

Abstract
Genetic variation in melanocortin-1 receptor (MC1R) is a known contributor to disease-free red hair in humans. Three loss-of-function single-nucleotide variants (rs1805007, rs1805008 and rs1805009) have been established as strongly correlated with red hair. The contribution of other loss-of-function MC1R variants (in particular rs1805005, rs2228479 and rs885479) and the extent to which other genetic loci are involved in red hair colour is less well understood. Here, we used the UK Biobank cohort to capture a comprehensive list of MC1R variants contributing to red hair colour. We report a correlation with red hair for both strong-effect variants (rs1805007, rs1805008 and rs1805009) and weak-effect variants (rs1805005, rs2228479 and rs885479) and show that their coefficients differ by two orders of magnitude. On the haplotype level, both strong- and weak-effect variants contribute to the red hair phenotype, but when considered individually, weak-effect variants show a reverse, negative association with red hair. The reversal of association direction in the single-variant analysis is facilitated by a distinguishing structure of MC1R, in which loss-of-function variants are never found to co-occur on the same haplotype. The other previously reported hair colour genes’ variants do not substantially improve the MC1R red hair colour predictive model. Our best model for predicting red versus other hair colours yields an unparalleled area under the receiver operating characteristic of 0.96 using only MC1R variants. In summary, we present a comprehensive statistically derived characterization of the role of MC1R variants in red hair colour and offer a powerful, economical and parsimonious model that achieves unsurpassed performance.
Introduction
Melanocortin-1 receptor (MC1R) is a seven-transmembrane G protein–coupled receptor, encoded by the gene MC1R on 16q24.3 (1). Endogenously activated by the melanocyte-stimulating hormone and the adrenocorticotropic hormone, this receptor is a critical component of skin and hair pigment biosynthesis. Upon ligand binding, it signals for the activation of adenylyl cyclase, which increases cyclic adenosine monophosphate (cAMP) production and leads to the assembly of a multi-protein complex, stabilized by the P gene protein (2). The multi-protein complex is directly responsible for the conversion of precursor DOPAquinone to eumelanin, otherwise preferentially converted to phaeomelanin (3,4). The eumelanin-to-phaeomelanin ratio determines skin and hair colour in humans and coat colour in other mammals (4–7).
A connection between non-synonymous polymorphisms in MC1R-encoding gene and human hair colour was first established by Valverde and colleagues in 1995 (6). In the compendium of literature on MC1R variants and human pigmentation published during the past 22 years, several conclusions are apparent: 1. all human MC1R functionally characterized nonsynonymous variants confer a loss of function (of varying degrees); 2. the variants with the highest functional effect demonstrated in vitro confer red hair colour and sun-sensitive skin with poor tanning ability; 3. these traits are expressed on different genetic backgrounds and skin pigmentation profiles indigenous to different geographic locations; 4. the model of inheritance for these pigmentation phenotypes is recessive with a dose-dependent effect, i.e. simple heterozygotes may exhibit a shade of red that lies between wild-type and variant homozygote or compound heterozygote extremes (8,9); 5. MC1R variants are necessary to express a disease-free red hair phenotype (10).
Although MC1R is an unusually polymorphic gene (11), only two of its high-penetrance variants, rs1805007 and rs1805008 (amino acid changes R151C and R160W, respectively), are prevalent across all populations studied for red hair (12–15). Additionally, rs1805009 (D294H) has a noticeable presence in the British Isles and in the Netherlands (6,12,16). Aside from these three variants, six rare high-penetrance variants have been observed in various populations: rs1805006 (D84E), rs11547464 (R142H), rs1110400 (I155T), rs312262906 (N29insA, merged into rs796296176), rs555179612 (179InsC) and rs201326893 (Y152OCH) (8,16,17). These nine variant alleles have been nicknamed ‘RHC’ or ‘R’ alleles to denote their high penetrance and strong association with the red hair colour (13), and individuals who are homozygous or compound heterozygous exhibit pure red hair in up to 96% of cases (18). Furthermore, three nonsynonymous low-penetrance common MC1R variants—rs1805005, rs2228479 and rs885479 (V60L, V92M and R163Q, respectively)—have been reported and designated ‘r’ alleles (19). These vary in their minor allele frequency (MAF) across different populations and have been found to have a correlation with red hair ranging from weak (19–21) to none (8,13). All 12 variants are marked in the MC1R schematic (Fig. 1).
Figure 1.
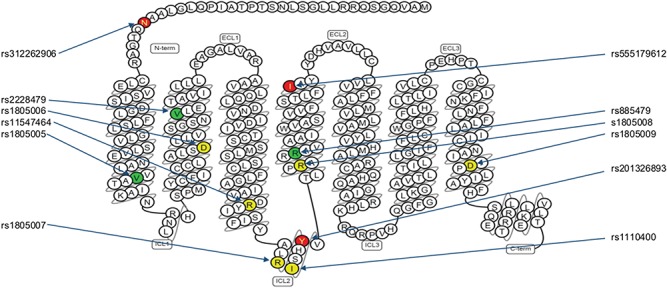
Previously reported variants in MC1R associated with red hair. The colour-coded circles in the figure correspond to the wild-type amino acid residues that are changed in the variants at the positions and to the resulting residues as specified here. High-penetrance R variants are in yellow: D84E (rs1805006), R142H (rs11547464), R151C (rs1805007), I155T (rs1110400), R160W (rs1805008) and D294H (rs1805009), and low-penetrance r variants are in green: V60L (rs1805005), V92M (rs2228479) and R163Q (rs885479). Frameshift variants N29insA (rs312262906), 179insC (rs555179612) and Y152OCH (rs201326893) are in red. Image courtesy of GPCRdb.org.
MC1R variant distribution differs widely between different parts of the world. The highest frequency of R alleles is observed in Northern Europe, whereas in more sun-exposed geographic regions, R alleles are very rare. Nevertheless, red-haired carriers of R alleles have been reported among European descendants in South Africa (22) and Australia (19), darker-skinned Southern Europeans (15), a darker-skinned Mongolian family (23) and black Jamaicans (14). On the other hand, r variants rs2228479 and rs885479, which are not known to have a strong effect on red hair, appear to be highly prevalent, reaching frequencies up to 73% in East Asia (24,25).
Several studies have reported cellular assays showing functional impairment for versions of MC1R carrying the common six variants: rs1805007, rs1805008, rs1805009, rs1805005, rs2228479 and rs885479 (26,27). While these studies diverge on the extent of some functional effects, there is consensus on the receptor’s signalling for cAMP production, in which the first three (R) variants confer considerable impairment and the latter three (r) show milder effects (27–29). The variants have also been classified in silico according to their cross-species conservation (SIFT) and according to their predicted structural alterations (PolyPhen) as tolerant and intolerant, with R alleles predictably falling in the latter category (30,31). The only two known complete loss-of-function (or null) variants are rs312262906 and rs555179612 (17).
Here, we sought to exploit the statistical power of the 500 000 individuals in the UK Biobank (UKBB) (32) to answer several outstanding questions regarding red hair genetics: 1. whether nonsynonymous (amino-acid-changing) coding-region MC1R variants are the primary effectors of the red hair phenotype, 2. whether r variants have any quantifiable contribution to this phenotype, 3. whether variants in other genes have any contribution to red hair beyond MC1R and 4. whether MC1R variants have an effect on other hair colours. In addition, we aimed to develop a high-powered statistical model trained on this data set using only MC1R variants as predictors.
Results
Association analysis for MC1R variants and red hair
First, we sought to confirm the previously reported variants as the primary effectors of the red hair phenotype. We used the minimum redundancy maximum relevance (mRMR) algorithm to determine whether the explanatory power for this phenotype (red versus dark hair) lay primarily in the coding region and with nonsynonymous variants. Although the algorithm was run on all imputed MC1R variants, the 10 top variants in the output (Table 1) were indeed nonsynonymous coding region variants, 8 missense (rs1805007, rs1805008, rs1805009, rs2228479, rs11547464, rs885479, rs1805006 and rs1805005) and 2 frameshift (rs312262906 and rs555179612) (Fig. 1). Seven of these variants (rs1805007, rs1805008, rs1805009, rs312262906, rs11547464, rs1805006 and rs555179612) have been previously reported as high penetrance (R) and the remaining three (rs2228479, rs885479 and rs1805005) as low penetrance (r). Thus, all the known common r variants and seven of nine known R variants were selected by our mRMR algorithm among the top discriminators of red hair.
Table 1.
mRMR-ranked MC1R variants in red versus dark hair colour
| mRMR rank | Variant | Function | MAF | Info score | Penetrance | mRMR score | |
|---|---|---|---|---|---|---|---|
| 1 | rs1805007 | Missense | 1.03e-2 | 1 | R | 1.28e-1 | |
| 2 | rs1805008 | Missense | 8.3e-2 | 1 | R | 3.58e-2 | |
| 3 | rs1805009 | Missense | 2.8e-2 | 0.93 | R | 2.17e-2 | |
| 4 | rs2228479 | Missense | 9.7e-2 | 1 | r | 8.4e-3 | |
| 5 | rs312262906 | Frameshift | 5.5e-3 | 0.82 | R | 7.1-e3 | |
| 6 | rs11547464 | Missense | 7.2e-3 | 1 | R | 4.4e-3 | |
| 7 | rs885479 | Missense | 4.6e-2 | 1 | r | 4.1e-3 | |
| 8 | rs1805006 | Missense | 1.22e-2 | 1 | R | 3.56e-3 | |
| 9 | rs555179612 | Frameshift | 1.93e-3 | 1 | R | 3.25e-3 | |
| 10 | rs1805005 | Missense | 1.11e-1 | 1 | r | 2.45e-3 |
The designations R, high-penetrance, and r, low-penetrance, are based on previously reported associations with red hair. Info score is a measure of imputation quality.
