was resuspended in an equal volume of TBS containing 1% Triton X-100 and mixed by gentle rotation at 4°C for 30 min. Samples were centrifuged at 100 000g for 30 min at 4°C, and the supernatant saved as the TBS-X soluble fraction. The pellet was resuspended in 50 mM Tris pH 6.8 containing 5 M guanidine hydrochloride and mixed by gentle rotation overnight at RT. Samples were centrifuged at 16 000g for 30 min at RT, and the supernatant saved as the guanidine soluble fraction.
Immunoblotting
A total of 7.5 μg of protein per sample was denatured in Laemmli sample buffer at 95°C for 5 min and electrophoresed on 4–15% Criterion Tris-HCl gels (Bio-Rad, #3450028). Proteins were transferred to nitrocellulose using the Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked in TBS containing 0.1% Tween-20 and 5% non-fat dry milk for 1 hr at RT and probed overnight at 4°C with primary antibody diluted in blocking solution: 6E10 (Biolegend, San Diego, CA, #SIG-39320, 1:5000), Y188 (Abcam, Cambridge, UK, #ab32136, 1:5000), 22C11 (Millipore, #MAB348, 1:3000), phospho-APP Thr668 (Cell Signaling, #6986, 1:1000), GSKβ (Cell Signaling #12456, 1:1000), phospho-GSK3β Ser9 (Cell Signaling #9322, 1:1000), PKCβII (ThermoFisher #PA5–13741, 1:250), PKCα (Abcam #ab32376, 1:5000) or GAPDH (Millipore #AB2302, 1:10,000). Primary antibodies were detected using anti-IgG secondary antibodies conjugated with IRDye (LI-COR, Lincoln, NE). Blots were imaged with an Odyssey Fc Imager and analyzed with Image Studio software (LI-COR).
Meso Scale Discovery assay
The concentration of human Aβ40 and 42 in guanidine cortical extracts was measured using Multiplex Aβ Peptide Panel 1 (Meso Scale Discovery, Rockville, MD, #K15200E). The assay was performed essentially as instructed by the manufacturer. Samples were diluted to stay within the linear range of the assay, requiring dilution of 1:1000 at 4 weeks of age, 1:5000 at 12 weeks and 1:10000 at 6 months. Initial dilution from the guanidine fraction was done in PBS containing 1% protease-free BSA (MP Biomedical, Santa Ana, CA #820451). The final working dilution was prepared in Diluent 35 included in the kit; Aβ blocking reagent was not used. Samples were read on a SECTOR Imager 6000 and concentrations calculated using Discovery Workbench software (Meso Scale Discovery).
Statistical Analysis
Statistical comparisons were done using Prism 6.0 (GraphPad, La Jolla, CA). Comparisons of three or more groups was done by 1-way or 2-way ANOVA followed by Bonferroni post hoc testing, while comparisons between two groups were done using 2-tailed Student’s t-test. All bar graphs display mean ± SEM.
Acknowledgements
We are grateful to Rebecca Corrigan and M. Danish Uddin for careful management of the APP/TTA mouse colony, Maxime Rousseaux, Paymaan Jafar-Nejad and Jeehye Park for their help with the cell-based screen, Kazuhiro Oka and Shuyun Deng and the Baylor College of Medicine Gene Vector Core for AAV production, Kira Chen for original artwork in Figure 4 and Gopal Thinakaran for the gift of N2a-APP cell lines.
Conflict of Interest statement. None declared.
Funding
Robert A. and Renee E. Belfer Family Foundation (to J.L.J., J.B. and H.Y.Z.); National Institutes of Health (R01 NS092615 to J.L.J., NIH T32 NS043124 support for C.H.H., NIH P30 CA125123 support for the mass spectroscopy core, and NIH U54 HD083092 to HYZ); BrightFocus Foundation (A2015016F to S.D.G., A2016151S to J.B. and H.Y.Z.); CPRIT Core Facility Award (RP120092).