Second, we passed each of these variants and the two other previously reported R alleles (rs201326893 and rs1110400) to a generalized linear model (GLM) to ascertain the probability of the dichotomous outcome—red versus dark hair—as well as determine the direction of their effect on red hair. The results are presented in Table 2. In confirmation of the well-established recessive model of inheritance for red hair, the effect size of R variants is substantially larger for the recessive model than for the additive model. While previous publications have disagreed on the effect of common r variants (rs1805005, rs2228479 and rs885479), with most reporting them as either silent or weakly associated with red hair, our results show for r alleles—surprisingly—a negative, significant correlation with red hair (and therefore positive correlation with dark hair), and for R alleles, the expected positive, significant correlation with red hair.
Table 2.
GLM output for single-variant associations with red versus dark hair
| Additive model | Recessive model | ||||||
|---|---|---|---|---|---|---|---|
| Variant | MAF | Info score | Penetrance | Effect (OR) | P-value | Effect (OR) | P-value |
| rs312262906 | 5.51e-03 | 0.82 | R | 9.95 | <2e-16 | NA | NA |
| rs1805005 | 1.11e-01 | 1 | r | 0.3446 | <2e-16 | 0.1036 | <2e-16 |
| rs1805006 | 1.22e-02 | 1 | R | 3.477 | <2e-16 | 10.63 | 2.81e-9 |
| rs2228479 | 9.72e-02 | 1 | r | 0.1086 | <2e-16 | 0.03357 | <2e-16 |
| rs11547464 | 7.16e-03 | 1 | R | 4.67 | <2e-16 | 346.7 | 2.30e-8 |
| rs1805007 | 1.03e-01 | 1 | R | 12.74 | <2e-16 | 272.1 | 0 |
| rs1805008 | 8.25e-02 | 1 | R | 5.119 | <2e-16 | 35.57 | 0 |
| rs885479 | 4.62e-02 | 1 | r | 0.163 | <2e-16 | 0.100 | 1.99e-8 |
| rs1805009 | 2.78e-02 | 0.93 | R | 6.658 | <2e-16 | 648.1 | <2e-16 |
| rs555179612 | 1.93e-03 | 1 | R | 10.56 | <2e-16 | NA | NA |
| rs201326893 | 2.56e-04 | 1 | R | 10.12 | <2e-16 | NA | NA |
| rs1110400 | 1.08e-02 | 0.98 | R | 1.320 | 4.96e-9 | 0.684 | 0.607 |
The designations R, high-penetrance, and r, low-penetrance, are based on previously reported associations with red hair. Effect size, here, OR > 1 denotes a positive association with red hair, and OR < 1 denotes a negative association with red hair. Association statistics are listed as NA, not available, for the recessive model for rare frameshift variants because there were no individuals homozygous for the minor allele at these variants. Info score is a measure of imputation quality.
Assessment of independent effects of r alleles
Next, we tested if the haplotypic structure of MC1R could be a possible explanation for the negative correlation between r alleles and red hair in the above analysis. The variants in the coding region have been reported to exhibit almost no pairwise linkage disequilibrium (LD, as measured by r-squared) in individuals with European ancestry (33). To reproduce and explore this finding, we visualized the LD pattern with variants included in Table 2 in Haploview (Fig. 2). Of note, frameshift variants rs312262906, rs555179612 and rs201326893 had insufficient MAFs (5.5e-3, 1.93e-3 and 2.56e-4) to be included in LD determination by Haploview. While, consistent with previous reports, we did not observe any correlation as measured by r-squared (diamond colour in the LD plot, Fig. 2), we did see strong LD between all included variants using the D’ measure (value inside the diamond, Fig. 2), which denotes the pairwise LD coefficient with the range unaffected by the difference in MAFs (r can range from −1 to 1 only when both MAFs are the same). Our results indicate that given all existing ≥1%-frequent haplotypes in this region (Table 3), no two variants’ minor alleles co-occur on the same haplotype/chromosome. It follows that having any one variant allele precludes the possibility of having another one on the same chromosome. By extension, being heterozygous for an r variant allele means at most being heterozygous for one R variant allele, and being homozygous for an r effectively nullifies the chance of having any R variants and therefore drastically reduces the chance of having red hair. This finding suggests that the negative association coefficient for r variants may be indicative of the absence of R minor alleles rather than of their own direct effect on red or dark hair.
Figure 2.
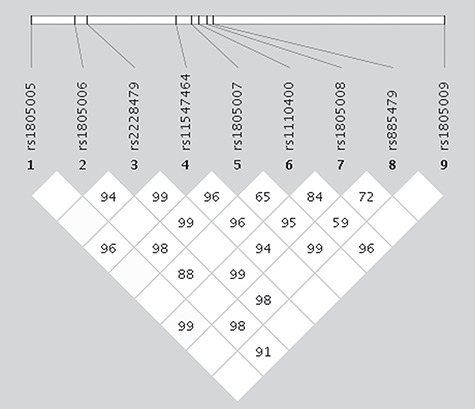
LD plot for 12 MC1R variants. Metric coding: value in diamonds = D′; colour scheme: r-squared (white = 0, red = 1 [here none]).
Table 3.
Haplotypes of MC1R variants above MAF 0.001
| Haplotype | rs312262906 | rs1805005 | rs1805006 | rs2228479 | rs11547464 | rs1805007 | rs1805008 | rs885479 | rs1805009 | rs555179612 | rS201326893 | rs1110400 | HF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 2 3 4 5 6 7 8 9 10 11 12 |
C C C C C C C C C C CA |
G T G G G G G G G G G |
C C C C C C C A C C C |
G G A G G G G G G G G |
G G G G G G G G G A G |
C C C T C C C C C C C |
C C C C T C C C C C C |
G G G G G A G G G G G |
G G G G G G C G G G G |
T T T T T T T T T T T |
C C C C C C C C C C C |
T T T T T T T T C T T |
5.0e-1 1.2e-1 9.5e-2 8.9e-2 7.3e-2 4.9e-2 2.2e-2 1.3e-2 1.1e-2 7.0e-3 4.2e-3 |
| C | G | C | G | G | C | C | G | G | TC | C | T | 1.6e-3 |
In each haplotype, the minor allele is highlighted in boldface-italics. The top haplotype is wild type. Every other haplotype carries only one variant’s minor allele. HF is haplotype frequency.
In this branch of investigation, it remained to answer the question whether the contribution of r alleles to red hair was truly negative or whether it was positive but dwarfed by that of R variants. To this end, we ran two types of analyses to test for the independent contribution of r variants to the red hair phenotype. First, we ran a haplotype association analysis, and second, we ran regression on r allele count while holding the count of R minor alleles constant. Haplotype association analysis showed that each haplotype was positively associated with red hair (Table 4). In other words, each variant’s minor allele, whether R or r, on a wild-type background of all other variants’ major alleles in this haploblock was correlated with red hair. However, based on odds ratios (ORs), r variant contribution to red hair is up to two orders of magnitude lower than R variant contribution.
Table 4.
Haplotype associations with red versus dark hair
| Variant | Frequency | Effect (OR) | P-value |
|---|---|---|---|
| 1 | 5.0e-1 | 0.89 | <2.0e-16 |
| 2 | 1.2e-1 | 3.36 | <2.0e-16 |
| 3 | 9.5e-2 | 0.91 | 0.070 |
| 4 | 8.9e-2 | 143.74 | <2.0e-16 |
| 5 | 7.3e-2 | 75.19 | <2.0e-16 |
| 6 | 4.9e-2 | 0.92 | 0.256 |
| 7 | 2.21e-2 | 105.11 | <2.0e-16 |
| 8 | 1.25e-2 | 56.26 | <2.0e-16 |
| 9 | 1.12e-2 | 18.12 | 9.65e-9 |
| 10 | 8.7e-3 | 5.98 | <2.0e-16 |
| 10 | 7.0e-3 | 82.43 | <2.0e-16 |
| 11 | 4.2e-3 | 776.66 | <2.0e-16 |
| 13 | 1.8e-3 | 1.20 | <2.0e-16 |
| 12 | 1.6e-3 | 1004.25 | <2.0e-16 |
Effect size, here, OR > 1 denotes a positive association with red hair. The most frequent haplotype (no.1 in Table 3) was used as the baseline against which all variant haplotypes were compared.
In the second set of analyses, we analysed for association of r variant minor alleles in the separate subsets of people with the total count of all R variant alleles equalling 0 and 1 and compared them to the full sample. The results (Fig. 3A) show that with ≤1 minor allele at all R variants considered together, r allele count is positively associated with red hair. In other words, r alleles mildly contribute to red hair on the background of a wild-type homozygous or a single heterozygous R genotype. In fact, the effect size of r variant count is higher on the background one R copy, suggesting that, expectedly, the contribution of r variants to red hair colour is stronger in individuals who already have one R allele. On the other hand, as single-variant analysis already shows (Table 2), in the whole sample r allele count is negatively associated with red hair.
Figure 3.
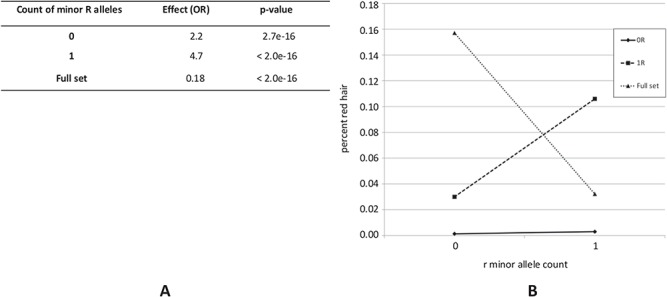
Interaction between r allele count and R allele count. Association for r allele count with red hair given an invariant R allele count in tabular (A) and graphical (B) format.
To visualize this relationship, we plotted the red hair frequency distribution as a function of r allele count separately in two collapsed R allele count groups, 0 and 1, and in the full sample (Fig. 3B). We can see that r allele count is positively correlated with red hair in both 0R and 1R groups but negatively correlated with red hair in the full sample.
Our results demonstrate that while individually the two variant classes contribute to red hair, the r variant contribution is substantially milder than that of R variants by comparison of the magnitude of their effect coefficients. We posit that the correlation structure between R and r variants, namely r-squared close to 0 and D′ close to 1, together with the high discrepancy in magnitude of effect, masks the true direction of association for the weaker-effect variants in single-variant analysis. The underlying direction of effect is revealed when all relevant variants are accounted for in haplotype analysis or, as we see below (Best predictive MC1R-based model for red hair), using multivariate regression analysis.
Best predictive MC1R-based model for red hair
Next, we sought to construct a model with the minimal number of MC1R variants in a GLM that would have the most predictive power in determining the expression of red hair. For comparison, we compiled a list of previous publications reporting red hair prediction models (Table 5). We performed mRMR in a holdout set of 150 000 individuals to make an initial selection of variables, the top 10 of which were used in the most parsimonious GLMs (Table 6 and Supplementary Material, Table S1 for red versus dark). The area under the receiver operating characteristic (AUROC) curve values were 0.95 for red versus other and 0.97 for red versus dark. (To compare directly, we used the same 12 variants from 5 genes proposed in (41), which gave us an AUROC of 0.93 for red versus other and 0.96 for red versus dark.) Interestingly, while for the red versus dark comparison all MC1R variants have a positive association (OR > 1) with red hair, (Supplementary Material, Table S1) in red versus other, the association for the two r variants (rs2228479 and rs885479) is negative (OR < 1, Table 6), signalling an effect flip when lighter hair colours—blonde and light brown—are grouped together with dark. We also performed least absolute shrinkage and selection operator (LASSO) regression on the complete set of imputed MC1R variants to take advantage of the innate attribute selection and coefficient penalization of LASSO to minimize overfitting and maximize predictive performance. The LASSO models demonstrate the best performance we achieved in predictive modelling of the hair colour phenotype (Table 7 and Supplementary Material, Table S2 for red versus dark). The AUROC values for LASSO models were 0.96 for red versus other and 0.98 for red versus dark. In both non-LASSO GLMs, the top three parameters are still the common R variants—rs1805007, rs1805008 and rs1805009—followed by a combination of rarer R variants, three common r variants (rs2228479, rs885479 and rs1805005) and two frameshift R mutations (rs312262906 and rs555179612). In LASSO models, we see all the known R and r variants as well as several more nonsynonymous variants and variants from 5′ and 3′ untranslated regions (UTRs).
Table 5.
Previous models for red hair prediction
| Publication | Year | Phenotype | N | P | MC1R variants | Other variants | AUROC | Other metric |
|---|---|---|---|---|---|---|---|---|
| Grimes et al. (18) | 2001 | Red and auburn | 197 | 0.274 | 14 | NA | NA | 20.960 |
| Branicki et al. (34) | 2007 | Red and blonde-red | 184 | 0.410 | 32 | NA | NA | 20.975 |
| Branicki et al. (35) | 2007 | Red | 390 | 0.240 | 43 | NA | NA | 20.960 |
| Sulem et al. (75) | 2007 | Red | 56918 | 0.055 | 32 | NA | NA | 60.700 |
| Walsh et al. (56) | 2013 | Red, blonde-red and auburn | 1551 | 0.088 | 74 | 811 | NA | 90.800 |
| Branicki et al. (37) | 2011 | Red, blonde-red and auburn | 385 | 0.249 | 102 | 1111 | 0.90 | 12 |
| Walsh et al. (6) | 2014 | Red, blonde-red and auburn | 1601 | 0.085 | 1311 | 144 | 0.92 | NA |
| Sochtig et al. (41) | 2015 | Red, blonde-red and auburn | 605 | 0.14 | 155 | 167 | 0.94 | 17 |
| Caliebe et al. (39) | 2016 | Red tint | 400 | 0.31 | 3 | NA | 0.75 | 18 |
| Siewierska-Gorska et al. (42) | 2017 | Red and blonde-red | 186 | 0.24 | 3 | 19 | 0.84 | 20 |
| Hysi et al. (40) | 2018 | Red | 2115 015 | 22 | 238 | 24268 | 250.87; 260.84 | 270.35 |
N, sample size; P, red hair prevalence in the sample.
1rs312262906, rs555179612, rs1805006, rs11547464
2Precision for variant homozygous or compound heterozygous redheads
3rs1805007, rs1805008
4rs1805007, rs1805008, rs11547464
55704 Icelanders and 1214 Dutch
6Precision at 0.50 classification threshold
7rs201326893, rs312262906, rs1805006, rs11547464
8rs1042602 (TYR), rs4959270 (EXOC2), rs28777 (SLC45A2), rs683 (TYRP1), rs2402130 (SLC24A4), rs12821256 (KITLG), rs2378249 (ASIP), rs12913832 (HERC2), rs1800407 (OCA2), rs16891982 (SLC45A2), rs12203592 (IRF4)
9Multiple linear regression highest probability hair colour category + a model for binary hair colour shade (light/dark) prediction; these two models used to make the final prediction, red-hair prediction accuracy, reported here.
10Combined minor allele count (max. 2) at any of the high-penetrance ‘R’ variants (rs201326893, rs312262906, rs1805006, rs11547464, rs1805007, rs1805008, rs1805009) or low-penetrance ‘r’ variants (rs1805005, rs2228479, rs1110400, rs885479)
11rs12913832 (HERC2), rs12203592 (IRF4), rs1042602 (TYR), rs4959270 (EXOC2), rs28777 (SLC45A2), rs683 (TYRP1), rs1800407 (OCA2), rs2402130 (SLC24A4), rs12821256 (KITLG), rs16891982 (SLC45A2), rs2378249 (ASIP)
12Sensitivity 0.78, specificity 0.95, precision 0.84, negative predictive value 0.93; 0.86 AUC for LASSO model
13rs201326893, rs312262906, rs1805006, rs11547464, rs1805007, rs1805008, rs1805009, rs1805005, rs2228479, rs1110400, rs885479
14rs1042602 (TYR), rs4959270 (EXOC2), rs28777 (SLC45A2), rs683 (TYRP1)
15rs11547464, rs1805006, rs1805007, rs1805008, rs1805009
16rs28777 (SLC45A2), rs35264875 (TPCN2), rs1129038, rs12913832 (HERC2), rs4778138, rs7495174 (OCA2), rs12931267 (FANCA)
17Bayes classification
18Sensitivity 0.19; specificity 0.09; accuracy 0.74; heritability for rs1805007 0.14 and for rs1805008 0.07
19rs16891982 (SLC45A2), rs12913832 (HERC2), rs1800401 (OCA2))
20Sensitivity 0.67; specificity 0.93; accuracy 0.87; positive predictive value (PPV) 0.74; negative predictive value (NPV) 0.90
217291 QIMR (Brisbane Twin Nevus Study, Australian Twin Registry, and Tasmanian Eye Study) and 7724 RS (Rotterdam Study)
22QIMR 0.054, RS 0.031, UKBB 0.047
23rs1805006, rs11547464, rs1805007, rs1805008, rs1805009, rs1805005, rs2228479, rs1110400
25QIMR
26RS
27Heritability
Table 6.
Predictive multivariate GLM for red versus other hair colours
| SNP ID | Function | MAF | Info score | Penetrance | Effect (OR) | P-value |
|---|---|---|---|---|---|---|
| rs1805007 | Missense | 1.03e-1 | 1 | R | 78.78 | <2.0e-16 |
| rs1805008 | Missense | 8.3e-2 | 1 | R | 35.32 | <2.0e-16 |
| rs1805009 | Missense | 2.78e-2 | 0.93 | R | 92.36 | <2.0e-16 |
| rs2228479 | Missense | 4.7e-3 | 1 | r | 0.80 | 2.6e-04 |
| rs312262906 | Frameshift | 9.7e-2 | 0.82 | R | 192.76 | <2.0e-16 |
| rs11547464 | Missense | 7.2e-3 | 1 | R | 46.93 | <2.0e-16 |
| rs885479 | Missense | 4.6e-2 | 1 | r | 0.78 | 6.1e-04 |
| rs1805006 | Missense | 1.22e-2 | 1 | R | 31.37 | <2.0e-16 |
| rs555179612 | Frameshift | 1.93e-3 | 1 | R | 225.45 | <2.0e-16 |
| rs76337330 | 5′ UTR | 4.9e-3 | 0.98 | ND | 0.63 | 1.03e-10 |
The designations R, high-penetrance, and r, low-penetrance, are based on previously reported associations with red hair. Effect size, here, OR > 1 denotes a positive association with red hair, and OR < 1 denotes a negative association with red hair. Info score is a measure of imputation quality.