References
- 1. Cerasoli E., Ryadnov M.G. and Austen B.M. (2015) The elusive nature and diagnostics of misfolded Aβ oligomers. Front. Chem., 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kummer M.P. and Heneka M.T. (2014) Truncated and modified amyloid-beta species. Alzheimers Res. Ther., 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar D., Ganeshpurkar A., Kumar D., Modi G., Gupta S.K. and Singh S.K. (2018) Secretase inhibitors for the treatment of Alzheimer's disease: long road ahead. Eur. J. Med. Chem., 148, 436–452. [DOI] [PubMed] [Google Scholar]
- 4. Tamayev R. and D'Adamio L. (2012) Memory deficits of British dementia knock-in mice are prevented by Aβ-precursor protein haploinsufficiency. J. Neurosci., 32, 5481–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zheng H., Minghao J., Trumbauer M.E., Sirinathsinghji D.J.S., Hopkins R., Smith D.W., Heavens R.P., Dawson G.R., Boyce S., Conner M.W. et al. (1995) β-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell, 81, 525–531. [DOI] [PubMed] [Google Scholar]
- 6. Muller U., Cristina N., Li Z.W., Wolfer D.P., Lipp H.P., Rulicke T., Brandner S., Aguzzi A. and Weissmann C. (1994) Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell, 79, 755–765. [DOI] [PubMed] [Google Scholar]
- 7. Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M. et al. (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet., 38, 24–26. [DOI] [PubMed] [Google Scholar]
- 8. Park J., Al-Ramahi I., Tan Q., Mollema N., Diaz-Garcia J.R., Gallego-Flores T., Lu H.C., Lagalwar S., Duvick L., Kang H. et al. (2013) RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature, 498, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lasagna-Reeves C.A., Haro M., Hao S., Park J., Rousseaux M.W., Al-Ramahi I., Jafar-Nejad P., Vilanova-Velez L., See L., De Maio A. et al. (2016) Reduction of Nuak1 decreases tau and reverses phenotypes in a tauopathy mouse model. Neuron, 92, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rousseaux M.W., Haro M., Lasagna-Reeves C.A., De Maio A., Park J., Jafar-Nejad P., Al-Ramahi I., Sharma A., See L., Lu N. et al. (2016) TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. eLife, 5, e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rousseaux M.W.C., Vazquez-Velez G.E., Al-Ramahi I., Jeong H.H., Bajic A., Revelli J.P., Ye H., Phan E.T., Deger J.M., Perez A.M. et al. (2018) A druggable genome screen identifies modifiers of α-synuclein levels via a tiered cross-species validation approach. J. Neurosci., 38, 9286–9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mochly-Rosen D., Das K. and Grimes K.V. (2012) Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov., 11, 937–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B. et al. (2008) Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U. S. A., 105, 5868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin J.N., Wolken N., Brown T., Dauer W.T., Ehrlich M.E. and Gonzalez-Alegre P. (2011) Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther., 18, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gestel M.A., Erp S., Sanders L.E., Brans M.A., Luijendijk M.C., Merkestein M., Pasterkamp R.J. and Adan R.A. (2014) shRNA-induced saturation of the microRNA pathway in the rat brain. Gene Ther., 21, 205–211. [DOI] [PubMed] [Google Scholar]
- 16. Silva J.M., Li M.Z., Chang K., Ge W., Golding M.C., Rickles R.J., Siolas D., Hu G., Paddison P.J., Schlabach M.R. et al. (2005) Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet., 37, 1281–1288. [DOI] [PubMed] [Google Scholar]
- 17. Meerbrey K.L., Hu G., Kessler J.D., Roarty K., Li M.Z., Fang J.E., Herschkowitz J.I., Burrows A.E., Ciccia A., Sun T. et al. (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A., 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Passini M.A. and Wolfe J.H. (2001) Widespread gene delivery and structure-specific patterns of expression in the brain after intraventricular injections of neonatal mice with an adeno-associated virus vector. J. Virol., 75, 12382–12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim J.Y., Ash R.T., Ceballos-Diaz C., Levites Y., Golde T.E., Smirnakis S.M. and Jankowsky J.L. (2013) Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. Eur. J. Neurosci., 37, 1203–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim J.-Y., Grunke S.D., Levites Y., Golde T.E. and Jankowsky J.L. (2014) Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J. Vis. Exp., 91, e51863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gandy S.E., Caporaso G.L., Buxbaum J.D., Cruz Silva O., Iverfeldt K., Nordstedt C., Suzuki T., Czernik A.J., Nairn A.C. and Greengard P. (1993) Protein phosphorylation regulates relative utilization of processing pathways for Alzheimer beta/A4 amyloid precursor protein. Ann. N. Y. Acad. Sci., 695, 117–121. [DOI] [PubMed] [Google Scholar]