Table 7.
MC1R variants selected by the LASSO model for red versus other hair colour
| Variant | Function | MAF | Info score | RH association |
|---|---|---|---|---|
| rs1110400 | Missense | 1.08e-02 | 0.98 | Yes |
| rs11547464 | Missense | 7.16e-03 | 1 | Yes |
| rs148003355 | 5′ UTR | 2.72e-04 | 0.68 | No |
| rs1805005 | Missense | 1.11e-01 | 1 | Yes |
| rs1805006 | Missense | 1.22e-02 | 1 | Yes |
| rs1805007 | Missense | 1.03e-01 | 1 | Yes |
| rs1805008 | Missense | 8.25e-02 | 1 | Yes |
| rs1805009 | Missense | 2.78e-02 | 0.93 | Yes |
| rs199920775 | Synonymous | 1.76e-04 | 0.66 | No |
| rs200000734 | Missense | 5.44e-04 | 1 | Yes |
| rs200050206 | Missense | 6.50e-04 | 0.55 | No |
| rs201326893 | Frameshift | 2.56e-04 | 1 | Yes |
| rs202197434 | Frameshift | 1.47e-04 | 0.45 | No |
| rs2228478 | Nonsynonymous | 1.15e-01 | 0.996 | No |
| rs2228479 | Missense | 9.72e-02 | 1 | Yes |
| rs312262906 | Frameshift | 5.51e-03 | 0.82 | Yes |
| rs3212359 | 5′ UTR | 3.15e-01 | 0.99 | No |
| rs3212361 | 5′ UTR | 2.43e-01 | 0.99 | No |
| rs3212371 | 5′ UTR | 1.14e-01 | 0.99 | No |
| rs3212379 | 5′ UTR | 7.46e-03 | 0.87 | No |
| rs34158934 | Missense | 2.61e-04 | 0.63 | No |
| rs34474212 | Missense | 4.80e-05 | 0.92 | No |
| rs34490506 | Synonymous | 1.89e-04 | 0.53 | No |
| rs367985661 | Synonymous | 8.00e-06 | 0.09 | No |
| rs368507952 | Missense | 4.83e-04 | 1 | Yes |
| rs374423188 | Missense | 4.53e-05 | 0.42 | No |
| rs376670171 | Missense | 4.00e-05 | 0.31 | No |
| rs555179612 | Frameshift | 1.93e-03 | 1 | Yes |
| rs572754025 | 3′ UTR | 3.47e-05 | 0.41 | No |
| rs577907985 | 5′ UTR | 7.54e-04 | 0.53 | No |
| rs765283788 | 3′ UTR | 2.88e-04 | 0.86 | No |
| rs868197501 | 5′ UTR | 4.00e-05 | 0.75 | No |
| rs885479 | Missense | 4.62e-02 | 1 | Yes |
Effect size, here, OR > 1 denotes a positive association with red hair. UTR is untranslated region. Info score is a measure of imputation quality, and the ‘RH association’ column shows whether a red hair association had been previously published.
Use of MC1R genotypes to discriminate between non-red hair colours
Next, we constructed a series of models over pairwise dichotomous hair colour classes to determine whether MC1R genotype could predict other hair colours with appreciable power. GLMs were run starting with the top-ranked variant and successively adding other variants from the MC1R locus in decreasing order of mRMR score (data not shown). AUROC convergence plots are shown in Figure 4. While in all pairwise comparisons with red hair, models reached 0.90 AUROC with 10 or fewer variants, discrimination between other hair colours was poor, ranging from 0.55 to 0.68 AUROC.
Figure 4.
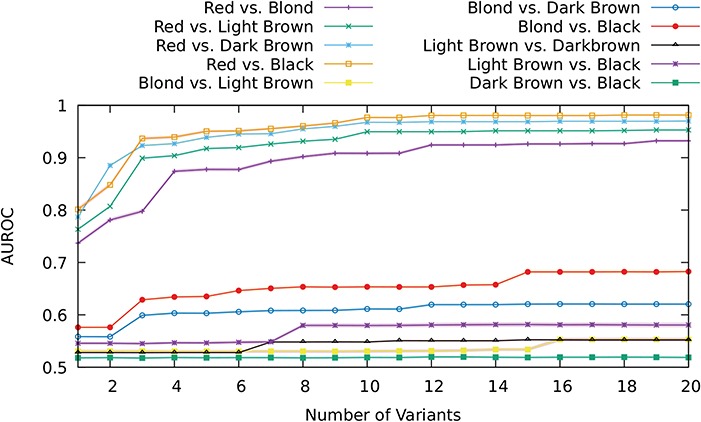
AUROC curves for all pairwise hair colour comparison GLMs with MC1R variants as predictors.
Contribution of other genes to red hair
To test for the contribution of other previously reported hair pigmentation genes above and beyond MC1R variants, we took the top 10 mRMR-scored MC1R predictor variants and combined them with all of the variants from these other genes (Table 8), one gene at a time. mRMR was then run on that combined set. The resulting top 10 variants from MC1R and from each other gene were used to produce Figure 5A (red versus other) and Supplementary Material, Figure S1A (red versus dark). Figure 5B (red versus other) and Supplementary Material, Figure S1B (red versus dark) were generated by taking the top 10 variants from MC1R and the top 100 variants from the other genes and passing them to LASSO, which further performed its own inherent subspace selection. The subspace of variants from the other genes was restricted only to improve LASSO computational time, but because of the low information of the remaining variants, excluding them from LASSO had no effect on the final models. In all cases, attribute selection was preformed strictly without knowledge of the testing set.
Table 8.
Previously reported hair colour genes
| Gene | Gene name | Citation |
|---|---|---|
| MC1R alone | Melanocortin-1 receptor | NA |
| ASIP | Agouti signaling protein | (53–54,56,71,75) |
| DCT | Dopachrome tautomerase | (58,76) |
| EDNRB | Endothelin receptor type B | (77) |
| HERC2 | HECT and RLD domain containing E3 ubiquitin protein ligase 2 | (37,38,39,47,48) |
| IRF4 | Interferon regulator factor 4 | (37,39,45,56) |
| KITLG | KIT ligand | (58,37,56,45) |
| MYO5A | Myosin VA | (58,76) |
| OCA2 | OCA2 melanosomal transmembrane protein | (37,48,38,42,47) |
| SLC24A4 | Solute carrier family 24 member 4 | (44,33,71,37,56,45,39) |
| SLC24A5 | Solute carrier family 24 member 5 | (76) |
| SLC45A2 | Solute carrier family 45 member 2 | (51,58,52,76,71,37,56,41) |
| TPCN2 | Two pore segment channel 2 | (75,45,41) |
| TYR | Tyrosinase | (37,48,56,72,10,58,71,36,48) |
| TYRP1 | Tyrosinase-related protein 1 | (37,73,74) |
Figure 5.
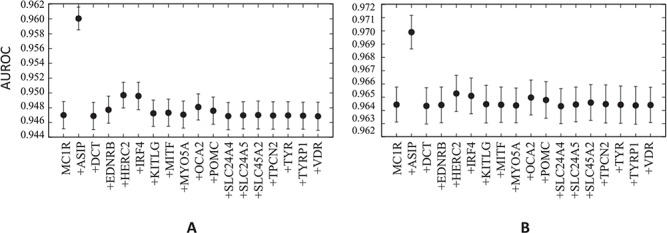
AUROC curves for (A) mRMR and (B) LASSO models using 10 top MC1R genetic variants and 10 top genetic variants from each gene for mRMR and 100 top genetic variants from each gene for LASSO. Red versus other hair colour. For both mRMR and LASSO, the model performance for all genes is statistically significantly different from the model using only MC1R variants. Despite the 10 iterations of 10-fold cross-validation to obtain an estimate of mean ROC performance, error bars for the 95% confidence interval are based on a standard error of the mean assuming a sample size of 10 rather than 100 due to lack of test set independence in folds between cross-validation iterations.
Figure 6 and Supplementary Material, Figure S2 show the relative importance of other genes over and above MC1R in determining red hair. Based on the statistically highly powered paired-sample t-test, we can confidently reject the null hypothesis that model performance with variants from other genes is the same as MC1R. In order to determine whether this difference is meaningful in terms of real predictive capacity, we compared the performance of our LASSO models to models using top 10 MC1R variants plus top 100 variants from 1000 randomly selected genes to measure predictive performance in red versus other hair colours (Fig. 6) and red versus dark hair colour (Supplementary Material, Fig. S2). The same subset of genes composes the data for both figures. It was constructed by taking the complete list of identified genes from the US National Center for Biotechnology Information (NCBI) database and performing a completely unbiased, pseudorandom selection of 1000 members. Several of the genes with a previously reported role in hair colour perform worse than the MC1R-only model, and their statistically significant deficiency is due to overfitting irrelevant noise in the training sets within the cross-validation procedure. Several that perform better lie within the 2 confidence interval for the distribution based on random genes. In red versus other hair colour prediction, ASIP, HERC2, OCA2 and IRF4, and to a lesser extent, POMC, SLC45A2 (and in red versus dark hair colour, ASIP, HERC2, OCA2, and to a lesser extent POMC, SLC45A2 and TYR), provide a lift to the models’ AUROC that lies outside the 2
confidence interval for the distribution based on random genes. In red versus other hair colour prediction, ASIP, HERC2, OCA2 and IRF4, and to a lesser extent, POMC, SLC45A2 (and in red versus dark hair colour, ASIP, HERC2, OCA2, and to a lesser extent POMC, SLC45A2 and TYR), provide a lift to the models’ AUROC that lies outside the 2 confidence interval. Even the best of the models including variants from another gene, MC1R with ASIP, is only a 0.57% improvement in AUROC for red hair versus other hair colour and 0.44% improvement in AUROC for red hair versus dark hair colour, which we deemed insufficient to sacrifice parsimony. In short, almost the entirety of the variation in the disease-free red hair phenotype is explained by MC1R variants alone; only 10 MC1R variants are sufficient to obtain the best predictive capacity yet reported, and 30 MC1R variants in a LASSO model can perform even better.
confidence interval. Even the best of the models including variants from another gene, MC1R with ASIP, is only a 0.57% improvement in AUROC for red hair versus other hair colour and 0.44% improvement in AUROC for red hair versus dark hair colour, which we deemed insufficient to sacrifice parsimony. In short, almost the entirety of the variation in the disease-free red hair phenotype is explained by MC1R variants alone; only 10 MC1R variants are sufficient to obtain the best predictive capacity yet reported, and 30 MC1R variants in a LASSO model can perform even better.
Figure 6.
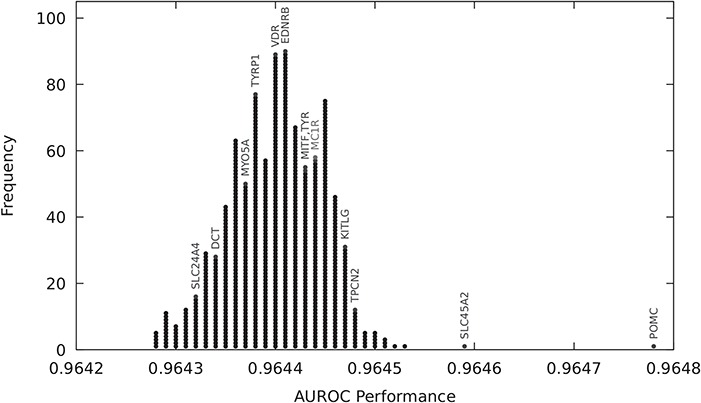
Red versus other hair colour prediction using LASSO models with 10 MC1R and 100 top mRMR-ranked variants from 1000 randomly selected genes. All the genes shown fall within 2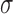 . ASIP, OCA2, IRF4 and HERC2 (not shown) have AUROC values 0.970, 0.965, 0.965 and 0.965, respectively, and are the only genes whose variants improve predictive performance above and beyond MC1R variants. The variants of these four genes and two other genes outperform MC1R-alone models with a statistically significant difference (t-test P-values: ASIP, <1e-16; HERC2, <1e16; OCA2, <1e-16; IRF4, <1e-16; POMC, 5.7e-10; SLC45A2, 8.3e-3).
. ASIP, OCA2, IRF4 and HERC2 (not shown) have AUROC values 0.970, 0.965, 0.965 and 0.965, respectively, and are the only genes whose variants improve predictive performance above and beyond MC1R variants. The variants of these four genes and two other genes outperform MC1R-alone models with a statistically significant difference (t-test P-values: ASIP, <1e-16; HERC2, <1e16; OCA2, <1e-16; IRF4, <1e-16; POMC, 5.7e-10; SLC45A2, 8.3e-3).
Discussion
Here, we present an in-depth analysis of the relationship between MC1R variants and the red hair phenotype, as well as report on an important caveat regarding the relativity of direction of genetic associations for single variants in the presence of a strong haplotypic structure, as exemplified by the MC1R gene. Testing common and rare variants across the entire gene locus has confirmed previous reports of nonsynonymous missense variants as the primary effectors of the red hair phenotype with additional contribution from frameshift mutations. However, although all prior studies agreed on the contribution of R alleles to red hair colour, the contribution of r variants has seen conflicting evidence. Most reports have shown either a weak association with red hair or no impact on hair colour for r alleles. Additionally, association between r alleles and dark hair (55,56) and darker skin (57) has been documented. Lastly, a negative association between r alleles and red hair also has precedents. One group reported that a comparison between r allele carriers (rs1805005 and rs2228479) and R allele carriers/wild-type group showed a correlation with lower red colour component in hair for the former (58), and two groups reported an OR < 1 for r with red hair: first Raimondi et al. in 2008 (46) and most recently, during the preparation of this manuscript, Morgan et al. (59), who also noted that this OR changed if R variants were included in the regression model.
Addressing conflicting previous reports regarding R and r variants, we determined that r variant alleles do contribute to red hair, although their contribution is much weaker by comparison to R variants. Haplotype analysis demonstrated that no two variant alleles among all R and r variants effectively co-occur; therefore, because a higher count of r alleles lowers the chance of R allele presence in a particular individual, regression on just r allele count misleadingly results in a negative association with red hair.
This illustrates an important drawback of variant-centered analysis in the presence of a strong haplotypic structure. A variant that is marginally protective relative to the rest of the sample population may in reality be deleterious on the background of ancestral haplotypes. The high LD between R and r variants, together with the high discrepancy in magnitude of effect, contribute to the observed effect reversal when diplotypes of the entire MC1R locus are reduced to essentially single variants with one of the alleles being r. This effect reversal is known as the flip-flop phenomenon (60,61), which may take place whenever there is a joint effect of multiple variants acting on a phenotype but only a subset of them is analysed for single-variant genetic association with the phenotype (62). Therefore, an important outcome of our study is the discovery that small-effect variants of the MC1R locus have their direction of effect flipped in single-variant association, which is rectified in multivariate analysis. This study thus exemplifies a phenomenon emergent in a large population with many rare genetic variants of strong effect size, in which the population phenotypic mean may become sufficiently elevated for the weaker rare-susceptibility variants to appear protective on the background of the overall prevalence of the phenotype. Caution is always advisable in interpreting the direction of effect in genome-wide association analyses without considering that joint effects of many loci may be at work.
We also tested MC1R variants in pairwise comparisons for all available hair colour phenotypes to determine their possible contribution to colours other than red. In addition to the associations with darker hair and skin for rs1805005 and rs2228479 mentioned above, prior publications have reported weak association for rs1805005 with blonde hair in (34,36,63). However, our results show that compared to red hair, the predictive power of MC1R variants for other hair colours is very weak (Fig. 4) and could not be reliably used for the purposes of identifying a missing individual’s phenotype. The higher AUROC for light–dark hair colour models compared to light–light and dark–dark could be explained by some overlap between strawberry blonde and blonde, as well as auburn with light brown, thereby giving some discriminatory power to the model for the red component in the latter hair colour in each pair. MC1R variant alleles are, expectedly, least informative in discriminating between dark brown and black hair, neither of which is likely to be contaminated by red hair colour.
Since 2001, MC1R variants have been exploited in forensic science to predict hair colour of missing individuals in police investigations. Of the relevant publications summarized in Table 5, the first two (18,34) relied on exclusively MC1R variants and contingency tables for red hair colour prediction. While their precision of 96% and 97.5%, respectively, for R homozygous or compound heterozygous genotypes predicting red hair is high, it is notable that their sample was enriched for red-haired individuals (27% and 41%, respectively) and not representative of the general population (2–5%). Thereafter, focus shifted from predicting only red hair to determining hair colour, and other genes were included (37,40–42,56,64). Among these reports, ones that used a more representative proportion of red-haired individuals (40,41,44,56,64) and AUROC as the performance metric (37,39,40,42,64), the best-performing model gave an AUROC of 0.94 for red versus other hair colour (41).
Harnessing the high-powered UKBB sample, we attempted to improve the predictive model for discriminating between red and non-red hair colour using only MC1R variants. While all previously published predictive models included variants from other genes and were nevertheless only able to obtain an AUROC of 0.94 for the red versus all other at best (41), our parsimonious GLM, which took only 10 MC1R variants as predictors, yielded an AUROC of 0.95 for red versus all other hair colours (Fig. 5A) and an AUROC of 0.97 for the most distinct class comparison, red versus dark (Supplementary Material, Fig. S1A). Our less parsimonious but still only MC1R-based LASSO model yielded an AUROC of 0.96 for red versus other (Fig. 5B) and 0.98 for red versus dark (Supplementary Material, Fig. S1B). Thus, our results show that it is possible to construct a model with near-perfect predictive capacity on MC1R variants alone.
Notwithstanding the AUROC values of 0.95 and 0.96 obtainable from MC1R variants in the red hair colour prediction using GLM and LASSO, respectively, we also checked whether adding variants from other genes might improve discrimination between red and dark hair. The addition of mRMR-ranked top variants from ASIP, HERC2, OCA2 and IRF4 did provide additional predictive capacity, while the addition of variants from other candidate genes was no better than randomly selected genes. The additional predictive capacity, although statistically significant, represented a mere 0.57% increase in AUROC in the best case (ASIP), which we do not interpret as phenotypically meaningful. A recent hair colour genome-wide association study, also done on the UKBB, by Morgan et al. (59) stipulates that including variants from eight other loci throughout the genome improves by 17% the heritability estimate obtained using only MC1R variants. These estimates use narrow-sense heritability (h2) and are therefore only sensitive to additive effects, which account for a fraction of the explanatory power of recessive and negatively linked MC1R variants.