- 22. Gandy S., Czernik A.J. and Greengard P. (1988) Phosphorylation of Alzheimer disease amyloid precursor peptide by protein kinase C and Ca2+/calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. U. S. A., 85, 6218–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caporaso G.L., Gandy S.E., Buxbaum J.D., Ramabhadran T.V. and Greengard P. (1992) Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A., 89, 3055–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki T., Nairn A.C., Gandy S.E. and Greengard P. (1992) Phosphorylation of Alzheimer amyloid precursor protein by protein kinase C. Neuroscience, 48, 755–761. [DOI] [PubMed] [Google Scholar]
- 25. Thinakaran G., Teplow D.B., Siman R., Greenberg B. and Sisodia S.S. (1996) Metabolism of the ``Swedish’’ amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the ``beta-secretase” site occurs in the golgi apparatus. J. Biol. Chem., 271, 9390–9397. [DOI] [PubMed] [Google Scholar]
- 26. Porter D.C., Moy G.W. and Vacquier V.D. (1988) CAMP-dependent protein kinase of sea urchin sperm phosphorylates sperm histone H1 on a single site. J. Biol. Chem., 263, 2750–2755. [PubMed] [Google Scholar]
- 27. Rodgers S.P., Born H.A., Das P. and Jankowsky J.L. (2012) Transgenic APP expression during postnatal development causes persistent locomotor hyperactivity in the adult. Mol. Neurodegener., 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chiang A.C.A., Fowler S.W., Reddy R., Pletnikova O., Troncoso J.C., Sherman M.A., Lesne S.E. and Jankowsky J.L. (2018) Discrete pools of oligomeric amyloid-β track with spatial learning deficits in a mouse model of Alzheimer amyloidosis. Am. J. Pathol., 188, 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deyts C., Thinakaran G. and Parent A.T. (2016) APP receptor? To be or not to be. Trends Pharmacol. Sci., 37, 390–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Su Y., Ryder J., Li B., Wu X., Fox N., Solenberg P., Brune K., Paul S., Zhou Y., Liu F. et al. (2004) Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry, 43, 6899–6908. [DOI] [PubMed] [Google Scholar]
- 31. Rockenstein E., Torrance M., Adame A., Mante M., Bar-on P., Rose J.B., Crews L. and Masliah E. (2007) Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci., 27, 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qing H., He G., Ly P.T., Fox C.J., Staufenbiel M., Cai F., Zhang Z., Wei S., Sun X., Chen C.H. et al. (2008) Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J. Exp. Med., 205, 2781–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dimitriadi M., Sleigh J.N., Walker A., Chang H.C., Sen A., Kalloo G., Harris J., Barsby T., Walsh M.B., Satterlee J.S. et al. (2010) Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet., 6, e1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rousseaux M.W., Revelli J.P., Vazquez-Velez G.E., Kim J.Y., Craigen E., Gonzales K., Beckinghausen J. and Zoghbi H.Y. (2018) Depleting Trim28 in adult mice is well tolerated and reduces levels of alpha-synuclein and tau. eLife, 7, e36768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Y.A., Zhou B., Wernig M. and Sudhof T.C. (2017) ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell, 168, 427–441.e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo C., Yang Z.H., Zhang S., Chai R., Xue H., Zhang Y.H., Li J.Y. and Wang Z.Y. (2017) Intranasal lactoferrin enhances alpha-secretase-dependent amyloid precursor protein processing via the ERK1/2-CREB and HIF-1alpha pathways in an Alzheimer's disease mouse model. Neuropsychopharmacology, 42, 2504–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho H.J., Kim S.K., Jin S.M., Hwang E.M., Kim Y.S., Huh K. and Mook-Jung I. (2007) IFN-gamma-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia, 55, 253–262. [DOI] [PubMed] [Google Scholar]
- 38. Zhang H., Gao Y., Dai Z., Meng T., Tu S. and Yan Y. (2011) IGF-1 reduces BACE-1 expression in PC12 cells via activation of PI3-K/Akt and MAPK/ERK1/2 signaling pathways. Neurochem. Res., 36, 49–57. [DOI] [PubMed] [Google Scholar]
- 39. Kim S.K., Park H.J., Hong H.S., Baik E.J., Jung M.W. and Mook-Jung I. (2006) ERK1/2 is an endogenous negative regulator of the gamma-secretase activity. FASEB J, 20, 157–159. [DOI] [PubMed] [Google Scholar]
- 40. Silva O.A., Iverfeldt K., Oltersdorf T., Sinha S., Lieberburg I., Ramabhadran T.V., Suzuki T., Sisodia S.S., Gandy S. and Greengard P. (1993) Regulated cleavage of Alzheimer beta-amyloid precursor protein in the absence of the cytoplasmic tail. Neuroscience, 57, 873–877. [DOI] [PubMed] [Google Scholar]
- 41. Argellati F., Domenicotti C., Passalacqua M., Janda E., Melloni E., Marinari U.M., Pronzato M.A. and Ricciarelli R. (2009) Protein kinase C-dependent alpha-secretory processing of the amyloid precursor protein is mediated by phosphorylation of myosin II-B. FASEB J, 23, 1246–1251. [DOI] [PubMed] [Google Scholar]