A limitation of our study is that the phenotype of interest, hair colour, was obtained by self-report, and its identification could be refined by more objective, quantitative methods. However, by relying on subjective human determination of hair colour, we approximate a real-life situation in which this information would be based on observation rather than an objective pigment quantification method.
In conclusion, our findings may be summarized in five parts. First, we have identified an effect reversal in conventional single-variant analysis that could occur given multi-locus effects, high LD and large differences in effect size. Second, we have confirmed a positive independent association for each of the previously reported nonsynonymous MC1R variants with red hair and discovered the contribution of several synonymous variants to red hair colour. Third, we offer for the purposes of red hair colour identification—for example in a forensic setting—a robust and parsimonious predictive model with a superior performance metric of AUROC 0.95 for which only 10 MC1R variant loci are needed. An even better performance metric of 0.96 is obtainable by still only using LASSO-derived genetic variants within the short MC1R locus. Fourth, we have shown that MC1R does not contribute significantly to hair colours other than red. Lastly, we conclude that contribution from other hair-colour–related genes to red hair colour is negligible and posit MC1R as the sole substantial genetic contributor to red hair colour.
Materials and Methods
UKBB cohort
Our study cohort comes from the UKBB, a repository of genotypes and phenotypes from 500 000 participants aged 40–69, recruited between 2006 and 2010 (application 20802). Genotyping was done on one of two 95%-overlapping arrays—Affymetrix UK BiLEVE Axiom and Affymetrix UK Biobank Axiom—containing 820 000 single-nucleotide variants. For all analyses, we used the imputed genotypes for Caucasian individuals, as specified in the UKBB Data Field, hereafter DF, 22006. Quality control filters for heterozygosity rate (DF22010) as well as sex mismatch, variant call rate, unintended duplicates and outliers of >10 standard deviations in ancestry principal component analysis (PCA) (DF22051) were applied, and individuals who withdrew from the study were removed, yielding an effective number of 402 000 participants. Hair colour (DF1747) was provided by self-report. Participants were asked to select one of five choices to describe their natural hair colour before greying: blonde, red, light brown, dark brown or black.
Statistical analyses
Model parameters: genotypes, phenotypes and covariates
For the analyses described below, we used all available variants (post-imputation) for each gene, and gene locus boundaries were defined according to chromosomal boundaries provided in the Gene Database hosted by the US NCBI (65) Genome Reference Consortium Human genome build 37. Genotypes (one or more genetic variant minor allele counts) were used as independent variables and the phenotype (hair colour) as the dependent variable. Covariates were used as described below, and regression coefficients were transformed into ORs.
Association analysis: GLM
We used two different modelling paradigms consistent with distinct goals. Given that the first goal was to demonstrate the statistical significance and effect magnitude and direction of associations, we applied GLMs. Without a model evaluation step, there was no need for a testing set; therefore, we used the entire cohort (500000). Additionally, we used covariates (age, sex, recruitment site and 40 ancestry PC vectors) to account for population stratification and dichotomized the hair colour phenotype, ‘red versus dark’, where the ‘dark’ category comprised dark brown and black hair colours. Given that the goal for this association analysis was to isolate the effect of genetic variants on red hair, blonde and light brown were withheld to maximize phenotype homogeneity, thereby avoiding possible overlap between red and strawberry blonde or auburn brown hair colour, which have likewise been reported to be mediated by MC1R variants.
Predictive capacity assessment: mRMR and LASSO
For the second goal, which was to demonstrate the predictive capacity of our models, we ran all possible pairwise hair colour comparisons, as well as ‘red versus other’. For these analyses, we did not use covariates, given the intention to determine how well a restricted set of genetic variants alone could discriminate between possible hair colours or determine the donor’s hair colour to be red, with no other information provided, as may be the case in a forensic investigation. We performed attribute selection using two different methods: the mRMR algorithm (66) and the LASSO (67). mRMR is valuable as an attribute ranking algorithm, ordering potential predictors by mutual information with the class variable penalized by average mutual information with previously selected attributes. LASSO performs variable selection and regularization to combat over-fitting and produce parsimonious models with the aim to select an optimal covariate subspace. We used mRMR to demonstrate performance convergence of the prediction task with an increasing number of variants from MC1R and to filter the space of candidate variants to use in LASSO to decrease computational workload.
Training and testing sets
To avoid information leakage between attribute selection and subsequent model construction, we divided our data set consistently with the UKBB genotype release schedule. The initial release (2016)—data for 150 000 individuals—was used as a holdout set for mRMR analysis, and the model was constructed and evaluated using the remaining 350 000 individuals from the full release (2017). Specifically, we split the remaining 350 000 individuals into 10 different sets of 10 mutually exclusive folds and alternately used every agglomeration of 9 folds in each set to predict hair colour in the remaining fold. LASSO feature selection was done exclusively within its training data. For each class under consideration, cross-validation folds were invariant across models, so the repeated cross-validation provided 100 paired samples of any given performance metric. Though these samples were not strictly independent, they can be used for confidence interval construction and model comparison tests. The confidence intervals in Figure 5 and Supplementary Material, Figure S1 were thus derived, and comparisons between models using only MC1R variants and models also incorporating variants from other genes are based on a dependent t-test for paired samples across these 100 prediction subsets.
Model performance metric
In contrast to the studies that report threshold-dependent metrics, such as accuracy or precision, we use the AUROC curve as a model performance metric for the following reasons. First, the class balance ratio changes across predictive tasks depending on the implicated hair colours. The AUROC metric is invariant to the class balance ratio and can therefore be used as a common interpretable performance characteristic of models to produce informative visualizations, such as Figure 4, that meaningfully demonstrate the relative difficulty of the predictive task irrespective of class imbalance. Second, commonly used measures such as accuracy can be misleading, since trivial predictors can report near-optimal performance on highly imbalanced classes by always predicting the majority class. Hair colour class balance ratios can approach 50:1 and are therefore subject to apparent distortion in the accuracy metric when reporting binary class predictive performance. The third and final reason is that measures such as accuracy, precision and recall are derived from a single instantiation of the classification confusion matrix, and there are as many such legitimate instantiations as discrete probabilities in the predictive output. While it is typical to use the confusion matrix resulting from a P(+) > 0.5 probability threshold, we offer instead AUROC as a measure of performance that does not require us to make a potentially suboptimal choice in trade-offs such as sensitivity versus specificity, a choice best left in the hands of potential users of this information for practical purposes, such as in forensic investigations.
Software
Software to perform these analyses included R version 3.4.4 with ‘caret’ (68) and ‘glmnet’ packages (50). mRMR analyses were conducted with validated software written by R.N.L. to improve the mRMR (43,66) reference implementation and FastmRMR (69) for efficient and flexible usage on the UKBB data set. This software is publicly available on GitHub [https://github.com/rlichtenwalter/mRMR].
Haplotype association analysis was performed using the haplo.stats R (49) package. LD was visualized using the Haploview software (70).
Supplementary Material
Acknowledgements
The authors would like to thank Dr Samar Khoury for help with data extraction from the UK Biobank.
Conflict of Interest statement. None declared.
Funding
The Canada Excellence Research Chair (CERC) Program; the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
References
- 1. Gantz I., Yamada T., Tashiro T., Konda Y., Shimoto Y., Miwa H. and Trent J.M. (1994) Mapping of the gene encoding the melanocortin-1 (α-melanocyte stimulating hormone) receptor (MC1R) to human chromosome 16q24.3 by fluorescence in situ hybridization. Genomics, 19, 394–395. [DOI] [PubMed] [Google Scholar]
- 2. Akey J.M., Wang H., Xiong M., Wu H., Liu W., Shriver M.D. and Jin L. (2001) Interaction between the melanocortin-1 receptor and P genes contributes to inter-individual variation in skin pigmentation phenotypes in a Tibetan population. Hum. Genet., 108, 516–520. [DOI] [PubMed] [Google Scholar]
- 3. Ito S. and Wakamatsu K. (2008) Chemistry of mixed melanogenesis-pivotal roles of dopaquinone. Photochem. Photobiol., 84, 582–592. [DOI] [PubMed] [Google Scholar]
- 4. Robbins L.S., Nadeau J.H., Johnson K.R., Kelly M.A., Roselli-Rehfuss L., Baack E., Mountjoy K.G. and Cone R.D. (1993) Pigmentation phenotypes of variant extension locus alleles result from point mutations that alter MSH receptor function. Cell, 72, 827–834. [DOI] [PubMed] [Google Scholar]
- 5. Thody A.J. and Graham A. (1998) Does α-MSH have a role in regulating skin pigmentation in humans? Pigment Cell Melanoma Res., 11, 265–274. [DOI] [PubMed] [Google Scholar]
- 6. Valverde P., Healy E., Jackson I., Rees J.L. and Thody A.J. (1995) Variants of the melanocyte–stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat. Genet., 11, 328–330. [DOI] [PubMed] [Google Scholar]
- 7. Marklund L., Moller M.J., Sandberg K. and Andersson L. (1996) A missense mutation in the gene for melanocyte-stimulating hormone receptor (MC1R) is associated with the chestnut coat color in horses. Mamm. Genome, 7, 895–899. [DOI] [PubMed] [Google Scholar]
- 8. Flanagan N., Healy E., Ray A., Philips S., Todd C., Jackson I.J., Birch-Machin M.A. and Rees J.L. (2000) Pleiotropic effects of the melanocortin 1 receptor (MC1R) gene on human pigmentation. Hum. Mol. Genet., 9, 2531–2537. [DOI] [PubMed] [Google Scholar]
- 9. Branicki W., Kupiec T., Wolańska-Nowak P. and Brudnik U. (2006) Determination of forensically relevant SNPs in the MC1R gene. Int. Congr. Ser., 1288, Elsevier, pp, 816–818. [Google Scholar]
- 10. Pośpiech E., Wojas-Pelc A., Walsh S., Liu F., Maeda H., Ishikawa T., Skowron M., Kayser M. and Branicki W. (2014) The common occurrence of epistasis in the determination of human pigmentation and its impact on DNA-based pigmentation phenotype prediction. Forensic. Sci. Int. Genet., 11, 64–72. [DOI] [PubMed] [Google Scholar]
- 11. Ezzedine K., Mauger E., Latreille J., Jdid R., Malvy D.,Gruber F., Galan P., Hercberg S., Tschachler E. and Guinot C. (2013) Freckles and solar lentigines have different risk factors in Caucasian women. J. Eur. Acad. Dermatol. Venereol., 27, e345–e356. [DOI] [PubMed] [Google Scholar]
- 12. Smith R., Healy E., Siddiqui S., Flanagan N., Steijlen P.M., Rosdahl I., Jacques J.P., Rogers S., Turner R., Jackson I.J. et al. (1998) Melanocortin 1 receptor variants in an Irish population. J. Invest. Dermatol., 111, 119–122. [DOI] [PubMed] [Google Scholar]
- 13. Box N.F., Chen W., Sturm R.A., Duffy D.L., Irving R.E., Russell A., Griffyths L.R., Parsons P.G. and Green A.C. (2001) Melanocortin-1 receptor genotype is a risk factor for basal and squamous cell carcinoma. J. Invest. Dermatol., 116, 224–229. [DOI] [PubMed] [Google Scholar]
- 14. Harding R.M., Tomlinson J.B., Ray A.J., Wakamatsu K., Rees J.L. and McKenzie C.A. (2003) Phenotypic expression of melanocortin-1 receptor mutations in Black Jamaicans. J. Invest. Dermatol., 121, 207–208. [DOI] [PubMed] [Google Scholar]
- 15. Pastorino L., Cusano R., Bruno W., Lantieri F., Origone P., Barile M., Gliori S., Shepherd G.A., Sturm R.A. and Scarra G.B. (2004) Novel MC1R variants in Ligurian melanoma patients and controls. Hum. Mutat., 24, 103–103. [DOI] [PubMed] [Google Scholar]
- 16. Mengel-Jørgensen J., Eiberg H., Børsting C. and Morling N. (2006) Genetic screening of 15 SNPs in the MC1R gene in relation to hair colour in Danes. Int. Congr. Ser., 1288, Elsevier, pp, 55–57. [Google Scholar]
- 17. Beaumont K.A., Shekar S.N., Cook A.L., Duffy D.L. and Sturm R.A. (2008) Red hair is the null phenotype of MC1R. Hum. Mutat., 29. [DOI] [PubMed] [Google Scholar]
- 18. Grimes E.A., Noake P.J., Dixon L. and Urquhart A. (2001) Sequence polymorphism in the human melanocortin-1 receptor gene as an indicator of the red hair phenotype. Forensic Sci. Int., 122, 124–129. [DOI] [PubMed] [Google Scholar]
- 19. Sturm R., Duffy D., Box N., Newton R., Shepherd A., Chen W., Marks L., Leonard J. and Martin N. (2003) Genetic association and cellular function of MC1R variant alleles in human pigmentation. Ann. N. Y. Acad. Sci., 994, 348–358. [DOI] [PubMed] [Google Scholar]
- 20. Duffy D.L., Box N.F., Chen W., Palmer J.S., Montgomery G.W., James M.R., Hayward N.K., Martin N.G. and Sturm R.A. (2004) Interactive effects of MC1R and OCA2 on melanoma risk phenotypes. Hum. Mol. Genet., 13, 447–461. [DOI] [PubMed] [Google Scholar]
- 21. Cook A.L., Chen W., Thurber A.E., Smit D.J., Smith A.G., Bladen T.G., Brown D.L., Duffy D.L., Pastorino L., Bianchi-Scarra G. et al. (2009) Analysis of cultured human melanocytes based on polymorphisms within the SLC45A2/MATP, SLC24A5/NCKX5, and OCA2/P loci. J. Invest. Dermatol., 129, 392–405. [DOI] [PubMed] [Google Scholar]
- 22. John P.R. and Ramsay M. (2002) Four novel variants in MC1R in red-haired South African individuals of European descent, S83P, Y152X, A171D, P256S. Hum. Mutat., 19, 461–462. [DOI] [PubMed] [Google Scholar]
- 23. Araki Y., Okamura K., Munkhbat B., Tamiya G., Erdene-Ochir B., Nemekhbaatar L., Hozumi Y. and Suzuki T. (2016) Whole-exome sequencing confirmation of multiple MC1R variants associated with extensive freckles and red hair: analysis of a Mongolian family. J. Dermatol. Sci., 84, 216–219. [DOI] [PubMed] [Google Scholar]
- 24. Peng S., Lu X.M., Luo H.R., Xiang-Yu J.-G. and Zhang Y.P. (2001) Melanocortin-1 receptor gene variants in four Chinese ethnic populations. Cell Res., 11, 81–84. [DOI] [PubMed] [Google Scholar]
- 25. Motokawa T., Kato T., Hashimoto Y., Hongo M., Ito M., Takimoto H. and Katagiri T. (2006) Characteristic MC1R polymorphism in the Japanese population. J. Dermatol. Sci., 41, 143–145. [DOI] [PubMed] [Google Scholar]
- 26. Beaumont K.A., Newton R.A., Smit D.J., Leonard J.H., Stow J.L. and Sturm R.A. (2005) Altered cell surface expression of human MC1R variant receptor alleles associated with red hair and skin cancer risk. Hum. Mol. Genet., 14, 2145–2154. [DOI] [PubMed] [Google Scholar]
- 27. Beaumont K.A., Shekar S.L., Newton R.A., James M.R., Stow J.L., Duffy D.L. and Sturm R.A. (2007) Receptor function, dominant negative activity and phenotype correlations for MC1R variant alleles. Hum. Mol. Genet., 16, 2249–2260. [DOI] [PubMed] [Google Scholar]
- 28. Schiöth H.B., Phillips S.R., Rudzish R., Birch-Machin M.A., Wikberg J.E. and Rees J.L. (1999) Loss of function mutations of the human melanocortin 1 receptor are common and are associated with red hair. Biochem. Biophys. Res. Commun., 260, 488–491. [DOI] [PubMed] [Google Scholar]
- 29. Ringholm A., Klovins J., Rudzish R., Phillips S., Rees J.L. and Schiöth H.B. (2004) Pharmacological characterization of loss of function mutations of the human melanocortin 1 receptor that are associated with red hair. J. Invest. Dermatol., 123, 917–923. [DOI] [PubMed] [Google Scholar]
- 30. Kumar P., Henikoff S. and Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073. [DOI] [PubMed] [Google Scholar]
- 31. Feng M.-S., Juan C., Jia Q.-H., Wang Q.-Y., Liu X.-R. and Pan S.-M. (2011) A comprehensive in silico analysis of functional and structural impact SNPs in the MC1R gene. J. Anim. Vet. Adv., 10, 928–931. [Google Scholar]
- 32. Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M. et al. (2015) UK Biobank, an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med., 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han J., Kraft P., Nan H., Guo Q., Chen C., Qureshi A., Hankinson S.E., Hu F.B., Duffy D.L., Zhao Z.Z. et al. (2008) A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet., 4, e1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Branicki W., Brudnik U., Kupiec T., Wolañska-Nowak P. and Wojas-Pelc A. (2007) Determination of phenotype associated SNPs in the MC1R gene. J. Forensic Sci., 52, 349–354. [DOI] [PubMed] [Google Scholar]
- 35. Branicki W., Wolańska-Nowak P., Brudnik U., Kupiec T., Szymańska K. and Wojas-Pelc A. (2007) Forensic application of a rapid test for red hair colour prediction and sex determination. Problems Forensic Sci., 69, 37–51. [Google Scholar]
- 36. Vaughn M. (2010) Blonde hair colour, classification, characterisation, and genetic associations for use in forensic science. Ph.D. thesis, Victoria University.