- 42. Wang L., Shim H., Xie C. and Cai H. (2008) Activation of protein kinase C modulates BACE1-mediated beta-secretase activity. Neurobiol. Aging, 29, 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu H., Greengard P. and Gandy S. (1995) Regulated formation of Golgi secretory vesicles containing Alzheimer beta-amyloid precursor protein. J. Biol. Chem., 270, 23243–23245. [DOI] [PubMed] [Google Scholar]
- 44. Lai A., Sisodia S.S. and Trowbridge I.S. (1995) Characterization of sorting signals in the beta-amyloid precursor protein cytoplasmic domain. J. Biol. Chem., 270, 3565–3573. [PubMed] [Google Scholar]
- 45. Vieira S.I., Rebelo S., Domingues S.C., Silva E.F. and Silva O.A. (2009) S655 phosphorylation enhances APP secretory traffic. Mol. Cell Biochem., 328, 145–154. [DOI] [PubMed] [Google Scholar]
- 46. Vieira S.I., Rebelo S., Esselmann H., Wiltfang J., Lah J., Lane R., Small S.A., Gandy S., Cruz e Silva E.F. and Cruz e Silva O.A. (2010) Retrieval of the Alzheimer's amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol. Neurodegener., 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu-Zhang A.X. and Newton A.C. (2013) Protein kinase C pharmacology: refining the toolbox. Biochem. J., 452, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kinouchi T., Sorimachi H., Maruyama K., Mizuno K., Ohno S., Ishiura S. and Suzuki K. (1995) Conventional protein kinase C (PKC)-alpha and novel PKC epsilon, but not -delta, increase the secretion of an N-terminal fragment of Alzheimer's disease amyloid precursor protein from PKC cDNA transfected 3Y1 fibroblasts. FEBS Lett., 364, 203–206. [DOI] [PubMed] [Google Scholar]
- 49. Choi D.S., Wang D., Yu G.Q., Zhu G., Kharazia V.N., Paredes J.P., Chang W.S., Deitchman J.K., Mucke L. and Messing R.O. (2006) PKCepsilon increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. U. S. A., 103, 8215–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Du Y., Zhao Y., Li C., Zheng Q., Tian J., Li Z., Huang T.Y., Zhang W. and Xu H. (2018) Inhibition of PKCdelta reduces amyloid-beta levels and reverses Alzheimer disease phenotypes. J. Exp. Med., 215, 1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Canu N., Amadoro G., Triaca V., Latina V., Sposato V., Corsetti V., Severini C., Ciotti M.T. and Calissano P. (2017) The intersection of NGF/TrkA signaling and amyloid precursor protein processing in Alzheimer's disease neuropathology. Int. J. Mol. Sci., 18, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Diaz de la Loza M.C. and Thompson B.J. (2017) Forces shaping the Drosophila wing. Mech. Dev., 144, 23–32. [DOI] [PubMed] [Google Scholar]
- 53. Li Y., Xu M., Ding X., Yan C., Song Z., Chen L., Huang X., Wang X., Jian Y., Tang G. et al. (2016) Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol., 18, 1065–1077. [DOI] [PubMed] [Google Scholar]
- 54. Etcheberrigaray R., Tan M., Dewachter I., Kuiperi C., Auwera I., Wera S., Qiao L., Bank B., Nelson T.J., Kozikowski A.P. et al. (2004) Therapeutic effects of PKC activators in Alzheimer's disease transgenic mice. Proc. Natl. Acad. Sci. U. S. A., 101, 11141–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hongpaisan J., Sun M.K. and Alkon D.L. (2011) PKC epsilon activation prevents synaptic loss, Abeta elevation, and cognitive deficits in Alzheimer's disease transgenic mice. J. Neurosci., 31, 630–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Savage M.J., Trusko S.P., Howland D.S., Pinsker L.R., Mistretta S., Reaume A.G., Greenberg B.D., Siman R. and Scott R.W. (1998) Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci., 18, 1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Q., Yu S., Zhao W., Qin S., Chu Q. and Wu K. (2018) EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer, 17, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park K.W., Webster D.A., Stark B.C., Howard A.J. and Kim K.J. (2003) Fusion protein system designed to provide color to aid in the expression and purification of proteins in Escherichia coli. Plasmid, 50, 169–175. [DOI] [PubMed] [Google Scholar]
- 59. Ayuso E., Mingozzi F. and Bosch F. (2010) Production, purification and characterization of adeno-associated vectors. Curr. Gene Ther., 10, 423–436. [DOI] [PubMed] [Google Scholar]
- 60. Jankowsky J.L., Slunt H.H., Gonzales V., Savonenko A.V., Wen J.C., Jenkins N.A., Copeland N.G., Younkin L.H., Lester H.A., Younkin S.G. et al. (2005) Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer's disease. PLoS Med., 2, e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mayford M., Bach M.E., Huang Y.Y., Wang L., Hawkins R.D. and Kandel E.R. (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science, 274, 1678–1683. [DOI] [PubMed] [Google Scholar]
- 62. Kim J.Y., Grunke S.D. and Jankowsky J.L. (2016) Widespread neuronal transduction of the rodent CNS via neonatal viral injection. Methods Mol. Biol., 1382, 239–250. [DOI] [PubMed] [Google Scholar]