- 37. Branicki W., Liu F., Duijn K., Draus-Barini J., Pośpiech E., Walsh S., Kupiec T., Wojas-Pelc A. and Kayser M. (2011) Model-based prediction of human hair color using DNA variants. Hum. Genet., 129, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sitek A., Rosset I., Żądzińska E., Siewierska-Górska A., Pietrowska E. and Strapagiel D. (2016) Selected gene polymorphisms effect on skin and hair pigmentation in Polish children at the prepubertal age. Anthropol. Anz., 73, 283–293. [DOI] [PubMed] [Google Scholar]
- 39. Caliebe A., Harder M., Schuett R., Krawczak M., Nebel A. and Wurmb-Schwark N. (2016) The more the merrier? How a few SNPs predict pigmentation phenotypes in the Northern German population. Eur. J. Hum. Genet., 24, 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hysi P.G., Valdes A.M., Liu F., Furlotte N.A., Evans D.M., Bataille V., Visconti A., Hemani G., McMahon G., Ring S.M. et al. (2018) Genome-wide association meta-analysis of individuals of European ancestry identifies new loci explaining a substantial fraction of hair color variation and heritability. Nat. Genet., 50, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Söchtig J., Phillips C., Maroñas O., Gómez-Tato A., Cruz R., Alvarez-Dios J., Cal M.-Á. C., Ruiz Y., Reich K., Fondevila M. et al. (2015) Exploration of SNP variants affecting hair colour prediction in Europeans. Int. J. Legal Med., 129, 963–975. [DOI] [PubMed] [Google Scholar]
- 42. Siewierska-Gorska A., Sitek A., Żądzińska E., Bartosz G. and Strapagiel D. (2017) Association of five SNPs with human hair colour in the Polish population. Homo, 68, 134–144. [DOI] [PubMed] [Google Scholar]
- 43. Ding C. and Peng H. (2005) Minimum redundancy feature selection from microarray gene expression data. J. Bioinf. Comput. Biol., 3, 185–205. [DOI] [PubMed] [Google Scholar]
- 44. Sulem P., Gudbjartsson D.F., Stacey S.N., Helgason A., Rafnar T., Magnusson K.P., Manolescu A., Karason A., Palsson A., Thorleifsson G. et al. (2007) Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet., 39, 1443–1452. [DOI] [PubMed] [Google Scholar]
- 45. Lin B.D., Mbarek H., Willemsen G., Dolan C.V., Fedko I.O., Abdellaoui A., Geus E.J., Boomsma D.I. and Hottenga J.-J. (2015) Heritability and genome-wide association studies for hair color in a Dutch twin family-based sample. Genes, 6, 559–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raimondi S., Sera F., Gandini S., Iodice S., Caini S., Maisonneuve P. and Fargnoli M.C. (2008) MC1R variants, melanoma and red hair color phenotype: a meta-analysis. Int. J. Cancer, 122, 2753–2760. [DOI] [PubMed] [Google Scholar]
- 47. Andrade E.S., Fracasso N.C., Júnior P.S.S., Simões A.L. and Mendes-Junior C.T. (2017) Associations of OCA2-HERC2 SNPs and haplotypes with human pigmentation characteristics in the Brazilian population. Leg. Med., 24, 78–83. [DOI] [PubMed] [Google Scholar]
- 48. Kastelic V. and Drobnič K. (2012) A single-nucleotide polymorphism (SNP) multiplex system, the association of five SNPs with human eye and hair color in the Slovenian population and comparison using a Bayesian network and logistic regression model. Croat. Med. J., 53, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sinnwell J. and Schaid D.. R package version 1.2. 2.
- 50. Friedman J., Hastie T. and Tibshirani R. (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw., 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- 51. Branicki W., Brudnik U., Draus-Barini J., Kupiec T. and WojasPelc A. (2008) Association of the SLC45A2 gene with physiological human hair colour variation. J. Hum. Genet., 53, 966–971. [DOI] [PubMed] [Google Scholar]
- 52. Nan H., Kraft P., Hunter D.J. and Han J. (2009) Genetic variants in pigmentation genes, pigmentary phenotypes, and risk of skin cancer in Caucasians. Int. J. Cancer, 125, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanetsky P.A., Swoyer J., Panossian S., Holmes R., Guerry D. and Rebbeck T.R. (2002) A polymorphism in the agouti signaling protein gene is associated with human pigmentation. Am. J. Hum. Genet., 70, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Meziani R., Descamps V., Gerard B., Matichard E., Bertrand G., Archimbaud A., Ollivaud L., Saiag P., Lebbé C., Basset-Seguin N. et al. (2005) Association study of the g. 8818A> G polymorphism of the human agouti gene with melanoma risk and pigmentary characteristics in a French population. J. Dermatol. Sci., 40, 133–136. [DOI] [PubMed] [Google Scholar]
- 55. Kastelic V. and Drobnič K. (2011) Single multiplex system of twelve SNPs: validation and implementation for association of SNPs with human eye and hair color. Forensic Sci. Int. Genet. Suppl. Ser., 3, e216–e217. [Google Scholar]
- 56. Walsh S., Liu F., Wollstein A., Kovatsi L., Ralf A., Kosiniak-Kamysz A., Branicki W. and Kayser M. (2013) The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci. Int. Gen., 7, 98–115. [DOI] [PubMed] [Google Scholar]
- 57. Candille S.I., Absher D.M., Belez S., Bauchet M., McEvoy B., Garrison N.A., Li J.Z., Myers R.M., Barsh G.S., Tang H. et al. (2012) Genome-wide association studies of quantitatively measured skin, hair, and eye pigmentation in four European populations. PLoS One, 7, e48294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mengel-From J., Wong T.H., Morling N., Rees J.L. and Jackson I.J. (2009) Genetic determinants of hair and eye colours in the Scottish and Danish populations. BMC Genet., 10, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morgan M.D., Pairo-Castineira E., Rawlik K., Canela-Xandri O., Rees J., Sims D., Tenesa A. and Jackson I.J. (2018) Genome-wide study of hair colour in UK biobank explains most of the SNP heritability. Nat. Commun., 9, 5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin P.-I., Vance J.M., Pericak-Vance M.A. and Martin E.R. (2007) No gene is an island, the flip-flop phenomenon. Am. J. Hum. Genet., 80, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zaykin D.V. and Shibata K. (2008) Genetic flip-flop without an accompanying change in linkage disequilibrium. Am. J. Hum. Genet., 82, 794–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shibata K., Diatchenko L. and Zaykin D.V. (2009) Haplotype associations with quantitative traits in the presence of complex multilocus and heterogeneous effects. Genet. Epidemiol., 33, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Box N.F., Wyeth J.R., O’Gorman L.E., Martin N.G. and Sturm R.A. (1997) Characterization of melanocyte stimulating hormone receptor variant alleles in twins with red hair. Hum. Mol. Genet., 6, 1891–1897. [DOI] [PubMed] [Google Scholar]
- 64. Walsh S., Chaitanya L., Clarisse L., Wirken L., Draus-Barini J., Kovatsi L., Maeda H., Ishikawa T., Sijen T., Knijff P. et al. (2014) Developmental validation of the HIrisPlex system, DNA-based eye and hair colour prediction for forensic and anthropological usage. Forensic Sci. Int. Gen., 9, 150–161. [DOI] [PubMed] [Google Scholar]
- 65. Wheeler D.L., Barrett T., Benson D.A., Bryant S.H., Canese K., Chetvertin V., Church D.M., DiCuccio M., Edgar R., Federhen S. et al. (2007) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 35, D5–D12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Peng H., Long F. and Ding C. (2005) Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell., 27, 1226–1238. [DOI] [PubMed] [Google Scholar]
- 67. Tibshirani R. (1996) Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Series B Stat. Methodol., 267–288. [Google Scholar]
- 68. Kuhn M. (2008) Caret package. J. Stat. Softw., 28, 1–26.27774042 [Google Scholar]
- 69. Ramirez-Gallego S., Lastra I., Martinez-Rego D., Bolon-Canedo V., Benitez J.M., Herrera F. and Alonso-Betanzos A. (2017) Fast-mRMR, Fast minimum redundancy maximum relevance algorithm for high-dimensional big data. Int. J. Intell. Syst., 32, 134–152. [Google Scholar]
- 70. Barrett J.C., Fry B., Maller J. and Daly M.J. (2004) Haploview, analysis and visualization of LD and haplotype maps. Bioinformatics, 21, 263–265. [DOI] [PubMed] [Google Scholar]
- 71. Eriksson N., Macpherson J.M., Tung J.Y., Hon L.S., Naughton B., Saxonov S., Avey L., Wojcicki A., Pe’er I. and Mountain J. (2010) Web-based, participant-driven studies yield novel genetic associations for common traits. PLoS Genet., 6, e1000993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. John P.R. (2014) DNA sequence variation in normal pigmentation Ph.D. thesis. University of the Witwatersrand, Faculty of Medicine.
- 73. Kenny E.E., Timpson N.J., Sikora M., Yee M.-C., Moreno-Estrada A., Eng C., Huntsman S., Burchard E.G., Stoneking M., Bustamante C.D. et al. (2012) Melanesian blond hair is caused by an amino acid change in TYRP1. Science, 336, 554–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Norton H.L., Correa E.A., Koki G. and Friedlaender J.S. (2014) Distribution of an allele associated with blond hair color across Northern Island Melanesia. Am. J. Phys. Anthropol., 153, 653–662. [DOI] [PubMed] [Google Scholar]
- 75. Sulem P., Gudbjartsson D.F., Stacey S.N., Helgason A., Rafnar T., Jakobsdottir M., Steinberg S., Gudjonsson S.A., Palsson A., Thorleifsson G. et al. (2008) Two newly identified genetic determinants of pigmentation in Europeans. Nat. Genet., 40, 835–837. [DOI] [PubMed] [Google Scholar]
- 76. Valenzuela R.K., Henderson M.S., Walsh M.H., Garrison N., Kelch J.T., Cohen-Barak O., Erickson D.T., John Meaney F., Bruce Walsh J., Cheng K.C. et al. (2010) Predicting phenotype from genotype, normal pigmentation. J. Forensic Sci., 55, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang M., Song F., Liang L., Nan H., Zhang J., Liu H., Wang L.E., Wei Q., Lee J.E., Amos C.I. et al. (2013) Genome-wide association studies identify several new loci associated with pigmentation traits and skin cancer risk in European Americans. Hum. Mol. Genet., 22, 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.